

Abstract
Mouse models provide valuable opportunities for probing the underlying pathology of human birth defects. Employing an ENU-based screen for recessive mutations affecting craniofacial anatomy we isolated a mouse strain, Dogface-like (DL), with abnormal skull and snout morphology. Examination of the skull indicated that these mice developed craniosynostosis of the lambdoid suture. Further analysis revealed skeletal defects related to the pathology of basal cell nevus syndrome (BCNS) including defects in development of the limbs, scapula, ribcage, secondary palate, cranial base, and cranial vault. In humans, BCNS is often associated with mutations in the Hedgehog receptor PTCH1 and genetic mapping in DL identified a point mutation at a splice donor site in Ptch1. Using genetic complementation analysis we determined that DL is a hypomorphic allele of Ptch1, leading to increased Hedgehog signaling. Two aberrant transcripts are generated by the mutated Ptch1DL gene, which would be predicted to reduce significantly the levels of functional Patched1 protein. This new Ptch1 allele broadens the mouse genetic reagents available to study the Hedgehog pathway and provides a valuable means to study the underlying skeletal abnormalities in BCNS. In addition, these results strengthen the connection between elevated Hedgehog signaling and craniosynostosis.
Keywords: Hedgehog, Craniosynostosis, Polydactyly, Craniofacial Defects, Omphalocele
Introduction
Forward genetic screens in mice have yielded considerable insight into the genetics underlying the molecular mechanisms responsible for normal development (Caspary and Anderson, 2006). In particular, N-ethyl-N-nitrosourea (ENU) mutagenesis has provided an effective means to introduce a high frequency of random point mutations into the mouse genome. Subsequently, phenotype-driven dominant and recessive screens, coupled with mapping and high-throughput sequencing approaches, have provided an unbiased means to identify novel genes critical for multiple aspects of development. Even in instances where the mutation maps to a gene previously linked to a developmental pathway, these ENU-induced mutations can yield new alleles that can provide valuable information on gene function during development.
We recently conducted a recessive ENU screen focused on identifying mutations altering the morphology of the craniofacial skeleton. In humans, birth defects affecting the head, face and oral tissues are frequent dysmorphologies with severe medical consequences (Gritli-Linde, 2008). While clefting of the lip and/or palate are common defects, there are many other syndromic and non-syndromic conditions that alter the shape and growth of the face and skull. One notable human facial defect, bossing of the forehead, is a feature of Basal Cell Nevus Syndrome (BCNS), previously termed Gorlin Syndrome (Evans and Farndon, 2010). BCNS is a dominantly inherited condition caused by mutations in one allele of PTCH1, the gene encoding the transmembrane receptor and negative regulator of the Hedgehog (Hh) signaling pathway (Lindstrom et al., 2006; Quijada et al., 2006; Robbins et al., 2012; Wicking et al., 1997). Many aspects of BCNS manifest during childhood and into adulthood, including basal cell carcinomas, nevi, palmar and plantar pits, jaw keratocysts, and medulloblastoma (Evans and Farndon, 2010; Kimonis et al., 1997; Kimonis et al., 2013; Lo Muzio, 2008). In addition, there are several notable congenital defects associated with the head and orofacial complex including macrocephaly, mandibular prognathism, coronoid hyperplasia, hyperpneumatization of the sphenoid, bridging of the sella turcica, high arched palate and occasional orofacial clefting (Evans and Farndon, 2010; Iwanaga et al., 1998; Kimonis et al., 1997; Kimonis et al., 2013; Lo Muzio, 2008). Skeletal defects in the trunk affect the sternum and ribcage, the shoulder, the vertebrae, and the limbs, the latter typified by syndactyly and polydactyly.
Studies using the mouse model system have provided insight into the function of the Ptch1 protein in development as well as in the etiology of BCNS. Ptch1-null mice die around embryonic day 9 (E9) and have neural tube closure defects (Goodrich et al., 1997; Hahn et al., 1998). Conditional Ptch1 gene knockout analyses have also been employed to study bone development and physiology and have indicated important roles for Ptch1 in formation of the long bones and calvaria, and in bone homeostasis (Mak et al., 2008; Mak et al., 2006) as well as in patterning the limb bud (Bruce et al., 2010; Butterfield et al., 2009). Ptch1 heterozygous mice develop problems associated with BCNS including medulloblastoma and basal cell carcinoma (Goodrich et al., 1997; Hahn et al., 1998). Additionally, such mice show variable defects associated with aberrant embryogenesis including large size, syndactyly and polydactyly as well as accelerated osteogenesis that affects pre- and post-natal bone formation (Goodrich et al., 1997; Hahn et al., 1998; Ohba et al., 2008). Despite the various models that can be utilized to study Ptch1 function in the mouse, analyses focused on development of the craniofacial skeleton relevant to BCNS are hampered by early embryonic lethality and exencephaly (Butterfield et al., 2009; Goodrich et al., 1997; Hahn et al., 1998; Milenkovic et al., 1999; Ngan et al., 2011). Here we describe the isolation and characterization of a new ENU-induced recessive mouse model, Ptch1DL that displays many of the skeletal hallmarks of BCNS. This mutation adds a valuable new hypomorphic allele of Ptch1 that can be used to assess the role of the Hh signaling pathway in multiple developmental events, particularly those regulating facial shape in both a clinical and evolutionary context (Roberts et al., 2011).
Results
Identification of the Dogface-like (DL) mutant
We used ENU-treated founder C57BL/6J male mice and a three-generation cross to conduct a recessive screen to identify mutations that affect late embryonic development, focusing in part on the embryonic day 18.5 (E18.5) craniofacial skeleton. From this, we identified a mutant line that we termed DL (Dogface-like) in which ∼25% of the offspring from heterozygous matings were larger than their littermates and displayed a more rounded skull with an abnormal angle between the cranium and the snout (Figure 1a, b). Examination of the external morphology indicated that these mice also exhibited preaxial polydactyly associated with all four limbs (Figure 1d and Table 1). When mice were allowed to develop to term, the mutant animals did not survive until weaning.
Figure 1.

Gross external developmental defects in the Ptch1DL homozygous mutants.
E18.5 wild type (a, c) and Ptch1DL mutants (b, d) showing abnormal flexure at skull and nasal juncture (red arrow, b) and forelimb preaxial polydactyly (black arrow, d).
Table 1.
Phenotypes of PtchDL homozygous mice and negative complementation with the Ptch1null allele.
| Phenotype* | Genotype DL/DL | +/+ and DL/+ and Ptch1-LacZ/+ | DL/Ptch1-LacZ |
|---|---|---|---|
| Dead | 0 | 0 | 17 |
| Exencephaly | 0 | 0 | 34 |
| Skull vault hypoplasia | 100 | 0 | 100 |
| Lambdoid suture synostosis | 100 | 0 | 100# |
| Narrow basisphenoid | 100 | 0 | 100 |
| Missing presphenoid | 100 | 0 | 100 |
| Hypoplastic alisphenoid | 100 | 0 | 100 |
| Hypoplastic palatal shelves | 100 | 0 | 100 |
| Coronoid hypoplastic | 37.5 | 0 | 100 |
| Kinked Tail | 0 | 0 | 34 |
| Abnormal sternum | 100 | 0 | 100$ |
| Defective scapulas | 12.5 | 0 | 50 |
| Affected forelimbs | 100 | 0 | 100 |
| Affected hindlimbs | 100 | 0 | 78 |
Numbers represent the percentage of a particular genotype showing an abnormal phenotype for the structure described.
Lambdoid suture synostosis was only scored in DL/Ptch1-LacZ embryos that underwent normal neural tube closure and had sufficient development of the calvaria for visualization.
Bifurcated and fused ribs occur in association with sternal defects in some embryos.
Skeletal staining of affected E18.5 mutants showed features apparent in the intact embryos, such as the domed cranial vault along with the aberrant angle between this region and the snout in lateral view compared with controls (Figure 2a, b). Additionally, dorsal views of Ptch1DL mutant skulls revealed large areas lacking skeletal components in regions where the bones of the skull vault normally juxtapose, particularly near the midline (Figure 2c, d red arrows). Ventral views of the skull (Figure 2e-h) showed defects of the cranial base, especially in components of the sphenoid bone. Notably, the presphenoid was absent, the basisphenoid was narrower along its rostral caudal axis, and the alisphenoids were hypoplastic. The palatal shelves were underdeveloped compared to wild-type and were absent in ∼10% of mutants in association with a cleft secondary palate (2/15 embryos). Examination of the mandible indicated that the coronoid process also failed to extend caudally (Figure 2j). Surprisingly, despite the apparent hypoplastic or perhaps delayed intramembranous ossification of the frontal, parietal and interparietal bones near the midline, there were consistent defects associated with the sutures. In particular, the lambdoid sutures, between the parietal and interparietal bones, were consistently malformed (Figure 2l). Whereas there was a gap present between these two bones in control embryos, Ptch1DL mutants showed consistent fusion of these two bones, especially towards the more ventral aspect of the skull, so that they became contiguous regions of ossification. Defects in suture formation were less apparent for other calvarial bones at this stage of development, and in comparison to the parietal and interparietal bones, such bones could readily be teased apart with fine forceps. Finally, interfrontal bone ossification was often seen at the rostral portion of the calvaria, associated with the abnormally wide distance between the two frontal bones (T. Williams, data not shown).
Figure 2.

Craniofacial skeletal abnormalities in Ptch1DL homozygous mutants.
E18.5 control (a, c, e, g, i, k) and Ptch1DL mutant skulls (b, d, f, h, j, l) shown in lateral (a, b), dorsal (c, d) and ventral views with mandibles removed (e-h). (g) and (h) show detail of cranial base with hyoid bone removed. (Note that the pila postopitca (Ppso), which forms a component of the orbitosphenoid, was occasionally present in the mutants. It can also be absent in control embryos at this stage). (i, j) lateral views of control dentary bone (i) and dentary bones from two separate Ptch1DL mutants (j). (k, l) show detail of lambdoid suture between parietal and interparietal bones. Midline is to left and lateral to right. Asterisk shows expected junction between bones.
Key. (b) Abnormal flexure at skull and nasal juncture is shown by a black arrow. (d) Red arrows demonstrate delayed or abnormal development of the bones of the cranial vault in the midline in Ptch1DL mutants. (f) White asterisk highlights the absence of the presphenoid bone and the underdevelopment of the palatal shelves, red arrow shows the abnormal morphology of the alisphenoid bone in Ptch1DL mutants. Bo, basioccipital; Bs, basisphenoid; cdp, condylar process; crp, coronoid process; Fr, frontal bone; Hy, hyoid; Ip, interparietal bone; Mx, palatine process of the maxilla; N, nasal bone; Pl, palatine; Ppso, pila postoptica; Pr, parietal bone; Ps, presphenoid; Px, premaxilla.
Examination of the trunk and appendicular skeleton revealed additional defects in Ptch1DL mice. The sternum was disorganized with irregular ossification associated with asymmetric insertion of individual pairs of ribs in all mutants examined (Figure 3a, b). The manubrium and xiphoid process were also thicker and/or irregular. Examination of the Ptch1DL scapulas showed that these were mostly normal, although one contained a large central foramen (Figure 3c, d). In addition, limb skeletons confirmed the underlying skeletal defects associated with preaxial polydactyly (Figure 3e-h). All hindlimbs examined had six digits, whereas the forelimbs were more variable, sometimes possessing six or seven digits but more often having an abnormally broad or syndactylous first digit.
Figure 3.
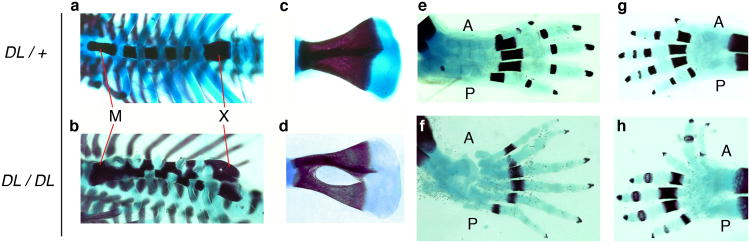
Additional skeletal abnormalities in Ptch1DL homozygous mutants.
E18.5 wild-type (a, c, e, g) and Ptch1DL mutants (b, d, f, h) showing ribcage in ventral view (a, b), scapula (c, d), hindlimb (e, f) and forelimb (g, h). Note the ribs have been cut by the investigators on one side of the sternum in the mutant sample. A, anterior; M, manubrium; P, posterior; X, xiphoid process.
DL corresponds to a point mutation in Ptch1
The ENU-mutagenized C57BL/6J founder male was outcrossed onto a wild type 129S1/SvImJ background, as were subsequent generations. The mutant phenotype has remained fairly consistent despite the change in genetic background. Meiotic recombination mapping enabled the approximate position of the mutation to be identified using polymorphic markers that distinguished between these two mouse strains. Initially, using markers spanning ∼10 mega base pair (Mb) intervals throughout the mouse autosomes, the mutation was mapped to an ∼12 Mb region on chromosome 13 (between polymorphic markers D13Mit13 and D13Mit 281). Subsequently, markers D13Mit283 and D13Mit 311 were used to refine the position of the mutation. Inspection of the ∼350,000 bp interval indicated that it contained Ptch1, encoding a receptor for Hedgehog ligands, a gene known to be responsible for controlling body size, limb patterning and bone development (Bruce et al., 2010; Goodrich et al., 1997; Hahn et al., 1998; Mak et al., 2008; Mak et al., 2006; Makino et al., 2001; Milenkovic et al., 1999; Ohba et al., 2008; Sweet et al., 1996). Therefore, Ptch1 genomic DNA from Ptch1DL mutants was sequenced and this revealed a single nucleotide change at the 3′-end of exon 13. This mutation altered a guanine present in all wild-type controls to an adenine in all Ptch1DL mice (arrow in Figure 4a). This nucleotide change is located within the splice donor sequence for exon 13, and would change the consensus sequence AG/GT to AAGT. Therefore, RT-PCR was performed to determine if Ptch1 mRNA splicing was altered in the Ptch1DL mutants. When primers located in Ptch1 exons 12 and 15 were employed for amplification of control RNA samples the expected 759-base pair (bp) product was generated (Figure 4b). However, Ptch1DL mutant RNA samples generated two PCR products: an abundant band shorter than the control and a weaker band that was a similar size as the control. Sequencing of the shorter Ptch1DL-b cDNA product demonstrated that it lacked 119 bp of the wild-type transcript due to skipping of exon 13. As a result of the aberrant joining of exon 12 to exon 14 there is a frame shift resulting in a premature stop codon 9 amino acids into the exon 14 sequence (see Figure 4 legend for sequence information). This alteration would result in a significantly truncated Ptch1 protein, Ptch1DL-b, lacking ∼870 C-terminal amino acids required for Hedgehog interaction and signal transduction (Figure 4c). This short product is presumably functionally inactive given these significant alterations. Sequence analysis of the longer Ptch1DL-a cDNA product also showed that this cDNA was mis-spliced despite its similar size to the control. In this instance, there was read-through across the normal position of splicing at the end of exon 13, with the cDNA continuing into the adjacent intron for an extra 36 bp before a cryptic splice donor sequence, GGGT, was used to splice to exon 14. This alteration would result in a Ptch1 protein, Ptch1DL-a, which would have an in-frame insertion of 12 amino acids that would be situated in the largest intracellular loop of the protein (Figure 4c; see Figure 4 legend for sequence information). Since the Ptch1DL homozygous embryos survive longer than Ptch1-null mice, we predict that the protein encoded by the Ptch1DL-a transcript retains some functionality in the Hedgehog signal transduction pathway.
Figure 4.

Ptch1DL harbors a point mutation in Ptch1. (a) Chromatograph sequencing result for Ptch1DL mutant showing Ptch1 genomic sequences at the junction of exon 13 and intron 13. The G to A point mutation in the Ptch1DL mutant is labeled with an arrow. (b) RT-PCR results for wild-type (lanes 1 and 2) and Ptch1DL mutant embryos (lanes 3 and 4) obtained using Ptch1 primers Pt2F and Pt2R. “M” = markers. (c) Graphic representation of wild-type Ptch1 protein and two putative Ptch1 proteins based on their mRNA sequences in Ptch1DL mutant embryos. The sterol sensing domain is shown by the red box in the wild-type protein. The DL-a form changes the normal sequence from DIFCC FTSPC VSRVI to DIFCC FTKYI SWCLG PVFFG PCVSR VI i.e. the addition of 36 nucleotides, with a new in-frame splice at a GGGT donor sequence, the change of S to K followed by the addition of 12 new amino acids. The DL-b form changes the normal sequence from FSLQA AVVVV FN to FSLQP LCQQG DSS* as exon 13, which has the mutated splice donor sequence, is skipped. The resulting splice from exon 12 to 14 is out of frame and creates a premature stop codon after 9 amino acids.
Allelic complementation studies demonstrate DL is an allele of Ptch1
The assignment of DL mutation to the Ptch1 locus was tested using a complementation analysis using two Ptch1 null alleles. The first allele, Ptch1tm1Zim (Hahn et al., 1998), has a deletion of exons 6 and 7, and the second, Ptch1-LacZ (Ptch1tm1Mps), has a knock-in of the LacZ reporter into exon 2 (Goodrich et al., 1997). Following mating of Ptch1DL and Ptch1 heterozygous mice, we isolated embryos at E18.5 and performed gross morphological analysis, skeletal staining and genotype analysis. Mice carrying one Ptch1DL allele and either of the Ptch1 null alleles were morphologically distinct from their littermates. Ptch1tm1Zim and Ptch1-LacZ gave similar results in this assay, although there was some variability in the severity of the mutant phenotype for both null alleles (Figures 5, 6 Table 1, and data not shown). At E18.5, viable mice with one Ptch1DL allele and one Ptch1 null allele were larger than their littermates and had a domed skull with distinct angular separation from the snout (Figure 5). Mice often had kinked tails and a small omphalocele, a body wall closure defect in which several loops of the gut enclosed in amniotic sac occurred outside the body cavity (Figure 5). All mutant mice also had pre-axial polydacytly of the forelimbs and hindlimbs (Figure 6). Several embryos presented with the neural tube closure defect exencephaly, while other mutants failed to develop and were dead upon collection at E18.5 (Table 1 and data not shown). Examination of the craniofacial skeleton in mutant mice with normal neural tube closure revealed similar defects as seen in Ptch1DL homozygous mice (Figure 6). Dorsal views of the skull revealed a major defect in the formation of the endochondrial bones of the calvaria, which was more severe than in homozygous Ptch1DL embryos (Figure 6d). There was also consistent fusion of the parietal and interparietal bones with absence of the intervening lambdoid suture (black arrow). Ventral views revealed absence of the presphenoid, a narrow basisphenoid, and hypoplasia of the alisphenoid. The palatal shelves were also underdeveloped (Figure 6f) as was the coronoid process of the mandible (Figure 6h). In the trunk, the sternum of Ptch1DL/Ptch1-LacZ mice was disorganized, thicker and more ossified than littermates (Figure 6j). There was a higher incidence of a foramen present in the scapula than in Ptch1DL homozygotes (5/10) and preaxial polydactyly and/or syndactyly was observed for all forelimbs and the majority of hindlimbs (Figure 6l and data not shown). In contrast, none of the control mice presented with these axial or appendicular skeletal defects. The genetic non-complementation observed between the Ptch1DL allele and either of the Ptch1-null alleles confirmed that Ptch1DL was a novel mutant allele for Ptch1. Moreover, previous studies have demonstrated that mice lacking Ptch1 die around E9.5 (Goodrich et al., 1997; Hahn et al., 1998). Therefore, since the homozygous Ptch1DL mice and Ptch1DL/Ptch1-null survive until E18.5, the Ptch1DL allele must be hypomorphic and produce sufficient functional Ptch1 protein to rescue the early developmental defects that result from the complete loss of Ptch1.
Figure 5.
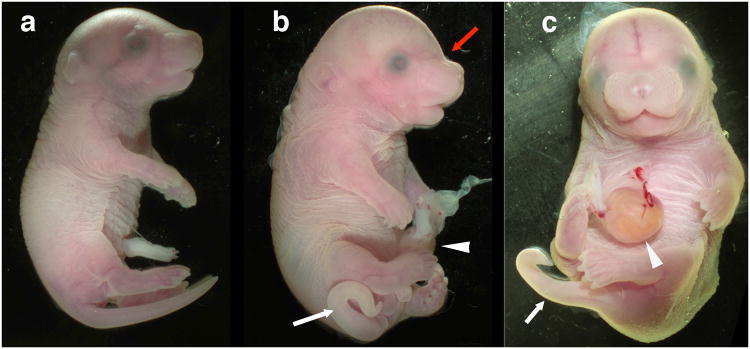
Allelic non-complementation between Ptch1DL and Ptch1tm1Zim at E18.5.
Whole mount images of wild-type (a) and two Ptch1DL/Ptch1tm1Zim embryos (b, c) in lateral (a, b) or ventral (c) view. In the mutant embryos, the kinked tail (white arrow), omphalocele (white arrowhead) and abnormal skull shape (red arrow) are illustrated.
Figure 6.

Skeletal staining illustrating non-complementation between Ptch1DL and Ptch1-LacZ mutant alleles.
E18.5 Ptch1DL heterozygotes (a, c, e, g, i, k) and Ptch1DL/Ptch1-LacZ mutants (b, d, f, h, j, l). Lateral views of skulls (a, b) with red arrow showing abnormal flexure at skull and nasal juncture. Dorsal view of skulls (c, d). Black arrow in (d) illustrates lack of lambdoid suture. Ventral view of skull with mandibles removed (e, f) with the absence of the presphenoid bone and the underdevelopment of the palatal shelves (red asterisk), and the aberrant morphology of the alisphenoid bone (white arrow) marked in the Ptch1DL/Ptch1-LacZ mutants. (g, h) Lateral view of dentary bones. (i, j) Ribcage in ventral view. (k, l) forelimbs.
Key: A, anterior; ap, angular process; Bo, basioccipital; Bs, basisphenoid; cdp, condylar process; crp, coronoid process; Fr, frontal bone; Hy, hyoid; Ip, interparietal bone; M, manubrium; Mx, palatine process of the maxilla; N, nasal bone; P, posterior; Pl, palatine; Pr, parietal bone; Ps, presphenoid; Px, premaxilla; X, xiphoid process.
Discussion
Previous sequence analyses of BCNS patients have identified multiple mutations that map to PTCH1 (Lindstrom et al., 2006; Quijada et al., 2006; Wicking et al., 1997). These types of mutations are concentrated in the N-terminal cytoplasmic region, the two large extracellular loops and the large intracellular loop. A majority of these mutations would be predicted to function as null alleles as they introduce premature nonsense codons causing significant truncations of the protein. Although there are over 100 different PTCH1 mutations associated with BCNS, to date there has been no evidence of any genotype-phenotype correlations. Here, we describe the identification of a new hypomorphic allele of Ptch1, Ptch1DL, which was isolated in an ENU-based screen for recessive mouse mutations affecting craniofacial morphology. The splice site mutation present in the Ptch1DL allele produces two mutant forms of the protein. The more prevalent Ptch1 DL-b transcript would produce a significantly truncated protein, with a disruption of the sterol sensing domain (Lindstrom et al., 2006) and loss of all further C-terminal regions. This form would be similar to at least one of the human mutations identified to date. The remaining DL-a transcripts would introduce an additional 12 amino acids into the large intracellular loop of the protein. This loop interacts with cyclin B1 and may function in Ptch1-mediated regulation of cell cycle progression (Barnes et al., 2001). However, since Ptch1DL homozygous and Ptch1DL/Ptch1null trans-heterozygotes live until birth, compared to the death of Ptch1-homozygous null animals around E9.5, the amount and function of the DL-a protein must be sufficient to fulfill critical aspects of early Ptch1 function.
Further support for essential levels of functional Ptch1 protein in Ptch1DL mutants is also suggested by an examination of heterozygous Ptch1DL animals. To date, we have not noted general overgrowth, brain tumors, or limb abnormalities in these mice (data not shown) compared to the occurrence of such phenotypes in some of the Ptch1null heterozygous strains (Goodrich et al., 1997; Hahn et al., 1998). At the same time, we note that abnormal heterozygous phenotypes have not been consistently reported, and that genetic background can have a large influence over the phenotypes associated with Ptch1 mutations (Ellis et al., 2003; Wakabayashi et al., 2007). Ptch1DL does appear to be a stronger recessive allele than the previously identified Ptch1mes mutation (Makino et al., 2001; Sweet et al., 1996). This latter allele arose spontaneously, and is caused by a 32nt deletion resulting in loss of the last 200 amino acids from the C-terminal intracellular tail of Ptch1 and its replacement by a novel 68 amino acid region following the frame-shift (Makino et al., 2001). In contrast to Ptch1DL homozygous mice, Ptch1mes homozygous mice are viable, although they have similar defects in the shape of the skull, and the patterning of the sternum and limbs (Sweet et al., 1996). Further, the Ptch1DL/Ptch1null mice have a stronger cranial base phenotype than trans-heterozygous Ptch1mes/Ptch1null mice. Both allelic combinations die at birth, are larger in size compared to control littermates, and exhibit polydactyly of all four limbs (Makino et al., 2001). However, unlike in Ptch1DL/Ptch1null mice, skeletal staining of Ptch1mes/Ptch1null embryos reveals that the sphenoid is of normal size, the presphenoid is present, and there is a long coronoid process associated with the dentary (data not shown). The Ptch1DL allele therefore provides a valuable new resource to study the function of Ptch1 in many developmental processes.
In this report, we have focused on the skeletal defects observed in Ptch1DL homozygous embryos and Ptch1DL/Ptch1null trans-heterozygotes. The phenotype of these animals provides mouse models to understand aspects of BCNS that influence the shape of the craniofacial complex, the ribcage, the shoulders, and the limbs (Evans and Farndon, 2010; Iwanaga et al., 1998; Kimonis et al., 1997; Kimonis et al., 2013; Leonardi et al., 2002; Lo Muzio, 2008). Both Ptch1DL–based mouse models display preaxial polydactyly, foramens of the scapula, and sternal dysmorphology. In the head, the prominent domed skull shape, similar to the forehead bossing in BCNS patients, may result from a combination of defective cranial base formation and hypoplastic development of the calvaria. Overt clefting of the secondary palate was uncommon in these allelic combinations, but the delayed and/or hypoplastic development of the palatal shelves may be linked to the high arched palate phenotype present in BCNS patients. The defects of the skeletal components that will eventually fuse to form the mature sphenoid bone is also intriguing given the observation of defects in the sella turcica and hyperpneumatization of the sphenoid reported in BCNS (Iwanaga et al., 1998). However, the morphology of the mandible appears to differ significantly between the mouse and human conditions. In particular, there is no clear prognathism in the mouse, and the coronoid process of the dentary is hypoplastic in the mouse models in contrast to the reports of hyperplasia of this structure in human (Leonardi et al., 2002). We hypothesize that these differences reflect either species-specific arrangements of the craniofacial skeleton or discrepancies in the age at which these structures were examined between these two species. Nevertheless, the observed prognathism and coronoid pathology in human, coupled with the coronoid hypoplasia in the Ptch1DL embryos, indicates that reduction of Ptch1 expression can have profound affects on the shape of the jaw and it's joints.
The Ptch1DL mutant is also relevant in understanding how other alterations in the Hedgehog signaling pathway, including ciliopathies, influence skeletal development and patterning. In this regard, aberrant Hedgehog signaling may underlie some instances of human craniosynostosis, a condition more commonly associated with defects in the Fgf pathway (Holmes, 2012; Klopocki et al., 2011; Yuksel-Apak et al., 2012). The strongest association is with RAB23, a negative regulator of Hedgehog signaling that is mutated in Carpenter Syndrome, a condition that includes synostosis of the metopic and sagittal sutures (Jenkins et al., 2007). Two reports have documented craniosynostosis in patients with large chromosomal duplications or deletions in the vicinity of IHH that are postulated to cause ectopic expression of this gene (Klopocki et al., 2011; Yuksel-Apak et al., 2012). Lastly, in rare instances, inappropriate fusion of the midline sutures can also occur in Grieg cephalopolysyndactyly, which is caused by mutations in GLI3, a transcription factor which acts downstream of PTCH1 to repress Hedgehog signaling (Hurst et al., 2011). However, analysis of how Hedgehog pathway genes influence craniosynostosis in the mouse has been complicated by exencephaly, early lethality and the function of such genes in ossification of the calvaria (Goodrich et al., 1997; Hahn et al., 1998; Rice et al., 2010; St-Jacques et al., 1999). The function of Ihh in intramembranous ossification has been controversial, with reports indicating that both loss and gain of Ihh function can cause hypoplastic cranial bone development (Abzhanov et al., 2007; Lenton et al., 2011; St-Jacques et al., 1999). We note that previous studies targeting Ptch1 function in chondrocytes, osteoblasts and osteocytes resulted in similar defects in formation and maintenance of the calvaria as observed in Ihhnull skulls (Mak et al., 2008; Mak et al., 2006; St-Jacques et al., 1999). Therefore it may be that normal development of the bones of the skull vault requires a critical range of Hedgehog activity. Nevertheless, the failure of the skull bones to juxtapose at the midline in mouse models that exhibit either increased or decreased Hedgehog activity has made it difficult to assess how the genes in this network impact craniosynostosis. One exception has been the Gli3Xt-J/Xt-J mouse model that displays lambdoid suture craniosynostosis (Rice et al., 2010). We find that the PtchDL models exhibit similar premature fusion of the parietal and interparietal bones towards the ventral side of the skull. These findings are consistent with the expression of Ptch1 in the osteogenic fronts of the calvaria, coincident with Ihh, Gli3 and Runx2 (Mak et al., 2008; Rice et al., 2010). Further analysis will be required to determine if the juxtaposition of other calvarial bones, particularly at the coronal suture, will result in synostosis in PtchDL mice. We note that the regions of the parietal and interparietal bones involved in suture formation are both derived from the mesoderm, whereas other bones involved in suture formation have at least one neural crest component (Jiang et al., 2002). Therefore, the embryonic origin of the lambdoid suture could also render it more sensitive to Hedgehog signaling levels. Taken together, though, the findings from the Gli3Xt-J/Xt-J and Ptch1DL indicate that a reduction of these two Hedgehog pathway inhibitors both result in lambdoid suture craniosynostsis. Furthermore, our findings raise the possibility that examples of human craniosynostosis could result from alterations in PTCH1 and that a degree of premature suture fusion may occur in BCNS.
Overall, the phenotypes we observe in PtchDL embryos are likely caused by a combination of reduced repression of molecular pathways regulated by both Shh and Ihh signaling. Loss of repression of the Hedgehog pathway downstream of Ihh likely causes ossification defects in the calvaria, sternum and scapula. With respect to the Hedgehog pathway, similar scapula and sternal defects are present in the Gli3Xt-J/Xt-J mouse model as seen with the Ptch1DL embryos (Johnson, 1967; Kuijper et al., 2005). A second link between the Hedgehog pathway and defects in the scapula comes from the examination of the Doublefoot (Dbf) mouse model (Hayes et al., 1998). Mice heterozygous for Dbf have polydactyly and scapula foramina, as well as defects in ossification of the calvaria (Hayes et al., 1998). Subsequent studies indicated that Ihh was ectopically expressed in the developing limbs and branchial arches of Dbf embryos as a result of a large chromosomal deletion in the vicinity of this gene (Babbs et al., 2008; Yang et al., 1998). In contrast, the polydactyly and syndactyly apparent in Ptch1DL homozygous or Ptch1DL/Ptch1null mice presumably result from deregulation of Shh signaling. We also predict that excess Shh signaling is responsible for the omphalocele observed in the Ptch1DL background, given the presence of this body wall defect in Gli3 mutants and its rescue by lowering Shh levels in this genetic background (Matsumaru et al., 2011). Despite the anticipated activation of the Hedgehog signaling pathway, our preliminary examination of the Ptch1DL/Ptch1-LacZ limbs has not shown an expansion of β-galactosidase expression, a readout of activation of Ptch1 transcription (data not shown). We therefore postulate that levels of functional Ptch1 protein required for this feedback loop to be activated must be lower than present in the Ptch1DL/Ptch1null mice.
With respect to the alteration of the skeletal elements of the cranial base, aberrant responses to both Shh and Ihh are likely involved in the defects seen in Ptch1DL homozygous and Ptch1DL/Ptch1null embryos (Balczerski et al., 2012; McBratney-Owen et al., 2008; Young et al., 2006). The cranial base is derived from both mesodermal and neural crest derivatives with the basisphenoid bone acting as a boundary in association with the rostral extent of the notochord. The notochord acts as a source of Shh and disruption of either Shh or Ihh can alter the development of the cranial base (Balczerski et al., 2012; Nagayama et al., 2008; St-Jacques et al., 1999; Young et al., 2006). Moreover, genetic manipulations that increase Hh signaling, including mice with Mks1 or Tmem107 mutations as well as a K14-Shh transgene, produce similar defects in the cranial base to the Ptch1DL mutants (Christopher et al., 2012; Cobourne et al., 2009; Cui et al., 2011; Weatherbee et al., 2009). Mks1 and Tmem107 mutants, which affect primary cilia function, also have comparable limb and sternal defects to the Ptch1DL mice, reinforcing the connection of these skeletal defects to ciliopathy and increased Hedgehog signaling (Christopher et al., 2012; Cui et al., 2011; Weatherbee et al., 2009). Finally, note that other genetic changes are also associated with loss, reduction or alteration of the presphenoid and basisphenoid bones including mutations in components of the Fgf, Wnt/β-catenin and TGF-β signaling pathways (Caparros-Martin et al., 2013; Dudas et al., 2006; Mai et al., 2010; Meester-Smoor et al., 2005; Nagayama et al., 2008; Pacheco et al., 2012; Reid et al., 2011; Winnier et al., 1997). Determining how these signal transduction pathways and their transcriptional effectors are integrated to control cranial base development will be important to understand the regulation of human facial morphology. In conclusion, the Ptch1DL allele adds a valuable new resource to probe the molecular control of development and patterning, especially with respect to the craniofacial skeleton.
Materials and Methods
Mice
Male C57BL/6J mice treated with ENU were kindly provided by Monica Justice (Baylor College of Medicine). Additional C57BL/6J mice, as well as 129S1/SvImJ, Ptch1tm1Mps/J (Ptch1-LacZ), and B6C3Fe a/a-Ptch1mes/J (Ptch1mes) were obtained from the Jackson Laboratory (Goodrich et al., 1997; Sweet et al., 1996). B6.129-Ptch1tm1Zim/Cnrm mice (Hahn et al., 1998) were obtained from Emma (The European Mouse Mutant Archive). This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee of the University of Colorado Denver. The Ptch1DL mutation on the C57BL/6J background was distinguished from 129S1/SvImJ in the same region using the two sets of primer pairs D13Mit283F (5′- GGA AGC AGT CTC CTG CCT C -3′) with D13Mit283R (5′- GAG AGG TGG CAC ATG AGG TT -3′); and D13mit311F (5′-TGG CTC CTC ATG TTC TAC CC -3′) with D13mit311R (5′-CCA GGT TGT TGC TGC ATT C -3′). PCR was performed using the following parameters: 94°C 12 min; 56 cycles of (94°C 20 sec, 55°C 30 sec, 72°C 45 sec); 72°C 7 min; 4°C hold. Products were resolved on 4% agarose gels and compared to C57BL/6J and 129S1/SvImJ standards. The D13Mit283 primers produce a band of 114bp from C57BL/6J genomic DNA, and a slightly larger band from 129S1/SvImJ. The D13Mit311 primers produce a band of 122bp from C57BL/6J genomic DNA, and a slightly smaller band from 129S1/SvImJ. After the position of the mutation was identified, genotype could be followed using the primers DLF (5′- GGT AAG TCC TTA CTG GTA TCA C -3′) and DLR (5′- CAC CAA ACT GTT CAT TGA TAA AC -3′) for PCR, followed by standard sequence analysis. PCR genotyping for the Ptch1-LacZ allele was performed using a protocol provided by the Jackson Laboratory using primers oIMR0204, oIMR0221, oIMR0716, and oIMR0717 (Jax, 2011). Ptch1mes and Ptch1tm1Zim genotyping was according to protocols recorded in associated publications (Hahn et al., 1998; Makino et al., 2001). Heterozygous PtchDL mice will be made available to the research community upon acceptance of the manuscript.
Skeletal staining
Skeletal staining with alcian blue (Sigma A3157) and alizarin red (Sigma A5533) was performed as described previously (Brewer et al., 2004). Briefly, embryos at 18.5 days post coitum (E18.5) were fixed in 95% ethanol at room temperature for 3 days and stained by 0.15% alcian blue and 0.005% alizarin red for 24 hours. Stained embryos were next cleared with 2% KOH and stored in 50% glycerol until photographed.
Reverse transcription-PCR (RT-PCR)
A detailed RT-PCR protocol was described previously (Feng and Williams, 2003). Briefly, 1ug of total RNA from limb was reverse transcribed into cDNA with SuperScript III (Invitrogen, Cat# 18080-093). Resulting cDNA was used to perform PCR using the AccuPrime Taq DNA polymerase System (Invitrogen, Cat# 12339-016). The primers used to amplify Ptch1 cDNA were Pt2F (5′- GCA TTC AGT GAA ACA GGA CAG AAT AAG AG -3′) and Pt2R (5′- CGT CTC TCA CTC GGG TGG TCC CAT AAA GG – 3′). PCR was performed using the following parameters: 94 °C 3 min; 35 cycles of (94°C 45 sec, 56°C 45 sec, 68°C 1 min); 68°C 10 min and 4°C hold.
Acknowledgments
We thank Dr. Vida Melvin for valuable comments on the manuscript. We also thank Lori Bulwith, Dr. Lei Zhang, Dr. Martin G. Hanson and Dr. Jian Huang for their involvement in the ENU mutagenesis screen, and Dr. Monica Justice for the gift of mutagenized male mice. We appreciate technical support from V. Bleu Knight for performing the genomic scan to identify the chromosomal region containing the Ptch1DL mutation. Funding from a Cancer Center Support Grant (P30CA046934) was used for our sequence analyses and we thank members of this core for their assistance. This research was also supported by National Institutes of Health Grants DE019843 (T.W.) and DE014181 (D.E.C). L.N. is an Investigator of the Howard Hughes Medical Institute.
Funding: This research was also supported by National Institutes of Health Grants DE019843 (T.W.) and DE014181 (D.E.C). L.N. is an Investigator of the Howard Hughes Medical Institute. Sequence analysis was performed using funding from a Cancer Center Support Grant (P30CA046934).
References
- Abzhanov A, Rodda SJ, McMahon AP, Tabin CJ. Regulation of skeletogenic differentiation in cranial dermal bone. Development. 2007;134:3133–3144. doi: 10.1242/dev.002709. [DOI] [PubMed] [Google Scholar]
- Babbs C, Furniss D, Morriss-Kay GM, Wilkie AO. Polydactyly in the mouse mutant Doublefoot involves altered Gli3 processing and is caused by a large deletion in cis to Indian hedgehog. Mechanisms of development. 2008;125:517–526. doi: 10.1016/j.mod.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balczerski B, Zakaria S, Tucker AS, Borycki AG, Koyama E, Pacifici M, Francis-West P. Distinct spatiotemporal roles of hedgehog signalling during chick and mouse cranial base and axial skeleton development. Developmental biology. 2012;371:203–214. doi: 10.1016/j.ydbio.2012.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes EA, Kong M, Ollendorff V, Donoghue DJ. Patched1 interacts with cyclin B1 to regulate cell cycle progression. EMBO J. 2001;20:2214–2223. doi: 10.1093/emboj/20.9.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer S, Feng W, Huang J, Sullivan S, Williams T. Wnt1-Cre-mediated deletion of AP-2alpha causes multiple neural crest-related defects. Dev Biol. 2004;267:135–152. doi: 10.1016/j.ydbio.2003.10.039. [DOI] [PubMed] [Google Scholar]
- Bruce SJ, Butterfield NC, Metzis V, Town L, McGlinn E, Wicking C. Inactivation of Patched1 in the mouse limb has novel inhibitory effects on the chondrogenic program. The Journal of biological chemistry. 2010;285:27967–27981. doi: 10.1074/jbc.M109.091785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield NC, Metzis V, Mcglinn E, Bruce SJ, Wainwright BJ, Wicking C. Patched 1 is a crucial determinant of asymmetry and digit number in the vertebrate limb. Development. 2009;136:3515–3524. doi: 10.1242/dev.037507. [DOI] [PubMed] [Google Scholar]
- Caparros-Martin JA, Valencia M, Reytor E, Pacheco M, Fernandez M, Perez-Aytes A, Gean E, Lapunzina P, Peters H, Goodship JA, Ruiz-Perez VL. The ciliary Evc/Evc2 complex interacts with Smo and controls Hedgehog pathway activity in chondrocytes by regulating Sufu/Gli3 dissociation and Gli3 trafficking in primary cilia. Human molecular genetics. 2013;22:124–139. doi: 10.1093/hmg/dds409. [DOI] [PubMed] [Google Scholar]
- Caspary T, Anderson KV. Uncovering the uncharacterized and unexpected: unbiased phenotype-driven screens in the mouse. Dev Dyn. 2006;235:2412–2423. doi: 10.1002/dvdy.20853. [DOI] [PubMed] [Google Scholar]
- Christopher KJ, Wang B, Kong Y, Weatherbee SD. Forward genetics uncovers Transmembrane protein 107 as a novel factor required for ciliogenesis and Sonic hedgehog signaling. Developmental biology. 2012;368:382–392. doi: 10.1016/j.ydbio.2012.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobourne MT, Xavier GM, Depew M, Hagan L, Sealby J, Webster Z, Sharpe PT. Sonic hedgehog signalling inhibits palatogenesis and arrests tooth development in a mouse model of the nevoid basal cell carcinoma syndrome. Developmental biology. 2009;331:38–49. doi: 10.1016/j.ydbio.2009.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui C, Chatterjee B, Francis D, Yu Q, SanAgustin JT, Francis R, Tansey T, Henry C, Wang B, Lemley B, Pazour GJ, Lo CW. Disruption of Mks1 localization to the mother centriole causes cilia defects and developmental malformations in Meckel-Gruber syndrome. Dis Model Mech. 2011;4:43–56. doi: 10.1242/dmm.006262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudas M, Kim J, Li WY, Nagy A, Larsson J, Karlsson S, Chai Y, Kaartinen V. Epithelial and ectomesenchymal role of the type I TGF-beta receptor ALK5 during facial morphogenesis and palatal fusion. Developmental biology. 2006;296:298–314. doi: 10.1016/j.ydbio.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis T, Smyth I, Riley E, Graham S, Elliot K, Narang M, Kay GF, Wicking C, Wainwright B. Patched 1 conditional null allele in mice. Genesis. 2003;36:158–161. doi: 10.1002/gene.10208. [DOI] [PubMed] [Google Scholar]
- Evans GD, Farndon PA. Nevoid Basal Cell Carcinoma Syndrome. In: Pagon RA, Bird TD, Dolan CR, Stephens K, editors. GeneReviews. Seattle (WA): Univrsity of Washington, Seattle; 2010. [PubMed] [Google Scholar]
- Feng W, Williams T. Cloning and characterization of the mouse AP-2 epsilon gene: a novel family member expressed in the developing olfactory bulb. Mol Cell Neurosci. 2003;24:460–475. doi: 10.1016/s1044-7431(03)00209-4. [DOI] [PubMed] [Google Scholar]
- Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- Gritli-Linde A. The Etiopathogenesis of Cleft Lip and Cleft Palate:: Usefulness and Caveats of Mouse Models. Current topics in developmental biology. 2008;84:37–138. doi: 10.1016/S0070-2153(08)00602-9. [DOI] [PubMed] [Google Scholar]
- Hahn H, Wojnowski L, Zimmer AM, Hall J, Miller G, Zimmer A. Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of Gorlin syndrome. Nat Med. 1998;4:619–622. doi: 10.1038/nm0598-619. [DOI] [PubMed] [Google Scholar]
- Hayes C, Lyon MF, Morriss-Kay GM. Morphogenesis of Doublefoot (Dbf), a mouse mutant with polydactyly and craniofacial defects. Journal of anatomy. 1998;193(Pt 1):81–91. doi: 10.1046/j.1469-7580.1998.19310081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes G. The role of vertebrate models in understanding craniosynostosis. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2012;28:1471–1481. doi: 10.1007/s00381-012-1844-3. [DOI] [PubMed] [Google Scholar]
- Hurst JA, Jenkins D, Vasudevan PC, Kirchhoff M, Skovby F, Rieubland C, Gallati S, Rittinger O, Kroisel PM, Johnson D, Biesecker LG, Wilkie AO. Metopic and sagittal synostosis in Greig cephalopolysyndactyly syndrome: five cases with intragenic mutations or complete deletions of GLI3. European journal of human genetics : EJHG. 2011;19:757–762. doi: 10.1038/ejhg.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwanaga S, Shimoura H, Shimizu M, Numaguchi Y. Gorlin syndrome: unusual manifestations in the sella turcica and the sphenoidal sinus. AJNR American journal of neuroradiology. 1998;19:956–958. [PMC free article] [PubMed] [Google Scholar]
- Jax. 2011 Protocol for genotyping: http://jaxmice.jax.org/protocolsdb/f?p=116:2:1406893156903556::NO:2:P2_MASTER_PROTOCOL_ID,P2_JRS_CODE:1687,003081. In.
- Jenkins D, Seelow D, Jehee FS, Perlyn CA, Alonso LG, Bueno DF, Donnai D, Josifova D, Mathijssen IM, Morton JE, Orstavik KH, Sweeney E, Wall SA, Marsh JL, Nurnberg P, Passos-Bueno MR, Wilkie AO. RAB23 mutations in Carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity. American journal of human genetics. 2007;80:1162–1170. doi: 10.1086/518047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Iseki S, Maxson RE, Sucov HM, Morriss-Kay GM. Tissue origins and interactions in the mammalian skull vault. Developmental biology. 2002;241:106–116. doi: 10.1006/dbio.2001.0487. [DOI] [PubMed] [Google Scholar]
- Johnson DR. Extra-toes: anew mutant gene causing multiple abnormalities in the mouse. J Embryol Exp Morphol. 1967;17:543–581. [PubMed] [Google Scholar]
- Kimonis VE, Goldstein AM, Pastakia B, Yang ML, Kase R, DiGiovanna JJ, Bale AE, Bale SJ. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1997;69:299–308. [PubMed] [Google Scholar]
- Kimonis VE, Singh KE, Zhong R, Pastakia B, Digiovanna JJ, Bale SJ. Clinical and radiological features in young individuals with nevoid basal cell carcinoma syndrome. Genetics in medicine : official journal of the American College of Medical Genetics. 2013;15:79–83. doi: 10.1038/gim.2012.96. [DOI] [PubMed] [Google Scholar]
- Klopocki E, Lohan S, Brancati F, Koll R, Brehm A, Seemann P, Dathe K, Stricker S, Hecht J, Bosse K, Betz RC, Garaci FG, Dallapiccola B, Jain M, Muenke M, Ng VC, Chan W, Chan D, Mundlos S. Copy-number variations involving the IHH locus are associated with syndactyly and craniosynostosis. American journal of human genetics. 2011;88:70–75. doi: 10.1016/j.ajhg.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuijper S, Beverdam A, Kroon C, Brouwer A, Candille S, Barsh G, Meijlink F. Genetics of shoulder girdle formation: roles of Tbx15 and aristaless-like genes. Development. 2005;132:1601–1610. doi: 10.1242/dev.01735. [DOI] [PubMed] [Google Scholar]
- Lenton K, James AW, Manu A, Brugmann SA, Birker D, Nelson ER, Leucht P, Helms JA, Longaker MT. Genesis. New York, NY: 2011. Indian hedgehog positively regulates calvarial ossification and modulates bone morphogenetic protein signaling; p. 2000. [DOI] [PubMed] [Google Scholar]
- Leonardi R, Caltabiano M, Lo Muzio L, Gorlin RJ, Bucci P, Pannone G, Canfora M, Sorge G. Bilateral hyperplasia of the mandibular coronoid processes in patients with nevoid basal cell carcinoma syndrome: an undescribed sign. American journal of medical genetics. 2002;110:400–403. doi: 10.1002/ajmg.10432. [DOI] [PubMed] [Google Scholar]
- Lindstrom E, Shimokawa T, Toftgard R, Zaphiropoulos PG. PTCH mutations: distribution and analyses. Human mutation. 2006;27:215–219. doi: 10.1002/humu.20296. [DOI] [PubMed] [Google Scholar]
- Lo Muzio L. Nevoid basal cell carcinoma syndrome (Gorlin syndrome) Orphanet journal of rare diseases. 2008;3:32. doi: 10.1186/1750-1172-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai S, Wei K, Flenniken A, Adamson SL, Rossant J, Aubin JE, Gong SG. The missense mutation W290R in Fgfr2 causes developmental defects from aberrant IIIb and IIIc signaling. Dev Dyn. 2010;239:1888–1900. doi: 10.1002/dvdy.22314. [DOI] [PubMed] [Google Scholar]
- Mak KK, Bi Y, Wan C, Chuang PT, Clemens T, Young M, Yang Y. Hedgehog signaling in mature osteoblasts regulates bone formation and resorption by controlling PTHrP and RANKL expression. Dev Cell. 2008;14:674–688. doi: 10.1016/j.devcel.2008.02.003. [DOI] [PubMed] [Google Scholar]
- Mak KK, Chen MH, Day TF, Chuang PT, Yang Y. Wnt/beta-catenin signaling interacts differentially with Ihh signaling in controlling endochondral bone and synovial joint formation. Development. 2006;133:3695–3707. doi: 10.1242/dev.02546. [DOI] [PubMed] [Google Scholar]
- Makino S, Masuya H, Ishijima J, Yada Y, Shiroishi T. A spontaneous mouse mutation, mesenchymal dysplasia (mes), is caused by a deletion of the most C-terminal cytoplasmic domain of patched (ptc) Dev Biol. 2001;239:95–106. doi: 10.1006/dbio.2001.0419. [DOI] [PubMed] [Google Scholar]
- Matsumaru D, Haraguchi R, Miyagawa S, Motoyama J, Nakagata N, Meijlink F, Yamada G. Genetic analysis of Hedgehog signaling in ventral body wall development and the onset of omphalocele formation. PloS one. 2011;6:e16260. doi: 10.1371/journal.pone.0016260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBratney-Owen B, Iseki S, Bamforth SD, Olsen BR, Morriss-Kay GM. Development and tissue origins of the mammalian cranial base. Developmental biology. 2008;322:121–132. doi: 10.1016/j.ydbio.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meester-Smoor MA, Vermeij M, van Helmond MJL, Molijn AC, van Wely KHM, Hekman ACP, Vermey-Keers C, Riegman PHJ, Zwarthoff EC. Targeted disruption of the Mn1 oncogene results in severe defects in development of membranous bones of the cranial skeleton. Molecular and Cellular Biology. 2005;25:4229–4236. doi: 10.1128/MCB.25.10.4229-4236.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milenkovic L, Goodrich LV, Higgins KM, Scott MP. Mouse patched1 controls body size determination and limb patterning. Development. 1999;126:4431–4440. doi: 10.1242/dev.126.20.4431. [DOI] [PubMed] [Google Scholar]
- Nagayama M, Iwamoto M, Hargett A, Kamiya N, Tamamura Y, Young B, Morrison T, Takeuchi H, Pacifici M, Enomoto-Iwamoto M, Koyama E. Wnt/beta-catenin signaling regulates cranial base development and growth. Journal of Dental Research. 2008;87:244–249. doi: 10.1177/154405910808700309. [DOI] [PubMed] [Google Scholar]
- Ngan ES, Garcia-Barcelo MM, Yip BH, Poon HC, Lau ST, Kwok CK, Sat E, Sham MH, Wong KK, Wainwright BJ, Cherny SS, Hui CC, Sham PC, Lui VC, Tam PK. Hedgehog/Notch-induced premature gliogenesis represents a new disease mechanism for Hirschsprung disease in mice and humans. The Journal of clinical investigation. 2011;121:3467–3478. doi: 10.1172/JCI43737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohba S, Kawaguchi H, Kugimiya F, Ogasawara T, Kawamura N, Saito T, Ikeda T, Fujii K, Miyajima T, Kuramochi A, Miyashita T, Oda H, Nakamura K, Takato T, Chung Ui. Patched1 haploinsufficiency increases adult bone mass and modulates Gli3 repressor activity. Developmental cell. 2008;14:689–699. doi: 10.1016/j.devcel.2008.03.007. [DOI] [PubMed] [Google Scholar]
- Pacheco M, Valencia M, Caparros-Martin JA, Mulero F, Goodship JA, Ruiz-Perez VL. Evc works in chondrocytes and osteoblasts to regulate multiple aspects of growth plate development in the appendicular skeleton and cranial base. Bone. 2012;50:28–41. doi: 10.1016/j.bone.2011.08.025. [DOI] [PubMed] [Google Scholar]
- Quijada L, Callejo A, Torroja C, Guerrero I. The Patched Receptor: Switching On/Off the Hedgehog Signaling Pathway. In: Ruiz i Altaba A, editor. Hedgehog-gli signaling in human disease. Austin, Tx: Landes Bioscience; 2006. [Google Scholar]
- Reid BS, Yang H, Melvin VS, Taketo MM, Williams T. Ectodermal Wnt/beta-catenin signaling shapes the mouse face. Developmental biology. 2011;349:261–269. doi: 10.1016/j.ydbio.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice DP, Connor EC, Veltmaat JM, Lana-Elola E, Veistinen L, Tanimoto Y, Bellusci S, Rice R. Gli3Xt-J/Xt-J mice exhibit lambdoid suture craniosynostosis which results from altered osteoprogenitor proliferation and differentiation. Human molecular genetics. 2010;19:3457–3467. doi: 10.1093/hmg/ddq258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins DJ, Fei DL, Riobo NA. The Hedgehog signal transduction network. Science signaling. 2012;5:re6. doi: 10.1126/scisignal.2002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RB, Hu Y, Albertson RC, Kocher TD. Craniofacial divergence and ongoing adaptation via the hedgehog pathway. Proc Natl Acad Sci U S A. 2011;108:13194–13199. doi: 10.1073/pnas.1018456108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes & development. 1999;13:2072–2086. doi: 10.1101/gad.13.16.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet HO, Bronson RT, Donahue LR, Davisson MT. Mesenchymal dysplasia: a recessive mutation on chromosome 13 of the mouse. The Journal of heredity. 1996;87:87–95. doi: 10.1093/oxfordjournals.jhered.a022981. [DOI] [PubMed] [Google Scholar]
- Wakabayashi Y, Mao JH, Brown K, Girardi M, Balmain A. Promotion of Hras-induced squamous carcinomas by a polymorphic variant of the Patched gene in FVB mice. Nature. 2007;445:761–765. doi: 10.1038/nature05489. [DOI] [PubMed] [Google Scholar]
- Weatherbee SD, Niswander LA, Anderson KV. A mouse model for Meckel syndrome reveals Mks1 is required for ciliogenesis and Hedgehog signaling. Human molecular genetics. 2009;18:4565–4575. doi: 10.1093/hmg/ddp422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicking C, Shanley S, Smyth I, Gillies S, Negus K, Graham S, Suthers G, Haites N, Edwards M, Wainwright B, Chenevix-Trench G. Most germ-line mutations in the nevoid basal cell carcinoma syndrome lead to a premature termination of the PATCHED protein, and no genotype-phenotype correlations are evident. American journal of human genetics. 1997;60:21–26. [PMC free article] [PubMed] [Google Scholar]
- Winnier GE, Hargett L, Hogan BL. The winged helix transcription factor MFH1 is required for proliferation and patterning of paraxial mesoderm in the mouse embryo. Genes & development. 1997;11:926–940. doi: 10.1101/gad.11.7.926. [DOI] [PubMed] [Google Scholar]
- Yang Y, Guillot P, Boyd Y, Lyon MF, McMahon AP. Evidence that preaxial polydactyly in the Doublefoot mutant is due to ectopic Indian Hedgehog signaling. Development. 1998;125:3123–3132. doi: 10.1242/dev.125.16.3123. [DOI] [PubMed] [Google Scholar]
- Young B, Minugh-Purvis N, Shimo T, St-Jacques B, Iwamoto M, Enomoto-Iwamoto M, Koyama E, Pacifici M. Indian and sonic hedgehogs regulate synchondrosis growth plate and cranial base development and function. Developmental biology. 2006;299:272–282. doi: 10.1016/j.ydbio.2006.07.028. [DOI] [PubMed] [Google Scholar]
- Yuksel-Apak M, Bogershausen N, Pawlik B, Li Y, Apak S, Uyguner O, Milz E, Nurnberg G, Karaman B, Gulgoren A, Grzeschik KH, Nurnberg P, Kayserili H, Wollnik B. A large duplication involving the IHH locus mimics acrocallosal syndrome. European journal of human genetics : EJHG. 2012;20:639–644. doi: 10.1038/ejhg.2011.250. [DOI] [PMC free article] [PubMed] [Google Scholar]