

Significance
Hybridization of two epigenetically distinct Arabidopsis accessions is associated with an altered methylome in the F1 hybrid brought about by the processes of Trans Chromosomal Methylation and deMethylation. In the loci studied, the altered F1 cytosine methylation (mC) patterns are inherited to the F2. These observations of trans-mC events are reminiscent of paramutation described in maize and may be common in hybridization events. In one locus, the altered mC states are associated with changed mRNA levels in the F1 hybrid and F2 plants. These events may contribute to F1 hybrid vigor and to the phenotypic heterogeneity and loss of vigor in the F2 through segregation of genetic and epigenetic determinants.
Keywords: heterosis, chromatin, histones, RdDM
Abstract
Hybridization in plants leads to transinteractions between the parental genomes and epigenomes that can result in changes to both 24 nt siRNA and cytosine methylation (mC) levels in the hybrid. In Arabidopsis the principle processes altering the hybrid methylome are Trans Chromosomal Methylation (TCM) and Trans Chromosomal deMethylation (TCdM) in which the mC pattern of a genomic segment attains the same mC pattern of the corresponding segment on the other parental chromosome. We examined two loci that undergo TCM/TCdM in the Arabidopsis C24/Landsberg erecta (Ler) F1 hybrids, which show patterns of inheritance dependent on the properties of the particular donor and recipient chromosomal segments. At At1g64790 the TCM- and TCdM-derived mC patterns are maintained in the F2 generation but are transmitted in outcrosses or backcrosses only by the C24 genomic segment. At a region between and adjacent to At3g43340 and At3g43350, the originally unmethylated Ler genomic segment receives the C24 mC pattern in the F1, which is then maintained in backcross plants independent of the presence of the parental C24 segment. In backcrosses to an unmethylated Ler allele, the newly methylated F1 Ler segment may act as a TCM source in a process comparable to paramutation in maize. TCM-derived mC patterns are associated with reduced expression of both At3g43340 and At3g43350 in F1 and F2 plants, providing support for such events influencing the transcriptome. The inheritance of the F1 mC patterns and the segregation of other genetic and epigenetic determinants may contribute to the reduced hybrid vigor in the F2 and subsequent generations.
Hybrid plants are used extensively in agriculture due to their superior productivity and biomass (1). The gains are assumed to be a consequence of changes to the transcriptome in the hybrid that alter the phenotype of the plant. In 1948, Shull proposed that heterosis had non-Mendelian features (2), Hollick and Chandler (3) have suggested an epigenetic basis to a case of single locus heterosis, and recent studies on the molecular biology of hybrids have suggested that epigenetic systems contribute to the mechanisms generating heterosis. Hybrids have altered small RNA (sRNA) frequencies and cytosine methylation (mC) patterns that can affect mRNA abundance (4, 5). In the nuclei of the hybrid plants, the two parental genomes and epigenomes interact, altering the patterns of mC across both genomes (6–8). These changes are localized to particular chromosomal segments and occur where the parents have differentially methylated regions (DMRs). Of the nonadditively methylated clusters in the hybrid, 86% are due to Trans Chromosomal Methylation (TCM) and Trans Chromosomal deMethylation (TCdM) (6). TCM involves a less methylated parental segment attaining the same mC pattern as the homologous highly methylated parental segment, whereas in TCdM the more highly methylated parental genomic segment shows a reduction in mC levels so as to resemble the pattern in the less methylated parental segment.
An mC is an epigenetic modification of DNA that has a role in silencing transposable elements and repetitive sequences and in regulating gene expression (reviewed in refs. 9, 10). Despite its repressive action, mC in genic regions does not always result in the silencing of gene activity. In mutants defective in mC, only a low number of genes with changed mC levels have altered transcriptional activity, implying a lack of a tight correlation of mC and gene expression (11–13). This means that although TCM and TCdM are the major epigenetic processes driving changes in the hybrid methylome, their roles in altering transcriptional activity, which may be important for generating the heterotic phenotype, remain to be determined.
The 24 nt short interfering RNAs (siRNAs) operating through the RNA-directed DNA methylation system are involved in de novo mC (reviewed in ref. 14). The presence of 24 nt siRNAs in regions undergoing TCM/TCdM suggests that they mediate transchromosomal interactions (6, 15). Not all loci that have different levels of mC in segments in the two parents and have associated 24 nt siRNAs undergo TCM/TCdM (6), indicating that other factors such as histone modifications that influence the state of chromatin may also be important for TCM/TCdM processes. Some histone modifications such as histone 3 lysine 9 acetylation/histone 4 acetylation (H3K9ac/H4ac) and di/trimethylation of histone 3 lysine 4 (H3K4me2/3) promote gene activity, whereas other modifications such as histone 3 lysine 9 dimethylation (H3K9me2) and histone 3 lysine 27 trimethylation (H3K27me3) repress gene expression (reviewed in refs. 16, 17). Histone modifications can change during plant development and influence the level of mC (18, 19).
Hybrid vigor is restricted to the F1 generation, with the F2 and subsequent generations showing reduced vigor and increased heterogeneity of heterotic traits such as yield and biomass. The traditional view has been that loss of vigor occurs through segregation of the genetic determinants of the vigor phenotype, but it may also be linked to segregation or further alterations to the hybrid epigenome. In relation to this possibility, we followed the inheritance of mC patterns produced by TCM/TCdM events at three genes in the C24/Landsberg erecta (Ler) F1 hybrid. We found that in the hybrids, these TCM-derived mC patterns can be accompanied by changes to histone acetylation levels and altered gene expression.
Results
In hybrids between Arabidopsis C24 and Ler accessions, DMRs in the first intron of At1g64790 and a genomic segment spanning At3g43340 and At3g43350 (referred to as At3g43340/50) act as recipient segments in Ler, acquiring the mC patterns present on C24 segments. The Ler segments gain methylation at CG, CHG, and CHH sites not methylated in the Ler parent (TCM), changing mC levels at these three genes in the hybrid. A TCdM event also occurs at an adjacent segment at At1g64790. We tracked At3g43340/50 and At1g64790 in the F2 generation and in progeny of the F1 hybrids (C24*/Ler*; asterisk denotes the segment derived from the F1 hybrid) backcrossed to the original unmethylated Ler parent or outcrossed to another accession unmethylated at this region of interest (SI Appendix, Figs. S1 and S2). This enabled us to determine whether the newly acquired mC pattern on the F1 Ler* segment is maintained in the following generation and is capable of promoting a new TCM event on the unmethylated chromosome in that generation.
The TCM-derived methylation pattern at the At3g43340/50 segment spans from Chr3: 15,285,707 to Chr3: 15,286,347, covering the central upstream sequence and extending into the first intron of each of the two genes, which are transcribed in opposite directions (Fig. 1A). Within this region, C24 and Ler are differentiated by seven SNPs and one small insertion/deletion, enabling us to distinguish between the two parental segments in the hybrid and look for changes in mC patterns. The DMR between parents and the mC pattern of the hybrid TCM event may extend further into the genes, but because of the multicopy characteristics of both genes, we are not able to define the termini of the TCM event. The same DMR occurs between Columbia (Col) and Ler, and hybrids between these two accessions also demonstrate TCM at this segment (SI Appendix, Fig. S3).
Fig. 1.

Inheritance of mC patterns at At3g43340/50. (A) Region in which TCM was observed, overlapping At3g43340 and At3g43350. Exons are indicated as gray and blue boxes (B) McrBC qRT-PCR of preanthesis floral buds from parents, F1 hybrids, F2s, Ler × F1 backcrosses, and F1 × Cvi outcrosses. Parental and F1 columns are derived from three biological replicates, with each replicate containing ≥4 plants. Each F2 column represents the combined result from ≥4 individual plants. The Ler × F1 (Ler/Ler combination) column is derived from six individuals, and the Ler × F1 (C24/Ler combination) and Cvi × F1 columns are derived from two individuals each. Column colors denote genotypes of plants. Black dotted line represents MPV. Error bars, SEM. (C) Bisulphite PCR of parents, F1 hybrids, and Ler × F1 backcrosses. For parents and F1 hybrids, bisulphite results are derived from a pool of ≥4 plants. For backcrosses (Ler*/Ler), six individuals were analyzed (SI Appendix, Fig. S4). Asterisk denotes a segment derived from F1, with individual plants labeled A and B. The box demonstrates how the individuals were derived. C24 and F1 graphs were modified from ref. 6. The mC levels are represented by blue (mCG), red (mCHG), and green (mCHH) columns.
In F2 plants derived from the C24/Ler F1 plants, the TCM-derived mC pattern at At3g43340/50 is present in all progeny genotypes (C24*/C24*, C24*/Ler*, and Ler*/Ler*), indicating transgenerational transmission of the TCM-derived mC pattern (Fig. 1B). In backcrosses of the F1 hybrid (C24*/Ler*) to the Ler parent, both heterozygous (C24*/Ler) and homozygous (Ler*/Ler) Ler progeny have an mC pattern similar to the F1 (Fig. 1 B and C and SI Appendix, Fig. S4A). Although in the Ler homozygotes the two Ler segments (Ler* and Ler) cannot be differentiated by SNPs, the level of methylated cytosines approaches 100% for the CG and CHG contexts, well above the midparent value (MPV) and consistent with both Ler segments having the C24 mC pattern (Fig. 1 B and C and SI Appendix, Fig. S4A). Digestion with the restriction enzyme McrBC followed by quantitative RT-PCR (qRT-PCR) analysis of the homozygous Ler individuals also showed high levels of mC, consistent with both segments being methylated (SI Appendix, Fig. S4B). The ability to maintain and initiate a new TCM event in the F1 × Ler backcrosses was observed in all Ler homozygous individuals studied (six plants; Fig. 1 B and C and SI Appendix, Fig. S4 A and B). Although we cannot rule out other cis or transacting factors mediating the TCM event, the fact that all six Ler*/Ler individuals analyzed showed similar mC patterns at the locus suggests that the trans signal is linked to the TCM region. These data show that the C24 mC pattern transferred to the Ler* segment by the original TCM event in the F1 is maintained in the Ler* chromosome in the next generation, where it results in a new TCM event transferring the C24 mC pattern to the unmethylated Ler chromosome.
In a cross of the F1 hybrid to a Cape Verde Islands (Cvi) accession that, like Ler, lacks mC at the At3g43340/50 region (SI Appendix, Fig. S2), the Ler* segment maintained its F1 mC pattern, whereas the Cvi segment remained unmethylated (SI Appendix, Fig. S5). In C24*/Cvi progeny, there was a similar outcome, with the Cvi segment remaining unmethylated and C24* maintaining its mC pattern (SI Appendix, Fig. S5). The Cvi region may have a property that prevents it from becoming methylated.
The TCM event at At3g43340/50 in F1 hybrids was present in preanthesis floral buds but not in the vegetative tissue of 15 days after sowing (DAS) seedlings (Figs. 1B and 2 and SI Appendix, Fig. S4C). In F2 progeny, the mC state of the Ler* segment was present in both preanthesis floral buds and 15 DAS vegetative tissue (Figs. 1B and 2 and SI Appendix, Fig. S4C). The different results between the F1 and F2 generations imply the mC on the Ler* segment in the F1 is not developmentally or tissue specific but may be the result of an mC-inducing signal that accumulates over time. Once the Ler* segment has become methylated, it retains the mC pattern in the next generation.
Fig. 2.
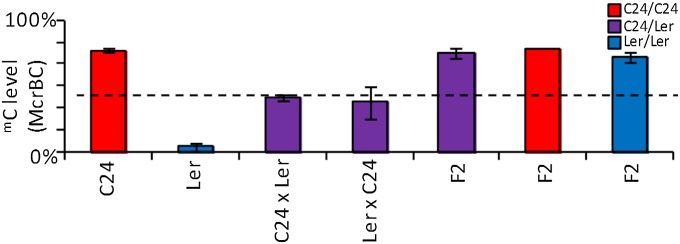
TCM-derived mC patterns at At3g43340/50 are present at different times in F1 and F2 15 DAS seedlings. Each column represents at least four individuals of each allelic combination. Column colors denote allelic combinations. Black dotted line represents MPV. Error bars, SEM.
To determine if the mC pattern on the Ler segment can be maintained into a third generation, we analyzed At3g43340/50 in progeny from selfed Ler*/Ler second-generation backcrossed plants where cytosines within both segments were methylated (SI Appendix, Fig. S6A). We also examined third-generation plants in which the same methylated Ler*/Ler backcross plants had been used in a second backcross to the Ler accession carrying the unmethylated Ler segment (SI Appendix, Fig. S6A). The third-generation plants, derived from selfing second-generation backcross plants, maintained the mC pattern and level. The third-generation plants derived from the second backcross to a Ler plant containing an unmethylated segment showed a reduction in mC levels and variation between individual progeny, with some showing low mC levels (Fig. 3 and SI Appendix, Fig. S6).
Fig. 3.

The mC levels at At3g43340/50 in third-generation plants derived from two backcrosses or one backcross followed by a selfing (SI Appendix, Fig. S6A). McrBC qRT-PCR was used to analyze mC levels of preanthesis floral bud material from third-generation plants. Individuals (Ler × F1) –A and –B are presented on the graph, as they are the plants from which the third-generation plants are derived and represent the average level of mC of each individual from two independent McrBC digests. Results of third-generation plants are derived from a pool of individuals (n ≥ 5). SP, progeny of selfed plants. Column colors denote allelic constitution of plants. Black dotted line represents MPV. Error bars, SEM.
TCM at At3g43340/50 Is Associated with Decreased mRNA Levels in the F1 and F2 Generations.
We examined the mRNA levels of At3g43340 and At3g43350 in the F1 and F2 generations. In both 15 DAS seedlings and in preanthesis floral buds, the unmethylated Ler parent had higher mRNA levels for each gene compared with the methylated C24 parent, suggesting that the mC at this region is associated with the decreased expression of both genes (Fig. 4 A and C). In F1 15 DAS seedlings, in which TCM has not yet occurred, the mRNA levels were at MPV, consistent with only the unmethylated Ler alleles being expressed. In preanthesis floral buds, where TCM has occurred, the hybrids show an expression level below MPV for both genes (Fig. 4A). In the F2 generation, we analyzed the mRNA and mC levels of both genes. In eight F2 individuals, which represented all possible genotype combinations (Fig. 4 B and C), seven had low mRNA levels and high levels of mC. One plant (F2-2) had higher mRNA levels coinciding with lower levels of mC (Fig. 4 B and C). These results suggest that these F1 TCM-derived mC patterns are associated with changes to mRNA levels in both the F1 and F2 plants.
Fig. 4.

TCM events at At3g43340/50 are associated with altered mRNA levels in F1 and F2 plants. (A) mRNA levels of parental and F1 hybrids for At3g43340 and At3g43350 in 15 DAS seedlings and preanthesis floral buds (Dataset S1, Table S1). (B) mRNA levels in buds of eight individual F2 plants (Dataset S1, Table S1). (C) McrBC qRT-PCR of the same F2 plants used in the expression analysis. Column colors denote genotype. Black dotted line represents MPV. Error bars, SEM. Red and black asterisks indicate statistically significant differences (P ≤ 0.05) in expression levels between parents and between hybrids and MPV, respectively.
TCM/TCdM-Derived mC Patterns at At1g64790 Are Maintained in Selfed Progeny but Not in Backcrosses or Outcrosses.
In C24/Ler hybrids, TCM and TCdM regions are positioned close together in the first intron of At1g64790 (Fig. 5). The C24 and Ler segments can be differentiated by nine SNPs, three of which are in the TCM/TCdM region. Both the TCM and TCdM events occur on the Ler* segment. The TCdM event involves the loss of CHH methylation, and the TCM event results in the gain of methylation of the only CG site and of several CHH sites, together with increases in CHG methylation levels. An SNP in the 24 nt siRNA reads from the TCM region showed that in the F1, in addition to those reads produced by the C24* segment, the Ler* segment produces 24 nt siRNAs (6). The mC at the TCM region appears to be dependent on the presence of 24 nt siRNAs, as C24 RNA polymerase IV mutants, which have reduced 24 nt siRNAs, lack CHH methylation and have decreased CHG methylation (20–21) (SI Appendix, Fig. S7).
Fig. 5.

TCM-derived mC patterns at At1g64790 in F1 and second-generation plants. Bisulphite PCR results of mC levels in parents, F1 hybrids, Ler × F1 backcross, and an F2 individual. For parents and F1 hybrids, bisulphite results are derived from a pool of ≥4 plants. SNPs were used to differentiate alleles. Asterisk denotes a segment derived from F1, with individual plants labeled F and A. The box demonstrates how the individuals were derived. Regions that undergo TCdM (blue) and TCM (red) are highlighted in boxes. C24, Ler, and F1 graphs are modified from ref. 6. The mC levels are represented by blue (mCG), red (mCHG), and green (mCHH) columns. Error bars, SEM.
The mC patterns observed in F2 plants indicated that TCM/TCdM-derived patterns found on the F1 Ler* segment are inherited (Fig. 5), similar to the inheritance pattern seen at the At3g43340/50 segment. In an outcross of the F1 C24 × Ler hybrid to Col, which has an mC pattern at At1g64790 resembling the Ler parent (SI Appendix, Fig. S2), the C24*/Col progeny have the same TCM- and TCdM-derived mC patterns as the C24 × Ler F1 hybrid (Fig. 5 and SI Appendix, Fig. S8). However, in the Ler*/Col progeny, the Ler* segment no longer has the TCM/TCdM-derived mC patterns, and has the original Ler parental mC pattern except for the single CG methylation site gained from the F1 TCM event. In the Ler*/Ler progeny of a Ler × F1 backcross, the Ler* genomic segment had the Ler parental mC pattern and not the TCM/TCdM F1 mC pattern (Fig. 5). This result is consistent with the observations we made in individuals derived from a Col × F1 outcross. This failure of inheritance in the outcross and backcross is similar to the loss of the TCM-derived mC pattern at the At3g43340/50 segment in the second backcross to a Ler plant unmethylated at the At3g43340/50 segment. mRNA levels of At1g64790 are similar in C24 and Ler and do not change in response to the TCM and TCdM events (Dataset S1, Table S1).
TCM Events Are Associated with Changes to Histone H3K9 Acetylation.
Because histone modifications influence the presence of mC (22), we analyzed key histone marks at At3g43340/50 and At1g64790. At At3g43340/50, H3K9ac showed an inverse correlation with mC in the parental accessions, with Ler showing high H3K9ac levels and low mC and C24 having low H3K9ac levels and high mC (Fig. 6). In the hybrids, there was a significant decrease in H3K9ac to a level indicating that the Ler* segment has lost its H3K9ac modifications. This demonstrates that increased mC levels, due to TCM, can be accompanied by changes in histone epigenetic marks and changes in mRNA levels (Fig. 4). Concordant changes in mC levels and histone marks may not always occur in TCM/TCdM regions. At At1g64790 we found a small but insignificant increase in the H3K9ac levels at the TCdM region and only small changes for H3K9ac and H3K9me2 at the TCM region (SI Appendix, Fig. S9).
Fig. 6.

TCM events correlate with decreases in H3K9ac levels. Chromatin immunoprecipitation (ChIP) analyses for H3K9ac and H3K9me2 levels at At3g43340/50 are from three independent ChIP experiments using qRT-PCR. The bisulphite graph from the C24 At3g43340/50 segment (Fig. 1) (6) is used as a reference and indicates regions analyzed by ChIP (blue, green, and purple bars shown underneath graph). For the ChIP graphs, columns represent the fold change relative to C24. H3K9ac levels at the TCM region were normalized against ACTIN, whereas H3K9me2 levels were normalized against TA3. The blue, green, and purple columns represent primer sets 1, 2, and 3, respectively, with primer set positions highlighted on the bisulphite PCR graph. The black dotted line represents MPV. Error bars, SEM. Red and black asterisks indicate statistically significant differences (P ≤ 0.05) in immunoprecipitation between parents and between hybrids and MPV, respectively.
Discussion
The Arabidopsis C24 × Ler reciprocal hybrids exhibit a strong vigor phenotype even though the parents have similar genomes (23). Given the need for diversity between parents to generate heterosis, we have suggested that the differences between the two parental epigenomes contribute to the hybrid vigor phenotype (reviewed in refs. 4, 5).
In the F1 hybrids, 24 nt siRNAs show a marked reduction in frequency and coincide with a parallel reduction in mC of homologous genomic regions (15) (reviewed in ref. 24). The reduced frequencies of 24 nt siRNAs occur at genomic segments that have different levels of 24 nt siRNAs in the two parents. At regions of the F1 genome where cytosines are differentially methylated between parents and where siRNAs are not reduced, increases in mC levels occur (6, 7, 15). These processes result in the altered F1 mC patterns and are termed TCM and TCdM events. In TCM and TCdM events, the mC pattern of one parental segment attains the same mC pattern as the homologous chromosomal segment of the other parent. TCM and TCdM occur most frequently in genes and flanking regulatory regions and are sometimes associated with changes in mRNA levels that may contribute to the gene activity pattern determining heterosis (6, 7, 15). The altered mC pattern in the genomic segment encompassing the upstream and 5′ regions of At3g43340 and At3g43350 is associated with reduced mRNA levels of both genes in F1 and F2 plants. Alterations to mC and 24 nt siRNA levels have also been reported in interspecific Arabidopsis hybrids and in hybrids of rice and maize (6–8, 15, 25–28).
A feature of F1 hybrid vigor is that the heterotic increase in yield, relative to the parents, is reduced in subsequent generations—a result assumed to be due to genetic segregation. The increased heterogeneity for biomass and seed yield traits in the F2 plants may also in part be due to changes in siRNA levels, mC patterns, and histone modifications. We found that the new F1 mC patterns at At3g43340/50 and At1g64790 are inherited into the F2 regardless of the genotype combination (i.e., C24*/Ler* or Ler*/Ler*). At At1g64790, in backcrosses or outcrosses to plants carrying an unmethylated homologous segment, only progeny carrying the C24* mC segment have a TCM event. The F1 mC pattern of the Ler At1g64790 segment is only maintained in a subsequent generation if inherited together with a methylated homologous segment. The genomic segment loses the F1 mC pattern in the next generation if exposed to an unmethylated homologous segment. This shows that although the F1 C24* and Ler* segments have near-identical mC patterns, there must be underlying differences between the two segments that enable only the C24* At1g64790 segment to maintain the mC pattern and also carry out TCM and TCdM events. No changes were observed in mRNA levels at At1g64790 in F1 hybrids, which may be due to the positioning of the TCM events in the intron of the gene, which unlike promoter mC may have no impact on gene expression. Also, the TCM/TCdM event is restricted to a 200 bp region that is directly adjacent to many methylated CG and CHG sites present in both parental accessions. Changing the expression of At1g64790 may require the alteration of these cytosine methylated sites rather than just those of the TCM/TCdM region.
At the At3g43340/50 region, C24* and Ler* segments of the F1 hybrid can act as a TCM source in a backcross to unmethylated Ler segments for one generation. This shows that unlike At1g64790, the F1 At3g43340/50 Ler* segment can maintain its F1 mC pattern when exposed to an unmethylated allele and is itself able to promote a TCM event. The outcross progeny of Ler*/Cvi also show that mC can be maintained by the Ler segment but does not necessarily result in a TCM event.
The difference in inheritance of mC between the At3g43340/50 and At1g64790 regions may be due to the strength of the TCM signal. This signal may involve 24 nt siRNAs, as they may be produced at only one chromosome segment but are capable of associating with both parental chromosome segments to establish mC patterns. Consistent with this, we have noted previously that TCM and TCdM events are associated with 24 nt siRNAs (6, 15), with levels of siRNA likely to be important for initiating these events (reviewed in refs. 4 and 5). The fact that Ler* At3g43340/50 can maintain and also promote TCM in a backcross whereas Ler* At1g64790 cannot suggests that the TCM signal is stronger in the Ler* At3g43340/50 case. The TCM-derived mC region at At3g43340/50 is longer than that at At1g64790 (∼6×), and contains many more methylated cytosine residues that may facilitate, through an increased production of 24 nt siRNAs, a stronger mC condition, enabling the Ler* segment to maintain the methylation state for a generation longer than at At1g64790. Another possible factor is the juxtaposition of a number of transposable elements within At3g43350, with their chromatin state possibly being more favorable for perpetuating the TCM pattern.
Our observations suggest that TCM events may not occur in the hybrid nucleus immediately after fertilization. The TCM event at At3g43340/50 was found late in F1 development (in floral buds), coinciding with decreased mRNA levels. In F2 plants, the TCM-derived mC pattern was present in 15 DAS seedlings as well as in the floral buds. The timing of the TCM event may be dependent on the strength of the signal initiating TCM/TCdM and may depend on the concentration of 24 nt sRNAs in the segment. Such factors could vary between individual loci, as signatures of TCM events are recognizable earlier in development than observed for At3g43340/50 (7).
We have suggested that siRNAs from the donor segment are shared with the recipient segment (reviewed in ref. 4) and that this could dilute siRNA frequencies on a per-segment basis to levels insufficient to initiate mC on the recipient segment or to maintain methylation on the donor segment. Consistent with a dilution effect, the newly methylated cytosine residues of the Ler* segment, at both loci, can lose its TCM-derived mC pattern when exposed to an unmethylated segment through backcrossing (At1g64790 1× backcross; At3g43340/50 2× backcross). In contrast, this new mC pattern is maintained in F2 plants, which are homozygous for the methylated Ler* segments.
The trans-chromosomal interactions changing mC patterns in F1 hybrids and subsequent generations have parallels to the interactions of alleles described in paramutation in maize (reviewed in refs. 29, 30). Both At3g43340/50 and the booster1 locus in maize involve a donor segment suppressing a recipient segment through siRNA activity, mC, and histone modification (31–33). The late onset of TCM at At3g43340/50 is paralleled by the mC associated with the B locus, where the mC level increases up until the 10 leaf stage (33). Paramutation can be sequence specific, with the B K55 and B-peru alleles being “immune” to paramutation (31), similar to our observations that the unmethylated At3g43340/50 locus of Cvi is not able to be involved in TCM. In the case of Cvi At3g43340/50, the lack of TCM could be due to sequence variation or to the chromatin state, which may include gene activity or unknown transacting factors (reviewed in refs. 4, 5). In the F1 hybrids, the level of mC induced by a TCM event varies between segments (6), as does the silencing ability of alleles that facilitate paramutation events in maize (reviewed in ref. 34). The ability to maintain the newly methylated state in subsequent generations varies in different cases of paramutation (reviewed in ref. 34), similar to the inheritance patterns at the two loci we have studied.
In our Arabidopsis hybrids, there are thousands of localized regions of TCM/TCdM (6). These events may be a common result of hybridization and could have implications for the transcriptome of the hybrid plant. Although paramutation was initially described at only a small number of loci in maize, recent evidence indicates it occurs more frequently, with both mC and gene expression in maize hybrids being altered in many genes through paramutation-like events (35–37). The two loci examined here maintained the F1 mC patterns into the F2 generation, whereas other TCM events may show variable penetrance in subsequent generations. Further analysis will be needed to investigate how many TCM events are stably maintained and how many are absent from the next generation. Adding further to the complexity is that the timing of the TCM event in the F1 can differ from that in the F2. Given the dynamic patterns of epigenetic changes being uncovered in hybrids, it is possible that a proportion of the altered gene expression patterns may only occur in the F1 generation. Although, at this time, the importance of TCM/TCdM events in generating the heterotic phenotype in Arabidopsis is unknown, their frequency, and consequences for gene expression suggest such events could make significant contributions to hybrid vigor.
Materials and Methods
Plant material, genotyping, and detailed experimental design are described in SI Appendix, SI Materials and Methods. Genomic DNA was bisulphite converted using the MethylEasy Xceed Kit (Human Genetic Signatures). Bisulphite PCR was carried out using unbiased bisulphite primers, allowing amplification of both converted and unconverted fragments (Dataset S1, Table S2). For McrBC digestion, 25–50 ng of genomic DNA were digested for 16 h at 37 °C. Results were obtained by qRT-PCR by comparing digested DNA with an undigested control. ChIP experiments were carried out as described in ref. 38. mRNA levels were ascertained from RNA-seq run on a Illumina HI-seq platform (Dataset S1, Table S1).
Supplementary Material
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1323656111/-/DCSupplemental.
References
- 1.Birchler JA, Yao H, Chudalayandi S, Vaiman D, Veitia RA. Heterosis. Plant Cell. 2010;22(7):2105–2112. doi: 10.1105/tpc.110.076133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shull GH. What is “heterosis”? Genetics. 1948;33(5):439–446. doi: 10.1093/genetics/33.5.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hollick JB, Chandler VL. Epigenetic allelic states of a maize transcriptional regulatory locus exhibit overdominant gene action. Genetics. 1998;150(2):891–897. doi: 10.1093/genetics/150.2.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Groszmann M, et al. Epigenetics in plants-vernalisation and hybrid vigour. Biochim Biophys Acta. 2011;1809(8):427–437. doi: 10.1016/j.bbagrm.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 5.Greaves IK, Groszmann M, Dennis ES, Peacock WJ. Trans-chromosomal methylation. Epigenetics. 2012;7(8):800–805. doi: 10.4161/epi.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greaves IK, et al. Trans chromosomal methylation in Arabidopsis hybrids. Proc Natl Acad Sci USA. 2012;109(9):3570–3575. doi: 10.1073/pnas.1201043109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen H, et al. Genome-wide analysis of DNA methylation and gene expression changes in two Arabidopsis ecotypes and their reciprocal hybrids. Plant Cell. 2012;24(3):875–892. doi: 10.1105/tpc.111.094870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chodavarapu RK, et al. Transcriptome and methylome interactions in rice hybrids. Proc Natl Acad Sci USA. 2012;109(30):12040–12045. doi: 10.1073/pnas.1209297109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Michalak P. Epigenetic, transposon and small RNA determinants of hybrid dysfunctions. Heredity (Edinb) 2009;102(1):45–50. doi: 10.1038/hdy.2008.48. [DOI] [PubMed] [Google Scholar]
- 10.Law JA, Jacobsen SE. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet. 2010;11(3):204–220. doi: 10.1038/nrg2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang XY, et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell. 2006;126(6):1189–1201. doi: 10.1016/j.cell.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 12.Lister R, et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133(3):523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kurihara Y, et al. Identification of the candidate genes regulated by RNA-directed DNA methylation in Arabidopsis. Biochem Biophys Res Commun. 2008;376(3):553–557. doi: 10.1016/j.bbrc.2008.09.046. [DOI] [PubMed] [Google Scholar]
- 14.Matzke M, Kanno T, Huettel B, Daxinger L, Matzke AJM. Targets of RNA-directed DNA methylation. Curr Opin Plant Biol. 2007;10(5):512–519. doi: 10.1016/j.pbi.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 15.Groszmann M, et al. Changes in 24-nt siRNA levels in Arabidopsis hybrids suggest an epigenetic contribution to hybrid vigor. Proc Natl Acad Sci USA. 2011;108(6):2617–2622. doi: 10.1073/pnas.1019217108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 17.Roudier F, Teixeira FK, Colot V. Chromatin indexing in Arabidopsis: An epigenomic tale of tails and more. Trends Genet. 2009;25(11):511–517. doi: 10.1016/j.tig.2009.09.013. [DOI] [PubMed] [Google Scholar]
- 18.Zilberman D, Coleman-Derr D, Ballinger T, Henikoff S. Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature. 2008;456(7218):125–129. doi: 10.1038/nature07324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sasaki TKA, Kobayashi A, Saze H, Kakutani T. RNAi-independent de novo DNA methylation revealed in Arabidopsis mutants of chromatin remodeling gene DDM1. Plant J. 2012;70(5):750–758. doi: 10.1111/j.1365-313X.2012.04911.x. [DOI] [PubMed] [Google Scholar]
- 20.Hamilton A, Voinnet O, Chappell L, Baulcombe D. Two classes of short interfering RNA in RNA silencing. EMBO J. 2002;21(17):4671–4679. doi: 10.1093/emboj/cdf464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herr AJ, Jensen MB, Dalmay T, Baulcombe DC. RNA polymerase IV directs silencing of endogenous DNA. Science. 2005;308(5718):118–120. doi: 10.1126/science.1106910. [DOI] [PubMed] [Google Scholar]
- 22.Qian W, et al. A histone acetyltransferase regulates active DNA demethylation in Arabidopsis. Science. 2012;336(6087):1445–1448. doi: 10.1126/science.1219416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schneeberger K, et al. Reference-guided assembly of four diverse Arabidopsis thaliana genomes. Proc Natl Acad Sci USA. 2011;108(25):10249–10254. doi: 10.1073/pnas.1107739108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Groszmann M, Greaves IK, Fujimoto R, James Peacock W, Dennis ES. The role of epigenetics in hybrid vigour. Trends Genet. 2013;29(12):684–690. doi: 10.1016/j.tig.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 25.Ha M, et al. Small RNAs serve as a genetic buffer against genomic shock in Arabidopsis interspecific hybrids and allopolyploids. Proc Natl Acad Sci USA. 2009;106(42):17835–17840. doi: 10.1073/pnas.0907003106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He GM, et al. Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant Cell. 2010;22(1):17–33. doi: 10.1105/tpc.109.072041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barber WT, et al. Repeat associated small RNAs vary among parents and following hybridization in maize. Proc Natl Acad Sci USA. 2012;109(26):10444–10449. doi: 10.1073/pnas.1202073109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y, Varala K, Moose SP, Hudson ME. The inheritance pattern of 24 nt siRNA clusters in arabidopsis hybrids is influenced by proximity to transposable elements. PLoS ONE. 2012;7(10):e47043. doi: 10.1371/journal.pone.0047043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chandler VL. Paramutation’s properties and puzzles. Science. 2010;330(6004):628–629. doi: 10.1126/science.1191044. [DOI] [PubMed] [Google Scholar]
- 30.Hollick JB. Paramutation: A trans-homolog interaction affecting heritable gene regulation. Curr Opin Plant Biol. 2012;15(5):536–543. doi: 10.1016/j.pbi.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 31.Stam M, Belele C, Dorweiler JE, Chandler VL. Differential chromatin structure within a tandem array 100 kb upstream of the maize b1 locus is associated with paramutation. Genes Dev. 2002;16(15):1906–1918. doi: 10.1101/gad.1006702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nobuta K, et al. Distinct size distribution of endogeneous siRNAs in maize: Evidence from deep sequencing in the mop1-1 mutant. Proc Natl Acad Sci USA. 2008;105(39):14958–14963. doi: 10.1073/pnas.0808066105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haring M, et al. The role of DNA methylation, nucleosome occupancy and histone modifications in paramutation. Plant J. 2010;63(3):366–378. doi: 10.1111/j.1365-313X.2010.04245.x. [DOI] [PubMed] [Google Scholar]
- 34.Pilu R. Paramutation: Just a curiosity or fine tuning of gene expression in the next generation? Curr Genomics. 2011;12(4):298–306. doi: 10.2174/138920211795860099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li L, et al. Mendelian and non-Mendelian regulation of gene expression in maize. PLoS Genet. 2013;9(1):e1003202. doi: 10.1371/journal.pgen.1003202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Regulski M, et al. The maize methylome influences mRNA splice sites and reveals widespread paramutation-like switches guided by small RNA. Genome Res. 2013;23(10):1651–1662. doi: 10.1101/gr.153510.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eichten SR, et al. Epigenetic and genetic influences on DNA methylation variation in maize populations. Plant Cell. 2013;25(8):2783–2797. doi: 10.1105/tpc.113.114793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Helliwell CA, Wood CC, Robertson M, James Peacock W, Dennis ES. The Arabidopsis FLC protein interacts directly in vivo with SOC1 and FT chromatin and is part of a high-molecular-weight protein complex. Plant J. 2006;46(2):183–192. doi: 10.1111/j.1365-313X.2006.02686.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.