


Abstract
The yeast Snu13p protein and its 15.5K human homolog both bind U4 snRNA and box C/D snoRNAs. They also bind the Rsa1p/NUFIP assembly factor, proposed to scaffold immature snoRNPs and to recruit the Hsp90-R2TP chaperone complex. However, the nature of the Snu13p/15.5K–Rsa1p/NUFIP interaction and its exact role in snoRNP assembly remained to be elucidated. By using biophysical, molecular and imaging approaches, here, we identify residues needed for Snu13p/15.5K–Rsa1p/NUFIP interaction. By NMR structure determination and docking approaches, we built a 3D model of the Snup13p–Rsa1p interface, suggesting that residues R249, R246 and K250 in Rsa1p and E72 and D73 in Snu13p form a network of electrostatic interactions shielded from the solvent by hydrophobic residues from both proteins and that residue W253 of Rsa1p is inserted in a hydrophobic cavity of Snu13p. Individual mutations of residues in yeast demonstrate the functional importance of the predicted interactions for both cell growth and snoRNP formation. Using archaeal box C/D sRNP 3D structures as templates, the association of Snu13p with Rsa1p is predicted to be exclusive of interactions in active snoRNPs. Rsa1p and NUFIP may thus prevent premature activity of pre-snoRNPs, and their removal may be a key step for active snoRNP production.
INTRODUCTION
During the last decades, numerous ribonucleoprotein particles (RNPs) containing small non-coding RNAs and proteins have been discovered in eukaryotes. Several of them share the common property to contain a primary RNA-binding protein which belongs to the L7Ae family of protein (‘L7Ae-like’ proteins) (1–3). They will be denoted here L7Ae RNPs. Members of this L7Ae RNP family play several essential roles in eukaryotic cells: (i) small nucleolar RNPs (box C/D and box H/ACA snoRNPs) are required for generation of mature ribosomal RNAs starting from pre-ribosomal RNA transcripts (maturation and post-transcriptional modifications) (4,5); (ii) the spliceosomal U4 small nuclear RNP (U4 snRNA) is an essential component of the pre-mRNA splicing machinery (6); (iii) small Cajal body-specific RNPs (box C/D and H/ACA scaRNPs) catalyse the post-transcriptional modifications of spliceosomal UsnRNPs (7); (iv) the vertebrate telomerase holoenzyme is needed for telomere synthesis (8,9); and (v) specific RNA–protein complexes which are assembled on the 3′ UTR regions of selenoprotein mRNAs (SECIS mRNPs) are required for selenocysteine incorporation (1,10). In their active forms, these various L7Ae RNPs consist of an RNA molecule containing a recognition motif for the ‘L7Ae-like’ protein and a set of specific proteins. The L7Ae RNPs can be divided into different classes depending on the identity of the ‘L7Ae-like’ protein they contain: vertebrate’s protein 15.5K (Snu13p in yeast) is a component of the U4 snRNP, box C/D snoRNPs and box C/D scaRNPs (3,11,12); vertebrate’s protein NHP2 (Nhp2p in yeast) is found in H/ACA snoRNPs, H/ACA scaRNPs and the telomerase RNP (13,14); finally, vertebrate’s protein SBP2 is specific for selenocysteine mRNPs (1,10).
The 3D structures established for human 15.5K and yeast Snu13p proteins are very similar (15,16), showing a high conservation throughout evolution. These two proteins recognize very similar K-turn motifs characterized by the presence of two helices, I and II, separated by a 3-nt long bulge-loop (3,16). One main feature of the K-turn 3D structure is the sharp angle formed by the backbone of the bulge-loop (16,17). It is generated by stacking of residues at positions 1 and 2 in the bulge on helices I and II, respectively. As a consequence, the U residue at position 3 in the loop, which is highly conserved in K-turn motifs and recognized by proteins of the L7Ae family, is protruding outside of the motif and plays a key role in the RNA–protein interaction (16). The two non-canonical sheared G:A base pairs at the extremity of helix II are also involved in this interaction (3,16,18,19). Binding of 15.5K/Snu13p allows further recruitment of the other specific core proteins: PRP31 for U4 snRNP (20), U3-55K (Rrp9p in yeast) on the U3 B/C motif (18,21), and NOP56, NOP58 and Fibrillarin (Nop1p in yeast) on box C/D snoRNAs as well as box C/D scaRNAs (22,23). The variable elements in K-turn motifs, i.e. length of helix I and sequence of helix II are key determinants for the selective recognition of these specific proteins (18,23,24). The determination of the crystal structure of the human PRP31 fragment encompassing amino acids 78–333 (PRP3178–333) in complex with 15.5K and a fragment of U4 snRNA (25), as well as the crystal structure of archaeal box C/D RNPs (26–28) have provided some clues to understand how diversity is generated on a common theme: namely, how very similar architecture formed by a K-turn motif bound to an ‘L7Ae-like’ protein can interact specifically with different sets of proteins. Although, it is well admitted that the 15.5K/Snu13p primary binding proteins play a key role in initiation of the L7Ae RNP biogenesis, one key remaining question is to know how these RNP complexes are assembled in vivo with a high degree of specificity. Indeed, whereas, assembly of the archaeal counterparts of snoRNPs seems to be mainly based on the capacity of their components to interact together (29–32), recent studies revealed an unexpected complexity of the snoRNP biogenesis pathways in eukaryotes (2,33–39). Eukaryotic RNAs are most generally produced in a precursor form that is subsequently processed and assembled with proteins. Up to now, only the biogenesis of vertebrate’s spliceosomal UsnRNPs is completely deciphered (37). Progresses were also made in the identification of cellular factors which are specifically required for H/ACA snoRNP biogenesis and determination of their roles. These factors interact with the nascent precursors of H/ACA RNAs (40,41). Some of them, like NAF1/Naf1p, function by mimicking and replacing a core protein in the immature particle, rather than as an external scaffold as in the case of the snRNA and the SMN complex (42). The Shq1p assembly factor mimics the RNA-binding surface, to prevent association of the H/ACA proteins with non-specific RNAs (43,44).
As mentioned above, one fascinating remaining question is to understand how by recognition of very similar K-turn motifs and with the assistance of RNP assembly factors, Snu13p/15.5k specifically initiates the assembly of three different kinds of RNPs (box C/D snoRNPs, the U4 snRNP and the B/C protein complex of the U3 snoRNP) (3,18,20–23). We recently found that the vertebrate’s NUFIP protein and the yeast Rsa1p protein (45) play a key role in this process and more generally in assembly of RNPs of the L7Ae family (2,36,46). These platform proteins facilitate formation of a macro-complex containing both the core RNP proteins and the assembly factors. More precisely, NUFIP/Rsa1p is proposed (i) to hold together core proteins in the immature particles; and (ii) to recruit the Hsp90-R2TP chaperone complex (2,39). The yeast and human core R2TP complexes are composed of four proteins: Pih1p (PIH1D1/NOP17 in human), Tah1p (hSpagh also named RPAP3 in human), Rvb1p (TIP49, also known as Pontin or p55 in vertebrates), Rvb2 (TIP48 also known as Reptin or p50 in vertebrates) (2,47). Protein PIH1D1/Pih1p interacts with protein RPAP3/Tah1p within the R2TP complex (2,47,48). By its interaction with NUFIP/Rsa1p, it is proposed that PIH1D1 allows the tethering of the R2TP complex on the pre-RNP (2), whereas by its interaction with Hsp90 (Hsp82 in yeast), protein RPAP3/Tah1p allows the recruitment of the chaperone on the pre-RNP. The two other proteins of the R2TP complex, TIP48 and TIP49 are AAA+ ATPases. They interact together and with the PIH1D1(Pih1p)-RPAP3(Tah1p) heterodimer (2,39,47). The implication of Hsp90 in the biogenesis of L7Ae RNPs was shown by the disappearance of newly synthesized U4, box C/D and telomerase RNAs, as well as of 15.5K, NOP58 and NHP2 proteins, upon inhibition of Hsp90 activity, suggesting that these proteins are clients of the chaperone (2,39). Therefore, as compared to UsnRNP assembly, snoRNP assembly provides a very different paradigm for RNP assembly and is an excellent model to decipher mechanisms ensuring specificity of RNP assembly.
Although, association of NUFIP/Rsa1p to the ‘L7Ae-like’ proteins appears to be important for RNP assembly, NUFIP and Rsa1p show a poor degree of similarity. We previously identified a sequence of 37 amino acids that is highly conserved in the two proteins (hPEP in NUFIP and yPEP in Rsa1p), and showed that these sequences are essential for association of NUFIP and Rsa1p with proteins 15.5K and Snu13p, respectively (2). Here, we used yeast molecular genetics, biochemical assays, cell imaging, NMR spectroscopy and molecular modeling experiments to: (i) identify the interacting amino acids in the NUFIP-–15.5K and Rsa1p–Snu13p complexes; (ii) establish the solution 3D structure of a fragment of the PEP region containing the interacting amino acids and a 3D structure model of its interaction with Snu13p; (iii) determine the respective functional importance of the Rsa1p and Snu13p interacting amino acids. In vivo substitution of Snu13p and Rsa1p interacting amino acids in yeast demonstrated their requirement for cell growth and the stability of box C/D snoRNAs. The structural model that we provide can explain why NUFIP/Rsa1p maintains immature snoRNP particles in an inactive conformation. It suggests that removal of NUFIP/Rsa1p is a key event during snoRNP maturation.
MATERIALS AND METHODS
Recombinant plasmids used in this study
Plasmids were manipulated by standard techniques. Mutations were generated using the QuickChange mutagenesis system according to the manufacturer’s instructions (Stratagene) and appropriate primer pairs.
The bacterial expression vector pGEX-6P-1::SNU13 was already described (19). To express the C-terminal 152 amino acids fragment of Rsa1p in Escherichia coli, a BglII-XhoI restriction fragment was released by digestion of plasmid pACTII::Rsa1p230–381 (previously designated as pACTII::N3C1) (2), and cloned into plasmid pET30c (Novagen). A 111 bp fragment containing the Rsa1p coding sequence of amino acids 230–266 was obtained by PCR amplification using two primers that generated a NheI site at one end and an XhoI site at the other end. To obtain the E. coli expression vector pET28a::RSA1230–266, this fragment was inserted between the NheI and XhoI sites of plasmid pET28a (Novagen).
For protein co-expression in E. coli, DNA fragments encoding the full-length Rsa1p, the Rsa1p230–381 fragment or protein Pih1p were amplified by standard PCR procedures, using primers that generated NdeI and BamHI restriction sites, and they were inserted in the pnCS vector (49–51). On the other hand, the Snu13p and Tah1p ORFs were inserted in the pnEA-3CH vector in order to produce His-tagged proteins.
For expression of WT or mutated Snu13p or Rsa1p protein in yeast, the Saccharomyces cerevisiae SNU13 and RSA1 ORFs were PCR amplified using primers that generated a BamH1 site at the ORF 5′ end and an XhoI site at its 3′ end, and were cloned between the BamH1 and SalI sites of a centromeric version of the pG1 (TRP1) expression vector (52). The resulting plasmids were designated as pG1::SNU13 and pG1::RSA1.
The recombinant plasmids used for Y2H assays were already described (2). Briefly, the ORFs were cloned by the Gateway system (invitrogen), or the hybridized complementary oligonucleotides encoding the PEP region and its variants were cloned in plasmid pACTII downstream from the GAL4 transactivation domain coding sequence, or in plasmid pAS2 downstream from the GAL4 DNA-binding domain coding sequence.
The plasmids coding for mRFP–LacI–NUFIP and GFP–15.5K were generated with the Gateway technology with destination vectors previously described (53).
Production of recombinant proteins
The recombinant pGEX-6P-1::SNU13 plasmid was used to produce WT and mutated GST–Snu13p proteins (19) in the E. coli BL21-CodonPlus strain (Novagen). GST–Snu13p proteins were purified from cell extract under native conditions, using Glutathione Sepharose 4B as recommended by the manufacturer (GE Healthcare). They were cleaved overnight on the beads using the PreScission protease (GE Healthcare). Compared to native Snu13p, the cleaved proteins had two additional G and P residues at their N-terminus. Plasmids pET30c::RSA1230–381 and pET28a::RSA1230–266 were used to produce the Rsa1p230–381 and Rsa1p230–266 fragments with an His6 N-terminal tag, in the E. coli BL21-CodonPlus strain (Novagen). Note that the peptide produced by plasmid pET28a::RSA1230–266 contained a 15 amino acid sequence resulting from cloning into plasmid pET28a which is located between the His6 tag and the Rsa1p PEP N-terminus. The recombinant His6Rsa1p230–266 and the His6Rsa1p230–381 protein were, respectively, purified under native and denaturing/refolding conditions, using a 5 ml His-Trap HP column (GE Healthcare). Proteins were eluted with 0.5 M imidazole at pH 8. When necessary, the His6 tag was removed by cleavage with the thrombin protease. After cleavage, or imidazole elution, proteins were dialysed against buffer D (20 mM HEPES-KOH, pH 7.9; 150 mM KCl; 1.5 mM MgCl2; 0.2 mM EDTA; 10% glycerol) and stored at –80°C.
Test of protein–protein interactions by their co-expression in E. coli
Co-expression tests in E. coli were carried out using the standard procedure previously described (49). The pRARE2 E. coli host cells (Novagen) were co-transformed with plasmids pnEA-3CH and pnCS expressing, His6Snu13p and full-length Rsa1p or its Rsa1p230–381 fragment, respectively. Growth was performed in 500 ml of 2X concentrated LB medium at 37°C to an absorbance at 600 nm of 0.8. Then, the temperature was switched to 20°C and protein co-expression was induced by addition of isopropyl-β-D-thiogalactopyranoside (Euromedex) at a 1 mM final concentration. Cells were further grown overnight at 20°C, collected, resuspended in buffer A (20 mM HEPES-NaOH, pH 7.5; 400 mM NaCl; 5 mM β-mercaptoethanol), and lysed by sonication. The soluble fraction was mixed with 100 µl of Talon resin (Clontech), incubated for 1 h at 4°C , the supernatant was removed and the resin washed extensively with buffer A. Proteins were eluted from the resin using buffer A containing 500 mM imidazole. Eluted proteins were analysed by SDS-PAGE.
Electrophoresis mobility shift assays
Yeast U14 box C/D snoRNA was synthesized by in vitro transcription with T7 RNA polymerase using a PCR fragment carrying the S. cerevisiae snoRNA coding sequence and was 5′ end labeled in conditions previously described (19). Briefly, after transcription, the RNA was dephosphorylated by treatment with the CIP enzyme, and 5′ end labeled with T4 polynucleotide kinase in the presence of [γ-32P]ATP, and purified by gel electrophoresis. For electrophoresis mobility shift assays (EMSA), WT or variant Snu13p were mixed with His6Rsa1p230–266 or His6Rsa1p230–381 fragments each at a 0.5 µM final concentration in buffer D, and incubated for 20 min at 4°C with 5 fmol of 32P-radiolabeled U14 RNA. The RNA–protein complexes formed were resolved by native gel electrophoresis in conditions described previously (30).
CD spectroscopy
Circular dichroïsm (CD) spectroscopy analyses were carried out using a Jobin-Yvon Mark VI circular dichrograph. Quartz spare split-compartment cuvettes with 0.44 cm path-length per compartment were used for binding measurements. The relevant protein solution was placed in one compartment of the cuvette and the target protein solution in the other compartment. CD spectra were recorded before and after mixing the cuvette contents and blanks were run for each interaction and subtracted from the raw data. At least two spectra were collected before and after mixing, an average spectrum was established in order to increase the signal-to-noise ratio. Spectra were recorded from 202 to 260 nm to test protein–protein interaction with a scan speed of 0.2 nm s−1. Proteins were adjusted at a 4 µM final concentration in 10 mM Tris-HCl buffer, pH 7.8; 150 mM NaCl. The results are presented as normalized Δε values. For protein–protein interactions we used amino acid mean residue mass of 110 Da. Taking into account the residue concentration and the optical path-length of the cuvette, the precision of measurements was of δ(Δε) = +/– 0.03 dm3.mol−1.cm−1.
NMR Spectroscopy and structure calculation
For solubility reasons, a synthetic peptide (GeneCust) of Rsa1p (Rsa1p238–259) was used for NMR analyses. The NMR sample of Rsa1p238–259 contained 2 mM of peptide in 10 mM NaPi, pH 3.7; 95% H2O; 5% D2O, 150 mM NaCl. NMR data were acquired at 293 and 305 K on a Bruker Avance III spectrometer equipped with a Z-axis gradients TCI cryoprobe. For proton resonance assignment, TOCSY spectra using two mixing times (30 and 60 ms) and NOESY spectra using three mixing times (75, 150 and 300 ms) were recorded. Assignment of heteronuclei was achieved using 1H–15N HSQC, 1H–13C HSQC and 1H–13C HSQC-TOCSY (with a mixing time of 60 ms) experiments in natural abundance. All spectra were processed with Topspin 3.0 (Bruker BioSpin) and analysed with CARA (54). Chemical shifts were deposited in the Biological Magnetic Resonance Data Bank under the reference 18959.
For peptide structure calculation, torsion angle restraints for the Ψ, ϕ and χ1 angles were derived from PREDITOR analysis (55) and distance restraints were derived from NOESY spectra using the automated procedure of CYANA 2.1 (56). Then, distance and angle restraints were used for water-refinement of 200 structures calculated with CNS 1.21 (57). Finally, the 10 structures with the lowest energies were selected as the most representative structures of Rsa1p238–259.
Docking and molecular modeling
Our strategy was to dock the 10 NMR structures of Rsa1p238–259 with the lowest energies onto the Snu13p X-ray 3D structure (PDB code 2ALE) using the Haddock 2.1/CNS 1.21 protocol (58–60). To this end, we first identified the Snu13p active residues (according to the Haddock nomenclature) (Table 3) by (i) high prediction scores from both WHISCY and PROMATE programs (61,62); (ii) high solvent accessibility; (iii) spatial proximity; and (iv) taking into account residues found to play key roles in the interaction by experimental assays. Snu13p passive residues were solvent accessible residues whose side-chains point toward one or more active residues (Table 3). For Rsa1p238–259, residues on the same side of the helix than the experimentally identified key residues were considered as ‘active residues’ in Haddock (Table 3). The automated mode of Haddock 2.1 was used to define semi-flexible segments of Snu13p, whereas, due to its small size, the entire Rsa1p238–259 fragment was considered as semi-flexible. 4000 structures were calculated during the rigid body energy minimization step (it0) and the top 200 best structures were used for the semi-flexible simulated annealing (it1), and the subsequent flexible explicit H2O refinement steps (it1-water). The top 10 structures with the best haddock score were selected as possible models of the interaction.
Table 3.
List of interacting residues and structural analysis for the 10 best Snu13p-Rsa1p238–259 structures of the Haddock 2.1 results
| Snu13p | Rsa1p238–259 | |
|---|---|---|
| Active residues | P6 K7 F9 L65 L69 E72 D73 | V242 W245 R246 R249 K250 W253 K256 I257 |
| Passive residues | E62 P68 N75 | |
| Semi-flexible segments | Automated mode | T238–N259 |
| Docking results | |
|---|---|
| RMSD from mean structure (Å) | |
| All backbone atoms | 1.01 ± 0.40 |
| All heavy atoms | 1.36 ± 0.41 |
| Energies (kcal/mol) | |
| E.inter | –434 ± 33 |
| E.vdw | –33 ± 8 |
| E.elec | –417 ± 36 |
| Buried surface area (Å2) | 1223 ± 66 |
| Number of AIR violation (>0.3 Å) | 1 ± 0.4 |
Then, the structure with the best haddock score was placed in a water box containing 6074 water molecules and 3 chlorine ions for ensuring the electrostatic neutrality of the system. All Arg, Lys, Asp and Glu side-chains were considered ionic. The resulting system, consisting in a total number of 20 587 atoms, was energy refined. The MD program NAMD (63) was employed in combination with the CHARMM27 force field (64) in order to describe the whole molecular system. Coulomb forces were evaluated with the particle-mesh Ewald method. Chemical bonds between hydrogen and heavy atoms were constrained to their equilibrium value by means of the SHAKE algorithm. The refinement procedure was based on several energy minimization steps carried out by the conjugate gradient method: first, the backbone of the protein complex was fixed such that only the protein side-chains and water molecule movements were variable; next, the whole system was relaxed. Finally, a 5-ns MD simulation was carried out in the isobaric-isothermal ensemble, maintaining the pressure and the temperature at 1.0 atm and 300.0 K, respectively, by means of Langevin dynamics and the Langevin piston approach. The equations of motion were integrated with a 2-fs time step, using the r-RESPA algorithm to update short- and long-range contributions at different frequencies.
Y2H assays
For yeast two-hybrid assays (Y2H), appropriate recombinant pACTII and pAS2 plasmids were used to transform haploid cells (strain CG929 or Y190, and strain Y187, respectively), which were then crossed. Diploids were selected on media suitable for double selection (Leu−, Trp−) and then plated on media suitable for triple selection (Leu−, Trp−, His−). 3-Amino-1,2,4-triazol (3AT) was used to evaluate the strength of the interactions. Growth was assessed after 3 and 5 days of incubation at 30°C.
Tests of the functionality of variant Snu13p and Rsa1p proteins in yeast
Standard S. cerevisiae growth and handling techniques were employed. The S. cerevisiae strain YPH499-GAL::SNU13 (Mata; trp1-Δ63; his3-Δ200; ura3–52; lys2–801; ade2–101; leu2-Δ1; HIS3::GAL1::SNU13) (3) was used to test whether Snu13p variants can complement depletion of the endogenous Snu13p protein. To this end, strain YPH499-GAL::SNU13 was transformed with recombinant pG1::SNU13 plasmids encoding WT or mutated Snu13p. Cells were grown overnight at 30°C in the Trp− selective medium containing galactose as the carbon source. They were collected by centrifugation, washed and diluted in Trp− selective medium containing glucose as the carbon source. Cells were grown at 30°C and constantly maintained in exponential phase by dilution with pre-warmed Trp−/Glc medium. The doubling time was monitored by measuring the absorbance at 600 nm (OD600).
The S. cerevisiae Y190-ΔRSA1 (YPL193W::kanMX2) strain was obtained by homologous recombination. A DNA fragment carrying the kanamycin cassette flanked by sequences amplified from the RSA1 gene was used to transform the haploid Y190 strain. Insertion of the kanamycin cassette at the RSA1 locus was then verified by PCR amplifications. The RSA1 knock-out strain obtained displayed a slow growth phenotype as already observed (45). For functional assays, it was transformed with plasmid pG1::RSA1 encoding WT or variant Rsa1p, and grown in YPD medium.
Evaluation of snoRNA levels in yeast
Total RNAs were extracted from exponentially growing yeast cultures. Northern blot analyses were performed in standard conditions using 5 µg of total RNAs and specific 5′ end 32P radiolabeled antisense probes:
OG-U1 (5′-GACCAAGGAGTTTGCATCAATGA-3′);
OG-snR63 (5′-TTATGTTGGCCACTCATCAC-3′);
OG-snR190 (5′-CGAGGAAAGAAGAGACACCATTATC-3′);
OG-snR45 (5′-CCTCAGATCGCTCCGAGA-3′);
OG-U14 (5′-TCACTCAGACATCCTAG-3′);
OG-snR13 (5′-AGCTTGAGTTTTTCCACACC-3′);
OG-U18 (5′-TGTGATAGTCAACCACATCTC-3′).
His-pull down assays of in vitro assembled RNPs
100 fmol of 5′ end 32P radiolabeled U14 RNA prepared according to (19) were incubated in the presence of yeast tRNAs (2 µM) with various sets of recombinant proteins during 30 min in buffer D at 30°C: 1 µM of protein Snu13p and of the His6Tah1p–Pih1p complex were used, whereas the Rsa1p230–381 fragment was used at a 0.25, 0.5, 1 or 2 µM concentration. The RNP complexes formed were incubated with Nickel Sepharose beads (GE Healthcare) for 15 min at room temperature in 200 µl of binding buffer (10 mM Tris-HCl, pH 8.0; 150 mM NaCl; 0.1% IGEPAL). The beads were washed three times for 10 min with 1 ml of binding buffer. RNAs were extracted by phenol-chloroform treatments, ethanol precipitated and fractionated on a 7% denaturing polyacrylamide gel.
Subcellular localization assay of WT and mutated 15.5K proteins in HeLa cells
The co-recruitment assay was done as previously described (53).
RESULTS
Alignment of the NUFIP and Rsa1p amino acid sequences revealed a low level of sequence similarity, except for their 37 amino acid long PEP regions (Figure 1A). Our previous data (2) had emphasized a major role of these PEP regions for interaction of the NUFIP and Rsa1p platform proteins, with their respective 15.5K and Snu13p partners. Thus, we used various biochemical, biophysical and genetic methods to identify the 15.5K and Snu13p amino acids which interact with hPEP and yPEP, respectively, and also to determine which amino acids of the hPEP and yPEP regions are crucial for these interactions. Based on the high sequence similarity of the 15.5K and Snu13p proteins (Figure 1B), and of the hPEP and yPEP regions, we postulated similar modes of interaction of these two proteins with their PEP partner regions. Therefore, information on these interactions was gained by performing some of the experiments on yeast and other ones in human cells. As in this study we used several Rsa1p sub-fragments (Figure 1A), each of them are identified as Rsa1px–y, where x and y represent the positions in the Rsa1p amino acid sequence of the amino acids at the sub-fragment extremities. For homogenization of the nomenclature, the fragments previously denoted yeast and human PEP regions will be denoted here Rsa1p230–266 and NUFIP223–260, respectively.
Figure 1.

Residues in 15.5K helix α3 are involved in the 15.5K–NUFIP223–260 interaction. (A) Schematic representation of the human protein NUFIP and the yeast protein Rsa1p. The homologous region between the two proteins (named hPEP in human and yPEP in yeast) is colored in gray. The various fragments of NUFIP and Rsa1p used in this study are represented. (B) The RNA-binding domain shared by members of the L7Ae family. Multiple amino acid sequence alignments were constructed using CLUSTALW. Secondary structure elements of human protein 15.5K are labeled on top of the sequences. Numbering corresponds to residues of 15.5K. Identical residues are highlighted in dark gray and similar residues are in light gray. (C) Yeast two-hybrid interactions between 15.5K and NUFIP, NUFIP223–260 or Alix as a negative control. Tests were also performed with NHP2, a core protein of box H/ACA snoRNPs. The following residues of 15.5K were changed into alanines, except when indicated. MutA: H17L18T19K20K21; MutB: K33Q34L35R36; MutC: E61P62E64; MutD: E74D75K76; MutE: R84S85K86; MutF: K107E108K113A114; MutG: Q27Q28; MutH: N31Y32K33; MutI: E124S R125S; MutJ: N40T43N47.
15.5K–NUFIP association depends upon the 15.5K helix α3
To identify the NUFIP interacting domain of 15.5K, the surface residues in 15.5K were mutagenized into alanines. A series of ten 15.5K mutants was generated, and the resulting proteins were tested for association with NUFIP and the NUFIP223–260 segment (hPEP), by the use of Y2H assays. Mutations in 15.5K which had a negative effect on its interaction with the NUFIP223–260 fragment also had a negative effect on the interaction with full-length NUFIP (Figure 1C). However, due its lower stability the NUFIP223–260–15.5K interaction was more drastically affected by mutations in 15.5K than the full-length NUFIP–15.5K interaction. Nine of the tested variants still interacted with NUFIP and NUFIP223–260, whereas one (variant D) had lost these interactions. Variant D carried three amino acid substitutions (E74D75K76/AAA) in a highly conserved sequence of the C-terminal part of helix α3 (Figure 1B). Next, by using the same approach, we tested the importance of each residue in the EDK sequence. The E74/A mutation turned-out to be sufficient to abolish the Y2H interaction with both NUFIP and NUFIP223–260, whereas the D75/A and K76/A mutations had no adverse effects (Figure 1C). Interestingly, while several proteins of the L30 super-family are able to recognize K-turn motifs in RNAs, conservation of an E residue homologous to residue E74 in 15.5K is a specific feature of proteins of the L7Ae family, namely, 15.5K/Snu13p, NHP2/Nhp2p and SBP2 (Figure 1B).
We also tested the impact on interaction of mutations in other parts of the 15.5K helix α3. Residues E64, I65, H68 and L71 were of particular interest, since in the U4 snRNP, E64, I65 and H68 were found to establish contacts with the PRP31 Nop domain (25), and L71 is spatially close to E74 (Figures 1B and 3A). Variants E64/A, I65/A, H68/A and L71/A still interacted with NUFIP, although the interaction was weaker for the L71/A variant (Figure 1C and data not shown). When tested with NUFIP223–260, the E64/A and I65/A 15.5K variants interacted with this small motif, whereas the H68/A and L71/A variants had lost the interaction (Figure 1C), reflecting the generally weaker interactions of NUFIP223–260 compared to full-length NUFIP. The weaker interaction observed for the H68/A mutation might be due to destabilization of the protein structure, not to abolition of a direct contact of 15.5K with NUFIP. Indeed, in the 15.5K 3D structure, residue H68 points toward the center of the protein and establishes an hydrogen bond with residue T43 of helix α2 (Figure 3A) (16) in contrast to residue L71 that is free of interaction. Altogether, two main conclusions were drawn from these data: (i) residue E74 of the 15.5K helix α3 is essential for binding to NUFIP; and (ii) residue L71 likely has some indirect or direct modulator effect on this interaction.
Figure 3.

The E72D73K74/AAA mutation in Snu13p abolishes Snu13p–Rsa1p interaction without major alteration of the protein structure. (A) Comparison of the 15.5K and Snu13p protein structures. 3D structure representations of the complex formed between human protein 15.5K and the 5′ stem-loop of U4 snRNA (PDB accession number: 1E7K) (16), and of the S. cerevisiae Snu13p protein (PDB accession number: 2ALE) (15) were obtained by using the PyMOL software (66). The residues of the C-terminus of helix α3, which are conserved in the two proteins, are indicated by black letters. On the U4-–15.5K structure, residues in pink interact with PRP31 in the U4–15.5K–PRP3178–333 complex (25). Alanine substitutions in the Snu13p variant used in the experiments of this figure are displayed in the insert. (B) The E72D73K74/AAA mutation in Snu13p abolishes Snu13p–Rsa1p interaction in E. coli co-expression assays. The wild type or mutated His-tagged protein Snu13p was co-expressed in E. coli with full-length Rsa1p. His-pull down assays from the crude cellular extracts were performed using IMAC. The soluble fraction (Input = In) and the purified proteins (Eluate = E) were fractionated by SDS-PAGE and revealed by Coomassie blue staining. The molecular weight ladder (L) is shown on the left. (C) Comparative CD analysis of WT Snu13p and its E72D73K74/AAA variant reveals limited effects of the substitution on the protein structure. WT and variant Snu13p were dissolved at a 4 µM concentration in 10 mM Tris-Hcl buffer, pH 7.8, 150 mM NaCl and CD spectra were collected from 200 to 260 nm. (D) The E72D73K74/AAA substitution in Snu13p abolishes its interaction with Rsa1p230–266 as evidenced by CD. A two-compartment cuvette was used and CD spectra were recorded from 205 to 260 nm prior and after mixing the material in the two compartments, as described in ‘Materials and Methods’ section. The CD spectra recorded before mixing the samples are represented in dark blue, and the spectra recorded after mixing the samples are in dark green.
The 15.5K E74D75K76/AAA mutant has an altered intra-cellular localization and does not bind to NUFIP in cellulo
Having shown the functional importance of residue E74 in the E74D75K76 motif of 15.5K for formation of the 15.5K–NUFIP interaction in a heterologous system, we then used a spatial recruitment assay in HeLa cells to test its functional importance in an authentic in cellulo context (Figure 2). The approach used was based on the LacI/LacO system (41,65), which circumvents artifacts occurring during co-immunoprecipitation assays, in particular, the loss of weak interactions or on the contrary the re-association of proteins in the test tube. More precisely, a HeLa cell line containing a tandem array of LacO sites integrated in its genome was transfected with the mRFP1–LacI–NUFIP expression vector. In the U2OS parental cell line devoid of LacO sites, the fusion protein mRFP1–LacI–NUFIP and WT NUFIP both had a diffuse localization in the nucleoplasm and some weak nucleolar staining (Figure 2A). When the GFP–15.5K fusion was co-expressed with mRFP1–LacI–NUFIP in U2OS cells, it localized in the nucleoli, Cajal bodies and nucleoplasm as previously described, and no particular co-localization was observed with mRFP1–LacI–NUFIP. In contrast, in the cell line containing the LacO array, GFP–15.5K co-localized with the bright spots formed by the binding of mRFP1–LacI–NUFIP to the LacO sites. This indicated that, as expected, NUFIP interacts with 15.5K in vivo. We then tested the 15.5K E74D75K76/AAA variant in the same assay (Figure 2B). In parental U2OS cells, the localization of the mutant protein differed from its wild-type counterpart. Consistent with a defect in its incorporation into U4 and box C/D RNPs, the mutant 15.5K accumulated at a lower level in nucleoli compared to WT 15.5K and had a more diffuse localization throughout the nucleoplasm and the cytoplasm. Moreover, in cells containing mRFP1–LacI–NUFIP bound to the LacO array, GFP–15.5K E74D75K76/AAA was unable to accumulate at the LacO sites, indicating that it was defective for NUFIP binding in vivo. Therefore, the data demonstrated the importance of the extremity of helix α3 of 15.5K for in cellulo binding of protein 15.5K to NUFIP.
Figure 2.

15.5K E74A does not bind NUFIP in vivo and is not efficiently incorporated into RNPs. (A) U2OS and U2OS LacO cells were transfected with mRFP–LacI–NUFIP and either GFP–15.5K, (B) or its EDK->AAA variant. WT 15.5K accumulates in nucleoli, in Cajal bodies and is also diffusely present in the nucleoplasm (top left panel). When mRFP1–LacI–NUFIP accumulates at the LacO sites, WT 15.5K is also enriched there (see insets in the top panel). In contrast, while the mutant 15.5K is also present in nucleoli, Cajal bodies and in the nucleoplasm, it is also present in large amount in the cytoplasm, indicating a defect in RNP assembly (bottom-left panel). In addition, this mutant fails to be recruited to the LacO sites by mRFP–LacI–NUFIP (bottom panels).
Then, for further characterization of the 15.5K/Snu13p–NUFIP/Rsa1p interaction, we turned to S. cerevisiae because of our capability to produce soluble recombinant Snu13p and Rsa1p proteins in E. coli, and the possibility to use yeast genetics to investigate the functional importance of the Snu13p–Rsa1p interaction in snoRNP assembly.
Amino acids in the Snu13p E74D75K76 motif are directly involved in the Snu13p–Rsa1p interaction
We hypothesized that, like for 15.5K, some residues in the Snu13p E72D73K74 motif located in the C-terminal part of helix α3 (Figures 1 and 3A) were important for binding to Rsa1p. Having in mind to get a clear demonstration of the formation of a stoichiometric complex between the proteins Snu13p and Rsa1p, we first used co-expression assays in E. coli to test this hypothesis. The His6-tagged WT Snu13p and its His6-tagged E72D73K74/AAA variant were each co-expressed with either full-length Rsa1p or its C-terminal Rsa1p230–381 fragment, and complex formation was investigated by Immobilized Metal Ion Affinity Chromatography (IMAC) (Figure 3B). Fractionation of the IMAC purified proteins by SDS-PAGE revealed the formation of a His6Snu13p–Rsa1p complex. Due to its low level of structuration, Rsa1p was highly sensitive to proteolysis in E. coli, as evidenced by mass spectrometry identification of Rsa1p fragments migrating below full-length Rsa1p in the SDS-PAGE fractionation (Figure 3B, lane 2). However, taking into account these Rsa1p degradation products, we estimated that interaction of Rsa1p and its sub-fragments with Snu13p occurred with an apparent 1:1 molar stoichiometry (Figure 3B, lane 2). Snu13p also co-purified with Rsa1p230–381, but not with the Rsa1p N-terminal domain (1-224) (data not shown). No specific co-purification of Rsa1p was detected with the His6-tagged E72D73K74/AAA Snu13p variant (Figure 3B, lane 4), which revealed the importance of residue(s) within the EDK motif for the Snu13p–Rsa1p interaction. Accordingly, when recombinant GST–Snu13p or its E72D73K74/AAA variant were immobilized on Glutathione Sepharose beads and incubated with the recombinant His6Rsa1p230–266 protein, GST–Snu13p retained significant amounts of His6Rsa1p230–266 protein (Supplementary Figure S1, lane 6), whereas GST alone or the E72D73K74/AAA Snu13p variant did not (Supplementary Figure S1, lane 8).
At this stage of the study, two explanations could be proposed for these data: either mutation of the E72D73K74 motif altered the overall Snu13p conformation and consequently abrogated the interaction with Rsa1p, or the Snu13p E72D73K74 motif was directly involved in the interaction. By providing information on the α-helix content of proteins, far UV CD spectroscopy can reveal conformational changes occurring upon generation of mutations in proteins or upon their interaction with a partner. As Snu13p predominantly folds into α-helices (15), this approach was perfectly adapted to test whether the E72D73K74/AAA mutation modified the Snu13p secondary structure. Consistent with the hypothesis of a physical interaction of the Snu13p E72D73K74 motif with Rsa1p, the CD spectra of WT Snu13p and its E72D73K74/AAA variant recorded from 200 to 260 nm were very similar (Figure 3C). Another argument in favor of a direct interaction was obtained by using a two compartments CD cuvette (Figure 3D): one was filled with Snu13p or its variant and the other one with His6Rsa1p230–266 at a protein to peptide molar ratio of 1. CD spectra were recorded from 205 to 260 nm before and after mixing the contents of the two compartments. When using WT Snu13p, the intensity of the signal at 222 nm was modified after mixing, which was indicative for the formation of a direct interaction between the two partners (Figure 3D, left panel). In contrast, no variation at 222 nm was detected when the Snu13p E72D73K74/AAA variant was used (Figure 3D, right panel), reflecting its inability to interact physically with His6Rsa1p230–266. Therefore, by using biochemical and biophysical approaches, we demonstrated: (i) the formation of a direct Snu13p–Rsa1p interaction at a 1:1 molar ratio; (ii) the direct involvement of some amino acids in the E72D73K74 motif in this interaction. In addition, we developed a CD-based assay to test for the Snu13p–Rsa1p interaction.
Residue E72 in the Snu13p EDK motif plays a key role in the Snu13p–Rsa1p interaction
Then, to delineate which of the three EDK amino acids was/were important for the Snu13p–Rsa1p interaction, we came back to the Y2H approach (Figure 4A). As expected from previous results (2), WT Snu13p interacted with both full-length Rsa1p and its C-terminal half (Rsa1p230–381). No interaction was detected with either the Snu13p E72D73K74/AAA variant or the Snu13p E72/A variant, whereas the D73/A and K74/A mutations had no marked deleterious effects on the interaction (data not shown). Therefore, as found for protein 15.5K, the E72 residue in the Snu13p EDK motif is required for interaction with Rsa1p. We completed these data by performing gel-shift assays with the U14 snoRNA–Snu13p complex (Figure 4B). As previously observed (2), Rsa1p230–381 and Rsa1p230–266 super-shifted the complex formed by Snu13p and an in vitro transcribed U14 box C/D snoRNA (Figure 4B, lanes 3 and 4). In contrast, it did not super-shift this complex, when the Snu13p E72D73K74/AAA or the E72/A variants were used (Figure 4B, lanes 7, 8, 11 and 12). Therefore, this residue is necessary for interaction of both free and snoRNA-bound Snu13p with Rsa1p.
Figure 4.

Residue E72 in Snu13p plays a key role in the Snu13p–Rsa1p interaction. (A) The E72/A Snu13p variant does not interact with Rsa1p in Y2H assays. In the Y2H assays Snu13p or its variants were used as the bait, whereas Rsa1p or its minimal functional fragment Rsa1p230–381 was used as the prey. A control was performed with an empty pACT2 vector (–). Cell were grown on solid glucose medium lacking histidine, in the presence of 3-Amino-1,2,4-triazol (3AT) at a 20, 30, 40 or 50 mM concentration. 3AT is competitive inhibitor of the HIS3 reporter gene product. The presence of colonies is indicative of a protein–protein interaction. (B) The E72/A mutation in Snu13p abolishes the capability of Rsa1p230–381 and Rsa1p230–266 to interact with the Snu13p–U14 snoRNA complex. Radiolabeled U14 snoRNA was incubated with WT Snu13p or one of its α3 helix variants and His-tagged Rsa1p230–381 or Rsa1p230–266 in conditions described in ‘Materials and Methods’ section. GST was used as a negative control. The complexes formed were fractionated by gel electrophoresis. Positions of the Snu13p–U14 RNA (RNP1), Rsa1p230–266–Snu13p–U14 RNA (RNP2) and Rsa1p230–381–Snu13p–U14 RNA (RNP3) complexes, as well as, of the free U14 RNA (U14) are indicated on the left side of the autoradiogram.
As these experiments demonstrated the major role of residue E72 of the Snu13p EDK motif for interaction with Rsa1p, we decided to determine which amino acid(s) in Rsa1p can be the partner(s) of this residue.
Rsa1p conserved residues W245 and R249 are required for binding to Snu13p
Alignment of the human NUFIP223–260 and yeast Rsa1p230–266 amino acid sequences revealed the presence of only one strictly conserved amino acids stretch (W245R246E247E248R249) (Supplementary Figure S2). We guessed that it might be important for the Snu13p–Rsa1p interaction. To test this hypothesis, we substituted individually most of its residues by alanines in the Rsa1p230–266 protein (Figure 5A). The effect on the interaction with Snu13p was assessed (Figure 5B), using as reporter signal, the specific modification of the CD signal at 220 nm observed upon formation of the Snu13p–Rsa1p230–266 complex (Figure 3D). Although the decrease in the CD signal at 222 nm observed for association of the E247/A variant with Snu13p was similar to that found for WT Rsa1p230–266, no modification of this signal was detected when mixing the Rsa1p230–266 W245/A, or R246/A, or R249/A variants with Snu13p (Figure 5B). These data strongly suggested the direct involvement of residues W245, R246 and R249 in the Snu13p–Rsa1p230–266 interaction. Accordingly, when we tested the ability of these four Rsa1p230–266 variants to form trimeric complexes with Snu13p and snoRNA U14 by gel-shift assays, only the Rsa1p230–266 E247/A variant super-shifted the Snu13p–U14 RNA complex (Figure 5C, lane 6).
Figure 5.

The Rsa1p residues R249 and W245 are essential for Snu13p interaction with full-length Rsa1p. (A) The conserved W239R240E241E242R243 amino acid sequence of Rsa1p230–266, is conserved in NUFIP. Amino acid substitutions in the variant used in the experiments of this figure are shown in red. (B) Substitutions of residues W245, R246 or R247 by an alanine in His6Rsa1p230–266 abolish its interaction with Snu13p as evidenced by CD. As in Figure 3, a two-compartment cuvette was used and CD spectra were recorded from 205 to 260 nm prior (blue) and after mixing (green) the material in the two compartments. (C) Residues W245, R246 and R249 in Rsa1p are needed to bind the Rsa1p230–266 fragment to the Snu13p–snoRNA U14 complex. Radiolabeled U14 snoRNA was incubated with WT Snu13p and WT or mutated His-tagged Rsa1p230–266, in conditions described in ‘Materials and Methods’ section. The complexes formed were fractionated by gel electrophoresis. Positions of the Snu13p–U14 RNA (RNP1) and Rsa1p230–266–Snu13p–U14 RNA (RNP2) complexes, as well as, of free U14 RNA (U14) are indicated on the left side of the autoradiogram. (D) The Rsa1p residues W245 and R249 are required for interaction of Snu13p with the Rsa1p230–281 fragment in Y2H assays, but not residue R246. In the Y2H assays, Snu13p was used as the bait, and the WT or variant Rsa1p230–381 fragments as the prey. The 3AT inhibitor was used at concentrations comprised between 10 and 50 mM. (E) The R249/A mutation in full-length Rsa1p abolishes Snu13p–Rsa1p interaction in co-expression assays, whereas the Rsa1p R246/A variant retains some capability to interact with Snu13p in these assays. Co-expression and co-purification assays were performed in the same conditions as in Figure 3B, using either full-length Rsa1p or Rsa1p230–381 variants.
Interestingly, when the same amino acid substitutions in the WREER motif were generated in a longer Rsa1p fragment (Rsa1p230–381) and their ability to abolish the interaction with Snu13p was tested by Y2H assays, only the W245/A and R249/A substitutions were found to impair the Y2H interaction (Figure 5D). The R246/A substitution had no visible effects, suggesting that as found for the 15.5K–NUFIP interaction (Figure 1C), the Snu13p–Rsa1p230–266 interaction is less stable than the Snu13p–Rsa1p230-–381 interaction and that residue R246 may be involved in the interaction but is not essential. Similarly, when we co-expressed the WT or variant Rsa1p or Rsa1p230–381 proteins with His6Snu13p in E. coli, and co-purified His6Snu13p with its associated proteins by IMAC, the R249/A substitution in both full-length Rsa1p and Rsa1p230–381 abolished the specific interaction with Snu13p, whereas the R246/A variant retained binding activity (Figure 5E).
Hence, our data identified three residues in Rsa1p (W245, R246 and R249) that may be located at the interface with Snu13p, and revealed a key role of W245 and R249 and a less significant role for R246 in this interaction. As the important residue in Snu13p was found to be a glutamate residue (E72), it was reasonable to find that an arginine residue (R249), which can establish electrostatic interactions, plays a key role in the partner protein. It was then important to test whether the Snu13p and Rsa1p residues found to have a role in the Snu13p–Rsa1p interaction have an importance for yeast cell growth and for C/D box snoRNP assembly.
The amino acids essential for the Snu13p–Rsa1p interaction are important for cell growth
Deletion of the RSA1 gene in S. cerevisiae leads to a growth defect, especially at high temperature (45). Hence, we tested whether abolition of the Snu13p–Rsa1p interaction can generate a similar growth phenotype. To this end, we used a yeast strain containing the chromosomal SNU13 ORF placed under the control of the conditional GAL1 promoter. These cells were transformed with a centromeric pG1 plasmid allowing constitutive expression of the WT or mutant Snu13p protein and cell growth was monitored in liquid media (Table 1A). In the presence of glucose in the medium (absence of expression of the genomic SNU13 gene), a doubling time of 3.26 h was observed for the ectopic expression of WT Snu13p from the pG1 plasmid. This doubling time was increased to 3.56 and 3.58 h, respectively, when the E72D73K74/AAA and E72/A Snu13p variants were expressed. Although, the increase was not strong, it was reproducibly observed, and supported the idea of a functional importance of the identified Snu13p–Rsa1p interaction, and in particular, of the Snu13p residue E72 involved in this interaction. To complete the demonstration of a functional role of the Snu13–Rsa1p interaction, WT and variant Rsa1p proteins were expressed from a centromeric plasmid in an RSA1 KO strain, and growth rates were measured (Table 1B). The KO strain had a doubling time ∼44% slower compared to WT cells. Ectopic expression of Rsa1p restored the wild-type growth rates (2.6 h). In contrast, only a partial restoration of cell growth was obtained for the Rsa1p R249/A and W245/A variants that did not bind to Snu13p (doubling time of 3.24 and 3.06 h, respectively).
Table 1.
Residues E72 in Snu13p and residues W245 and R249 in Rsa1p are required for optimal yeast cell growth
| (A) | ||
|---|---|---|
| Strain Gal::SNU13 | G (h) | Growth defect (% WT) |
| pG1::SNU13-WT | 3.26±0.04 | |
| pG1::SNU13-E72D73K74/AAA | 3.56±0.05 | –9.2 |
| pG1::SNU13-E72/A | 3.58±0.09 | –9.8 |
| pG1::SNU13-L69/A | 3.22±0.09 | +1.2 |
| (B) | ||
|---|---|---|
| Strain ΔRSA1 | G (h) | Growth defect (% WT) |
| pG1::(-) | 3.74±0.18 | –44.2 |
| pG1::RSA1-WT | 2.6±0.13 | |
| pG1::RSA1-W245/A | 3.06±0.10 | –17.8 |
| pG1::RSA1-R246/A | 2.55±0.10 | +1.7 |
| pG1::RSA1-R249/A | 3.24±0.03 | –24.8 |
(A) Effects of ectopic expression of WT or variant Snu13p proteins from recombinant pG1::SNU13 plasmid on growth of YPH499-GAL::SNU13 yeast cell. YPH499-GAL::SNU13 cells were transformed with recombinant pG1::SNU13 plasmids expressing WT or mutated Snu13p. Transformed cells were grown at 30°C in YPD medium containing glucose in order to block genomic expression of Snu13p. Cells were maintained in exponential growth by successive dilutions. The effects of the ectopic expression of WT and mutated Snu13p on cell growth were monitored by measuring the absorbance at 600 nm. The cell doubling times (G) were calculated and are expressed in hours (second column). Estimated errors on the G-values were calculated from three distinct experiments performed in the same conditions. G-values were used to calculate a growth defect percentage for each variant Snu13p protein (third column). (B) Effects of the ectopic expressions of WT and variant Rsa1p from recombinant pG1::RSA1 plasmid on growth of an RSA1 KO strain. Same legend as in panel A.
The R246/A substitution in Rsa1p, which did not abolish binding to Snu13p, had a more limited effect on cell growth. Therefore, substitutions of the three amino acids which play a key role in the Snu13p–Rsa1p interaction all have a negative effect on yeast cell growth showing the functional importance of the identified interaction.
The Rsa1p residue R249, essential for the Snu13p–Rsa1p interaction, is important for box C/D snoRNA stability
As defects in snoRNA assembly are known to lead to partial degradation of snoRNAs (2,3), we guessed that if the Snu13p–Rsa1p interaction plays an important role for snoRNP assembly, its abolition in vivo should lead to a decreased level of cellular snoRNAs. To test this hypothesis, we used northern blot analyses to evaluate the steady-state levels of a selected number of box C/D snoRNAs in the ΔRSA1 S. cerevisiae strain Y190 carrying an RSA1 gene disruption, its isogenic strain with an active RSA1 gene, and in the ΔRSA1 strain complemented by ectopic expression of WT or R249/A Rsa1p protein. The selected snoRNAs included U14 which is involved in both pre-rRNA 2′-O methylation and its early endo-nucleolytic cleavage steps leading to 18S rRNA (67,68), and snoRNAs U18, snR63, snR190, snR45 and snR13, which are only involved in 2′-O methylations of the pre-rRNA (68). The results obtained confirmed that disruption of the RSA1 gene is associated to a drastic decrease of the amounts of most of the tested C/D box snoRNAs (snR63, snR190, snR45, snR13 and U18) (Figure 6). Ectopic expression of WT Rsa1p restored normal snoRNA amounts, whereas the expression of the variant R249/A Rsa1p protein did not. Among the tested RNAs, only U14 snoRNA showed a lower decrease in the absence of Rsa1p expression that is restored upon expression of the Rsa1p R249/A variant. Due to its essential role in 18S rRNA production, U14 may have a longer life span than the other box C/D snoRNAs tested, or the assembly of the U14 snoRNP may be less dependent upon the Snu13p–Rsa1p interaction. In contrast, the decreased levels of snoRNAs snR63, snR190, snR45, snR13 and U18 are likely indicative of a decreased efficiency of their assembly into snoRNPs. Therefore, our data strongly supported the idea of a functional importance of the Snu13p–Rsa1p interaction for box C/D snoRNP biogenesis and as a consequence for yeast cell growth. To go one step further in understanding the snoRNP assembly process, we tested whether abolition of this interaction may impair the recruitment of other components of the snoRNP assembly machinery.
Figure 6.

The Rsa1p residue R249 is required for full assembly of several yeast box C/D snoRNPs. The steady-state levels of a selected series of box C/D snoRNAs were measured by northern blot analysis in total extracts from RSA1 KO yeast cells expressing either WT Rsa1p or its R249/A variant from the pG1::RSA1 plasmid. The specific 5′ end labeled DNA probes used for each snoRNA are given in ‘Materials and Methods’ section.
Residue E72 in Snu13p is required for recruitment of the Pih1–Tah1p dimer on a snoRNA in in vitro assays
We had previously shown that Rsa1p230–381 interacts with Pih1p, an integral component of the R2TP complex (2), and had proposed that through this interaction with Pih1p and its interaction with Snu13p, Rsa1p tethers the R2TP complex onto Snu13p bound to the snoRNA. Our identification of a Snu13p residue required for the Snu13p–Rsa1p interaction (E72) opened us the possibility to test the importance of this interaction for the recruitment of Pih1p on a box C/D snoRNA in in vitro experiments. For this purpose, as protein Pih1 has a strong tendency to aggregate and to be degraded when produced alone in E. coli, we produced a His6Tah1p–Pih1p complex by co-expression of the two proteins in E. coli, and purified the complex on Ni-Sepharose beads (69). Then, we tested the capability of interaction of this complex with 32P labeled U14 snoRNA in the presence or the absence of the WT recombinant Rsa1p230–381 C-terminal domain and of the recombinant WT Snu13p protein or its E72/A variant. Retention of U14 snoRNA on the beads was detected by autoradiography after electrophoresis on a denaturing gel. As shown in Figure 7A, very little amounts of RNA were co-purified with the His6Tah1p–Pih1p in the absence or the presence of Snu13p alone, showing a weak capacity of the His6Tah1p–Pih1p to form a stable complex with U14 snoRNA alone or the Snu13p–U14 complex. In contrast, in the presence of the Rsa1p230–381 C-terminal domain, retention of the snoRNA was substantially improved, indicating that this domain of Rsa1p can mediate the recruitment of the Tah1p–Pih1p complex on the Snu13p–U14 complex. Accordingly, when using the E72/A Snu13p variant which does not interact with Rsa1p, only a background level of RNA was detected on the beads (Figure 7B). Thus, the Snu13p–Rsa1p interaction, and in particular the E72 Snu13p residue, is required for binding of the R2TP complex to pre-snoRNP complexes. This observation likely explains the decreased amounts of snoRNAs that we detected in yeast strains with an impaired Snu13p–Rsa1p interaction.
Figure 7.

Interaction of Snu13p with the Rsa1p230–281 fragment is required for recruitment of the Tah1p–Pih1p complex on the Snu13p–U14 snoRNA complex. (A) Rsa1p230–381 is required for efficient association of the Snu13p–U14 snoRNA complex on the His6Tah1p–Pih1p complex. The radiolabeled U14 snoRNA was incubated with the recombinant Snu13p protein and the His6Tah1p–Pih1p dimer bound to Ni-Sepharose beads, in the absence or the presence of an increasing concentration of Rsa1p230–381 fragment (from 0.25 to 2 µM). The RNAs retained on the beads were analysed by denaturing gel electrophoresis. The input corresponds to 10% of the total quantity of radiolabeled RNA used in the binding assays (left lane). A control assay was performed in the absence of Snu13p (second lane). (B) The E72 mutation in Snu13p which abolishes the Snu13p–Rsa1p230–281 interaction abrogates the recruitment of the Snu13p–U14 snoRNA complex on the Tah1p–Pih1p complex. His-pull down experiments and analysis of the retained U14 snoRNA were performed as in panel A, using the WT Rsa1p230–281 fragment at a 2 µM concentration, and either WT Snu13p or its E72/A variant at a 1 µM concentration.
Having shown that some residues involved in the Snu13p–Rsa1p interaction are required for yeast growth and more precisely for snoRNP assembly, we turned to structural approaches to better understand the assembly process.
Rsa1p includes an alpha helix able to bind Snu13p
The solution structure of Snu13p had been solved by X-ray crystallography (15). In contrast, no 3D structure was available for Rsa1p. As the Rsa1p230–266 fragment was not soluble enough for NMR studies, we looked for suitable Rsa1p230–266 minimal sub-fragments. Firstly, we found by in vivo Y2H approach that Rsa1p238–261 was sufficient for interaction with Snu13p (Table 2). Secondly, we show by isothermal titration calorimetry (ITC) that the synthetic peptide Rsa1p238–259 was able to bind Snu13p in vitro with a dissociation constant value of 18 ± 1 µM (Supplementary Figure S3). These data suggest that the two last residues of the previous determined Y2H fragment are not essential in the interaction. Finally, the solubility of this Rsa1p238–259 fragment was compatible with 3D structure analysis by NMR spectroscopy, and it contained the residues described above as being involved in Snu13p binding. Based on the clear NMR data collected, we could unambiguously assign most of the resonances obtained for this peptide and determine its 3D structure in solution. A schematic view of the NMR distance restraints and structural statistics for the calculation are displayed in Supplementary Figure S4 and Supplementary Table S1, respectively. As shown by the superimposition of the 10 structures having the lowest energies (Figure 8A), Rsa1p238–259 is folded into a α-helix.
Table 2.
Y2H assays between Snu13p and Rsa1p sub-fragments or their variants
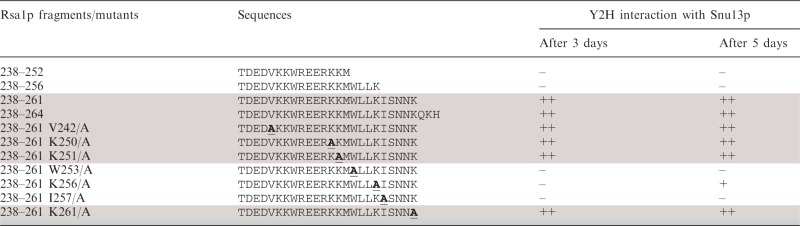 |
The sequences encoding fragments of Rsa1p (Partner A) and the sequence encoding Snu13p (Partner B) were fused to Gal4AD and Gal4BD, respectively. After 3 and 5 days of yeast growth on Leu–/Trp–/His– selective media, the interaction was scored by eye, using the number of positive clones.
Figure 8.

The Rsa1p238–259 NMR structure and modeling of the Snu13p–Rsa1p interaction show both electrostatic and hydrophobic components at the protein–protein interface. (A) Backbone superimposition of the 10 best structures of Rsa1p238–259. (B) Side-chains representation of the lowest energy Rsa1p238–259 structure. The helical conformation is highlighted using a cartoon representation. The hydrogen atoms are not represented. (C) Ribbon representation of the 10 3D models with the lowest haddock score resulting from the docking of the lowest energy NMR structure of Rsa1p238–259 on the X-ray structure of Snu13p. (D) Ionic interaction network between R246, R249 and K250 in Rsa1p238–259 and E72 and D73 in Snu13p. (E) Highlight of the Snu13p pocket hosting the W253 in Rsa1p238–259. The figures have been done with PyMOL software (66).
In agreement with our functional analysis, in the established structure, the side-chains of residues R246, and of the two residues W245 and R249 which play a key role in the Snu13p interaction, are positioned on the same face of the helix, in a configuration favorable for the establishment of interactions with Snu13p amino acids (Figure 8B).
As residues V242, K250, W253, K256 and I257 were also located on the same face of the Rsa1p helix (Figure 8B), some of them might also interact with Snu13p. Thus, we decided to test the effects of their substitutions into alanine on the stability of Snu13p–Rsa1p interaction by Y2H experiments (Table 2). As expected, the control substitution of K251 residue located on the opposite side of the helix had no effect on the Y2H signal (Table 2). In contrast, individual substitutions of residues W253, I257 abolished the Y2H signal, strongly suggesting that these residues also have an important role in the interaction. The K256/A substitution limited but did not abolished the Y2H signal, and substitutions of residues V242 and K250 had no detectable effects (Table 2). Therefore, the structure we solved for fragment Rsa1p238–259 helped us to identify two additional Rsa1p residues (W253 and I257) expected to be important for the Snu13p–Rsa1p interaction and they suggested that residue K256 may also be involved.
Unfortunately, the intermediate exchange regime between Snu13p and Rsa1p238–259 gave rise to low quality NMR spectra (data not shown). This low quality avoided us to perform an experimental determination of the 3D structure of the Snu13p–Rsa1p238–259 complex by the NMR approach. Nevertheless, one possibility to get some structural information on the Rsa1p–Snu13p interface was to use in silico docking of the established structures.
3D structure modeling of the Snu13p–Rsa1p238–259 interface reveals both electrostatic and hydrophobic interactions
To build a 3D model of the Snu13p–Rsa1p238–259 interaction, we docked the NMR structure established for Rsa1p238–259 onto the crystal structure of Snu13p by using Haddock 2.1/CNS 1.21 (58,59) (PDB accession number: 2ALE, (15)). We introduced the knowledge we gained from the various experiments described above to drive the docking experiment (see ‘Materials and Methods’ section). Results of the docking procedure are presented in Table 3. According to energy and RMSD values calculated for the 10 models with the lowest Haddock scores, Rsa1p238–259 is predicted to adopt a preferential positioning on Snu13p in which the N-terminal part of the peptide is close to the EDK motif of the protein, whereas the C-terminal part accommodates into the groove formed by helix α3 and the N-terminal part of Snu13p (Figure 8C).
The best model defined by the Haddock scores (Figure 8D and E) can explain the role in the Snu13p–Rsa1p interaction of the Snu13p E72 and the Rsa1p R249 residues found to be required for optimal yeast cell growth. Indeed, in this model, the carboxylate groups of residues E72 and D73 in Snu13p are involved in a network of electrostatic bonds with the side-chains of residues R246, R249 and K250 in Rsa1p238–259 and the position of R249 is frozen through hydrogen bonding with the hydroxyl group of residue Y78 in Snu13p (Figure 8D). According to our model, the Rsa1p W245 residue shields the electrostatic network of interactions around E72 from the solvent, which may explain its role in the interaction and its need for optimal cell growth. The last Y2H assays have pointed out the possible importance of the Rsa1p residues W253 and I257 and to a lesser extent K256 in the Snu13p–Rsa1p238–259 interaction. One explanation for the importance of residue W253 can be its participation to strong hydrophobic interactions. Indeed, according to our model, its indol ring points into a large pocket formed by the aliphatic side-chains of the Snu13p residues K7, F9, L65 and L69 (Figure 8E). Concerning residues K256 and I257, through their long side-chains they enclose residue W253 in a cage favorable for hydrophobic interactions with residues of the Snu13p hydrophobic pocket.
The stability of the proposed model in an aqueous medium as tested by a 5-ns Molecular Dynamic simulation (see ‘Materials and Methods’ section) shows that the overall 3D structure of the model does not undergo drastic changes. The network of ionic interactions still exists during the 5 ns of simulation (mean RMSD value of 1.41 ± 0.26 Å compared to the initial haddock structure) and the location of the side-chain of W253 in the Snu13p pocket described above is stable over time (analysis of the trajectory calculated with NAMD is available as Supplementary Table S2). Therefore, to assess the stabilizing effect generated by binding of W253 in the Snu13p hydrophobic pocket, we substituted one of the hydrophobic residues of this pocket, L69, by an alanine residue. The choice of L69 was dictated by our Y2H results with the human proteins and by its close vicinity to the Rsa1p W253 residue (Figure 8E). The effect of the mutation on the Snu13p–Rsa1p230–266 binding was tested by EMSA experiments. As illustrated in Figure 9, the L69/A mutation abolished the binding of Rsa1p230–266 to the Snu13p–U14 RNA complex, demonstrating the in vitro stabilization effect of the Snu13p residue L69 on the Snu13p–Rsa1p230–266 interaction. These data are in good agreement with our finding of a role of the human 15.5K residue L71 (the counterpart of L69 in Snu13p) in the NUFIP223–260–15.5K interaction (Figure 1C).
Figure 9.

Residue L69 in Snu13p participates in vitro in the interaction of the Rsa1p230–266 fragment with Snu13p. Formation of the U14 RNA–Snu13p–Rsa1p230–266 complexes and analyses by gel-shift assays were as in Figure 4B. Positions of the RNP1 and RNP2 complexes and of the free U14 RNA are indicated as in Figure 4B.
DISCUSSION
Several proteins have been proposed to participate in box C/D snoRNP biogenesis. However, little is known on their mechanism of action. NUFIP and its yeast homolog Rsa1p were identified as key factors in the biogenesis of RNPs containing proteins of the L7Ae family and were found to interact with proteins 15.5K and Snu13p, respectively (2). Based on previous data, the 15.5K–NUFIP and Snu13p–Rsa1p interactions were expected to facilitate recruitment of the HSP90-R2TP complex. Accordingly, in the absence of NUFIP and Rsa1p, a marked decrease of the box C/D snoRNP levels has been observed (2).
Here, by combination of molecular and cellular analyses, we were able to identify some of the amino acids located at the Snu13p–Rsa1p and 15.5K–NUFIP interfaces and their mutation highlighted for the first time the crucial role of these interactions for box C/D snoRNP assembly. This prompted us to use structural approaches to improve our knowledge on the Snu13p–Rsa1p interface and we propose a 3D structure model of this interaction. Our data bring insights on how NUFIP and Rsa1p can discriminate L7Ae members within the larger L30 protein super-family and suggest a function for NUFIP/Rsa1p in inhibiting activity of immature snoRNPs.
A stable Snu13p–Rsa1p interaction is required for efficient assembly of C/D box snoRNPs in yeast
For the first time, we show in cellulo effects of mutations abolishing the 15.5K–NUFIP and Snu13p–Rsa1p interactions. The 15.5K mutant protein unable to bind NUFIP shows a substantially altered intra-cellular localization, with a stronger cytoplasmic signal that suggests a partial defect in incorporation into U4 and C/D RNPs (Figure 2). In yeast, deletion of the RSA1 gene was known to induce a marked temperature sensitive phenotype (45). However, as Rsa1p had been proposed to have several functions (45), it was difficult to assign the cause of the observed growth defect. Here, based on our detailed analysis of the Snu13p–Rsa1p interaction, we could create point mutations in Snu13p and Rsa1p, and dissect the functional importance of electrostatic and Van der Waals interactions formed at the Snu13p–Rsa1p interface.
Point mutation of residue E72 in Snu13p increased the cell doubling time by a factor of ∼10%, whereas point mutations of residue R249 and W245 in Rsa1p increased the cell doubling time by factors of ∼25 and ∼18%, respectively. This indicates that the electrostatic interactions developed by residues E72 (Snu13p) and R249 (Rsa1p) play important roles in vivo. We can reasonably assume that one of these roles consists in enabling the recruitment of Rsa1p on pre-snoRNPs and therefore the association of the R2TP complex and other snoRNP core proteins on pre-snoRNPs. In agreement with this statement, we showed that in vitro binding of the Tah1p–Pih1p dimer to pre-snoRNP is dependent on the presence of Rsa1p and that mutation of residue E72 in Snu13p impairs this binding (Figure 7). Also consistent with this statement, the level of box C/D snoRNAs after R249/A substitution in Rsa1p was very similar to that found upon deletion of the RSA1 gene: the snR13 and snR63 snoRNAs were undetected in the two types of mutant cells. The stronger effect on yeast growth of mutations in Rsa1p compared to mutations in Snu13p suggests that the Rsa1p amino acids involved in the Snu13p binding may also be involved in the interaction with other target proteins.
Interestingly, however, the effect of deletion or mutations of the RSA1 gene on the U14 snoRNA steady-state was low, which may either results from a greater stability of this RNA or from the lower dependence of the U14 snoRNP assembly on Rsa1p. Therefore, we cannot exclude the possibility that some of the yeast snoRNAs can be efficiently assembled without the help of the assembly machinery. Accordingly, in plants, atNUFIP has an effect that is specific for box C/D snoRNAs encoded by polycistronic not monocistronic genes (70).
NUFIP and Rsa1p discriminate L7Ae members through helix α3
Prior to the present study, very little was known about amino acids required for discrimination of ‘L7Ae-like’ proteins by the platform proteins Rsa1p and NUFIP. Previous studies proposed the involvement of the 15.5K E124 and R125 residues in NUFIP recruitment (22,46). The present data invalidate this conclusion, because we show that the E124R125/SS mutant has the capability to interact efficiently with NUFIP and the small NUFIP223-–260 domain in Y2H assays (Mut I in Figure 1C). The absence of conservation of the E124 and R125 15.5K residues in protein Snu13p also makes it very unlikely that they participate to an interaction expected to be conserved from yeast to human. Thus, the effect previously observed was likely to be indirect.
The E74D75K76N77/RKMT mutation in Snu13p was also found to abrogate detection of the 15.5K–NUFIP association in a cellular extract (46), but this inability has been attributed to an improper folding of this 15.5K variant, possibly because of the large diversity of amino acid substitutions used (25). To avoid such structural defect, in the present study the amino acids in the three EDK residues were substituted by alanine residues, which are unable to establish stable interactions with other residues in the protein. In addition, the EDK residues in 15.5K and Snu13p (15,16) do not develop internal interactions, and no major modification of the protein structure was thus expected upon their mutation into AAA and this was confirmed by our CD data (Figure 3C). Thus, we are confident that the effects we observed are direct.
Our data point to an essential role of the helix α3 acidic residue, E74 in 15.5K and E72 in Snu13p, in the recognition of Snu13p and 15.5K by Rsa1p and NUFIP, respectively. Interestingly, residue E74 is also conserved in proteins NHP2 and SBP2 of the L7Ae family (Figure 1B), which all interact with NUFIP (2). In contrast, this residue is not conserved in the members of the L30 super-family which do not belong to the L7Ae family (Figure 1B) (71). Accordingly, co-expression and Y2H data failed to detect an interaction between NUFIP and other members of the L30 family that contain no counterparts of residue E74 ((2) and data not shown). This suggests that the presence of residue E74/E72 is a good predictor of the interaction with NUFIP or Rsa1p.
In contrast to 15.5K, NHP2 and SBP2 do not bind to the isolated PEP region of NUFIP (NUFIP223–260), suggesting that regions other than PEP help the binding of NUFIP to these proteins (Figure 1C and data not shown). This stronger implication of amino acid sequences outside of PEP may be linked to sequence variations within the PEP recognition regions of these proteins. For instance, amino acid sequence alignments show the replacement of L71 in 15.5K (L69 in Snu13p) by a valine residue in NHP2 (V96 in human; Figure 1B). Furthermore, while the L71/V mutation does not inhibit binding of 15.5K with full-length NUFIP, this mutation prevents binding to NUFIP223–260. Conversely, while NHP2 does not bind NUFIP223–260, the V96/L substitution in NHP2 allows formation of the interaction (Figure 1C). These observations reinforce the idea that L71 in 15.5K and its counterpart L69 in Snu13p, which are components of the hydrophobic pocket of these proteins, modulate their affinity for the isolated PEP region of Rsa1p and NUFIP, respectively. However, consistent with a lack of major effect of L71/V mutation in binding to full-length NUFIP, the similar L69/A substitution in Snu13 only had a limited effect on cell growth (Table 1). Therefore, for both Snu13p and 15.5K some residues other than PEP residues should contribute somehow to the stability of the interaction with Rsa1p/NUFIP, either by direct contact or by modulation of the PEP conformation. Nevertheless, the requirement of residues outside of PEP is quite lower for Snu13p/15.5K as compared to NHP2 and SBP2, suggesting that residues L69/L71 which have no counterpart in these two proteins may participate in the discrimination of Snu13p and 15.5K from other L7Ae-like proteins.
Our 3D structure model reveals intricate electrostatic and Van der Waals interactions at the Snu13p–Rsa1p interface
By combining data from Y2H assays, site-directed mutagenesis, co-expression assays, NMR analysis and computer modeling, we built a comprehensive 3D structure model of the Snu13p–Rsa1p interaction. According to this model, basic residues in Rsa1p (R246, R249 and K250) and acidic residues in Snu13p (E72 and D73) form a network of ionic interactions. However, we found that only one of the Snu13p acidic residues (E72) and one of the Rsa1p basic residues (R249) are indispensable for the interaction. Similarly, in Y2H assays, only the 15.5K E74 residue in protein 15.5K is essential for interaction of 15.5K with full-length NUFIP. Inspection of our model brings some insight into this observation: the interaction of E72 in Snu13p with the residue R249 in Rsa1p is likely more stable than the interaction of D73 in Snu13p with R246 and K250 residues in Rsa1p, probably because these latter interactions are more solvent-exposed. We assume that this explains why alanine substitutions of R246, and also K250, in Y2H experiments do not disrupt the Snu13p–Rsa1p interface. In contrast, according to our model, the E72–R249 interaction is stabilized by surrounding hydrophobic residues shielding it from the solvent. Among these hydrophobic residues, residue W245 in Rsa1p may play a crucial role explaining why its mutation disrupts the interaction and has a strong effect on yeast growth. The Snu13p residues F9, and L69 are also expected to protect the E72–R249 ionic interaction against the solvent. In addition, the side-chain of Y78 in Snu13p may be able to fix the guanidinium group of R249 in a conformation favorable for the interaction with residue E72 in Snu13p.
Altogether, this can explain why residues Snu13p E72, Rsa1p R249 and W245 are required for a stable Snu13p–Rsa1p interaction and for optimal yeast cell growth. The lower functional importance of the D73(Snu13p)–R246(Rsa1p) interaction as compared to the E72(Snu13p)–R249(Rsa1p) interaction is evidenced by the similar growth rate defects of cells expressing the E72A Snu13p variant and cells expressing the E72D73K74/AAA Snu13p variant. Accordingly, the low functional importance of the Rsa1p residue R246 is evidenced by the absence of growth phenotype of yeast cells expressing the Rsa1p R246A variant.
In our 3D structure model, the heterocyclic indol of residue W253 is located in a hydrophobic pocket formed by the aliphatic side-chains of Snu13p residues K7, F9, L65 and L69. We could reasonably assume that positioning of residue W253 in the Snu13p pocket anchors the PEP region of Rsa1p at the Snu13p surface. This can explain why disruption of residue W253 abolishes the Snu13p–Rsa1p238–261 interaction in Y2H assays. In addition, as shown by these Y2H experiments, anchoring of residue W253 seems to be particularly dependent from residues K256 and I257 in Rsa1p. Importantly, the Rsa1p residue W253 is quite well conserved in NUFIP (Y247), and the Snu13p hydrophobic building blocks of its binding pocket are conserved in the human 15.5K protein (Supplementary Figure S2 and Figure 1B). This strongly suggests that Van der Waals interactions similar to those established by the yeast Rsa1p W253 residue are formed at the 15.5K–NUFIP interface. Accordingly, mutation of residue L71 in 15.5K had a negative effect on the NUFIP223–260–15.5K interaction. Although in our in vitro assays the L69/A mutation in Snu13p also had a negative effect on the Snu13p–Rsa1p230–266 interaction, residue L69 is not essential for yeast cell growth (Table 1). The four other hydrophobic amino acids in the Snu13p pocket likely compensate for the L69/A mutation in vivo. Therefore, we propose that the Snu13p–Rsa1p and 15.5K–NUFIP interactions are stabilized by interdependent electrostatic and Van der Waals interactions, requiring the presence of four residues which are conserved from yeast to human: E72 in Snu13p and its E74 homolog in 15.5K, R249 in Rsa1p and its R243 counterpart in NUFIP, W245 in Rsa1p and its W239 homolog in NUFIP, and W253 in Rsa1p and its Y247 counterpart in NUFIP (Figure 1B and Supplementary Figure S2).
In Y2H assays fragment Rsa1p238–261 gave a strong positive signal with Snu13p. The K261A substitution had no effect on the Snu13p affinity (Table 2) suggesting that at least the side-chain of this residue is not involved in the interaction. The other missing residue in Rsa1p238–259, residue N260, is not expected to have an orientation allowing its interaction with Snu13p (Figure 8D and E). However, the proposed model is based on conservation of the helix fold of the peptide upon binding and we cannot exclude some mild modulation of the helical structure of the Rsa1p238–259 fragment by its surrounding sequences within full-length Rsa1p or upon complex formation with Snu13p. This might also explain why in our different assays, the full-length Rsa1p seemed to have a slight different behavior toward Snu13p compared to shorter Rsa1p fragments. Further experimental structure determination is needed to answer these questions.
One L7Ae residue in the archaeal counterpart of the Snu13p EDK triplet develops an electrostatic interaction with residue K289 in NOP5 and R135 in fibrillarin
NUFIP and Rsa1p bind to the 15.5K and Snu13p proteins on a face opposite to that needed for RNA binding (Figure 3A). This is consistent with the possibility to form a ternary complex containing the snoRNA, Snu13p (or 15.5K) and Rsa1p (or NUFIP) plus the other assembly factors. However, one important question is to know whether the core proteins Nop56p, Nop58p and Nop1p (or NOP56, NOP58 and Fibrillarin in human) recruited by the combined action of Rsa1p and the R2TP complex can form a stable mature snoRNP in the presence of Rsa1p or whether Rsa1p has to leave the pre-snoRNP complex in the ultimate steps of the assembly process. One way to get insight into this question is to know whether the Snu13p E72 and 15.5K E74 residues are available for interaction with Rsa1p and NUFIP in mature box C/D snoRNPs, or are involved in snoRNP internal protein–protein interactions, or are buried inside the snoRNPs.
Up to now, no 3D structure of eukaryotic snoRNPs is available to look for availability of residue E72/E74 in the mature particle. However, several crystal structures of archaeal box C/D sRNP have been recently established (26,27), and archaeal and eukaryotes box C/D RNPs are similar. Two types of substrate-bound archaeal structures have been resolved by crystallography. The first type corresponds to an inactive half-mer sRNP, which contains the three core proteins L7Ae, Fibrillarin, and NOP5 (the archaeal homolog of NOP56/Nop56p and NOP58/Nop58p) (27). The second type corresponds to a catalytically active complete bipartite sRNP with two copies of each core proteins on a same box C/D RNA molecule (26). The main difference between these structures occurs on the so-called catalytic module, which is composed of Fibrillarin and the Fibrillarin-binding domain in NOP5. In the half-mer structure, this module is located in an open configuration, far away from the RNA modification site, and it must undergo a large rotational movement to form the closed, active sRNP structure (27). A counterpart of the EDK motif is found in helix α3 of archaeal L7Ae proteins (i.e. D74E75K76 in the Sulfolobus solfataricus L7Ae protein) (Figure 10A). In the open and inactive configuration, the D74E75K76 motif of S. solfataricus L7Ae protein is readily accessible. Thus, if the eukaryotic motif has the same structure, it could interact with NUFIP/Rsa1p. Remarkably however, formation of the active structure and the necessary movement of the catalytic module towards the D74E75K76 motif of L7Ae creates an interaction between this motif and both NOP5 and Fibrillarin, which appear to maintain the catalytic module in the closed configuration required for catalysis (Figure 10C). Indeed, in the active closed structure, the L7Ae amino acids D74 and E75 of this motif and residue L71 corresponding to the L69 residue of Snu13p, are found at the interface between L7Ae and NOP5, and Fibrillarin (Figure 10B). Whereas, the L7Ae D74 residue appears to be free of interaction in the active C/D box sRNP structure, its neighbor residue E75 forms two stable electrostatic interactions with highly conserved amino acids: one with the residue K289 of NOP5 (K310 in human) and the other one with residue R135 in Fibrillarin (R218 in human) (Figure 10C–E). Therefore, it clearly participates in the sRNP architecture. In addition, residues D74 and L71 are located in a deep narrow cavity of the snoRNP limiting their capacity of interaction with a protein. Hence, if we assume similar interaction between Snu13p, Nop56p or Nop58p and Fibrillarin as those found for the archaeal proteins, we can conclude that the Snu13p–Rsa1p interaction can occur in the inactive, open snoRNP structure, but cannot take place in the active and closed snoRNP structure. This feature likely explains why NUFIP/Rsa1p specifically binds snoRNP precursors, and further indicates that removal of NUFIP/Rsa1p is a key event during snoRNP maturation. It also suggests how the presence of Rsa1p could prevent formation of the final active snoRNP structures, which may be important to prevent premature catalytic activity of incompletely assembled snoRNPs.
Figure 10.

L7Ae E75 residue, the archaeal counterpart of the D73 in Snu13p EDK motif makes electrostatic interactions with residue K289 in NOP5 and R135 in fibrillarin. (A) Partial alignment of the amino acid sequence of L7Ae proteins from several archaea species with the corresponding amino acid sequences of Homo sapiens 15.5K and S. cerevisiae Snu13p proteins reveals the conservation of two acidic residues at positions corresponding to residues E72 and D73 in Snu13p. The alignment was established using CLUSTALW. Identical and similar amino acids are in dark gray and light gray boxes, respectively. The conserved acidic residues corresponding to E72 and D73 and the more specific hydrophobic L69 of the W245 binding pocket in Snu13p are in red. (B) The L7Ae residues L71, D74 and E75 in the S. solfataricus L7Ae protein corresponding to residues L69, E72 and D73 in Snu13p, respectively, are located at the bottom of a narrow cavity in the active C/D box snRNP crystal structure. The PyMOL software was used to represent the complete S. solfataricus box C/D snRNP 3D structure (26) (PDB: 3PLA). L7Ae proteins are in pink, the NOP5 proteins in blue and Fibrillarin (FIB) in orange. The RNA is in green. The cluster of L71, D74 and E75 residues is represented in red. An enlargement of the part of the 3D structure containing these three residues is shown in the inset. (C) The carboxylate group of L7Ae residue E75 forms two ion pairs, one with NH3(+) group of NOP5 residue K289 and another one with guanidium group of residue R135 in Fibrillarin. Moreover, residue L71 in L7Ae is located at the interface between L7Ae, Fibrillarin and NOP5. Ribbon representation of the interactions between L7Ae, Fibrillarin and NOP5 in the crystal structure of a S. solfataricus box C/D sRNP (26) (PDB: 3PLA). (D) Residue K289 in S. solfataricus NOP5, which interacts with the L7Ae E75 residue in the active sRNP structure, is conserved in eukaryotic NOP56 and NOP58 proteins and in the U4 snRNP PRP31 protein. The alignment was performed as in panel A. Identical and similar amino acids are in dark gray and light gray boxes, respectively. The conserved K residue corresponding to K289 in S. solfataricus NOP5 is in red. (E) Residue R135 in S. solfataricus Fibrillarin, which interacts with the L7Ae E75 residue in the active sRNP structure, is conserved in eukaryotic Fibrillarin proteins. The alignment was performed as in panel A. Identical and similar amino acids are in dark gray and light gray boxes, respectively. The conserved R residue corresponding to R135 in S. solfataricus NOP5 is in red.
ACCESSION NUMBERS
The resonance assignments have been deposited in the BMRB with accession code 18959. The corresponding coordinates and restraints for structure calculations have been deposited in the PDB with accession code 2M3F.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Funding for open access charge: French ‘Agence Nationale de la Recherche’ (ANR-06-BLAN-0208, ANR-11-BSV8-01503); French ‘Association pour la Recherche contre le Cancer’ (SFI20101201793); French ‘Ligue Nationale contre le cancer’ Grand-Est (N° 30025555); the European Associated Laboratory (LEA)on pre-mRNA splicing created by CNRS, UL, UM1, UM2 and Max Planck Institut; French ‘Ministère de l’Enseignement Supérieur et de la Recherche’ (doctoral fellowships to B.R. and R.B.); ‘Centre National de la Recherche Scientifique; ‘Pôle de Recherche Scientifique et Technologique’ «Ingénierie Moléculaire, Thérapeutique et Santé (IMTS)» of ‘Région lorraine’.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
C. Bobo is acknowledged for her technical assistance. A. Krol and C. Allmang-Cura are thanked for sharing data on protein SBP2. B. Maigret is thanked for fruitful discussions on molecular dynamics. NMR, ITC and CD facilities of the «Service Commun de Biophysicochimie des Interactions Moléculaires» (SCBIM) of Université de Lorraine were deeply appreciated.
REFERENCES
- 1.Allmang C, Carbon P, Krol A. The SBP2 and 15.5 kD/Snu13p proteins share the same RNA binding domain: identification of SBP2 amino acids important to SECIS RNA binding. RNA. 2002;8:1308–1318. doi: 10.1017/s1355838202020034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boulon S, Marmier-Gourrier N, Pradet-Balade B, Wurth L, Verheggen C, Jady BE, Rothe B, Pescia C, Robert MC, Kiss T, et al. The Hsp90 chaperone controls the biogenesis of L7Ae RNPs through conserved machinery. J. Cell Biol. 2008;180:579–595. doi: 10.1083/jcb.200708110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Watkins NJ, Segault V, Charpentier B, Nottrott S, Fabrizio P, Bachi A, Wilm M, Rosbash M, Branlant C, Luhrmann R. A common core RNP structure shared between the small nucleoar box C/D RNPs and the spliceosomal U4 snRNP. Cell. 2000;103:457–466. doi: 10.1016/s0092-8674(00)00137-9. [DOI] [PubMed] [Google Scholar]
- 4.Fatica A, Tollervey D. Making ribosomes. Curr. Opin. Cell Biol. 2002;14:313–318. doi: 10.1016/s0955-0674(02)00336-8. [DOI] [PubMed] [Google Scholar]
- 5.Kiss T. Small nucleolar RNAs: an abundant group of noncoding RNAs with diverse cellular functions. Cell. 2002;109:145–148. doi: 10.1016/s0092-8674(02)00718-3. [DOI] [PubMed] [Google Scholar]
- 6.Wahl MC, Will CL, Luhrmann R. The spliceosome: design principles of a dynamic RNP machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 7.Jady BE, Darzacq X, Tucker KE, Matera AG, Bertrand E, Kiss T. Modification of Sm small nuclear RNAs occurs in the nucleoplasmic Cajal body following import from the cytoplasm. EMBO J. 2003;22:1878–1888. doi: 10.1093/emboj/cdg187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blackburn EH, Collins K. Telomerase: an RNP enzyme synthesizes DNA. Cold Spring Harb. Perspect. Biol. 2011;3 doi: 10.1101/cshperspect.a003558. doi: 10.1101/cshperspect.a003558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kiss T, Fayet-Lebaron E, Jady BE. Box H/ACA small ribonucleoproteins. Mol. Cell. 2010;37:597–606. doi: 10.1016/j.molcel.2010.01.032. [DOI] [PubMed] [Google Scholar]
- 10.Copeland PR, Fletcher JE, Carlson BA, Hatfield DL, Driscoll DM. A novel RNA binding protein, SBP2, is required for the translation of mammalian selenoprotein mRNAs. EMBO J. 2000;19:306–314. doi: 10.1093/emboj/19.2.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nottrott S, Hartmuth K, Fabrizio P, Urlaub H, Vidovic I, Ficner R, Luhrmann R. Functional interaction of a novel 15.5kD [U4/U6.U5] tri-snRNP protein with the 5′ stem-loop of U4 snRNA. EMBO J. 1999;18:6119–6133. doi: 10.1093/emboj/18.21.6119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darzacq X, Jady BE, Verheggen C, Kiss AM, Bertrand E, Kiss T. Cajal body-specific small nuclear RNAs: a novel class of 2'-O-methylation and pseudouridylation guide RNAs. EMBO J. 2002;21:2746–2756. doi: 10.1093/emboj/21.11.2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henras A, Henry Y, Bousquet-Antonelli C, Noaillac-Depeyre J, Gelugne JP, Caizergues-Ferrer M. Nhp2p and Nop10p are essential for the function of H/ACA snoRNPs. EMBO J. 1998;17:7078–7090. doi: 10.1093/emboj/17.23.7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang C, Meier UT. Architecture and assembly of mammalian H/ACA small nucleolar and telomerase ribonucleoproteins. EMBO J. 2004;23:1857–1867. doi: 10.1038/sj.emboj.7600181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oruganti S, Zhang Y, Li H. Structural comparison of yeast snoRNP and spliceosomal protein Snu13p with its homologs. Biochem. Biophys. Res. Commun. 2005;333:550–554. doi: 10.1016/j.bbrc.2005.05.141. [DOI] [PubMed] [Google Scholar]
- 16.Vidovic I, Nottrott S, Hartmuth K, Luhrmann R, Ficner R. Crystal structure of the spliceosomal 15.5kD protein bound to a U4 snRNA fragment. Mol. Cell. 2000;6:1331–1342. doi: 10.1016/s1097-2765(00)00131-3. [DOI] [PubMed] [Google Scholar]
- 17.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 18.Clery A, Senty-Segault V, Leclerc F, Raue HA, Branlant C. Analysis of sequence and structural features that identify the B/C motif of U3 small nucleolar RNA as the recognition site for the Snu13p-Rrp9p protein pair. Mol. Cell. Biol. 2007;27:1191–1206. doi: 10.1128/MCB.01287-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marmier-Gourrier N, Clery A, Senty-Segault V, Charpentier B, Schlotter F, Leclerc F, Fournier R, Branlant C. A structural, phylogenetic, and functional study of 15.5-kD/Snu13 protein binding on U3 small nucleolar RNA. RNA. 2003;9:821–838. doi: 10.1261/rna.2130503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nottrott S, Urlaub H, Luhrmann R. Hierarchical, clustered protein interactions with U4/U6 snRNA: a biochemical role for U4/U6 proteins. EMBO J. 2002;21:5527–5538. doi: 10.1093/emboj/cdf544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Granneman S, Pruijn GJ, Horstman W, van Venrooij WJ, Luhrmann R, Watkins NJ. The hU3-55K protein requires 15.5K binding to the box B/C motif as well as flanking RNA elements for its association with the U3 small nucleolar RNA in Vitro. J. Biol. Chem. 2002;277:48490–48500. doi: 10.1074/jbc.M206631200. [DOI] [PubMed] [Google Scholar]
- 22.Schultz A, Nottrott S, Watkins NJ, Luhrmann R. Protein-protein and protein-RNA contacts both contribute to the 15.5K-mediated assembly of the U4/U6 snRNP and the box C/D snoRNPs. Mol. Cell. Biol. 2006;26:5146–5154. doi: 10.1128/MCB.02374-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watkins NJ, Dickmanns A, Luhrmann R. Conserved stem II of the box C/D motif is essential for nucleolar localization and is required, along with the 15.5K protein, for the hierarchical assembly of the box C/D snoRNP. Mol. Cell. Biol. 2002;22:8342–8352. doi: 10.1128/MCB.22.23.8342-8352.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schultz A, Nottrott S, Hartmuth K, Luhrmann R. RNA structural requirements for the association of the spliceosomal hPrp31 protein with the U4 and U4atac small nuclear ribonucleoproteins. J. Biol. Chem. 2006;281:28278–28286. doi: 10.1074/jbc.M603350200. [DOI] [PubMed] [Google Scholar]
- 25.Liu S, Li P, Dybkov O, Nottrott S, Hartmuth K, Luhrmann R, Carlomagno T, Wahl MC. Binding of the human Prp31 Nop domain to a composite RNA-protein platform in U4 snRNP. Science. 2007;316:115–120. doi: 10.1126/science.1137924. [DOI] [PubMed] [Google Scholar]
- 26.Lin J, Lai S, Jia R, Xu A, Zhang L, Lu J, Ye K. Nature. 2011;469:559–563. doi: 10.1038/nature09688. [DOI] [PubMed] [Google Scholar]
- 27.Xue S, Wang R, Yang F, Terns RM, Terns MP, Zhang X, Maxwell ES, Li H. Structural basis for substrate placement by an archaeal box C/D ribonucleoprotein particle. Mol. Cell. 2010;39:939–949. doi: 10.1016/j.molcel.2010.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye K, Jia R, Lin J, Ju M, Peng J, Xu A, Zhang L. Structural organization of box C/D RNA-guided RNA methyltransferase. Proc. Natl Acad. Sci. USA. 2009;106:13808–13813. doi: 10.1073/pnas.0905128106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baker DL, Youssef OA, Chastkofsky MI, Dy DA, Terns RM, Terns MP. RNA-guided RNA modification: functional organization of the archaeal H/ACA RNP. Genes Dev. 2005;19:1238–1248. doi: 10.1101/gad.1309605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Charpentier B, Muller S, Branlant C. Reconstitution of archaeal H/ACA small ribonucleoprotein complexes active in pseudouridylation. Nucleic Acids Res. 2005;33:3133–3144. doi: 10.1093/nar/gki630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Omer AD, Ziesche S, Ebhardt H, Dennis PP. In vitro reconstitution and activity of a C/D box methylation guide ribonucleoprotein complex. Proc. Natl Acad. Sci. USA. 2002;99:5289–5294. doi: 10.1073/pnas.082101999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tran EJ, Zhang X, Maxwell ES. Efficient RNA 2'-O-methylation requires juxtaposed and symmetrically assembled archaeal box C/D and C'/D' RNPs. EMBO J. 2003;22:3930–3940. doi: 10.1093/emboj/cdg368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chari A, Paknia E, Fischer U. The role of RNP biogenesis in spinal muscular atrophy. Curr. Opin. Cell Biol. 2009;21:387–393. doi: 10.1016/j.ceb.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 34.Massenet S, Pellizzoni L, Paushkin S, Mattaj IW, Dreyfuss G. The SMN complex is associated with snRNPs throughout their cytoplasmic assembly pathway. Mol. Cell. Biol. 2002;22:6533–6541. doi: 10.1128/MCB.22.18.6533-6541.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matera AG, Terns RM, Terns MP. Non-coding RNAs: lessons from the small nuclear and small nucleolar RNAs. Nat. Rev. Mol. Cell Biol. 2007;8:209–220. doi: 10.1038/nrm2124. [DOI] [PubMed] [Google Scholar]
- 36.McKeegan KS, Debieux CM, Boulon S, Bertrand E, Watkins NJ. A dynamic scaffold of pre-snoRNP factors facilitates human box C/D snoRNP assembly. Mol. Cell. Biol. 2007;27:6782–6793. doi: 10.1128/MCB.01097-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Neuenkirchen N, Chari A, Fischer U. Deciphering the assembly pathway of Sm-class U snRNPs. FEBS Lett. 2008;582:1997–2003. doi: 10.1016/j.febslet.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 38.Pellizzoni L, Yong J, Dreyfuss G. Essential role for the SMN complex in the specificity of snRNP assembly. Science. 2002;298:1775–1779. doi: 10.1126/science.1074962. [DOI] [PubMed] [Google Scholar]
- 39.Zhao R, Kakihara Y, Gribun A, Huen J, Yang G, Khanna M, Costanzo M, Brost RL, Boone C, Hughes TR, et al. Molecular chaperone Hsp90 stabilizes Pih1/Nop17 to maintain R2TP complex activity that regulates snoRNA accumulation. J. Cell Biol. 2008;180:563–578. doi: 10.1083/jcb.200709061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ballarino M, Morlando M, Pagano F, Fatica A, Bozzoni I. The cotranscriptional assembly of snoRNPs controls the biosynthesis of H/ACA snoRNAs in Saccharomyces cerevisiae. Mol. Cell. Biol. 2005;25:5396–5403. doi: 10.1128/MCB.25.13.5396-5403.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Darzacq X, Kittur N, Roy S, Shav-Tal Y, Singer RH, Meier UT. Stepwise RNP assembly at the site of H/ACA RNA transcription in human cells. J. Cell Biol. 2006;173:207–218. doi: 10.1083/jcb.200601105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leulliot N, Godin KS, Hoareau-Aveilla C, Quevillon-Cheruel S, Varani G, Henry Y, Van Tilbeurgh H. The box H/ACA RNP assembly factor Naf1p contains a domain homologous to Gar1p mediating its interaction with Cbf5p. J. Mol. Biol. 2007;371:1338–1353. doi: 10.1016/j.jmb.2007.06.031. [DOI] [PubMed] [Google Scholar]
- 43.Li S, Duan J, Li D, Ma S, Ye K. Structure of the Shq1-Cbf5-Nop10-Gar1 complex and implications for H/ACA RNP biogenesis and dyskeratosis congenita. EMBO J. 2011;30:5010–5020. doi: 10.1038/emboj.2011.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walbott H, Machado-Pinilla R, Liger D, Blaud M, Rety S, Grozdanov PN, Godin K, van Tilbeurgh H, Varani G, Meier UT, et al. The H/ACA RNP assembly factor SHQ1 functions as an RNA mimic. Genes Dev. 2011;25:2398–2408. doi: 10.1101/gad.176834.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kressler D, Doere M, Rojo M, Linder P. Synthetic lethality with conditional dbp6 alleles identifies rsa1p, a nucleoplasmic protein involved in the assembly of 60S ribosomal subunits. Mol. Cell. Biol. 1999;19:8633–8645. doi: 10.1128/mcb.19.12.8633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McKeegan KS, Debieux CM, Watkins NJ. Evidence that the AAA+ proteins TIP48 and TIP49 bridge interactions between 15.5K and the related NOP56 and NOP58 proteins during box C/D snoRNP biogenesis. Mol. Cell. Biol. 2009;29:4971–4981. doi: 10.1128/MCB.00752-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao R, Houry WA. Hsp90: a chaperone for protein folding and gene regulation. Biochem. Cell Biol. 2005;83:703–710. doi: 10.1139/o05-158. [DOI] [PubMed] [Google Scholar]
- 48.Back R, Dominguez C, Rothe B, Bobo C, Beaufils C, Morera S, Meyer P, Charpentier B, Branlant C, Allain FH, et al. High-resolution structural analysis shows how Tah1 Tethers Hsp90 to the R2TP complex. Structure. 2013;21:1834–1847. doi: 10.1016/j.str.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 49.Diebold ML, Fribourg S, Koch M, Metzger T, Romier C. Deciphering correct strategies for multiprotein complex assembly by co-expression: application to complexes as large as the histone octamer. J. Struct. Biol. 2011;175:178–188. doi: 10.1016/j.jsb.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 50.Fribourg S, Romier C, Werten S, Gangloff YG, Poterszman A, Moras D. Dissecting the interaction network of multiprotein complexes by pairwise coexpression of subunits in E. coli. J. Mol. Biol. 2001;306:363–373. doi: 10.1006/jmbi.2000.4376. [DOI] [PubMed] [Google Scholar]
- 51.Romier C, Ben Jelloul M, Albeck S, Buchwald G, Busso D, Celie PH, Christodoulou E, De Marco V, van Gerwen S, Knipscheer P, et al. Co-expression of protein complexes in prokaryotic and eukaryotic hosts: experimental procedures, database tracking and case studies. Acta Crystallogr. D Biol. Crystallogr. 2006;62:1232–1242. doi: 10.1107/S0907444906031003. [DOI] [PubMed] [Google Scholar]
- 52.Bitter GA, Egan KM. Expression of heterologous genes in Saccharomyces cerevisiae from vectors utilizing the glyceraldehyde-3-phosphate dehydrogenase gene promoter. Gene. 1984;32:263–274. doi: 10.1016/0378-1119(84)90002-7. [DOI] [PubMed] [Google Scholar]
- 53.Boulon S, Pradet-Balade B, Verheggen C, Molle D, Boireau S, Georgieva M, Azzag K, Robert MC, Ahmad Y, Neel H, et al. HSP90 and its R2TP/Prefoldin-like cochaperone are involved in the cytoplasmic assembly of RNA polymerase II. Mol Cell. 2010;39:912–924. doi: 10.1016/j.molcel.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Keller RLJ. The Computer Aided Resonance Assignment Tutorial. 1st edn. Zürich: CANTINA; 2004. [Google Scholar]
- 55.Berjanskii MV, Neal S, Wishart DS. PREDITOR: a web server for predicting protein torsion angle restraints. Nucleic Acids Res. 2006;34:W63–W69. doi: 10.1093/nar/gkl341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guntert P. Automated NMR structure calculation with CYANA. Methods Mol. Biol. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- 57.Brunger AT. Version 1.2 of the Crystallography and NMR system. Nat. Protoc. 2007;2:2728–2733. doi: 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- 58.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 59.de Vries SJ, van Dijk AD, Krzeminski M, van Dijk M, Thureau A, Hsu V, Wassenaar T, Bonvin AM. HADDOCK versus HADDOCK: new features and performance of HADDOCK2.0 on the CAPRI targets. Proteins. 2007;69:726–733. doi: 10.1002/prot.21723. [DOI] [PubMed] [Google Scholar]
- 60.Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 61.de Vries SJ, van Dijk AD, Bonvin AM. WHISCY: what information does surface conservation yield? Application to data-driven docking. Proteins. 2006;63:479–489. doi: 10.1002/prot.20842. [DOI] [PubMed] [Google Scholar]
- 62.Neuvirth H, Raz R, Schreiber G. ProMate: a structure based prediction program to identify the location of protein-protein binding sites. J. Mol. Biol. 2004;338:181–199. doi: 10.1016/j.jmb.2004.02.040. [DOI] [PubMed] [Google Scholar]
- 63.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.MacKerell AD, Jr, Banavali N, Foloppe N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers. 2000;56:257–265. doi: 10.1002/1097-0282(2000)56:4<257::AID-BIP10029>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 65.Janicki SM, Tsukamoto T, Salghetti SE, Tansey WP, Sachidanandam R, Prasanth KV, Ried T, Shav-Tal Y, Bertrand E, Singer RH, et al. From silencing to gene expression: real-time analysis in single cells. Cell. 2004;116:683–698. doi: 10.1016/s0092-8674(04)00171-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.DeLano WL. New York: Schrödinger, LLC; 2010. The PyMOL Molecular Graphics System, Version 1.3r1. [Google Scholar]
- 67.Li HD, Zagorski J, Fournier MJ. Depletion of U14 small nuclear RNA (snR128) disrupts production of 18S rRNA in Saccharomyces cerevisiae. Mol. Cell. Biol. 1990;10:1145–1152. doi: 10.1128/mcb.10.3.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Piekna-Przybylska D, Decatur WA, Fournier MJ. New bioinformatic tools for analysis of nucleotide modifications in eukaryotic rRNA. RNA. 2007;13:305–312. doi: 10.1261/rna.373107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Eckert K, Saliou JM, Monlezun L, Vigouroux A, Atmane N, Caillat C, Quevillon-Cheruel S, Madiona K, Nicaise M, Lazereg S, et al. The Pih1-Tah1 cochaperone complex inhibits Hsp90 molecular chaperone ATPase activity. J. Biol. Chem. 2010;285:31304–31312. doi: 10.1074/jbc.M110.138263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rodor J, Jobet E, Bizarro J, Vignols F, Carles C, Suzuki T, Nakamura K, Echeverria M. AtNUFIP, an essential protein for plant development, reveals the impact of snoRNA gene organisation on the assembly of snoRNPs and rRNA methylation in Arabidopsis thaliana. Plant J. 2011;65:807–819. doi: 10.1111/j.1365-313X.2010.04468.x. [DOI] [PubMed] [Google Scholar]
- 71.Koonin EV, Bork P, Sander C. A novel RNA-binding motif in omnipotent suppressors of translation termination, ribosomal proteins and a ribosome modification enzyme? Nucleic Acids Res. 1994;22:2166–2167. doi: 10.1093/nar/22.11.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.