

Abstract
Induction of thrombospondin 1 (TSP-1) is generally assumed to suppress tumor growth through inhibiting angiogenesis; however, it is less clear how TSP-1 in dendritic cells (DCs) influences tumor progression. We investigated tumor growth and immune mechanism by downregulation of TSP-1 in dendritic cells. Administration of TSP-1 small hairpin RNA (shRNA) through the skin produced anticancer therapeutic effects. Tumor-infiltrating CD4+ and CD8+ T cells were increased after the administration of TSP-1 shRNA. The expression of interleukin-12 and interferon-γ in the lymph nodes was enhanced by injection of TSP-1 shRNA. Lymphocytes from the mice injected with TSP-1 shRNA selectively killed the tumor cells, and the cytotoxicity of lymphocytes was abolished by depletion of CD8+ T cells. Injection of CD11c+ TSP-1–knockout (TSP-1-KO) bone marrow–derived DCs (BMDCs) delayed tumor growth in tumor-bearing mice. Similarly, antitumor activity induced by TSP-1-KO BMDCs was abrogated by depletion of CD8+ T cells. In contrast, the administration of shRNAs targeting TSP-2, another TSP family member, did not extend the survival of tumor-bearing mice. Finally, TSP-1 shRNA functioned as an immunotherapeutic adjuvant to augment the therapeutic efficacy of Neu DNA vaccination. Collectively, the downregulation of TSP-1 in DCs produces an effective antitumor response that is opposite to the protumor effects by silencing of TSP-1 within tumor cells.
Introduction
Dendritic cells (DCs) are able to prevent overactive immune responses through several mechanisms, including the expression of immunomodulators and the promotion of regulatory T-cell (Treg) responses.1,2,3,4 For example, immunomodulator indoleamine-2,3-dioxygenase (IDO)–expressing DCs activate Tregs and induce immunosuppression in tumor and infection.5,6
Thrombospondin 1 (TSP-1) and TSP-2 belong to the TSP family and have multiple functional domains. Both TSPs contain thrombospondin type 1 repeats (TSRs), which interact with CD36 and β-1 integrins. The TSR domains have been shown to inhibit angiogenesis and thereby inhibit tumor progression.7,8 TSP-1 is a potent angiogenesis inhibitor9,10,11 and promotes tumor cytotoxicity by recruiting macrophages into tumor sites.12 An eight-amino-acid fragment in the second TSR of TSP-1 has been evaluated for cancer therapy as the drug ABT-510.12,13,14 TSP-1 also plays a role as a major activator of transforming growth factor-β (TGF-β) by releasing the active TGF-β from the latent TGF-β complex.15,16,17,18,19 The TSR repeats of TSP-1 are able to activate TGF-β; in contrast, the TSR repeats of TSP-2 cannot activate TGF-β because they lack the three-amino-acid sequence RFK that is between the first and second TSRs in TSP-1.20
TSP-1 functions as a suppressor in immune regulation through its interaction with CD47 on immune cells. The binding of TSP-1 to CD47 promotes the generation of CD4+ Foxp3+ Tregs from CD4+ CD25− human T cells.15 TSP-1 produced during the early activation of DCs downregulates the expression of TNF-α, interleukin (IL)-10, and IL-12 by interacting with CD47.21 During immunosuppression induced by extracellular adenosine triphosphate, human monocyte-derived DCs upregulate IDO and TSP-1 to inhibit T-cell proliferation.22 Taken together, these results demonstrate that TSP-1 plays an important role in the regulation of immune responses by directly affecting DCs and T cells.
Modulation of DCs is an effective strategy for cancer therapy by enhancing patient immune responses.23 Administration of DNA vaccines by gene gun has been shown to be an efficient method to deliver target genes to antigen-presenting cells in vivo.24,25,26,27 In agreement with these findings, our previous studies demonstrated that delivery of small hairpin RNA (shRNA)-targeting IDO, an immunosuppressive gene, into skin DCs via gene gun elicits antitumor responses in vivo.28,29 Because TSP-1 induces tolerogenic signals in DCs, we hypothesized that the downregulation of TSP-1 in skin DCs would induce antitumor effects. In this study, we demonstrate that the silencing of TSP-1 in DCs either by shRNA targeting TSP-1 or by gene deletion of TSP-1 exhibited anticancer therapeutic effects. In addition, the combination of Neu-DNA vaccine and TSP-1 shRNA further improved cancer therapeutic effects in vivo.
Results
Skin administration of TSP-1 shRNA exhibits therapeutic efficacy
To investigate whether silencing the expression of TSP-1 in skin DCs could produce antitumor effects, we used TSP-1 shRNA to downregulate TSP-1 expression in vivo with the method established in our laboratory before.29 Two types of animal tumor models were used, including the MBT-2 bladder tumor model in syngeneic C3H/HeN mice and the LL2 lung tumor model in C57BL/6 mice. TSP-1 shRNA was constructed and its in vitro knockdown efficiency was measured by cotransfecting COS-7 cells with TSP-1 shRNA and exogenous TSP-1. The decrease in TSP-1 expression was demonstrated with western blotting (Figure 1a). To measure the knockdown efficiency of TSP-1 shRNA in DCs, TSP-1 shRNA was administered to mice using a low-pressure gene gun. CD11c+ DCs were then isolated from inguinal lymph nodes (LNs) and examined for TSP-1 expression. Skin administration of TSP-1 shRNA significantly decreased the level of TSP-1 messenger RNA (mRNA) in CD11c+ DCs compared with that in DCs isolated from mice that received scramble TSP-1 shRNA (Figure 1b). Besides, the expression level of TSP-1 in CD11c+ DCs returned to normal value at day 6 after receiving TSP-1 shRNA (Supplementary Figure S1). To estimate the suitable dosage of TSP-1 shRNA in inducing antitumor response, 5–20 µg of TSP-1 shRNA were examined in MBT-2 tumor-bearing mice. The results exhibited that 10 or 20 µg of TSP-1 shRNA achieved similar optimal responses (Supplementary Figure S2); therefore, 10 µg of shRNA were used in subsequent experiments. The effects of TSP-1 shRNA on tumor growth were investigated in two animal tumor models: MBT-2 bladder cancer cells in C3H mice and LL2 lung tumor cells in C57BL/6 mice. Either TSP-1 shRNA or scramble shRNA plasmid was delivered weekly via gene gun into tumor-bearing mice, and the tumor growth was measured. In both LL2 and MBT-2 tumor models, tumor growth was significantly delayed in mice that were treated with skin administration of TSP-1 shRNA compared with mice that were treated with control or scramble shRNA (left panel of Figure 1c,d). The survival rate of mice that received TSP-1 shRNA was significantly higher than that of mice in the control group and of mice that received scramble TSP-1 shRNA (right panel of Figure 1c,d). Hence, skin administration of TSP-1 shRNA by a low-pressure gene gun promotes potent antitumor therapeutic effects in tumor-bearing mice.
Figure 1.

Skin administration of TSP-1 shRNA produced therapeutic effects in mouse tumor models. (a) The knockdown efficiency of TSP-1 shRNAs. Immunoblotting analysis of HA-TSP-1 in cells transfected with HA-TSP-1 and TSP-1 shRNA. (b) TSP-1 shRNA attenuated TSP-1 expression in skin dendritic cells (DCs). Real-time polymerase chain reaction analysis of TSP-1 in CD11c+ cells isolated from inguinal lymph nodes 3 days after the third administration of TSP-1 shRNA. HPRT served as an internal control (n = 3 mice per group, mean ± SEM). (c) TSP-1 shRNA exerted antitumor effects on MBT-2 bladder tumors. Mice bearing MBT-2 tumor cells were treated with TSP-1 shRNA or scramble shRNA. Left panel: MBT-2 tumor size in C3H/HeN mice. Right panel: lifespan of C3H/HeN mice after subcutaneous challenge with MBT-2 cells. (d) Antitumor effects of TSP-1 shRNA on LL2 lung tumor cells. Left panel: growth curve of LL2 tumor cells in C57BL/6 mice. Right panel: survival curve of LL2-tumor-bearing mice (*P < 0.05, **P < 0.01, control versus TSP-1 shRNA). Ctrl, control; mRNA, messenger RNA; Scr, scramble; shRNA, short hairpin RNA; TSP-1, thrombospondin 1.
Antitumor therapeutic effects of TSP-1 shRNA are not due to off-target effects
Because shRNA can have off-target effects, we examined the therapeutic efficacy of two other TSP-1 shRNAs, which targeted either TSP-1 sequence 1675 (TSP-1 shRNA-2) or sequence 1991 (TSP-1 shRNA-3). The relative silencing efficacies of each TSP-1 shRNA and of the scramble TSP-1 shRNA were evaluated by cotransfection with HA-truncated TSP-1 plasmid in vitro. Three different TSP-1 shRNAs were effective in silencing TSP-1 in vitro (Figure 2a). In the MBT-2 animal tumor model, tumor growth was delayed to a similar extent by the delivery of each of the three different TSP-1 shRNAs (Figure 2b). To further demonstrate the specificity of TSP-1 shRNA, TSP-1–deficient hosts were used for therapeutic experiments. As predicted, TSP-1 shRNA did not exhibit anticancer therapeutic effects in TSP-1-KO mice bearing LL2 lung tumor cells (Figure 2c). On the other hand, previous study reported that suppressive DCs upregulated both IDO and TSP-1 to inhibit T-cell proliferation.22 Downregulation of IDO in DCs also elicited an antitumor response in vivo.29 Therefore, we investigated mRNA levels of IDO in CD11c+ in DCs with real-time polymerase chain reaction (PCR) after skin administration of TSP-1 shRNA. Expression level of IDO in DCs that received TSP-1 shRNA was not altered when compared with the level of IDO in DCs without treatment (Supplementary Figure S3). These data indicated that TSP-1 shRNA induced antitumor responses by specifically silencing TSP-1 in vivo.
Figure 2.

The therapeutic effects of TSP-1 shRNA were not due to off-target effects. (a) Silencing induced by the three TSP-1 shRNAs. COS-7 cells were transfected with the HA-TSP-1 and three different TSP-1 shRNAs, and the HA-TSP-1 expression was detected by immunoblotting. (b) MBT-2 tumor-bearing C3H/HeN mice were treated with the indicated shRNA, and the tumor sizes were examined (*P < 0.05; control group versus the other three groups). (c) TSP-1 shRNA had no therapeutic effect in TSP-1 knockout (KO) mice. Growth of LL2 tumor sizes in TSP-1-KO mice treated with TSP-1 shRNA. NS, no statistical difference; shRNA, short hairpin RNA; TSP-1, thrombospondin 1.
Skin administration of TSP-1 shRNA does not influence tumor angiogenesis
Because TSP-1 was reported to affect angiogenesis, we investigated whether TSP-1 shRNA affected angiogenesis in tumor-bearing mice. The number of blood vessels was not significantly different either in mice that received scramble shRNA or in those that received TSP-1 shRNA, as demonstrated by immunostaining of CD31, an endothelial cell marker (Supplementary Figure S4). Thus, the skin delivery of TSP-1 shRNA via gene gun efficiently induced an immune response but did not affect tumor angiogenesis at the low dosage (10 µg) used.
Silencing TSP-1 in DCs promotes cellular immunity
To explore the immunological mechanisms underlying the antitumor responses induced by TSP-1 shRNA, tumor-infiltrating immune cells were analyzed by immunohistochemistry. The number of tumor-infiltrating CD4+ and CD8+ T cells in mice treated with TSP-1 shRNA was increased three to fivefold compared with mice in the control group or mice treated with scramble shRNA (Supplementary Table S1). The levels of Th1-related cytokine mRNAs, such as interferon-γ (IFN-γ) and interleukin-12 (IL-12), were significantly upregulated after the administration of TSP-1 shRNA (Figure 3a). The level of IFN-γ mRNA was three times higher in the LNs of mice that received TSP-1 shRNA than in the LNs of mice that received scramble shRNA. The mRNA levels of the Th2-related cytokines, IL-4 and IL-10, were not significantly altered. To investigate whether skin administration of TSP-1 shRNA induced cytotoxic cellular immunity, we used an in vitro cytotoxic T-lymphocyte (CTL) assay. The cytotoxic activity of lymphocytes from mice that received TSP-1 shRNA was significantly higher than that of lymphocytes from the control group of mice or from mice that received scramble shRNA (Figure 3b, upper panel). In addition, TSP-1 shRNA-activated splenocytes specifically lysed target MBT-2 tumor cells but not unrelated LL2 tumor cells (Figure 3b, upper panel). To investigate the significance of CD8+ T cells and natural killer (NK) cells in TSP-1 shRNA–induced cytotoxicity, CD8+ T cells or NK cells were depleted for in vitro CTL assay. Depletion of CD8+ T cells abolished the ability to kill target cells; in contrast, depletion of NK cells did not significantly alter cytotoxic activity against MBT-2 (Figure 3b, lower panel). To further confirm the essential role of CD8 T cells in mediating the antitumor effects induced by TSP-1 shRNA, we examined the therapeutic efficacy of TSP-1 shRNA in C3H/Hen mice with depletion of CD8+ T cells in vivo. Depletion of CD8+ T cells completely abrogated the therapeutic efficacy of TSP-1 shRNA (Figure 3c). In addition, TSP-1 shRNA did not exhibit antitumor effect in NOD-SCID mice (Figure 3d,e). The lack of adaptive immune cells rendered TSP-1 shRNA unable to delay the tumor growth or extend the survival of tumor-bearing mice. Therefore, adaptive cellular immunity, especially cytotoxic CD8+ T cells, is an essential mediator of the antitumor response induced by TSP-1 shRNA. Taken together, the downregulation of TSP-1 by gene deletion and the administration of TSP-1 shRNA produced antitumor effects through the generation of cytotoxic CD8+ T cells in the mouse tumor model. These data demonstrated the therapeutic potential of TSP-1depletion in DCs for inducing antitumor activity.
Figure 3.

The immunological mechanism of the antitumor response induced by TSP-1 shRNA. (a) Th1 cytokines were induced in the lymph nodes of mice that received TSP-1 shRNA. Expression levels of interferon-γ (IFNγ), interleukin (IL)-12, IL-10, and IL-4 in lymph nodes were measured with real-time polymerase chain reaction. The relative expression fold was compared with control group (*P < 0.05; n = 3 mice per group, mean ± SEM). (b) Splenocytes from mice treated with TSP-1 shRNA were cytotoxic against tumor cells. Effector cells were isolated from mice that received the indicated treatments. MBT-2 luciferase cells were used as target cells. Cytotoxicity was measured by detecting the release of luciferase. Upper left panel: cytotoxicity splenocytes against MBT-2 tumor cells in MBT-2 mouse tumor model. Upper right panel: cytotoxicity splenocytes against LL2 tumor cells in MBT-2 mouse tumor model. Lower left panel: cytotoxicity of CD8+ T-cell–depleted splenocytes against MBT-2 tumor cells in MBT-2 mouse tumor model. Lower right panel: cytotoxicity of NK-cell–depleted splenocytes against MBT-2 tumor cells in MBT-2 mouse tumor model (**P < 0.01, control versus TSP-1 shRNA; *P < 0.05, control versus TSP-1 shRNA). (c) Growth curve of MBT-2 tumor cells in C3H/HeN with CD8 depletion (*P < 0.05, shTSP-1 versus shTSP-1 + CD8 depletion). (d) MBT-2 tumor size in NOD-SCID mice with the indicated treatments. (e) Lifespan of MBT-2 tumor-bearing NOD-SCID mice. mRNA, messenger RNA; NS, no statistical difference; Scr, scramble; shRNA, short hairpin RNA; TSP-1, thrombospondin 1.
TSP-1–deficient DCs exhibit antitumor effects
To further investigate whether downregulation of TSP-1 gene in skin DCs produces antitumor effects, TSP-1–deficient (TSP-1-KO) or wild-type (WT) bone marrow–derived DCs (BMDCs) were generated and isolated. LL2 tumors were implanted subcutaneously (s.c.) into C57BL/6 mice, and TSP-1-KO and WT BMDCs were injected near the tumor sites at day 8 when the tumor is palpable. To increase the therapeutic efficacy, the injection was repeated 7 days after the first inoculation. Tumor growth was significantly delayed in mice that had received TSP-1-KO BMDCs compared with mice in the control group (Figure 4a). The optimal therapeutic effects induced by s.c. injection of TSP-1-KO BMDCs were also evaluated with dose–response (Supplementary Figure S5). Moreover, tumor-bearing mice treated with TSP-1-KO BMDCs had longer survival times (Figure 4b). The levels of Th1 and Th2 cytokine mRNA were also evaluated in the mice that received WT and TSP-1-KO BMDCs. The expression levels of IFN-γ in LN from mice that received TSP-1-KO BMDCs were significantly higher than in LN from the mice that received WT-BMDCs (Figure 4c). IL-12 was enhanced slightly in lymphocytes of the TSP-1-KO BMDCs group compared with that in the WT-BMDCs group (nonstatistically significant). The mRNA level of IL-4 and IL-10 were not altered in mice that received TSP-1-KO BMDCs group compared with that in mice that received WT-BMDCs (Figure 4d). To further explore the immunological mechanism of antitumor effects induced by TSP-1-KO BMDCs, cytotoxic cellular immunity was examined by using in vitro CTL assay. Splenocytes from mice that received TSP-1-KO BMDCs were effective against Lewis lung carcinoma cells but not against MBT-2 cells (Supplementary Figure S6). Since DCs are known to induce nonspecific NK cells,30 we wished to determine the roles of CD8+ T cells or NK cells in the tumor cytotoxic effects. Depletion of CD8+ T cells, but not that of NK cells, abrogated the cytotoxicity of splenocytes in vitro (Figure 4e). Taken together, these data demonstrated that TSP-1–deficient DCs cloud induce CTL response against cancer cells in the mouse tumor model.
Figure 4.

TSP-1–deficient dendritic cells displayed stronger antitumor effects than control dendritic cells. (a) TSP-1–deficient dendritic cells enhanced anticancer therapeutic effects. LL2 tumor-bearing mice were injected subcutaneously with 1 × 105 bone marrow–derived dendritic cells (BMDCs) isolated from TSP-1 knockout (KO) or wild-type (WT) mice. The tumor growth was measured (**P < 0.01, BMDC-TSP-1 KO versus BMDC-WT). (b) TSP-1–deficient DCs prolong the survival of mice. Kaplan–Meier analysis was performed on the mice survival data (*P < 0.05, BMDC-TSP-1 KO versus BMDC-WT). (c) Bar graphs represent the quantification of Th1 cytokines expression in the lymph nodes. (d) Th2 cytokines expression in the lymph nodes. Lymph node messenger RNA (mRNA) was isolated after immunization. Interferon-γ (IFN-γ), interleukin (IL)-12, IL-4, IL-10, and HPRT mRNA expression levels were measured by real-time polymerase chain reaction The relative expression fold was compared with control group. *P < 0.05, BMDC-TSP-1 KO versus BMDC-WT; n = 4 mice per group, mean ± SEM. (e) Splenocytes from mice that received TSP-1-KO BMDCs were cytotoxic against tumor cells. Effector cells were isolated from mice that received the indicated treatments. LL2 luciferase cells were used as target cells. Left panel: cytotoxicity splenocytes against LL2 tumor cells. Middle panel: cytotoxicity of CD8+ T-cell–depleted splenocytes against LL2 tumor cells. Right panel: cytotoxicity of NK-cell–depleted splenocytes against LL2 tumor cells (**P < 0.01, BMDCs-TSP-1 KO versus BMDCs-WT; *P < 0.05, BMDCs-TSP-1 KO versus BMDCs-WT). shRNA, short hairpin RNA; TSP-1, thrombospondin 1.
TSP-2 shRNA does not exhibit anticancer effects
To determine whether the observed anticancer effects were specific to the silencing of TSP-1, we assayed the antitumor effects of a shRNA targeting TSP-2. TSP-2 is similar to TSP-1 but may differ in its capacity to regulate TGF-β signaling. The knockdown efficiency of TSP-2 shRNA was evaluated by cotransfecting COS-7 cells with TSP-2 shRNA and HA-TSP-2. The administration of TSP-2 shRNA significantly attenuated the expression of TSP-2 in vitro (Figure 5a). The silencing of TSP-2 was tested for its antitumor effects by administering TSP-2 shRNA to the skin of MBT-2 syngeneic mice. TSP-2 shRNA was much less effective at attenuating tumor growth than TSP-1 shRNA and did not extend the survival of the mice (Figure 5b,c). To further compare TSP-1 shRNA and TSP-2 shRNA induced cytotoxic cellular immunity, we analyzed cytotoxic activity of splenocytes from mice after treatment with TSP-1 shRNA and TSP-2 shRNA. The result showed that splenocytes from mice that received TSP-1 shRNA had significantly higher cytotoxic activity than in the TSP-2 shRNA group (Figure 5d). In addition, TGF-β is able to influence Tregs; therefore, CD4+ Foxp3+ Tregs were analyzed in tumor-bearing mice. TSP-1 shRNA, but not TSP-2 shRNA, decreased the percentage of CD4+ Foxp3+ Tregs in the LNs (Figure 5e). To further explore the immunological mechanisms of the antitumor effects, the frequency of CD8+ IFN-γ+ T cells were evaluated with flow cytometry after the administration of TSP-1 shRNA. The ratio of CD8+ IFN-γ+ T cells per CD8+ T cells was significantly higher in mice that received TSP-1 shRNA than in mice from the control group (Supplementary Figure S7). In summary, although TSP-1 and TSP-2 had similar structure and function, only the silencing of TSP-1 expression in DCs altered local immune response but not the silencing of TSP-2.
Figure 5.
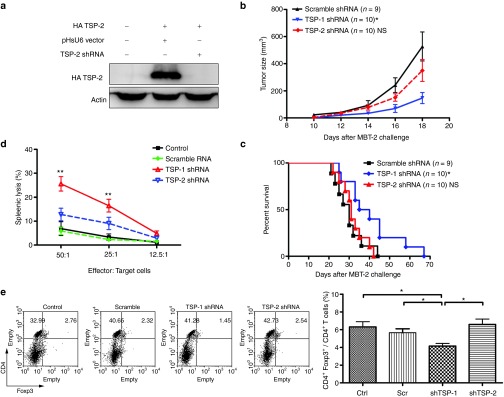
Therapeutic effects of TSP-2 shRNA in the MBT-2 mouse tumor model. (a) TSP-2 shRNA abolished the expression of TSP-2 in vitro. HA-TSP-2 expression was detected by immunoblotting. (b) Tumor-bearing mice were treated with the indicated shRNA, and the tumor sizes (*P < 0.05, TSP-1 shRNA versus TSP-2 shRNA) and (c) survival rates (*P < 0.05, TSP-1 shRNA versus TSP-2 shRNA) were determined. (d) TSP-1 shRNA induces stronger CTL response than TSP-2 shRNA. Effector cells were splenocytes isolated from mice with the indicated treatments. The target cells were MBT-2 luciferase cells (**P < 0.01, control versus TSP-1 shRNA in the ratio 25:1; **P < 0.01, TSP-1 shRNA versus TSP-2 shRNA in the ratio 50:1, n = 3). (e) Analysis of regulatory CD4+ Foxp3+ T cells in mice that received TSP-1 or TSP-2 shRNA. Left panel: representative flow cytometry analysis of the percentage of CD4+ Foxp3+ T cells in the lymph nodes. Right panel: the bar graph represents average + SEM of the percentage of CD4+ Foxp3+ T cells (*P < 0.05; n = 3 mice per group). NS, no statistical difference; Scr, scramble; shRNA, short hairpin RNA; TSP-1, thrombospondin 1.
TSP-1 shRNA enhances the antitumor therapeutic efficacy of Neu DNA vaccine
Finally, we wished to study whether TSP-1 shRNA functions as an adjuvant therapy for cancer treatment. We have previously demonstrated the therapeutic efficacy of the Neu DNA vaccine in the MBT-2 mouse tumor model31; therefore, we examined the adjuvant effects of TSP-1 shRNA when used with the Neu DNA vaccine. TSP-1 shRNA was inserted into the human cyto-Neu expression plasmid containing a U6 promoter. This plasmid construct was called human cyto-Neu_shTSP-1. The human cyto-Neu_shTSP-1 plasmid was able to downregulate cotransfected truncated TSP-1 and express the truncated Neu protein (Figure 6a). Skin administration of cyto-Neu_shTSP-1 fusion plasmid resulted in a better antitumor response than administration of either TSP-1 shRNA or cyto-Neu DNA vaccine alone (Figure 6b,c). Mice vaccinated with cyto-Neu_shTSP-1 survived longer than the mice that received either TSP-1 shRNA or cyto-Neu DNA vaccine alone. Therefore, TSP-1 shRNA may act as an adjuvant to further enhance the efficacy of Neu DNA vaccination in cancer therapy. In order to measure the immunological changes, tumor-infiltrating immune cells and cytokine expression level were analyzed. Tumor-infiltrating CD4+ and CD8+ T cells were significantly higher in cyto-Neu_shTSP-1 group than in other groups (Supplementary Table S2). Th1-related cytokine expression of the cyto-Neu_shTSP-1 group was enhanced 1.5-fold when compared with the cyto-Neu group (Figure 6d,e). These results supported the notion that TSP-1 shRNA functions as an immunotherapeutic adjuvant to boost antitumor immunity induced by the Neu DNA vaccine in the animal tumor model.
Figure 6.

TSP-1 shRNA augments the therapeutic effects of the Neu DNA vaccine. (a) A plasmid encoding TSP-1 shRNA and the intracellular region of Neu induced the repression of TSP-1 and the expression of the Neu protein. COS-7 cells were transfected with the indicated plasmids, and the expression levels of the extracellular domain of Neu and of TSP-1 were evaluated by immunoblotting. pRCMV was used as the vector control for human cyto-Neu. (b) MBT-2 tumor-bearing mice were administered with the indicated plasmids, and MBT-2 tumor volumes were measured at the indicated times (*P < 0.05, Neu_shTsp-1 versus Neu or TSP-1 shRNA). (c) Kaplan–Meier analysis was performed on the mice survival data (*P < 0.05, Neu_shTsp-1 versus Neu or TSP-1 shRNA). (d,e) Th1 cytokines were induced in the lymph nodes of mice that received TSP-1 shRNA, Neu, and Neu_shTsp-1. Bar graphs represent the quantification of Th1 and Th2 cytokine expression in lymph nodes. Expression levels of interferon γ (IFN-γ), interleuki (IL)-12, IL-10, and IL-4 were measured with real-time polymerase chain reaction. Relative gene expression was compared with the TSP-1 shRNA group (*P < 0.05; n = 4 mice per group, mean ± SEM). mRNA, messenger RNA; NS, no statistical difference; shRNA, short hairpin RNA; TSP-1, thrombospondin 1.
Discussion
Recent studies have demonstrated that activation of the immune response is effective against cancer progression in clinical trials.32,33 Here, we report that skin administration of TSP-1 shRNA or adoptive transfer of TSP-1–deficient DCs produces antitumor effects by modulating immune responses in animal tumor models. The adaptive immune response is essential, as demonstrated by the lack of anticancer therapeutic effects in depletion of CD8+ T cells. Delivery of TSP-1 shRNA or TSP-1–deficient BMDCs is able to influence the LN microenvironment, as demonstrated by the observation that the expression of IFN-γ and IL-12 are significantly upregulated in treated mice. In addition, the therapeutic efficacy of the Neu DNA vaccine was enhanced when it was codelivered with TSP-1 shRNA into tumor-bearing mice. The anticancer therapeutic effect is specific to the silencing of TSP-1, because the delivery of TSP-2 shRNA has a minimal effect on tumor burden. Altogether, these results indicate that the silencing of TSP-1 in DCs is an effective antitumor strategy. Furthermore, the adverse inhibitory effects of TSP-1 on host DC immunity may need to be reevaluated in cancer therapies that use TSP-1 agonists or that induce TSP-1.
It is intriguing that the therapeutic effect can occur by the injection of TSP-1 shRNA in a distal site from the tumor lesion. It is possible that skin-resident DCs may be partially activated by TSP-1 shRNA and activate immune responses to the tumor antigens when countering the tumor cells (live cells or apoptotic debris). Although the DC maturation is reported to accompany an attenuation of antigen capture ability, Drutman and Trombetta have shown that the capacity of DCs capture and present antigens does not to be significantly downregulated when DC mature in vivo.34 Furthermore, Carbone's group35 has shown that migratory DCs are able to transfer antigen to a LN-resident DC population. Communication between different DC populations may occur in LN. Therefore, DCs activated by TSP-1 shRNA may retain the ability to capture tumor antigens from tumor cells in circulation; on the other hand, DCs activated by silencing TSP-1 may communicate with other DCs in LN and activate specific antitumor immune responses.
TSP-1 functions in an autocrine manner to negatively regulate the secretion of some cytokines from DCs. The interaction between TSP-1 and its receptors, CD36 and CD47, decreases the secretion of IL-12, TNF-α, and IL-10. This phenomenon is reversed by the application of a neutralizing antibody against TSP-1.21 IL-12 and IFN-γ are key cytokines involved in Th1-mediated immunity, which is correlated with CTL responses. In our animal tumor studies, the expression of IL-12 and IFN-γ mRNA increased in the draining LNs of mice that received skin-administered TSP-1 shRNA and that therefore had DCs with downregulated expression of TSP-1. Adoptive transfer of TSP-1–deficient BMDCs also induced higher levels of IL-12 and IFN-γ mRNA in the LNs. Therefore, our data suggested that the immune response could be successfully modified by downregulating TSP-1 expression in DCs.
The structure and function of TSP-2 is quite similar to TSP-1.36 Both bind to the CD47 receptor, which is involved in the suppression of inflammatory responses in mice.37 Among the members of the TSP family, TSP-1 is the only protein that can activate TGF-β. TSP-1 has a TGF-β activation domain in a type-1-repeat domain, which may assist in converting inactive TGF-β to its active form. TSP-1–deficient mice and TGF-β–deficient mice exhibit similar adverse, multiorgan inflammatory responses.19 TSP-1 expressed by ocular pigment epithelium cells induces Tregs and suppresses bystander T cells in vitro.38 Our results support a model in which CD4+ Foxp3+ Tregs are suppressed by TSP-1 shRNA. In addition to TGF-β, TSP-2 may play an important role in regulating cardiomuscular function. In this role, its function also differs from that of TSP-1. Downregulation of TSP-1 in DCs is more effective against tumor progression than downregulation of TSP-2, which suggests that TSP-1 plays a more prominent role than TSP-2 in modulating the immune responses of skin DCs. The anti–TSP-1 antibody has been used to block the interaction between TSP-1 and CD47 or CD3621; however, TSP-1 shRNA may be used to simultaneously abrogate multiple interactions between TSP-1 and CD36, CD47, or integrins. Because CD47 has been implicated as an antitumor agent,39 it will be interesting to study whether the downregulation of CD47 results in similar anticancer therapeutic effects. Such studies may further clarify the role of the TSP-1-CD47 axis in regulating DC-mediated immune responses.
TSP-1 is known to inhibit angiogenesis and inflammatory responses, thereby suppressing tumor formation. Bocci et al.40 have reported on the antiangiogenic role of TSP-1 in metronomic chemotherapy. In contrast, we found that silencing TSP-1 in DCs by shRNA exerts an antitumor effect. Adoptive transfer of TSP-1–deficient BMDCs into mice bearing LL2 tumors also suppressed tumor progression. Our results suggest that the function of TSP-1 in endothelial cells and DCs may exert opposite effects during cancer treatment. Systemic treatment of TSP-1 may be a double-edged sword for cancer therapy. Treatment with ABT-510 has been shown to attenuate inflammation and angiogenesis in a mouse model.41 Our results suggest that the ABT-510 mimetic peptide may be a better choice for cancer therapy than full-length TSP-1, which may inhibit the immune response by interfering with the functionality of DCs.
Adenosine triphosphate has been shown to tolerize DCs by regulating IDO and TSP-1.22 We previously demonstrated that regulating IDO in DCs is an effective anticancer therapy. In that study, TSP-1 shRNA was able to inhibit tumor progression but that it might be less effective than IDO shRNA. Those results suggested that IDO may regulate additional pathways that are important in tumor immunity, such as the pathways involving dioxane receptor reactions.42 It is also possible that TSP-1 participates in multiple interactions with CD36, CD47, and integrin molecules. Together, these multiple interactions may elicit complex responses in DCs and lead to weaker antitumor effects. Because an anti-CD47 antibody has been shown to effectively treat tumors by altering tumor-associated macrophages,39 it will be interesting to investigate whether anti-CD47 exerts its antitumor effects by activating the DCs that surround tumors.
Recently, it has been shown that Escherichia coli alters the functions of DCs, and that these alterations can be abolished by TSP-1 shRNA or CD47 shRNA.43 These previous results, together with the results we presented here, suggest that TSP-1 shRNA targeting DCs may be useful as an adjuvant therapy for antibacterial or anticancer treatments in the future.
Materials and Methods
Cell lines and antibodies. The LL2 murine lung cancer cell line was a kind gift from Professor Chao-Liang Wu. The MBT-2 murine bladder carcinoma cell line has been described previously.29 The following antibodies were used in Western blotting: anti-Neu (clone Ab-20; Lab Vision Corp. Fremont, CA), which recognizes the extracellular domain of Neu; anti-HA (clone 3F10; Roche, Montreal, CA), which recognizes the HA-tag of HA-truncated TSP-1; and β-actin–specific mouse monoclonal antibody (clone mab1501; Chemicon, Temecula, CA). To evaluate knockdown efficiency of TSP-1 shRNA, COS-7 cells were cotransfected with the HA-TSP-1 (HA-tagged) plasmid and the pHsU6 vector or TSP-1 shRNA (TSP-1 shRNA-1, TSP-1 shRNA-2, or TSP-1 shRNA-3). To evaluate knockdown efficiency of TSP-2 shRNA, COS-7 cells were cotransfected with the HA-TSP-2 (HA-tagged) plasmid and the pHsU6 vector or TSP-2 shRNA.
Animals. Female, 8- to 10-week-old C57BL/6, C3H/HeN, and NOD-SCID mice were obtained from the Laboratory Animal Center at National Cheng Kung University (Tainan, Taiwan). Thbs1tm1Hyn transgenic mice (TSP-1-KO mice) were obtained from the Jackson laboratory (Bar Harbor, Maine). All protocols in this study involving mice were approved by the Animal Welfare Committee at National Cheng Kung University.
Plasmid construction and preparation of DNA vaccine. TSP-1 shRNAs were inserted into pHsU6 vectors as described previously.29 The target sequences of the TSP-1 shRNAs were the following: 5′-GCC AGA ACT CGG TTA CCA T-3′ (for TSP-1 shRNA-1), 5′-CCA ACA AAC AGG TGT GCA A-3′ (for TSP-1 shRNA-2), 5′-GCA ACT ACC TGG GTC ACT A-3′ (for TSP-1 shRNA-3), and 5′-GGT CCA ACA GTC GAA CTC T-3′ (for scramble TSP-1 shRNA). The target sequence of the TSP-2 shRNA was the following: 5′ -CCT GGT GTG TGC TAC TAA T-3′. Human cyto-Neu_shTSP-1 was constructed by subcloning the U6 promoter/TSP-1 shRNA fragment into a plasmid containing the extracellular domain of human Neu driven by the CMV promoter (Invitrogen, Carlsbad, CA).31 Truncated TSP-1 (from 1083 to 2642 bp of the coding sequence) and truncated TSP-2 (from 1999 to 3519 bp of the coding sequence) were cloned from C3H/HeN mouse LNs and subcloned into the HA6L plasmid as described before.44 The plasmid was named HA-TSP-1 and HA-TSP-2, respectively. All plasmid DNA was prepared using the EndoFree Qiagen Plasmid Mega Kit (Qiagen, Montreal, CA).
Measurement of the therapeutic efficacy on established tumors. MBT-2 cells (1 × 106 cells in 500 µl phosphate-buffered saline) were injected s.c. into C3H/HeN and NOD-SCID mice. For experiments using the LL2 tumor model, 4 × 105 LL2 cells in 250 µl phosphate-buffered saline were injected s.c. into C57BL/6 mice. For experiments using TSP-1 knockout mice, 1 × 105 LL2 cells in 250 µl phosphate-buffered saline were injected s.c. into Thbs1tm1Hyn transgenic mice. On day 8, 10 µg of plasmid DNA was administered to the abdominal area of the MBT-2 tumor-bearing mice.25,29 The plasmid was dissolved in 20 µl of water for treatment. The injection was performed on day 8, 15, and 22. The administration was performed using a low-pressure–accelerated gene gun (BioWare Technologies, Taipei, Taiwan) with 50 per square inch of helium gas pressure. Tumor size was measured using calipers. Tumor volume was calculated using the following formula: volume = (A2 × B × 0.5236), where A and B represent the shortest and longest diameters, respectively. Mice were sacrificed when the tumor volume exceeded 2,500 mm3 or when they were expected to become moribund shortly.
Immunohistochemistry. Tumor-bearing mice were sacrificed 4 days after the third treatment. Tumors were removed from mice, embedded in optimal cutting temperature compound (Sakura Finetek USA, Torrance, CA), and then cryosectioned to a thickness of 5 µm. The immune cells were detected with anti-CD4 (clone GK 1.5; BD Biosciences Pharmingen, San Diego, CA), anti-CD8 (clone 53–6.7; BD Biosciences Pharmingen), or anti-pan-NK (clone DX5; BD Biosciences Pharmingen) antibodies. Tumor vessels were detected with anti-CD31 (BD Biosciences Pharmingen). Tumor-infiltrating immune cells were counted at a magnification of ×400. Blood vessel was viewed at a magnification of ×200. Three randomly chosen fields/sample from three mice were evaluated.
Induction of CTLs and measurement of CTL activity assay in vitro. Tumor-bearing mice were injected with plasmid three times as previously described. Four days after the third DNA treatment, spleen cells were harvested and grown in RPMI 1640 with 25 mmol/l HEPES and l-glutamate (GibcoBRL, Rockville, MD). The media was supplemented with penicillin (100 U/ml), streptomycin (100 µg/ml), 10% FBS, and 50 mmol/l 2-mercaptoethanol. For induction, cells were treated with target cell lysate. After 3 days of incubation, nonadherent cells were harvested for use as effector cells. Effector cells were plated with MBT-2 luciferase cells or Lewis lung carcinoma luciferase cells, which were used as target cells. Target cells (5 × 103/well) were incubated for 8 hours in triplicate at 37 °C with serial dilutions (50:1, 25:1, and 12.5:1) of effector cells. After 8 hours, cells were pelleted by centrifugation, and supernatant was collected. Specific lysis was calculated based on the amount of luciferase released into the supernatant, as measured by a conventional luciferase detection system (Promega, Madison, WI). The test solution (100 µl) was mixed with 100 µl of the substrate (luciferin) and placed into a MiniLumat LB9506 luminometer (EG & G Berthold, Bad Wildbad, Germany). Light emission was recorded for 10 seconds. To clarify the roles of CD8+ T cells or NK cells in shTSP-1–induced cytolytic activity, anti-CD8 antibody (10 µg/ml, clone 53–6.7; BD Bioscience) or anti-Asialo GM1 antibody (1:100 dilution; Wako Pure Chemicals, Osaka, Japan) were used for depletion of CD8+ T cells or NK cells. 1 × 107 spleen cells were treated with antibody at 37 °C for 30 minutes before adding to target cells. At least 90% of CD8+ T cells and NK cells were depleted as determined by flow cytometry analysis.
Depletion of CD8+ T cells in vivo. For in vivo depletion of CD8+ T cells, 200 µg of anti-CD8 antibody (clone 53–6.7, BD Bioscience) or isotype control antibody were i.p. injected into mice at day 7, 8, 15, and 22. About 90% of CD8+ T cells were depleted as determined by flow cytometry analysis.
Generation of BMDCs and isolation of CD11c+ cells. Mouse BMDCs were isolated from bone marrow as previously described45 with minor modifications. Briefly, bone marrow cells were flushed from the femurs and tibiae of WT or TSP-1–deficient C57BL/6 mice and were cultured in RPMI with recombinant murine granulocyte-macrophage colony-stimulating factor (20 ng/ml) and IL-4 (20 ng/ml). On day 3 of culture, floating cells were removed, and fresh culture media was added. On day 6, both loosely adherent and nonadherent BMDC aggregates were collected. CD11c+ DCs were isolated using CD11c (N418) microbeads (Miltenyi Biotec, Auburn, CA). After separation, the purity of CD11c+ cells was >90%, as determined by flow cytometry.
Adoptive transfer of BMDCs into tumor-bearing mice. The protocol for the adoptive transfer of BMDCs was obtained from two published reports25,46 and was used with minor modifications. C57BL/6 mice were injected s.c. with 4 × 105 LL2 tumor cells on day 1. On days 8 and 15, 1 × 105 BMDCs in 200 µl phosphate-buffered saline were injected s.c. near the tumor site.
Real-time PCR. To measure the knockdown efficiency of TSP-1 shRNA in vivo, CD11c+ DCs were isolated by using CD11c (N418) microbeads (Miltenyi Biotec, Auburn, CA) at the indicated day. To measure the immune mechanism induced by TSP-1 shRNA, 3 days after the third treatment, inguinal LNs were collected from the tumor-bearing mice that received the control treatment, scramble shRNA or TSP-1 shRNA. Total RNA was extracted from lymphocytes or CD11c+ cells using TRIzol (Invitrogen). cDNA was synthesized using MMLV Reverse Transcriptase (Promega). The following previously described TSP-1 primer sequences for real-time PCR were used47: TSP-1 forward, 5′-GGA ACG GAA AGA CAA CAC TG-3′ and TSP-1 reverse, 5′-AGT TGA GCC CGG TCC TCT TG-3′. The following previously described HPRT, IL12p40, IFN-γ, IL-10, and IL-4 primer sequences for real-time PCR were used48: HPRT forward, 5′-GTT GGA TAC AGG CCA GAC TTT GTT G-3′; HPRT reverse, 5′-GAT TCA ACT TGC GCT CAT CTT AGG C-3′; IL12p40 forward, 5′-TGC TGG TGT CTC CAC TCA TGG C-3′; IL12p40 reverse, 5′-TTT CAG TGG ACC AAA TTC CAT T-3′; IFN-γ forward, 5′-AAC GCT ACA CAC TGC ATC TTG G-3′; IFN-γ reverse, 5′-CAA GAC TTC AAA GAG TCT GAG G-3′; IL-10 forward, 5′-CCA GTT TTA CCT GGT AGA AGT GAT G-3′; IL-10 reverse, 5′-TGT CTA GGT CCT GGA GTC CAG CAG ACT CAA-3′; IL-4 forward, 5′-GAA TGT ACC AGG AGC CAT ATC-3′; and IL-4 reverse, 5′-CTC AGT ACT ACG AGT AAT CCA-3′. The following previously described IDO primer sequences for real-time PCR were used.29 IDO forward, 5′-TGT GGC TAG AAA TCT GCC TGT-3′; and IDO reverse: 5′-CTG CGA TTT CCA CCA ATA GAG-3′. HPRT served as an internal control. Real-time PCR was performed on an Applied Biosystems 7900HT real-time PCR instrument (Applied Biosystems, Foster City, CA) using FastStart Universal SYBR Green I Master (Roche Diagnostics, Mannheim, Germany). The cycling conditions were 10 minutes at 95 °C and 45 cycles at 95 °C for 15 seconds and 60 °C for 60 seconds.
Flow cytometry analysis of lymphocytes. Lymphocytes were collected from the inguinal LNs of mice 3 days after the third administration of TSP-1 shRNA. Lymphocytes were stained with FITC-conjugated anti-CD4 (clone GK1.5; BD Bioscience) and pigment epithelium–conjugated anti-Foxp3 (clone MF23; BD Bioscience) or pigment epithelium–conjugated anti–IFN-γ (BD Bioscience) antibodies. BD Pharmingen Transcription Factor Buffer Set was used for intracellular staining of Foxp3 and IFN-γ. A FACS Calibur flow cytometer (BD Bioscience) was used to determine protein expression. The lymphocytes were gated based on the side- and forward-scatter characteristics of T cells.
Statistical analyses. Statistical analyses of the tumor curve were done by two-way ANOVA test. Kaplan–Meier analysis was performed on the survival rates of the mice. P values <0.05 were considered significant. All statistical analyses were done with GraphPad Prism 5 software.
SUPPLEMENTARY MATERIAL Figure S1. The expression of TSP-1 in CD11c+ DCs of LN after receiving TSP-1 shRNA. Figure S2. Antitumor response was significantly induced by using 10 µg of TSP-1 shRNA. Figure S3. Expression levels of IDO in CD11c+ DCs in inguinal LN after skin administration of TSP-1 shRNA. Figure S4. Angiogenesis at the tumor site is not enhanced by skin administration of TSP-1 shRNA. Figure S5. Therapeutic effects induced by treating increasing numbers of BMDCs in LL2 tumor-bearing mice. Figure S6. TSP-1-KO BMDCs induced cytotoxicity against LL2 tumor cells, but not MBT-2 tumor cells. Figure S7. IFN-γ–producing T cells were increased after treatment with TSP-1 shRNA in vivo. Table S1. Tumor-infiltrating CD4 T cells, CD8 T cells, and NK cells with TSP-1 shRNA treatment. Table S2. Tumor-infiltrating CD4 T cells, CD8 T cells, and NK cells with Neu_shTSP-1 treatment.
Acknowledgments
This study is supported by the grant NSC-102-2325-B-006-007 from National Science Council, Taiwan and NHRI-EX100-9927B1 from National Health Research Institute, Taiwan to M.-D.L. To establish Centers of Excellence for Cancer Research in Taiwan, funding was provided by the grant DOH101-TD-C-111-003, Department of Health, Executive Yuan, Taiwan.
Supplementary Material
References
- Akbari O, DeKruyff RH, Umetsu DT. Pulmonary dendritic cells producing IL-10 mediate tolerance induced by respiratory exposure to antigen. Nat Immunol. 2001;2:725–731. doi: 10.1038/90667. [DOI] [PubMed] [Google Scholar]
- Quaratino S, Duddy LP, Londei M. Fully competent dendritic cells as inducers of T cell anergy in autoimmunity. Proc Natl Acad Sci USA. 2000;97:10911–10916. doi: 10.1073/pnas.190204697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutella S, Danese S, Leone G. Tolerogenic dendritic cells: cytokine modulation comes of age. Blood. 2006;108:1435–1440. doi: 10.1182/blood-2006-03-006403. [DOI] [PubMed] [Google Scholar]
- Yamazaki S, Iyoda T, Tarbell K, Olson K, Velinzon K, Inaba K, et al. Direct expansion of functional CD25+ CD4+ regulatory T cells by antigen-processing dendritic cells. J Exp Med. 2003;198:235–247. doi: 10.1084/jem.20030422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn DH, Sharma MD, Hou D, Baban B, Lee JR, Antonia SJ, et al. Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J Clin Invest. 2004;114:280–290. doi: 10.1172/JCI21583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makala LH. The role of indoleamine 2, 3 dioxygenase in regulating host immunity to leishmania infection. J Biomed Sci. 2012;19:5. doi: 10.1186/1423-0127-19-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Galardi E, Duquette M, Lawler J, Parangi S. Antiangiogenic treatment with three thrombospondin-1 type 1 repeats versus gemcitabine in an orthotopic human pancreatic cancer model. Clin Cancer Res. 2005;11:5622–5630. doi: 10.1158/1078-0432.CCR-05-0459. [DOI] [PubMed] [Google Scholar]
- Zhang X, Lawler J. Thrombospondin-based antiangiogenic therapy. Microvasc Res. 2007;74:90–99. doi: 10.1016/j.mvr.2007.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Herndon ME, Lawler J. The cell biology of thrombospondin-1. Matrix Biol. 2000;19:597–614. doi: 10.1016/s0945-053x(00)00107-4. [DOI] [PubMed] [Google Scholar]
- Dameron KM, Volpert OV, Tainsky MA, Bouck N. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science. 1994;265:1582–1584. doi: 10.1126/science.7521539. [DOI] [PubMed] [Google Scholar]
- Iruela-Arispe ML, Bornstein P, Sage H. Thrombospondin exerts an antiangiogenic effect on cord formation by endothelial cells in vitro. Proc Natl Acad Sci USA. 1991;88:5026–5030. doi: 10.1073/pnas.88.11.5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Manso G, Galli S, Ridnour LA, Tsokos M, Wink DA, Roberts DD. Thrombospondin 1 promotes tumor macrophage recruitment and enhances tumor cell cytotoxicity of differentiated U937 cells. Cancer Res. 2008;68:7090–7099. doi: 10.1158/0008-5472.CAN-08-0643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebbinghaus S, Hussain M, Tannir N, Gordon M, Desai AA, Knight RA, et al. Phase 2 study of ABT-510 in patients with previously untreated advanced renal cell carcinoma. Clin Cancer Res. 2007;13 22 Pt 1:6689–6695. doi: 10.1158/1078-0432.CCR-07-1477. [DOI] [PubMed] [Google Scholar]
- Markovic SN, Suman VJ, Rao RA, Ingle JN, Kaur JS, Erickson LA, et al. A phase II study of ABT-510 (thrombospondin-1 analog) for the treatment of metastatic melanoma. Am J Clin Oncol. 2007;30:303–309. doi: 10.1097/01.coc.0000256104.80089.35. [DOI] [PubMed] [Google Scholar]
- Grimbert P, Bouguermouh S, Baba N, Nakajima T, Allakhverdi Z, Braun D, et al. Thrombospondin/CD47 interaction: a pathway to generate regulatory T cells from human CD4+ CD25- T cells in response to inflammation. J Immunol. 2006;177:3534–3541. doi: 10.4049/jimmunol.177.6.3534. [DOI] [PubMed] [Google Scholar]
- Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Annu Rev Immunol. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- Murphy-Ullrich JE, Poczatek M. Activation of latent TGF-beta by thrombospondin-1: mechanisms and physiology. Cytokine Growth Factor Rev. 2000;11:59–69. doi: 10.1016/s1359-6101(99)00029-5. [DOI] [PubMed] [Google Scholar]
- Narizhneva NV, Razorenova OV, Podrez EA, Chen J, Chandrasekharan UM, DiCorleto PE, et al. Thrombospondin-1 up-regulates expression of cell adhesion molecules and promotes monocyte binding to endothelium. FASEB J. 2005;19:1158–1160. doi: 10.1096/fj.04-3310fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz-Cherry S, Murphy-Ullrich JE. Thrombospondin causes activation of latent transforming growth factor-beta secreted by endothelial cells by a novel mechanism. J Cell Biol. 1993;122:923–932. doi: 10.1083/jcb.122.4.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young GD, Murphy-Ullrich JE. The tryptophan-rich motifs of the thrombospondin type 1 repeats bind VLAL motifs in the latent transforming growth factor-beta complex. J Biol Chem. 2004;279:47633–47642. doi: 10.1074/jbc.M404918200. [DOI] [PubMed] [Google Scholar]
- Doyen V, Rubio M, Braun D, Nakajima T, Abe J, Saito H, et al. Thrombospondin 1 is an autocrine negative regulator of human dendritic cell activation. J Exp Med. 2003;198:1277–1283. doi: 10.1084/jem.20030705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marteau F, Gonzalez NS, Communi D, Goldman M, Boeynaems JM, Communi D. Thrombospondin-1 and indoleamine 2,3-dioxygenase are major targets of extracellular ATP in human dendritic cells. Blood. 2005;106:3860–3866. doi: 10.1182/blood-2005-05-1843. [DOI] [PubMed] [Google Scholar]
- Melief CJ. Cancer immunotherapy by dendritic cells. Immunity. 2008;29:372–383. doi: 10.1016/j.immuni.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Condon C, Watkins SC, Celluzzi CM, Thompson K, Falo LD. DNA-based immunization by in vivo transfection of dendritic cells. Nat Med. 1996;2:1122–1128. doi: 10.1038/nm1096-1122. [DOI] [PubMed] [Google Scholar]
- Lin CC, Yen MC, Lin CM, Huang SS, Yang HJ, Chow NH, et al. Delivery of noncarrier naked DNA vaccine into the skin by supersonic flow induces a polarized T helper type 1 immune response to cancer. J Gene Med. 2008;10:679–689. doi: 10.1002/jgm.1183. [DOI] [PubMed] [Google Scholar]
- Porgador A, Irvine KR, Iwasaki A, Barber BH, Restifo NP, Germain RN. Predominant role for directly transfected dendritic cells in antigen presentation to CD8+ T cells after gene gun immunization. J Exp Med. 1998;188:1075–1082. doi: 10.1084/jem.188.6.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provinciali M, Barucca A, Pierpaoli E, Orlando F, Pierpaoli S, Smorlesi A. In vivo electroporation restores the low effectiveness of DNA vaccination against HER-2/neu in aging. Cancer Immunol Immunother. 2012;61:363–371. doi: 10.1007/s00262-011-1107-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TW, Lee JH, He L, Boyd DA, Hardwick JM, Hung CF, et al. Modification of professional antigen-presenting cells with small interfering RNA in vivo to enhance cancer vaccine potency. Cancer Res. 2005;65:309–316. [PubMed] [Google Scholar]
- Yen MC, Lin CC, Chen YL, Huang SS, Yang HJ, Chang CP, et al. A novel cancer therapy by skin delivery of indoleamine 2,3-dioxygenase siRNA. Clin Cancer Res. 2009;15:641–649. doi: 10.1158/1078-0432.CCR-08-1988. [DOI] [PubMed] [Google Scholar]
- Liu C, Lou Y, Lizée G, Qin H, Liu S, Rabinovich B, et al. Plasmacytoid dendritic cells induce NK cell-dependent, tumor antigen-specific T cell cross-priming and tumor regression in mice. J Clin Invest. 2008;118:1165–1175. doi: 10.1172/JCI33583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CC, Chou CW, Shiau AL, Tu CF, Ko TM, Chen YL, et al. Therapeutic HER2/Neu DNA vaccine inhibits mouse tumor naturally overexpressing endogenous neu. Mol Ther. 2004;10:290–301. doi: 10.1016/j.ymthe.2004.05.015. [DOI] [PubMed] [Google Scholar]
- Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. IMPACT Study Investigators Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- Drutman SB, Trombetta ES. Dendritic cells continue to capture and present antigens after maturation in vivo. J Immunol. 2010;185:2140–2146. doi: 10.4049/jimmunol.1000642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, Zhan Y, et al. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity. 2006;25:153–162. doi: 10.1016/j.immuni.2006.04.017. [DOI] [PubMed] [Google Scholar]
- Adams JC, Lawler J. The thrombospondins. Int J Biochem Cell Biol. 2004;36:961–968. doi: 10.1016/j.biocel.2004.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamy L, Foussat A, Brown EJ, Bornstein P, Ticchioni M, Bernard A. Interactions between CD47 and thrombospondin reduce inflammation. J Immunol. 2007;178:5930–5939. doi: 10.4049/jimmunol.178.9.5930. [DOI] [PubMed] [Google Scholar]
- Futagami Y, Sugita S, Vega J, Ishida K, Takase H, Maruyama K, et al. Role of thrombospondin-1 in T cell response to ocular pigment epithelial cells. J Immunol. 2007;178:6994–7005. doi: 10.4049/jimmunol.178.11.6994. [DOI] [PubMed] [Google Scholar]
- Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci USA. 2012;109:6662–6667. doi: 10.1073/pnas.1121623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocci G, Francia G, Man S, Lawler J, Kerbel RS. Thrombospondin 1, a mediator of the antiangiogenic effects of low-dose metronomic chemotherapy. Proc Natl Acad Sci USA. 2003;100:12917–12922. doi: 10.1073/pnas.2135406100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punekar S, Zak S, Kalter VG, Dobransky L, Punekar I, Lawler JW, et al. Thrombospondin 1 and its mimetic peptide ABT-510 decrease angiogenesis and inflammation in a murine model of inflammatory bowel disease. Pathobiology. 2008;75:9–21. doi: 10.1159/000113790. [DOI] [PubMed] [Google Scholar]
- Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478:197–203. doi: 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- Mittal R, Gonzalez-Gomez I, Prasadarao NV. Escherichia coli K1 promotes the ligation of CD47 with thrombospondin-1 to prevent the maturation of dendritic cells in the pathogenesis of neonatal meningitis. J Immunol. 2010;185:2998–3006. doi: 10.4049/jimmunol.1001296. [DOI] [PubMed] [Google Scholar]
- Lu TJ, Lai WY, Huang CY, Hsieh WJ, Yu JS, Hsieh YJ, et al. Inhibition of cell migration by autophosphorylated mammalian sterile 20-like kinase 3 (MST3) involves paxillin and protein-tyrosine phosphatase-PEST. J Biol Chem. 2006;281:38405–38417. doi: 10.1074/jbc.M605035200. [DOI] [PubMed] [Google Scholar]
- Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med. 2010;16:880–886. doi: 10.1038/nm.2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olerud J, Johansson M, Lawler J, Welsh N, Carlsson PO. Improved vascular engraftment and graft function after inhibition of the angiostatic factor thrombospondin-1 in mouse pancreatic islets. Diabetes. 2008;57:1870–1877. doi: 10.2337/db07-0724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang TT, Yen MC, Lin CC, Weng TY, Chen YL, Lin CM, et al. Skin delivery of short hairpin RNA of indoleamine 2,3 dioxygenase induces antitumor immunity against orthotopic and metastatic liver cancer. Cancer Sci. 2011;102:2214–2220. doi: 10.1111/j.1349-7006.2011.02094.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.