

Abstract
The central importance of epigenetics to the aging process is increasingly being recognized. Here we perform a methylome-wide association study (MWAS) of aging in whole blood DNA from 718 individuals, aged 25–92 years (mean = 55). We sequenced the methyl-CpG-enriched genomic DNA fraction, averaging 67.3 million reads per subject, to obtain methylation measurements for the ∼27 million autosomal CpGs in the human genome. Following extensive quality control, we adaptively combined methylation measures for neighboring, highly-correlated CpGs into 4 344 016 CpG blocks with which we performed association testing. Eleven age-associated differentially methylated regions (DMRs) passed Bonferroni correction (P-value < 1.15 × 10−8). Top findings replicated in an independent sample set of 558 subjects using pyrosequencing of bisulfite-converted DNA (min P-value < 10−30). To examine biological themes, we selected 70 DMRs with false discovery rate of <0.1. Of these, 42 showed hypomethylation and 28 showed hypermethylation with age. Hypermethylated DMRs were more likely to overlap with CpG islands and shores. Hypomethylated DMRs were more likely to be in regions associated with polycomb/regulatory proteins (e.g. EZH2) or histone modifications H3K27ac, H3K4m1, H3K4m2, H3K4m3 and H3K9ac. Among genes implicated by the top DMRs were protocadherins, homeobox genes, MAPKs and ryanodine receptors. Several of our DMRs are at genes with potential relevance for age-related disease. This study successfully demonstrates the application of next-generation sequencing to MWAS, by interrogating a large proportion of the methylome and returning potentially novel age DMRs, in addition to replicating several loci implicated in previous studies using microarrays.
INTRODUCTION
Human populations are living longer now than at any time in history, with world life expectancy more than doubling in the last two centuries (1). While mortality has been delayed, aging is still accompanied by significantly elevated risk for many diseases (2–4). With an increasingly long-lived population, there is a strong impetus to understand the biology of the aging processes and reduce age-related illness.
The effects of aging on the genome include telomere attrition and accumulation of mutations (5). In addition, epigenetic factors are increasingly being recognized as centrally important to the aging process (6). Among epigenetic modifications, the most intensively studied is the methylation of cytosine residues at the carbon 5 position—5mC (7). Reported age-related changes to DNA methylation have typically included a general loss of methylation, or hypomethylation, across the genome (6,8,9), accompanied by site-specific hypermethylation at several gene promoters (10,11).
The successful identification of differentially methylated regions (DMRs) associated with aging in studies of candidate loci (12–14) has stimulated interest in mapping all such sites genome-wide. As a step in this direction, several recent studies have used the Illumina Infinium 27 K microarray to investigate age-related methylation effects in fibroblasts (15), cultured human cells (16), buccal DNA (17), whole blood (18–21) and concordance of age-related change across multiple tissue types (22). To date, two studies on aging have also employed the higher density Infinium 450 K array (23,24). While these arrays enable the detection of methylation at ∼27 000 or 450 000 CpG sites, there are >28 million CpG sites in the human genome (25). Therefore, these array studies surveyed only a small fraction of the potentially methylated portion of the genome. Furthermore, much of the methylation relevant for individual differences occurs outside gene promoters and CpG islands (26).
As an alternative to arrays, next-generation sequencing (NGS) technology (27) can be applied to the study of methylation (28). The most comprehensive method for ascertaining 5mC status is bisulfite sequencing, where unmethylated cytosines are converted to uracil (29). This single-base resolution is attractive, but whole-genome bisulfite sequencing (WGBS) is not yet economically feasible in the large sample sizes required for adequate statistical power in association studies (28). For example, Heyn et al. (24) recently used WGBS to compare genome-wide methylation in peripheral blood DNA from a newborn infant and a centenarian but used Infinium 450 K arrays to confirm DMRs in additional individuals of intermediate ages.
Alternative NGS methods have been developed to perform genome-wide methylation analyses that are more cost-effective than WGBS (28). One such method is affinity-based capture. In two common variants of this approach, the methylated genomic fraction is captured using methyl-CpG binding domain (MBD) protein (30) or antibodies specific to 5mC (31), followed by sequencing on an NGS instrument. The number of fragments mapping to a locus approximates the level of methylation. While lacking single-base resolution, both MBD and antibody-based capture methods are capable of detecting DMRs (32) and have good genome-wide representation of the methylome (33). One previous study has used antibody-based capture and NGS to discover aging DMRs in blood DNA from a sample of 38 individuals (34).
Here we present a large-scale methylome-wide association study (MWAS) of aging in whole blood DNA from >700 individuals using MBD-based capture and sequencing (MBD-seq). Our use of NGS affords a more comprehensive view of aging DMRs across the genome compared with previous studies using microarrays. Furthermore, our sample size provides substantially more statistical power than any previous NGS study of DNA methylation and aging. With this approach, we identify multiple aging DMRs in DNA from blood that may provide insight into aging epigenetics in this tissue.
RESULTS
Primary MWAS with age
Our MWAS sequencing pipeline (35) and computational analysis methods (36,37) have been described previously, and the work flow for this project is summarized in Figure 1. After quality control (QC), our final study sample consisted of 718 subjects with, on average, 31.6 million reads per subject (SD = 13.4 million). These subjects had an average age of 55.2 years (SD = 11.8), ranging from 25 to 92 years. Additional demographic and sample information is provided in Supplementary Material, Table S1. Sequence read positional data and phenotype information have been submitted to dbGaP (http://www.ncbi.nlm.nih.gov/gap) for public release.
Figure 1.

Flow diagram of data processing and quality control (QC). MBD-seq, methyl-CpG binding domain (MBD) protein-enriched sequencing; PCA, principal component analysis; MWAS, methylome-wide association study.
Our use of MBD protein, which only binds to methylated CpGs (30), led us to focus on the ∼27 million autosomal CpG sites in the human genome. Methylation measurements were obtained for each CpG by estimating the number of sequenced fragments covering it (37). After QC and adaptively combining correlated coverage estimates at neighboring CpG sites to reduce the size of the data set (38), we obtained 4 344 016 CpG ‘blocks’. Each block comprised, on average, 3.1 highly-correlated CpGs and had a mean length of 73.7 bp (see Supplementary Material, Table S2).
We used the quantitative coverage estimates for the ∼4.3 million blocks as input for multiple regression analyses to test for association with age. We accounted for sex in the regression model, in addition to multiple technical confounders and five principal components (PCs) that captured unmeasured confounders. The quantile–quantile-plot for this analysis is shown in Figure 2. This plot indicates an enrichment of small P-values as compared with the expectation under the null. Eleven DMRs passed the conservative Bonferroni correction (P-value < 1.15 × 10−8). Alternatively, there were 70 aging DMRs with q-values of <0.1. As motivated previously, this false discovery rate (FDR) threshold provides a good balance between discovery of true effects and controlling for false positives (39) and is relatively robust to the effects of correlated tests (40–44). In our MWAS, q < 0.1 corresponds to P-value of <1.37 × 10−6. For concision, Table 1 shows details on the 11 aging DMRs passing Bonferroni correction only, whereas Figure 3 shows a circular plot of the genome-wide distribution of the top 70 DMRs. Figure 3 also integrates results from further analyses outlined later. Genomic co-ordinates and analysis statistics for all 70 DMRs are provided in Supplementary Material, Table S3. Overall, 42 DMRs showed decreased (hypo) methylation with age, whereas the remaining 28 showed hypermethylation.
Figure 2.
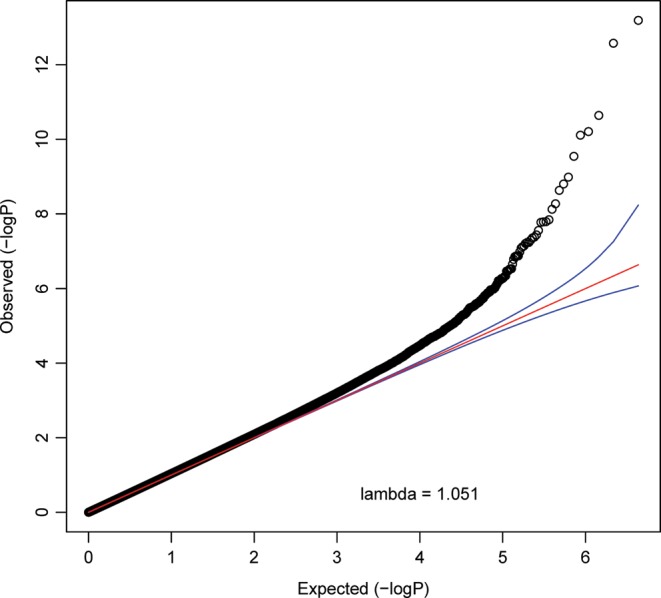
Quantile–quantile (QQ) plot of observed CpG block association P-values (y-axis) against expected P-values (x-axis). The negative logarithm in base 10 of the association P-value is plotted. The red line depicts the expectation under the null hypothesis. The deviation of many points above the 95% confidence interval (blue lines) in the right upper corner points to a considerable number of significant findings.
Table 1.
Aging DMRs with MWAS P-values passing Bonferroni correction
| chr | Start | End | CpGs | T-value | P-value | q-value | Gene name |
|---|---|---|---|---|---|---|---|
| 8 | 50937289 | 50937521 | 8 | −7.65 | 6.47E-14 | 3.28E-07 | SNTG1 |
| 3 | 115854624 | 115854638 | 3 | −7.45 | 2.66E-13 | 6.74E-07 | LSAMP |
| 2 | 145278477 | 145278615 | 5 | −6.79 | 2.29E-11 | 3.87E-05 | ZEB2 |
| 12 | 21928555 | 21928715 | 6 | −6.64 | 6.25E-11 | 7.88E-05 | KCNJ8 |
| 5 | 166514632 | 166514727 | 5 | −6.61 | 7.76E-11 | 7.88E-05 | 0 |
| 2 | 66654412 | 66654571 | 6 | −6.40 | 2.87E-10 | 0.0002 | MEIS1 |
| 2 | 49810159 | 49810380 | 5 | −6.19 | 1.04E-09 | 0.0008 | 0 |
| 20 | 41343306 | 41343427 | 4 | −6.12 | 1.58E-09 | 0.0010 | PTPRT |
| 1 | 24718513 | 24718583 | 5 | −6.05 | 2.38E-09 | 0.0013 | C1orf201 |
| 3 | 71478830 | 71478943 | 5 | −5.91 | 5.38E-09 | 0.0027 | FOXP1 |
| 10 | 103527522 | 103527659 | 4 | −5.85 | 7.57E-09 | 0.0035 | FGF8/NPM3/MGEA5 |
Chromosome (‘chr’), ‘start’ and ‘end’ positions for each CpG block are given, in addition to the number of ‘CpGs’ in the block. Also shown are the signed test statistic values for the regression (‘T-value’), ‘P-values’ and ‘q-values’. ‘Gene name’ indicates genes within 10 kb (plus or minus) of the block.
Figure 3.
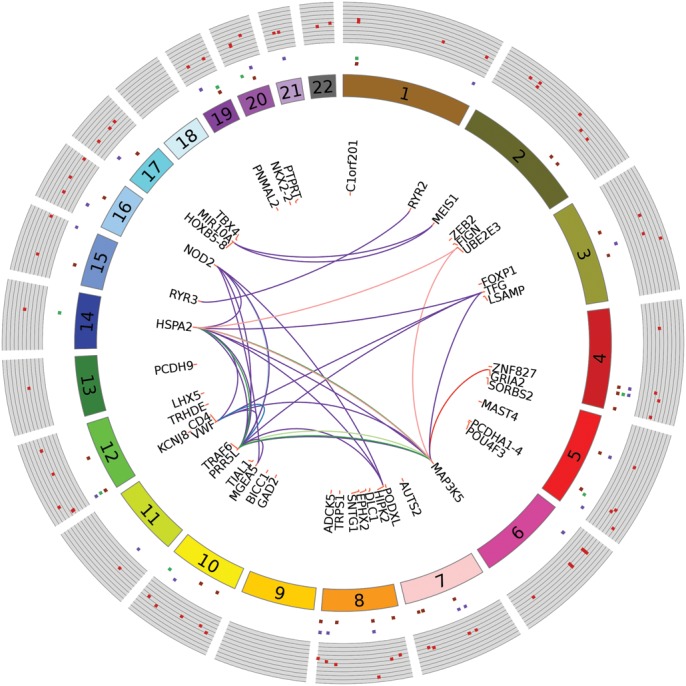
Circular plot with MWAS results and pathways/networks. Each dark-red spot in outermost track with gray background is −log10(MWAS P-value). Genomic features associated with these sites are shown by repeats (dark-purple), exons (green) and introns (orange), respectively, in the concentric circular tracks between the P-values and chromosome number. Of the 68 unique genes associated (±20 kb flaking region) with the top 70 DMRs (q-value < 0.1), the most relevant 45 genes for pathways and networks are shown. Links between genes indicate co-occurrence in pathways and protein–protein interactions networks (Table 3). Lines connecting genes in deep-purple represent protein–protein interactions. Genes co-occurring in reference pathways are linked as follows: c-Jun kinases (light blue), Downstream TCR signaling (deep blue), p38 MAPK signaling pathway (light green), MAPK signaling pathway (green), protein processing in endoplasmic reticulum (pink) and amyotrophic lateral sclerosis, ALS (red).
MWAS results and overlap with genomic features
For each genomic feature of interest, we examined enrichment among our top results compared with the genome-wide average, for all top 70 DMRs in addition to hyper- and hypo-methylated DMRs separately. Results are shown in Figure 4A and B, with significant (P < 0.05) deviations from the genome-wide average indicated by solid dots. Statistics and P-values for all tests are provided in Supplementary Material, Table S4.
Figure 4.

Bioinformatics plot of genomic features for the top sites. The horizontal axis shows the proportion of features (vertical axis) overlapping with the top sites (q-values < 0.1). Three groups of points are plotted: red circles corresponding to all 70 top DMRs, blue squares for hypermethylated age-DMRs and green triangles for hypomethylated age-DMRs. Where enrichment compared with the genome-wide average is significant (P < 0.05), points are shown as solid color blocks. In panel A, ‘Exon’, ‘Intron’ and ‘Gene’ designate overlap with RefSeq genes; ‘CGI’ denotes overlap with a CpG island; ‘Shore’ is ±2 kb flanking a CGI; ‘Upstream 2 kb/8kb’ indicates within 2 or 8 kb upstream of transcription start site; ‘DNase cluster’ indicates a genomic region hypersensitive to DNaseI; ‘Conserv’ indicates regions of high conservation across eutherian mammals; ‘TFBS’ indicates conserved motifs for transcription factor-binding sites in humans and rodents. In panel B, ‘CTCF’ is CCCTC-binding factor protein; ‘EZH2’ is enhancer of zeste homolog 2, a histone-lysine N-methyltransferase; ‘H2AZ’ is H2A histone family, member Z, whereas the remaining categories indicate histone modifications described using standard nomenclature.
Considering the first ten annotations for all DMRs (Fig. 4A), all but ‘Genes’ and ‘Introns’ were significantly enriched. Thus, we observe that our top findings are more likely to be in CGIs or CpG shores, situated upstream from the start of transcription in regions that are more likely to be evolutionarily conserved among eutherian mammals and that are enriched for predicted transcription factor binding sites. The most striking finding was with CGIs, which were enriched 20-fold among our top results compared with the genome-wide average (P = 7.71 × 10−10).
Considering hyper- and hypo-methylated DMRs separately, once again all but ‘Genes’ and ‘Introns’ were significantly enriched for both directions of effect, except for ‘Exons’ in the hypomethylated category that was also non-significant. Notably, among hypomethylated DMRs, none overlapped with a CGI. Thus, all significant DMRs in CGIs were hypermethylated with age. The enrichment of CGIs in the hypermethylated DMRs, relative to the genome-wide average, was over 50-fold (P = 1.04 × 10−13). Further comparison of hyper- and hypo-methylated DMRs indicated that hypermethylated DMRs tend to be more enriched for exons and upstream regions close to the start of transcription, whereas hypomethylated DMRs tended to be enriched for DNase clusters. The latter observation is noteworthy because the approximately fourfold enrichment is from a relatively high genome-wide baseline. That is, 15.4% of all blocks overlap with a DNase cluster, which increases to 66.7% among the hypomethylated DMRs. This enrichment was also highly significant (P = 9.16 × 10−14).
We next looked for overlap with binding sites for regulatory proteins and genomic regions associated with specific histone modifications. The results are shown in Figure 4B. In this analysis, hypermethylated DMRs were not significantly enriched for any of the features we studied. Therefore, all of the significant findings in this analysis are driven by the hypomethylated DMRs. These were significantly enriched for transcriptional repressor CTCF, histone-lysine N-methyltransferase EZH2 and histone 2A family, member Z. Hypomethylated DMRs were also significantly enriched for histone 3, lysine 27 acetylation (H3K27ac), histone 3, lysine 4 methylation, dimethylation and trimethylation (H3K4m1, m2 and m3) and histone 3, lysine 9 acetylation (H3K9ac).
Replication of top findings
We aimed to replicate five findings using pyrosequencing of bisulfite converted whole blood DNA from independent subjects. This targeted method has excellent quantitative accuracy for methylation studies (45). From the top five findings in genes (see Table 1), we were able to design good pyrosequencing assays for three DMRs, located close (within 20 kb) to the genes LSAMP, (limbic system-associated membrane protein), ZEB2 (zinc finger E-box binding homeobox 2) and MEIS1 (Meis homeobox 1). As all of these DMRs showed hypomethylation with age, we also selected the most significant hypermethylated DMR in a gene. This was in GRIA2 (glutamate receptor, ionotropic, AMPA 2), which passed FDR of <0.1 and had a MWAS P-value of 2.7 × 10−8 (see Supplementary Material, Table S3). Finally, we included a ‘negative control’ using a block that showed no association with age in the MWAS. This was located proximal to the gene FLI1 (Friend leukemia virus integration 1) and had a MWAS P-value of 0.76. Replication was carried out in an independent sample of 558 individuals from the same Swedish population as our main sample and had a similar age profile (mean = 57.4, SD = 10.9) (see Supplementary Material, Table S5 for additional sample information). Results are shown in Table 2. All of the hypomethylated DMRs replicated with several P-values in the range 10−20–10−30, whereas GRIA2 also replicated, albeit at a more modest level of significance. All effect directions in the replication sample were in the same direction as in the MWAS. Finally, the ‘negative control’ showed no association as expected.
Table 2.
Replication of selected top sites
| Gene | Chr | Position (bp) | n | Beta | T-value | P-value | Pearson r |
|---|---|---|---|---|---|---|---|
| LSAMP | 3 | 115854638 | 528 | −0.65 | −12.49 | 1.74E-31 | −0.47 |
| 115854632 | 514 | −0.50 | −12.71 | 2.72E-32 | −0.47 | ||
| 115854624 | 500 | −0.44 | −11.04 | 1.75E-25 | −0.43 | ||
| ZEB2 | 2 | 145278477 | 511 | −0.66 | −10.38 | 5.54E-23 | −0.41 |
| 145278485 | 511 | −0.59 | −10.54 | 1.32E-23 | −0.42 | ||
| 145278509 | 493 | −0.47 | −6.13 | 1.81E-09 | 0.27 | ||
| MEIS1 | 2 | 66654495 | 513 | −0.52 | −10.55 | 1.22E-23 | −0.41 |
| 66654515 | 506 | −0.47 | −10.87 | 7.96E-25 | −0.42 | ||
| 66654518 | 508 | −0.37 | −8.71 | 4.41E-17 | −0.35 | ||
| 66654531 | 415 | −0.47 | −9.26 | 1.18E-18 | −0.39 | ||
| GRIA2 | 4 | 158142838 | 539 | 0.26 | 2.37 | 1.81E-02 | 0.10 |
| 158142841 | 540 | 0.37 | 3.86 | 1.25E-04 | 0.17 | ||
| 158142863 | 455 | 0.35 | 2.22 | 2.67E-02 | 0.05 | ||
| Negative control | |||||||
| FLI1 | 11 | 128649311 | 121 | −0.02 | −0.17 | 0.87 | −0.01 |
| 128649306 | 121 | −0.06 | −0.58 | 0.56 | −0.04 | ||
| 128649270 | 121 | −0.01 | −0.09 | 0.93 | 0.01 | ||
Each block associated in the MWAS is comprised of several highly-correlated CpGs. In the replication study, which uses bisulfite sequencing, we have single-base resolution and so can interrogate the methylation levels for each individual CpG. Therefore, in the table, the position and association statistics for each individual CpG are shown. ‘Gene’, gene in which DMR is located; ‘Chr’, chromosome; ‘Position’, co-ordinate on hg19; ‘n’, number of samples, out of 558 total, with methylation measurements for that CpG; ‘Beta’, regression coefficient; ‘T-value’, test statistic value.
Pathways
The top 70 DMRs are located within 20 kb of 68 unique genes. To test for functional relationships between these genes, we looked for their co-occurrence within consensus reference pathways and protein–protein interaction networks (46). We also tested for co-regulation by specific microRNAs (47). The results of these analyses are shown in Table 3. We observed significant enrichment (q < 0.1) in five protein–protein interaction networks and six reference pathways. However, no microRNA was significant after controlling the FDR at the 0.1 level.
Table 3.
Network and pathway analysis
| Networks | |||||
|---|---|---|---|---|---|
| P-value | q-value | Network center | Input overlap | Size | Enrichment |
| 0.0002 | 0.047 | PBX3 (ID:5090) | HOXB8 (−); MEIS1 (−) | 7 | 2 (28.6%) |
| 0.0003 | 0.047 | RYR3 (ID:6293) | RYR3 (−); RYR2 (+) | 8 | 2 (25.0%) |
| 0.0006 | 0.083 | CYLD (ID:1540) | MGEA5 (−); TRAF6 (−); NOD2 (−) | 54 | 3 (5.5%) |
| 0.001 | 0.087 | MAP3K3 (ID: 4215) | HSPA2(+); VWF (−); TFG (−); TRAF6 (−); MAP3K5 (−) | 250 | 5 (2.0%) |
| 0.001 | 0.087 | TAB2 (ID:23118) | HSPA2 (+); HIPK2 (+); TRAF6 (−); NOD2 (−) | 149 | 4 (2.7%) |
| Pathways | |||||
| P-value | q-value | Pathway | Input overlap | Size | Enrichment |
| 0.0006 | 0.022 | JNK (c-Jun kinases) phosphorylation | TRAF6 (−); NOD2 (−) | 16 | 2 (12.5%) |
| 0.002 | 0.022 | Downstream TCR signaling | CD4 (−); TRAF6 (−) | 29 | 2 (7.1%) |
| 0.002 | 0.022 | p38 MAPK signaling | TRAF6 (−); MAP3K5 (−) | 29 | 2 (6.9%) |
| 0.003 | 0.027 | MAPK signaling pathway | HSPA2 (+); FGF8 (−); TRAF6 (−); MAP3K5 (−) | 268 | 4 (1.5%) |
| 0.007 | 0.034 | Protein processing in endoplasmic reticulum | HSPA2 (+); UBE2E3; MAP3K5 (−) | 166 | 3 (1.8%) |
| 0.007 | 0.034 | Amyotrophic lateral sclerosis (ALS) | GRIA2 (+); MAP3K5 (−) | 53 | 2 (3.8%) |
Reference network and pathway data were obtained from ConsensusPathDB, a meta-database of 30 different public sources of interaction data. The top 70 DMRs implicated 68 unique genes (±20 kb). Where an identical gene combination indicated two or more similar pathways, only the most significant is shown. The direction of age-related methylation association is indicated by the ‘+’ (hypermethylation) or ‘−’ (hypomethylation) in parentheses following the gene name.
The top network result centered on the PBX3 (pre-B-cell leukemia homeobox 3) protein and involved the developmental genes HOXB8 (homeobox B8) and MEIS1 (Meis homeobox 1). The second network centered on RYR3 (ryanodine receptor R3), and our results included the gene encoding this protein, plus the gene encoding ryanodine receptor 2. Other significant networks centered on CYLD [cylindromatosis (turban tumor syndrome)], MAP3K3 (mitogen-activated protein kinase kinase kinase 3) and TAB2 (TGF-beta-activated kinase 1/MAP3K7 binding protein 2).
MAPK-related results were also observed for reference pathways. In addition to a generic large pathway (MAPK signaling), a smaller and more specific MAPK sub-pathway (p38 MAPK signaling) was implicated. The most significant reference pathway overall was ‘JNK, (c-Jun kinases) phosphorylation by TAK1’. JNK refers to c-Jun N-terminal kinases, which also belong to the mitogen-activated protein kinase family and are activated by TAK1 (TGF-beta activated kinase 1, encoded by the gene MAP3K7) (48). Note that TAK1-interacting protein 2 (TAB2) was among the top network findings. Other pathways included ‘Downstream TCR signaling’, which involves gene expression following T-cell receptor activation, ‘protein processing in the endoplasmic reticulum’, and genes associated with ‘Amyotropic Lateral Sclerosis (ALS)’ etiology.
Sex differences
In our main analysis, sex was regressed out. To investigate sex differences, we carried out an MWAS to detect sites where the age-related methylation association differed most between the sexes. Results are shown in Supplementary Material, Table S7. No sites passed our FDR threshold for declaring significance (q < 0.1). One explanation is that power is reduced compared with the main analysis. We also performed the association analysis with age for each sex separately (see Supplementary Material, Tables S8 and S9). These analyses show somewhat more pronounced age-related methylation changes in males.
DISCUSSION
Our study successfully identified several DMRs associated with aging in blood DNA. Strengths of the study included a wide age range in the study population and a much larger sample size compared with previous NGS studies. Our use of NGS also results in an improvement in the number of sites interrogated over arrays. A limitation of the study was that we examined one tissue only. Tissues differ in methylation levels at many loci, so investigation of blood can only provide information on a subset of all possible methylation changes that occur with aging. Also, the DNA from whole blood is almost completely sourced from leucocytes. While several lineages exist, these all function within the immune system, so our observations may be limited to normal aging processes within this system. However, for the sample sets described, blood was the only DNA source available to us. We also did not examine the sex chromosomes, because methylation measurements on the sex chromosomes will be confounded by the mechanisms of X-inactivation in females and hemizygosity in males. These sex-specific effects require alternative analysis methods that are outside the scope of the current manuscript.
Results, summary and comparison with literature
Previous studies of blood DNA methylation and aging that used Illumina Infinium arrays found predominantly hypermethylation with age (19,20). In the current study, we found mostly hypomethylated sites. An explanation for this discrepancy is that the Infinium array focuses on gene promoters and CGIs. Among our top 70 results, all DMRs located in CGIs showed hypermethylation, indicating that our results are compatible if we focus on similar sites. The difference is that we interrogated many more sites, and many of these sites showed hypomethylation.
Our observation of purely age-related hypermethylation at CGIs, although striking, is by no means novel. Many previous studies had the ability to interrogate these features (19,20) and observed similar patterns. On the other hand, hypomethylated aging DMRs have been less well characterized. Based on our findings, this is probably because they tend to lie outside CGIs and the regions targeted by arrays. In our analysis, hypomethylated DMRs were enriched with binding sites for regulatory proteins or specific histone modifications. These regulatory proteins included transcriptional repressor CTCF, polycomb protein EZH2 and histone 2A member Z. A previous study by Koch et al., using pluripotent stem cells, linked regions of senescence-associated hypomethylation to binding sites for polycomb proteins (49). While we could not access data for all of the polycomb proteins they investigated, both our study and theirs observed focal hypomethylation at EZH2 binding sites. The molecular parallel between senescence in cell culture and aging has already been posited (20). However, the extent to which this observed overlap between different biological samples is indicative of a general aging mechanism will require additional work to verify experimentally.
Considering specific histone modifications, we observed enrichment for H3K27ac, H3K4m1, H3K4m2, H3K4m3 and H3K9ac at hypomethylated DMRs. These marks are typically associated with active chromatin (50), an observation consistent with our hypomethylated DMRs also being enriched for DNase clusters. These indicate open or active chromatin sensitive to digestion by DNaseI. Some of these marks have been implicated in aging or age-related outcomes. For example, levels of trimethylated lysine 4 of histone H3 (H3K4me3) have been associated with longevity in model organisms (51). While the links between these histone marks and aging remain to be fully elucidated, our results indicate that hypomethylated aging DMRs may be of functional relevance.
Consistency of specific findings with previous studies
The most similar study to ours, in terms of approach, was that by Salpea et al. (34), who used meDIP coupled with NGS to detect aging DMRs in blood DNA from a sample of 38 individuals. Their study identified seven main loci of interest, none of which showed precise positional overlap with our top 70 DMRs. Incomplete statistical power is the most likely explanation for this difference, although technical differences in the capture methods could also be relevant. However, one of their findings was with the PCDHG (protocadherin gamma) cluster, and we did observe DMRs at the related PCDHA (protocadherin alpha) cluster and PCDH9. Notably, the array studies by Bell et al. (19) and Rakyan et al. (20) also identified the PCDHA region among their significant aging DMRs, whereas several protocadherin genes were implicated by Hannum et al. (23). Therefore, convergent evidence from multiple studies of blood DNA indicates that protocadherins are subject to differential methylation with age. Protocadherins contribute to neural circuit development and provide individual cells with their specific identity (52). Methylation has been shown to regulate expression of the different isoforms encoded by the cluster (53). Protocadherins have been shown to interact with c-Jun N-terminal kinases (JNKs) and frizzled 7 in model organisms (54). The former was the top protein–protein interaction network finding in our results, and the latter was among the top aging loci identified by Salpea et al. (34).
We looked for overlap with other genes identified by Bell et al. (see Supplementary Material, Table S1) and genes proximal (±20 kb) to DMRs in our study. The overlap consisted of GRIA2 (glutamate receptor, ionotropic, AMPA 2), LAG3 (lymphocyte-activation gene 3) and LHX5 (LIM homeobox 5). GRIA2 was the most significant hypermethylated site in our MWAS and replicated using pyrosequencing in an independent sample. This gene was one of five loci found to be continuously hypermethylated with age in several different tissue types in a study by Koch et al. (22), with a mean correlation of GRIA2 methylation with age across tissues of 0.62. Genes that show consistent methylation effects with age across tissues appear to be in a minority compared with tissue-specific methylation changes, as far as can be ascertained on the basis of current published studies. However, these may provide starting points for future explorations of aging biology. Notably, genetic variation at the related gene GRIA1 was robustly associated with longevity in a genome-wide association study meta-analysis (55). Several other genes identified by Bell et al. were members of gene families represented among our top findings. The overlap with protocadherins was mentioned earlier, but Bell et al. also detected several HOX genes including HOXA9, HOXA13, HOXD11 and HOXD13. Several HOXB family members were implicated in our analysis, whereas another homeobox gene (MEIS1) was among our top findings that passed Bonferroni correction and replicated in an independent sample. Methylation changes at homeobox genes have previously been associated with aging and cellular senescence (16).
A study by Rakyan et al. (20) implicated three loci that overlapped with our findings (see Supplementary Material, Table S3). In addition to the protocadherin alpha cluster, these were MYO3B (myosin IIIB) and MAP3K5 (mitogen-activated protein kinase kinase kinase 5). While no studies have specifically linked the MYO3B gene to aging, levels of myosin heavy chain expression have been reported to drop in older individuals (56). MAP3K5 is a mitogen-activated protein kinase kinase kinase of the JNK and p38 MAPK pathways. It plays vital roles in the cellular response to these stressors, such as the regulation of apoptosis, and has been associated with neurodegenerative diseases (57). Stress-activated MAPK cascades that converge on JNKs play a vital role in the regulation of cellular senescence (58).
Finally, a recent study by Hannum et al. used the Illumina Infinium 450 K array to identify aging DMRs in blood DNA. The authors used their data to create a predictive model of aging and among the DMRs most relevant to their model were several at protocadherin and homeobox genes (see Supplementary Material, Table S3). However, they also examined overlap between aging DMRs and genes showing age-related changes in expression. This is of particular relevance because such an overlap suggests a possible age-related functional effect at these loci. We compared the 68 genes implicated by our top findings with those loci showing both altered methylation and expression in the Hannum et al. study (see Supplementary Material, Table S6). Specific genes that overlapped were EPHX2 (epoxide hydrolase 2), FOXP1 (forkhead box P1), NPM3 (nucleophosmin/nucleoplasmin 3) and PRR5L (proline rich 5 like). These genes have diverse functions and, while none of them have been explicitly linked to aging previously, their combined methylation and expression changes with aging would seem to make them high priority candidates for further characterization in future.
Among the potentially novel methylation findings to emerge from our study were the ryanodine receptors. While these have established links with aging and senescence and may play a role in the pathology of Alzheimer's disease (59), no previous studies have shown methylation changes with age at these genes, to our knowledge. Of potential interest is the central role played by RYR2 in the signaling cascade produced by Resveratrol, a drug that mimics the effects of dietary restriction (60), which is known to increase longevity.
In summary, our study combined large sample size and comprehensive assessment of age-related methylation using NGS to identify multiple aging DMRs. Our results indicate some consistency with aging DMRs reported in previous studies. These associations, while not guaranteeing functional relationships to aging and age-related pathology, provide an interesting starting point from which to consider future functional studies.
MATERIALS AND METHODS
Sample
Our initial sample consisted of 738 subjects from Sweden, average age = 55, and age range from 25 to 92 years. Subjects were controls collected as part of a larger project entitled ‘A Large-Scale Schizophrenia Association Study in Sweden’. This overarching project (61–63) is supported by grants from the National Institute of Mental Health and the Stanley Foundation and aims at understanding the etiology of schizophrenia and bipolar disorder plus their clinical and epidemiological correlates. Peripheral blood was donated at the local medical facilities of the participants. DNA was extracted from EDTA blood using the Gentra Puregene kit for automated extraction with the Autopure LS robot (Qiagen).
MBD-seq
Genomic DNA was first sheared to median fragment size of 125 bp using a Covaris E210. We used the MethylMiner kit from Invitrogen, which employs MBD protein-based enrichment, to capture fragments with one or more methylated CpGs. Dependent on sample availability, we used 2–5 µg (mean = 4.19 µg, SD = 0.99 µg) of DNA starting material and eluted the captured fragments in 500 mm NaCl to increase the relative number of fragments from CpG poor regions (35), which otherwise would not be well covered (64). Methylated fragments were prepared for multiplexed single end (50 bp) sequencing on the SOLiD (Life Technologies) following the manufacturer's standard protocols. Based on observations suggesting that 30–60 million total reads per sample may be sufficient for genome-wide methylation analysis (64,65), we aimed for an average of 60 million reads with a minimum of 40 million. Samples with <40 million reads were rerun to supplement reads.
Alignment and read quality control
The SOLiD system aligns in color space and uses 2-base encoding (66), producing two ‘color calls’ for each base. After deleting poor quality reads (>2 missing color calls), we obtained an average of 67.3 million (SD = 26.9 million) total reads per sample. The mean quality value (QV = −10log10(p) with P being the probability of an error) per color call was 21.4 (SD = 1.1). Reads were aligned to the human genome (build hg19/GRCh37) with BioScope 1.2 (Life Technologies), using a seed-and-extend approach combined with local alignment and multiple schemas. The percentage of mapped reads was 69.2% (SD = 6.2). All runs with <40% alignment or those producing a very small number of reads (<1 million total) were deleted. We also deleted reads with multiple poor quality alignments, whereas high copy number duplicate reads were collapsed to single reads. This led to the elimination of 32.1% of the mapped reads. After all QC, 19 subjects were excluded because they had <15 million reads remaining and one subject was excluded because s/he withdrew consent during the study.
MBD-seq methylation measures
MBD-seq returns an approximation of the amount of methylation at a locus by essentially summing the number of fragments covering each CpG site. However, methylation of any CpG in the entire fragment, not just the sequenced 50 bp, could lead to its capture by MBD protein. Therefore, we estimated the fragment size distribution for each sample from the sequencing data, based on the distribution of reads around isolated CpGs. This non-parametric method has been validated against paired-end libraries where fragment size is known (37). The sample-specific estimated fragment size distribution was then used to calculate the probability for each read that the fragment it is tagging covers the CpG under consideration. Coverage estimates for each of the 26 752 702 autosomal CpGs in the reference genome were then calculated for each subject by taking the sum of the probabilities that all fragments in its neighborhood cover the CpG (37).
To safeguard against errors arising from regions that are problematic to align, we previously performed an in silico alignment experiment, by fragmenting the reference genome and aligning it back to itself using standard alignment software (35). This experiment showed that 36% (10.5 million) of all genome-wide CpGs were in regions difficult to align, with the majority (71.8%) located in regions flagged as repetitive elements by RepeatMasker. These CpGs were dropped from further consideration. To reduce the size of the data set, the remaining ∼16 million CpGs were adaptively combined by collapsing highly inter-correlated coverage estimates at adjacent CpG sites into a single mean coverage estimate (38). This resulted in 5 074 538 CpG ‘blocks’. Supplementary Material, Table S2 provides additional descriptive statistics of the genome-wide block structure. Prior to association testing, we identified 730 522 blocks with very low (<99% of noise) levels of coverage. Eliminating these likely non-methylated sites left 4 344 016 blocks for the MWAS.
MWAS association testing
We used the block data obtained after the data reduction stage as input for multiple regression analyses to test for association with age. In the primary association analysis, we accounted for multiple potential confounders including sex and possible assay-related technical artifacts, such as the quantity of genomic DNA starting material for MethylMiner, quantity of methylation-enriched DNA captured and sample batch. We also performed a principal component analysis (PCA) of the methylation data, using our MethylPCA software (36), to identify unmeasured confounders to be regressed out. Based on a scree test (Supplementary Material, Fig. S1), we selected the first five PCs for this purpose. Such unmeasured confounders include, but are not limited to, environmental factors such as diet, lifestyle or medication use, in addition to biological factors such as cell heterogeneity in the blood samples used to provide genomic DNA. Considering specifically cell heterogeneity, as is true for most tissues, blood is a biological system consisting of a variety of cell types. This can produce many significant findings if (1) the relative abundance of cell types differs systematically with the outcome of interest (in our case, age), plus (2) methylation patterns differ across cell types. As these conditions are known to be true, cell heterogeneity has the potential to cause erroneous findings. However, cell type heterogeneity will be captured by the PCA because it causes differences across many methylation sites (67,68). Previous studies have shown that PCA could accurately distinguish and control for methylation profiles in peripheral blood cells and transformed B-lymphocytes (69) and PCA has been used previously to control for unmeasured confounders in methylation studies (70). Therefore, by regressing out PCs, we therefore control for unmeasured confounders, including for possible cell type heterogeneity.
An analysis of genome-wide SNP data on these subjects (63) suggested that, in this fairly homogeneous sample, ancestry did not substantially contribute to variation in the methylome and therefore was not included as a covariate in the MWAS. Finally, to account for multiple testing, we controlled the FDR (71) at the 0.1 level (39). Operationally, the FDR was controlled using q-values that are FDRs calculated using the P-values of the individual tests as thresholds for declaring significance (72,73).
To test for sex differences in age-related methylation changes, we used a likelihood ratio test with one degree of freedom that compared the explained variance of a multiple regression model that allowed for sex differences through and age–sex interaction term versus a model that only included main effects of age and sex. We also tested for aging effects within members of each sex individually, using the same analysis method as described for the main MWAS.
Bioinformatics of genomic features
Annotations for build hg19 were obtained via UCSC genome browser download. These included: (1) CpG islands (GC content of ≥50% or greater, length > 200 bp, CpG ratio > 0.6), (2) CpG shores defined as 2 kb flanking a CpG island (26), (3) repetitive elements from RepeatMasker (www.repeatmasker.org), (4) within RefSeq gene boundaries, exons and introns, (5) potential gene promoters (2 and 8 kb upstream from most 5′ transcription start site), (6) evolutionarily conserved regions based on sequence homology in 29 eutherian mammals, (7) predicted transcription factor-binding sites based on consensus sequence from human, mouse and rat as provided in the TransFac database version 7.0 and (8) DNase clusters from the University of Washington DNaseI hypersensitivity submission to ENCODE. For polycomb/regulatory proteins and histone modifications, we used ENCODE data for EBV-transformed B-cell line GM12878 (74,75). For each annotation, we calculated the percentage of significantly associated blocks (DMRs) that overlapped with these features and compared this to overlap for all blocks genome-wide, to calculate fold enrichment. Significance was calculated using Fisher's exact test, to account for occasional small cell numbers for rare annotations.
Pathway, network neighborhood and microRNA analysis
We used ConsensusPathDB (http://cpdb.molgen.mpg.de/), a human meta-database of 30 public repositories of biological interaction data hosted by the Max Planck Institute of Molecular Genetics in Berlin, Germany (46), plus data from TargetScan 5.1 for microRNA binding (47). To perform enrichment analysis, a hypergeometric test was performed to calculate the significance of the overlap between the genes from our input list (all genes ±10 kb from DMRs with q-values of <0.1 in the MWAS) and those present in each reference pathway, network or microRNA target list. If more than one gene was implicated by a single DMR, and these appeared in the same pathway, these were collapsed to a single observation and the P-value recalculated.
Replication
For replication purposes, we used pyrosequencing, which allows for targeted sequencing of bisulfite-converted DNA with high quantitative accuracy (45). Genomic DNA was bisulfite converted using Epitect 96 (Qiagen), and reactions were carried out using the PyroMark system from Qiagen according to standard protocols. Supplementary Material, Table S6 provides primer sequences. Standard curves including five DNA samples with known methylation levels (0, 25, 50 75 and 100% methylation, created using methylated (#59665) and unmethylated (#59655) EpiTect Control DNA) were run for each assay and at least two plate controls of known methylation levels were included on each plate. To test for association between age and methylation in the pyrosequencing data, age was regressed on percent methylation at each CpG site. Sex and plate indicator variables were included in the multiple regressions to control for sex differences and batch effects and extreme outlying observations were removed.
SUPPLEMENTARY MATERIAL
FUNDING
This study was supported by the National Institute of Mental Health (NIMH) grant RC2MH089996. It is part of a larger project entitled ‘A Large-Scale Schizophrenia Association Study in Sweden’ that is supported by grants from NIMH (MH077139) and the Stanley Foundation. Institutions involved in this project are: Karolinska Institutet, University of North Carolina at Chapel Hill, Virginia Commonwealth University, Broad Institute and the US National Institute of Mental Health.
Supplementary Material
ACKNOWLEDGEMENTS
MBD-enrichment, library construction and next-generation sequencing were done by EdgeBio, Gaithersburg, MD.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Riley J. Rising Life Expectancy: A Global History. Cambridge: Cambridge University Press; 2001. [Google Scholar]
- 2.Duron E., Hanon O. Hypertension, cognitive decline and dementia. Arch Cardiovasc. Dis. 2008;101:181–189. doi: 10.1016/s1875-2136(08)71801-1. [DOI] [PubMed] [Google Scholar]
- 3.Barzilai N., Huffman D.M., Muzumdar R.H., Bartke A. The critical role of metabolic pathways in aging. Diabetes. 2012;61:1315–1322. doi: 10.2337/db11-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Issa J.P. Epigenetic variation and human disease. J. Nutr. 2002;132:2388S–2392S. doi: 10.1093/jn/132.8.2388S. [DOI] [PubMed] [Google Scholar]
- 5.Sahin E., Depinho R.A. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature. 2010;464:520–528. doi: 10.1038/nature08982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gonzalo S. Epigenetic alterations in aging. J. Appl. Physiol. 2010;109:586–597. doi: 10.1152/japplphysiol.00238.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laird P.W. Principles and challenges of genome-wide DNA methylation analysis. Nat. Rev. Genet. 2010;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- 8.Calvanese V., Lara E., Kahn A., Fraga M.F. The role of epigenetics in aging and age-related diseases. Ageing Res. Rev. 2009;8:268–276. doi: 10.1016/j.arr.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 9.Fuke C., Shimabukuro M., Petronis A., Sugimoto J., Oda T., Miura K., Miyazaki T., Ogura C., Okazaki Y., Jinno Y. Age related changes in 5-methylcytosine content in human peripheral leukocytes and placentas: an HPLC-based study. Ann. Hum. Genet. 2004;68:196–204. doi: 10.1046/j.1529-8817.2004.00081.x. [DOI] [PubMed] [Google Scholar]
- 10.Fraga M.F. Genetic and epigenetic regulation of aging. Curr. Opin. Immunol. 2009;21:446–453. doi: 10.1016/j.coi.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 11.Fraga M.F., Esteller M. Epigenetics and aging: the targets and the marks. Trends Genet. 2007;23:413–418. doi: 10.1016/j.tig.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 12.So K., Tamura G., Honda T., Homma N., Endoh M., Togawa N., Nishizuka S., Motoyama T. Quantitative assessment of RUNX3 methylation in neoplastic and non-neoplastic gastric epithelia using a DNA microarray. Pathol. Int. 2006;56:571–575. doi: 10.1111/j.1440-1827.2006.02010.x. [DOI] [PubMed] [Google Scholar]
- 13.Choi E.K., Uyeno S., Nishida N., Okumoto T., Fujimura S., Aoki Y., Nata M., Sagisaka K., Fukuda Y., Nakao K., et al. Alterations of c-fos gene methylation in the processes of aging and tumorigenesis in human liver. Mutat. Res. 1996;354:123–128. doi: 10.1016/0027-5107(96)00056-5. [DOI] [PubMed] [Google Scholar]
- 14.Bornman D.M., Mathew S., Alsruhe J., Herman J.G., Gabrielson E. Methylation of the E-cadherin gene in bladder neoplasia and in normal urothelial epithelium from elderly individuals. Am. J. Pathol. 2001;159:831–835. doi: 10.1016/S0002-9440(10)61758-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gronniger E., Weber B., Heil O., Peters N., Stab F., Wenck H., Korn B., Winnefeld M., Lyko F. Aging and chronic sun exposure cause distinct epigenetic changes in human skin. PLoS Genet. 2010;6:e1000971. doi: 10.1371/journal.pgen.1000971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bork S., Pfister S., Witt H., Horn P., Korn B., Ho A.D., Wagner W. DNA Methylation pattern changes upon long-term culture and aging of human mesenchymal stromal cells. Aging Cell. 2010;9:54–63. doi: 10.1111/j.1474-9726.2009.00535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bocklandt S., Lin W., Sehl M.E., Sanchez F.J., Sinsheimer J.S., Horvath S., Vilain E. Epigenetic predictor of age. PLoS One. 2011;6:e14821. doi: 10.1371/journal.pone.0014821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teschendorff A.E., Menon U., Gentry-Maharaj A., Ramus S.J., Weisenberger D.J., Shen H., Campan M., Noushmehr H., Bell C.G., Maxwell A.P., et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20:440–446. doi: 10.1101/gr.103606.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bell J.T., Tsai P.C., Yang T.P., Pidsley R., Nisbet J., Glass D., Mangino M., Zhai G., Zhang F., Valdes A., et al. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet. 2012;8:e1002629. doi: 10.1371/journal.pgen.1002629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rakyan V.K., Down T.A., Maslau S., Andrew T., Yang T.P., Beyan H., Whittaker P., McCann O.T., Finer S., Valdes A.M., et al. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;20:434–439. doi: 10.1101/gr.103101.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alisch R.S., Barwick B.G., Chopra P., Myrick L.K., Satten G.A., Conneely K.N., Warren S.T. Age-associated DNA methylation in pediatric populations. Genome Res. 2012;22:623–632. doi: 10.1101/gr.125187.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koch C.M., Wagner W. Epigenetic-aging-signature to determine age in different tissues. Aging. 2011;3:1018–1027. doi: 10.18632/aging.100395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hannum G., Guinney J., Zhao L., Zhang L., Hughes G., Sadda S., Klotzle B., Bibikova M., Fan J.B., Gao Y., et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell. 2013;49:359–367. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heyn H., Li N., Ferreira H.J., Moran S., Pisano D.G., Gomez A., Diez J., Sanchez-Mut J.V., Setien F., Carmona F.J., et al. Distinct DNA methylomes of newborns and centenarians. Proc. Natl. Acad. Sci. USA. 2012;109:10522–10527. doi: 10.1073/pnas.1120658109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bibikova M., Le J., Barnes B., Saedinia-Melnyk S., Zhou L., Shen R., Gunderson K.L. Genome-wide DNA methylation profiling using infinium(R) assay. Epigenomics. 2009;1:177–200. doi: 10.2217/epi.09.14. [DOI] [PubMed] [Google Scholar]
- 26.Irizarry R.A., Ladd-Acosta C., Wen B., Wu Z., Montano C., Onyango P., Cui H., Gabo K., Rongione M., Webster M., et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Metzker M.L. Sequencing technologies—the next generation. Nat. Rev. Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 28.Rakyan V.K., Down T.A., Balding D.J., Beck S. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 2011;12:529–541. doi: 10.1038/nrg3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frommer M., McDonald L.E., Millar D.S., Collis C.M., Watt F., Grigg G.W., Molloy P.L., Paul C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Serre D., Lee B.H., Ting A.H. MBD-isolated genome sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res. 2010;38:391–399. doi: 10.1093/nar/gkp992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mohn F., Weber M., Schubeler D., Roloff T.C. Methylated DNA immunoprecipitation (MeDIP) Methods Mol. Biol. 2009;507:55–64. doi: 10.1007/978-1-59745-522-0_5. [DOI] [PubMed] [Google Scholar]
- 32.Nair S.S., Coolen M.W., Stirzaker C., Song J.Z., Statham A.L., Strbenac D., Robinson M.D., Clark S.J. Comparison of methyl-DNA immunoprecipitation (MeDIP) and methyl-CpG binding domain (MBD) protein capture for genome-wide DNA methylation analysis reveal CpG sequence coverage bias. Epigenetics. 2011;6:34–44. doi: 10.4161/epi.6.1.13313. [DOI] [PubMed] [Google Scholar]
- 33.Harris R.A., Wang T., Coarfa C., Nagarajan R.P., Hong C., Downey S.L., Johnson B.E., Fouse S.D., Delaney A., Zhao Y., et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat. Biotechnol. 2010;28:1097–1105. doi: 10.1038/nbt.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salpea P., Russanova V.R., Hirai T.H., Sourlingas T.G., Sekeri-Pataryas K.E., Romero R., Epstein J., Howard B.H. Postnatal development- and age-related changes in DNA-methylation patterns in the human genome. Nucleic Acids Res. 2012;40:6477–6494. doi: 10.1093/nar/gks312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aberg K.A., McClay J.L., Nerella S., Xie L.Y., Clark S.L., Hudson A.D., Bukszar J., Adkins D., Consortium S.S., Hultman C.M., et al. MBD-seq as a cost-effective approach for methylome-wide association studies: demonstration in 1500 case–control samples. Epigenomics. 2012;4: 605–621. doi: 10.2217/epi.12.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen W., Gao G., Nerella S., Hultman C.M., Magnusson P.K., Sullivan P.F., Aberg K.A., van den Oord E.J. MethylPCA: a toolkit to control for confounders in methylome-wide association studies. BMC Bioinformatics. 2013;14:74. doi: 10.1186/1471-2105-14-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van den Oord E.J., Bukszar J., Rudolf G., Nerella S., McClay J.L., Xie L.Y., Aberg K.A. Estimation of CpG coverage in whole methylome next-generation sequencing studies. BMC Bioinformatics. 2013;14:50. doi: 10.1186/1471-2105-14-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aberg K., Khachane A.N., Rudolf G., Nerella S., Fugman D.A., Tischfield J.A., van den Oord E.J. Methylome-wide comparison of human genomic DNA extracted from whole blood and from EBV-transformed lymphocyte cell lines. Eur. J. Hum. Genet. 2012;20:953–955. doi: 10.1038/ejhg.2012.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van den Oord E.J., Sullivan P.F. False discoveries and models for gene discovery. Trends Genet. 2003;19:537–542. doi: 10.1016/j.tig.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 40.Brown B.W., Russell K. Methods correcting for multiple testing: operating characteristics. Stat. Med. 1997;16:2511–2528. doi: 10.1002/(sici)1097-0258(19971130)16:22<2511::aid-sim693>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 41.Fernando R.L., Nettleton D., Southey B.R., Dekkers J.C., Rothschild M.F., Soller M. Controlling the proportion of false positives in multiple dependent tests. Genetics. 2004;166:611–619. doi: 10.1534/genetics.166.1.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Korn E.L., Troendle J.F., McShane L.M., Simon R. Controlling the number of false discoveries: application to high-dimensional genomic data. J. Stat. Plan. Infer. 2004;124:379–398. [Google Scholar]
- 43.Sabatti C., Service S., Freimer N. False discovery rate in linkage and association genome screens for complex disorders. Genetics. 2003;164:829–833. doi: 10.1093/genetics/164.2.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsai C.A., Hsueh H.M., Chen J.J. Estimation of false discovery rates in multiple testing: application to gene microarray data. Biometrics. 2003;59:1071–1081. doi: 10.1111/j.0006-341x.2003.00123.x. [DOI] [PubMed] [Google Scholar]
- 45.Reed K., Poulin M.L., Yan L., Parissenti A.M. Comparison of bisulfite sequencing PCR with pyrosequencing for measuring differences in DNA methylation. Anal Biochem. 2010;397:96–106. doi: 10.1016/j.ab.2009.10.021. [DOI] [PubMed] [Google Scholar]
- 46.Kamburov A., Pentchev K., Galicka H., Wierling C., Lehrach H., Herwig R. ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res. 2011;39:D712–D717. doi: 10.1093/nar/gkq1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Friedman R.C., Farh K.K., Burge C.B., Bartel D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang C., Deng L., Hong M., Akkaraju G.R., Inoue J., Chen Z.J. TAK1 Is a ubiquitin-dependent kinase of MKK and IKK. Nature. 2001;412:346–351. doi: 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 49.Koch C.M., Reck K., Shao K., Lin Q., Joussen S., Ziegler P., Walenda G., Drescher W., Opalka B., May T., et al. Pluripotent stem cells escape from senescence-associated DNA methylation changes. Genome Res. 2012;23:248–259. doi: 10.1101/gr.141945.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Azuara V., Perry P., Sauer S., Spivakov M., Jorgensen H.F., John R.M., Gouti M., Casanova M., Warnes G., Merkenschlager M., et al. Chromatin signatures of pluripotent cell lines. Nat. Cell Biol. 2006;8:532–538. doi: 10.1038/ncb1403. [DOI] [PubMed] [Google Scholar]
- 51.Han S., Brunet A. Histone methylation makes its mark on longevity. Trends Cell Biol. 2012;22:42–49. doi: 10.1016/j.tcb.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hirayama T., Yagi T. The role and expression of the protocadherin-alpha clusters in the CNS. Curr. Opin. Neurobiol. 2006;16:336–342. doi: 10.1016/j.conb.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 53.Kawaguchi M., Toyama T., Kaneko R., Hirayama T., Kawamura Y., Yagi T. Relationship between DNA methylation states and transcription of individual isoforms encoded by the protocadherin-alpha gene cluster. J. Biol. Chem. 2008;283:12064–12075. doi: 10.1074/jbc.M709648200. [DOI] [PubMed] [Google Scholar]
- 54.Medina A., Swain R.K., Kuerner K.M., Steinbeisser H. Xenopus paraxial protocadherin has signaling functions and is involved in tissue separation. EMBO. J. 2004;23:3249–3258. doi: 10.1038/sj.emboj.7600329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Walter S., Atzmon G., Demerath E.W., Garcia M.E., Kaplan R.C., Kumari M., Lunetta K.L., Milaneschi Y., Tanaka T., Tranah G.J., et al. A genome-wide association study of aging. Neurobiol Aging. 2011;32:2109.e15–2109.e28. doi: 10.1016/j.neurobiolaging.2011.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nair K.S. Aging muscle. Am. J. Clin. Nutr. 2005;81:953–963. doi: 10.1093/ajcn/81.5.953. [DOI] [PubMed] [Google Scholar]
- 57.Takeda K., Noguchi T., Naguro I., Ichijo H. Apoptosis signal-regulating kinase 1 in stress and immune response. Annu. Rev. Pharmacol. Toxicol. 2008;48:199–225. doi: 10.1146/annurev.pharmtox.48.113006.094606. [DOI] [PubMed] [Google Scholar]
- 58.Maruyama J., Naguro I., Takeda K., Ichijo H. Stress-activated MAP kinase cascades in cellular senescence. Curr. Med. Chem. 2009;16:1229–1235. doi: 10.2174/092986709787846613. [DOI] [PubMed] [Google Scholar]
- 59.Mattson M.P. ER calcium and Alzheimer's disease: in a state of flux. Sci. Signal. 2010;3:pe10. doi: 10.1126/scisignal.3114pe10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Park S.J., Ahmad F., Philp A., Baar K., Williams T., Luo H., Ke H., Rehmann H., Taussig R., Brown A.L., et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell. 2012;148:421–433. doi: 10.1016/j.cell.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schizophrenia Psychiatric Genome-Wide Association Study Consortium. Genome-wide association study of schizophrenia identifies five novel loci. Nat. Genet. 2011;43:969–976. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.International Schizophrenia Consortium. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bergen S.E., O'Dushlaine C.T., Ripke S., Lee P.H., Ruderfer D.M., Akterin S., Moran J.L., Chambert K.D., Handsaker R.E., Backlund L., et al. Genome-wide association study in a Swedish population yields support for greater CNV and MHC involvement in schizophrenia compared with bipolar disorder. Mol. Psychiatry. 2012;17:880–886. doi: 10.1038/mp.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bock C., Tomazou E.M., Brinkman A.B., Muller F., Simmer F., Gu H., Jager N., Gnirke A., Stunnenberg H.G., Meissner A. Quantitative comparison of genome-wide DNA methylation mapping technologies. Nat. Biotechnol. 2010;28:1106–1114. doi: 10.1038/nbt.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chavez L., Jozefczuk J., Grimm C., Dietrich J., Timmermann B., Lehrach H., Herwig R., Adjaye J. Computational analysis of genome-wide DNA methylation during the differentiation of human embryonic stem cells along the endodermal lineage. Genome Res. 2010;20:1441–1450. doi: 10.1101/gr.110114.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McKernan K.J., Peckham H.E., Costa G.L., McLaughlin S.F., Fu Y., Tsung E.F., Clouser C.R., Duncan C., Ichikawa J.K., Lee C.C., et al. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res. 2009;19:1527–1541. doi: 10.1101/gr.091868.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu Y., Aryee M.J., Padyukov L., Fallin M.D., Hesselberg E., Runarsson A., Reinius L., Acevedo N., Taub M., Ronninger M., et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat. Biotechnol. 2013;31:142–147. doi: 10.1038/nbt.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Houseman E.A., Accomando W.P., Koestler D.C., Christensen B.C., Marsit C.J., Nelson H.H., Wiencke J.K., Kelsey K.T. DNA Methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sun Y.V., Turner S.T., Smith J.A., Hammond P.I., Lazarus A., Van De Rostyne J.L., Cunningham J.M., Kardia S.L. Comparison of the DNA methylation profiles of human peripheral blood cells and transformed B-lymphocytes. Hum. Genet. 2010;127:651–658. doi: 10.1007/s00439-010-0810-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bell J.T., Pai A.A., Pickrell J.K., Gaffney D.J., Pique-Regi R., Degner J.F., Gilad Y., Pritchard J.K. DNA Methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 2011;12:R10. doi: 10.1186/gb-2011-12-1-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Benjamini Y., Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. B Met. 1995;57:289–300. [Google Scholar]
- 72.Storey J.D. The positive false discovery rate: A Bayesian interpretation and the q-value. Ann. Stat. 2003;31:2013–2035. [Google Scholar]
- 73.Storey J.D., Tibshirani R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ram O., Goren A., Amit I., Shoresh N., Yosef N., Ernst J., Kellis M., Gymrek M., Issner R., Coyne M., et al. Combinatorial patterning of chromatin regulators uncovered by genome-wide location analysis in human cells. Cell. 2011;147:1628–1639. doi: 10.1016/j.cell.2011.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ernst J., Kheradpour P., Mikkelsen T.S., Shoresh N., Ward L.D., Epstein C.B., Zhang X., Wang L., Issner R., Coyne M., et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.