

Abstract
The mitochondrial DNA base modification 8-hydroxy 2′-deoxyguanine (8-OHdG) is one of the most common DNA lesions induced by reactive oxygen species (ROS) and is considered an index of DNA damage. High levels of mitochondrial 8-OHdG have been correlated with increased mutation, deletion, and loss of mitochondrial (mt) DNA, as well as apoptosis. 8-Oxoguanosine DNA glycosylase-1 (OGG1) recognizes and removes 8-OHdG to prevent further DNA damage. We evaluated the effects of OGG1 on mtDNA damage, mitochondrial function, and apoptotic events induced by oxidative stress using H9C2 cardiac cells treated with menadione and transduced with either Adv-Ogg1 or Adv-Control (empty vector). The levels of mtDNA 8-OHdG and the presence of apurinic/apyrimidinic (AP) sites were decreased by 30% and 35%, respectively, in Adv-Ogg1 transduced cells (P < 0.0001 and P < 0.005, respectively). In addition, the expression of base excision repair (BER) pathway members APE1 and DNA polymerase γ was upregulated by Adv-Ogg1 transduction. Cells overexpressing Ogg1 had increased membrane potential (P < 0.05) and decreased mitochondrial fragmentation (P < 0.005). The mtDNA content was found to be higher in cells with increased OGG1 (P < 0.005). The protein levels of fission and apoptotic factors such as DRP1, FIS1, cytoplasmic cytochrome c, activated caspase-3, and activated caspase-9 were lower in Adv-Ogg1 transduced cells. These observations suggest that Ogg1 overexpression may be an important mechanism to protect cardiac cells against oxidative stress damage.
Keywords: 8-oxoguanine glycosylase, oxidative stress, mitochondrial DNA damage, mitochondrial function, apoptosis
heart disease is still the major contributor to morbidity and mortality in the United States and Worldwide. Some of the hallmarks of heart failure are increased oxidative stress, high-energy phosphate depletion, fibrosis, inflammation, and cell death (29, 37, 38). During heart failure mitochondria produce more oxygen radicals than normal, resulting in a stressful oxidative environment (37). Due to its proximity to the major site of mitochondrial reactive oxygen species (ROS) generation, mitochondrial DNA (mtDNA) is one of the cellular components most affected by oxidative stress. This is a significant risk factor as quantitative and qualitative defects in mtDNA have been associated with cardiac cell death (26). mtDNA damage can trigger a series of events such as decreased transcription and incomplete assembly of the electron transport chain (ETC) complexes, which leads to decreased ETC activity and thus lower ATP synthesis, increased ROS production, and apoptosis (9, 14). 8-Hydroxy 2′-deoxyguanine (8-OHdG) is one of the most common ROS-induced DNA lesions and is considered an index of DNA damage (19, 27). High levels of mitochondrial 8-OHdG have been correlated with increased mutation, deletion, fragmentation, and loss of mtDNA (28, 31, 42). 8-Oxoguanine DNA glycosylase 1 (OGG1), an enzyme in the initial steps of the base excision repair (BER) pathway, is present in the nucleus as well as in the mitochondria. OGG1 recognizes and removes 8-OHdG to prevent further lesions in DNA (1, 2). Yeast studies provide evidence that OGG1 protects the mitochondrial genome from spontaneous as well as induced oxidative damage (36). Vongsamphanh et al. (40) showed that mitochondrial overexpression of Ogg1 reduces the levels of 8-OHdG and decreases the formation of abasic sites, thereby preventing poly(GT) tract instability triggered by oxidative stress formation of lesions in mtDNA. Several studies have reported that mitochondrial OGG1 overexpression in conditions of oxidative stress protects pulmonary artery endothelial, oligodendrocytes, and HeLa cells against apoptosis (10–12). These findings indicate that mtDNA integrity is a key factor in cell survival, and OGG1 may be a critical player in maintaining mtDNA integrity.
With respect to heart disease, one recent study showed that transgenic mice with cardiac-specific overexpression of active mitochondrial OGG1 showed reduced cardiac fibrosis following transaortic constriction (41). Despite this protection, the authors did not find significant reduction in cardiac hypertrophy or improvement in cardiac function as measured by echocardiography 13 wk after surgery. However, the study did not evaluate whether mitochondrial overexpression of Ogg1 affected mitochondrial function or apoptosis. Any protective effect of increased expression of mitochondrial Ogg1 on cardiac mitochondria under conditions of oxidative stress remains unclear. Thus the goal of our study was to evaluate the effects of mitochondrial Ogg1 overexpression on mtDNA repair, mitochondrial dysfunction, and apoptosis in H9C2 cardiac muscle cells under conditions of oxidative stress.
MATERIALS AND METHODS
Adenovirus vector.
The full-length mouse Ogg1 was obtained from IMAGE clone ID 3498005 (ResGen, an Invitrogen) where it was unidirectionally inserted into pCMV-Sport6 (Invitrogen) SalI/NotI. Ogg1 was removed by digestion with SalI (blunt)/NotI and inserted into pENTR1A (Invitrogen) cut with EcoRI (blunt)/NotI to create the entry clone pENTR1A-mOGG1. pENTR1A-mOGG1 was recombined with pAd/CMV/V5-DEST (Invitrogen) via the LR recombination reaction using LR clonase II to generate the final construct pAD-mOGG1, a transfection-ready plasmid encoding the E1- and E3-deleted adenoviral genome and Ogg1 driven by the human immediate early CMV promoter. This plasmid was transfected into 293 cells to produce replication-incompetent adenovirus particles and was propagated to achieve high titer. Adenoviral particles were CsCl-purified and quantified by plaque titer assay. Empty vector (control) adenoviral particles were produced similarly.
Treatment.
H9C2 cells were plated in 15-mm Petri dishes with DMEM (4.5 g/l glucose) (Invitrogen) supplemented with 10% fetal bovine serum (FBS, Invitrogen) and 1% penicillin/streptomycin/fungizone and incubated at 5% CO2 and 37°C. At 80% confluence H9C2 cells were transduced with either Adv-Ogg1 or an empty vector (Adv-Control) and incubated for 24 h. The next day, the medium was removed, the cells were washed 3× with 1× PBS and fresh medium was added to maintain the cells for an additional 48 h incubation. Cells were then treated with 50 μM menadione (Sigma Aldrich) dissolved in DMEM with no FBS for 1 h. After menadione treatment, cells were washed 3× with 1× PBS, fresh medium with 10% FBS was added, and the cells were kept at 5% CO2 and 37°C for an additional 2 h. Finally the cells were harvested and collected for the different assays.
Total protein extract.
Harvested cells were washed twice with cold 1× PBS. Then cells were lysed with a Teflon-glass homogenizer in a lysis buffer containing 0.25 M sucrose, 10 mM Tris-HCl (pH 7.5), 3 mM MgCl2, 0.1 mM EDTA, 0.05% NP40, and a mix of protease and phosphatase inhibitors (Thermo Scientific, Waltham, MA). The homogenate was centrifuged at 1,000 g for 10 min at 4°C, and the supernatant was recovered for protein determination and Western blot analysis.
Mitochondria isolation.
For mitochondria isolation, cells were treated similarly as for total protein extraction. The lysis buffer consisted of 0.25 M sucrose, 10 mM Tris-HCl (pH 7.5), 3 mM MgCl2, 0.1 mM EDTA, and a mix of phosphatase and protease inhibitors (Thermo Scientific, Waltham, MA). After homogenization, the homogenate was centrifuged at 1,000 g for 10 min at 4°C. The supernatant was recovered and centrifuged at 12,000 g for 15 min at 4°C. The supernatant was saved for mitochondrial quality analysis, and the pellet was washed in lysis buffer and centrifuged again at 12,000 g for 15 min at 4°C. Finally, the pellet (mitochondria) was recovered and suspended in 500 μl of lysis buffer and used for protein determination and Western blot analysis.
Immunoblotting.
Total and mitochondrial protein extracts were separated by SDS-polyacrylamide gel electrophoresis (NuPAGE 4–12% polyacrylamide Bis-Tris, Invitrogen) and transferred to PVDF membranes (Immobilon-P, Millipore).
Primary antibodies used for immunoblotting included Ogg1 (Novus Biologicals); total OXPHOS cocktail, COXI and TATA binding protein (TBP) (Abcam); cytochrome c, ND4, Bax, Caspase-3, Fis1, Mfn1, Mfn2, Opa1 and actin (Santa Cruz Biotechnology); cleaved caspase-9, pDrp1 (ser616), pDrp1 (ser637), VDAC (Cell Signaling, Danvers, MA.); and Drp1 (BD Biosciences).
Membranes were incubated overnight at 4°C with primary antibodies and then with their corresponding horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. The immunoblots were detected with Western Lightning enhanced chemiluminescence detection reagent (PerkinElmer LAS, Norton, OH). Band intensity was quantified with ImageJ software and expressed in arbitrary units (35). Actin and VDAC were used as loading controls for total and mitochondrial extracts, respectively.
RNA extraction and quantitative real-time PCR.
Total RNA was extracted from H9C2 cells using Trizol reagent (Invitrogen, Carlsbad CA), and transcript levels were measured by qRT-PCR. Gene-specific primers are listed in Table 1. Primers were designed using PerlPrimer software (30). First-strand cDNA was synthesized using the SuperScript III kit (Invitrogen, Carlsbad, CA). Synthesized cDNA was mixed with SYBR green-DyNAMO (New England Biolabs) and amplified using a 7300 Real Time PCR System (Applied Biosystems, Foster City, CA). All reactions were performed in triplicate. The relative amounts of mRNAs were calculated by using the comparative CT method (User Bulletin no. 2, Applied Biosystems). Actin was used as a control, and all results were normalized to the abundance of actin mRNA.
Table 1.
Gene-specific primers
| Primer | Forward | Reverse | Accession No. |
|---|---|---|---|
| PPARγ coactivator 1α (PGC1α) | ATGAATGCAGCGGTCTTAGC | AACAATGGCAGGGTTTGTTC | NM_031347.1 |
| Cytochrome oxidase subunit I (COX1) | AGCCGGAAACCTAGCCCATGC | CACCCCGGCTAGGTGGAGGG | |
| NADH dehydrogenase subunit (ND4) | TCCCTCACAACACACCCCCT | TGGAGCTTCCACGTGGGCTT | |
| DNA polymerase gamma (DNA polymerase γ) | TGGGATGCATGGCTGCACGG | CAAAGGACTGCCCAGCCCCG | NM_053528.1 |
| Apurinic/Apyrimidinic endonuclease 1 (APE1) | TGCCGAAGCGGGGGAAGAGA | CCCTCTCCTGCGGCCTCCTT | NM_024148.1 |
| Actin | GTCCACCCGCGAGTACAACCT | CGACGAGCGCAGCGATATCG | NM_031144.2 |
Mitochondrial and nuclear DNA isolation.
mtDNA and nuclear DNA (nDNA) extraction was performed using kits from Wako Chemicals and Biovision Research Products (Mountain View, CA), respectively. The mtDNA and nDNA concentrations were measured using a Nanodrop Spectrophotometer ND-1000 (Thermo Scientific, Waltham, MA).
Mitochondria DNA content.
The mitochondria DNA content was determined in a similar fashion as Ciapaite et al. (8). Briefly, DNA was extracted using a kit from Biovision Research Products (Mountain View, CA). The mtDNA content was determined by qRT-PCR. The relative amount of mtDNA to nuclear DNA was calculated by using the comparative CT method (User Bulletin no. 2, Applied Biosystems) and was represented as a fold change compared with the control. The primers for mtDNA (COX1 and ND4) and nuclear DNA (PGC1α) are shown in Table 1.
8-OHdG determination.
mtDNA and nDNA samples were prepared similarly to Kakimoto et al. (25) with slight modifications. Briefly, 60 μg of either mtDNA or nDNA were mixed with 15 μl of 200 mM sodium acetate (pH 5.3) and 6 units of nuclease P1 in 10 mM sodium acetate. The mix was incubated for 1 h at 37°C to digest the DNA into nucleotides. Then a mix of 15 μl of 1 M Tris-HCl (pH 8.5), 5 mM EDTA and 6 units of alkaline phosphatase was added to the samples and the mixtures were incubated for 1 h at 37°C to hydrolyze the nucleotides to nucleosides. The nucleoside samples were used for the determination of 8-OHdG by an ELISA kit from Oxis Health Products (Portland, OR), following the manufacturer's protocol.
Abasic sites determination.
The abasic sites [apurinic/apyrimidinic (AP) sites] in mtDNA were determined using a kit from Biovision Research Products (Mountain View, CA) following the manufacturer's instructions.
Mitochondrial DNA deletion analysis.
mtDNA deletions were determined by PCR using a set of primers published by Kakimoto et al. (25) and Fukagawa et al. (15) (Table 2). The set of primers L782 and H1309, L768 and H1296, and L797 and H1309 allowed us to determine the 4,834-bp deletion, which is the common deletion detected in mtDNA under oxidative stress conditions and that has been correlated with the levels of 8-OHdG (23). The sequence primers L1576 and H1626 were used for control amplification of wild-type mtDNA. Sequencing and numbering are based on the published rat mtDNA sequence (13, 16, 45). Briefly, PCR reactions contained 0.2 mM dNTPs, 0.3 mM of each primer, 1 unit of KOD Hot Start DNA polymerase (EMD biosciences) and 0.1 μg of mtDNA template. The thermal cycling conditions were as follows: 95°C for 2 min, followed by 35 cycles at 95°C for 20 s, 60°C for 30 s, 70°C for 1 min., and 70°C for final extension for 5 min. PCR products were resolved on a 1% agarose gel. The gel was then stained with 0.5 μg/ml ethidium bromide in 1× Tris-borate-EDTA buffer. The bands were visualized on a UV transilluminator followed by Polaroid photography and the band intensities of the PCR products were quantified using ImageJ software (35). The levels of mtDNA deletion were expressed as the band intensities relative to that of the control wild-type and normalized to the empty vector-treated cell mtDNA.
Table 2.
Primers used for mtDNA deletion analysis
| Name | Sequence |
|---|---|
| L588 | AGCCGTCCTACTACTTCTCTCACTG |
| L768 | GCTTAGAGCGTTAACCTTTTAAG |
| L782 | TTTCTTCCCAAACCTTTCCT |
| L797 | GTTCCCATCAATTCTATTCC |
| L1576 | GGTTCTTACTTCAGGGGCCATC |
| H1296 | CAGCAGTTATGGATGTGGCG |
| H1309 | AAGCCTGCTAGGATGCTTC |
| H1511 | GGCCTCCGATTCATGTTAAGACTA |
| H1626 | GTGGAATTTTCTGAGGGTAGGC |
From Kakimoto (25).
Mitochondrial membrane potential measurement.
The mitochondrial membrane potential was measured by JC-1 as described previously (17). Briefly, H9C2 cells were loaded with 0.5 mM JC-1 at 37°C for 15 min and washed three times with media. The cells were visualized under a Nikon Diaphot epi-fluorescence microscope equipped with a 40 × fluor objective interfaced with a Photon Technologies, dual emission system, with the excitation wavelength set to 485 nm via a monocromator. Fluorescence emission was split and directed to two photomultiplier tubes through 20 nm band-pass filters centered at 531 and 584 nm, respectively. In addition, an aperture mechanism allowed fluorescence to be collected from a selected area, which was always positioned over the cytoplasmic region of individual cells. Data was shown as a ratio (F584/F531). Loss of mitochondrial membrane potential is indicated by a decrease in the red (F584)/green (F531) fluorescence intensity ratio.
Mitochondrial fragmentation.
After menadione treatment, the mitochondrial morphology was observed. Cells were stained with 10−7 M MitoTracker Green FM for 30 min to visualize mitochondrial morphology and then washed 3 times with media (17). Images were captured with a DeltaVision deconvolution microscope system (Applied Precision) located at the University of California, San Diego Cancer Center microscope facility. Using a 60× (numerical aperture 1.4) lens, images of ∼50 serial optical sections, spaced by 0.2 μm, were acquired. The data sets were deconvolved using SoftWorx software (Applied Precision) on a Silicon Graphics Octane workstation. The morphological changes are described using the parameter of roundness [(perimeter length)2/(4 × π × area)] using ImagePro-PLUS software (Media Cybernetics).
Statistical analysis.
Statistical analysis was performed using the GraphPad Prism version 5.02 for Windows (GraphPad Software). The results were evaluated using a t-test analysis and P < 0.05 was considered statistically significant.
RESULTS
Our results indicate that beneficial effects of OGG1 occur only when cells were exposed to increased ROS levels generated by menadione treatment. There were no differences in the parameters measured when H9C2 cells were not treated with menadione (data not shown). These observations indicated that mitochondrial Ogg1 overexpression does not confer additional protection in nonoxidative stress conditions.
Western blot analysis showed that the mitochondrial OGG1 protein level was increased by 100% in cells transduced with Adv-Ogg1 compared with cells transduced with Adv-Control (Fig. 1). There was ∼35% increase in the nuclear OGG1 protein level in Adv-Ogg1 cells.
Fig. 1.
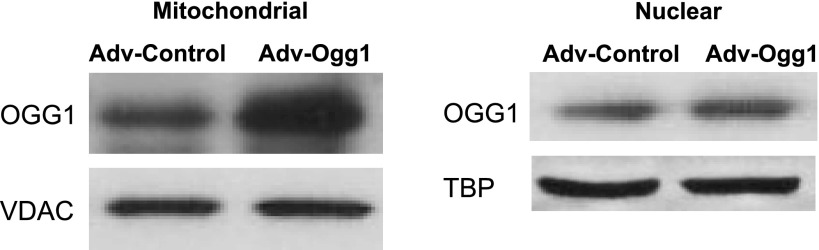
Representative Western blots of mitochondrial and nuclear OGG1 protein levels. Cells transduced with either an empty vector (Adv-Control) or a vector carrying the sequence of mitochondrial Ogg1 (Adv-Ogg1) were treated with menadione (50 μM) for 1 h. After menadione treatment, cells were washed with 1× PBS and incubated in fresh DMEM for 2 h at 37°C and 5% CO2. Mitochondrial and nuclear proteins were isolated and used for Western blot analysis. The results demonstrated increased mitochondrial and nuclear Ogg1 expression in H9C2 cells and are representative of 3 independent experiments.
Mitochondrial DNA damage and base excision repair enzymes.
In H9C2 cells not transduced with Adv, menadione treatment caused a 70% increase in the 8-OHdG compared with non-menadione-treated cells (data not shown). In contrast, Adv-Ogg1 menadione-treated cells showed ∼30% lower levels of mitochondrial 8-OHdG compared with Adv-Control menadione-treated cells. (P < 0.0001, Fig. 2A). Nuclear 8-OHdG was not detected in either Adv-Control or Adv-Ogg1-transduced cells (data not shown), despite the higher nuclear OGG1 protein levels in Adv-Ogg1 cells. The AP sites were also decreased by elevated levels of mitochondria OGG1 (P < 0.005, Fig. 2B). The mRNA levels of Ape1 and DNA polymerase γ (Fig. 2, C and D), downstream enzymes of the BER pathway, were increased in Adv-Ogg1-transduced cells (P < 0.005 and P < 0.001, respectively). These results suggest that elevated levels of mitochondrial OGG1 may upregulate the expression of APE1 and DNA polymerase γ and stimulate the global BER pathway leading to an improvement in DNA repair activity.
Fig. 2.

Markers of DNA damage and mRNA levels of the base excision repair (BER) pathway enzymes were measured in cells exposed to oxidative stress. Ogg1-transduced cells showed significantly reduced mitochondrial DNA (mtDNA) damage and increased expression of BER pathway enzymes. A: mitochondrial 8-OHdG levels normalized to total mtDNA. B: frequency of abasic (AP) sites in mtDNA. C and D: fold changes in the mRNA levels of APE1 and DNA γ polymerase compared with control. Bars represent means ± SE. Experiments were repeated 3 times, and assays were done in triplicate. *P < 0.001, **P < 0.005, #P < 0.0001.
Mitochondrial DNA fragmentation, mtDNA content, and levels of ETC proteins.
High concentrations of mitochondrial 8-OHdG have been correlated with increased mtDNA fragmentation (20). In the current study, mtDNA fragmentation was evaluated by measuring the frequency of a 4,834-bp deletion that has been reported to be one of the most frequent deletions resulting from oxidative damage (13, 16, 45). Figure 3, A and B, shows that cells transduced with Adv-Ogg1 had ∼50% lower levels of mtDNA deletion products (P < 0.05).
Fig. 3.
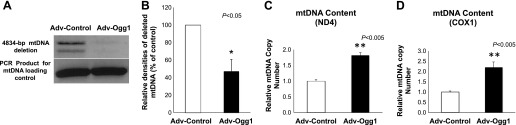
mtDNA deletions and mtDNA content. A: PCR products amplified from mtDNA with a 4,834-bp deletion. PCR products representing a 4,834-bp deletion (top bands) and PCR product of the control primers (bottom bands). B: levels of the 4,834-deleted mtDNA were determined as the ratio of the signal intensity relative to that for the control mtDNA and were expressed as a percentage of levels measured in Adv-Control-treated mtDNA. Ogg1 overexpression significantly reduced mtDNA deletions under oxidative stress conditions. C: mtDNA content was determined by measuring the levels of two genes encoded by mtDNA, (ND4 and COXI) relative to the levels of a nuclear DNA gene (PGC1α), and found to be significantly more abundant in Adv-Ogg1-transduced cells. Graph represents the fold change of mtDNA content relative to control. Bars represent means ± SE. Experiments were repeated 3 times, and assays were done in triplicate. *P < 0.05, **P < 0.005.
Increased mtDNA deletions induced by oxidative damage could cause loss of mtDNA content and affect the expression of the ETC subunits. We determined the mtDNA content in cells transduced with either Adv-control or Adv-Ogg1. Figure 3, C and D, shows that the mtDNA copy number, using two different mitochondrial DNA encoded genes (ND4 and COX1), was higher in the Adv-Ogg1-transduced cells than Adv-control-transduced cells (P < 0.005). In a similar fashion the levels of ND4 and COX1 protein were increased by ∼30% and ∼70%, respectively, in the Adv-Ogg1 transduced cells (Fig. 4, B and C, respectively). The levels of the ETC complexes, as determined using the total OXPHOS antibody cocktail, showed increases in CIII and CIV in Adv-Ogg1 cells (Fig. 4, E and F, respectively, P < 0.05).
Fig. 4.
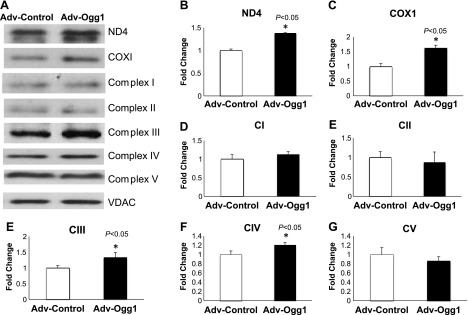
Mitochondrial Ogg1 overexpression prevents loss of mitochondrial DNA-encoded proteins under oxidative stress conditions. A: representative Western blots of ND4 and COX1 mtDNA-encoded subunits and total OXPHOS proteins [from electron transport chain (ETC) complexes I–V]. B–G: graphs represent the fold change of protein levels relative to control. Bars indicated means ± SE. Experiments were repeated 3 times, and assays were done in triplicate. Signal intensities were normalized to VDAC. *P < 0.05 vs. control.
Membrane potential.
Cells overexpressing mitochondrial Ogg1 had an increase in several key ETC proteins. This would presumably increase the oxidative phosphorylation capacity of the mitochondria. Hence, we determined the membrane potential as an indirect measurement of the oxidative phosphorylation (Fig. 5). The cells transduced with Adv-Ogg1 showed higher mitochondrial membrane potential than cells transduced with Adv-Control (P < 0.05).
Fig. 5.

Increased mitochondrial OGG1 protein levels protect against loss of mitochondrial membrane potential under oxidative stress conditions. After menadione treatment cells were loaded with JC-1 for analysis of membrane potential. Graph shows relative mitochondrial membrane potential (red/green fluorescence ratio). n = 30 H9C2, *P < 0.05 vs control. Data are expressed as means ± SE.
Mitochondria fragmentation and fission-fusion proteins.
Oxidative stress induces mitochondrial fragmentation (43). Cells with increased expression of Ogg1 exhibited less mitochondrial fragmentation compared with Adv-Control cells (Fig. 6, P < 0.05).
Fig. 6.

Mitochondrial fragmentation is reduced by mitochondrial Ogg1 overexpression under oxidative stress conditions. After treatment, cells were loaded with Mitotracker Green FM for mitochondria staining. A: representative images of mitochondrial morphology are shown. B: mitochondrial morphological change is analyzed by the mean roundness. n = 15 H9C2 cells, *P < 0.05 vs. control. Data are expressed as means ± SE. The scale bar is 10 μm.
We measured the levels of mitochondrial fission and fusion proteins as evidence of mitochondrial fragmentation (4). Immunoblot analysis shows that proteins inducing mitochondrial fission were reduced in Adv-Ogg1 cells compared with control cells (Fig. 7). For instance, the phosphorylation status of DRP1(ser616) was significantly reduced in cells overexpressing mitochondrial Ogg1 (Fig. 7B, P < 05). In a similar fashion the protein levels of FIS1 (Fig. 7D) and BAX (Fig. 7H) were lower in the Adv-Ogg1 cells (P < 0.05 and P < 0.01, respectively). However, mitochondrial fusion proteins were increased by elevated mitochondrial Ogg1 expression (Fig. 7). Increased phosphorylation of DRP1 on serine 637 is associated with reduced fission events. Adv-Ogg1 cells showed increased phosphorylation of DRP1 (Ser637) compared with Adv-control cells (Fig. 7C, P < 0.01). Similarly the protein levels of OPA1 (Fig. 7G) and MFN2 (Fig. 7E) were higher in Adv-Ogg1 (P < 0.01 and P < 0.05, respectively), which suggested increased fusion events. The higher levels of fusion proteins and the lower levels of fission proteins were in accordance with the decreased mitochondrial fragmentation seen in Adv-Ogg1-transduced cells (Fig. 6).
Fig. 7.
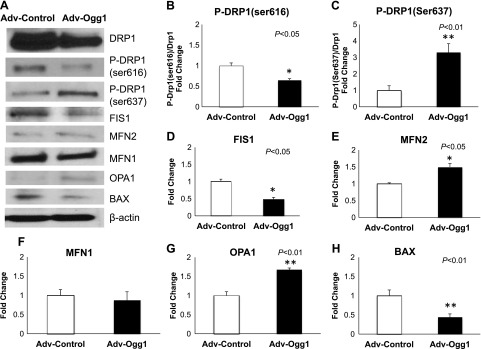
Effects of mitochondrial Ogg1 overexpression on fission and fusion proteins under oxidative stress conditions. A: representative Western blots of fission proteins [DRP1, p-DRP1(ser616), FIS1, and Bax] and fusion proteins [OPA1, MFN1, MFN2, p-DRP1(ser637)]. Immunoblot analysis showed that mitochondrial fission and mitochondrial fusion proteins were reduced and increased, respectively, in Adv-Ogg1 transduced cells. B–H: graphs represent the fold change of protein levels relative to control. Bars indicated means ± SE. Experiments were repeated 3 times, and assays were done in triplicate. *P < 0.05, **P < 0.01.
Apoptosis.
The results shown above indicate that protecting mtDNA against oxidative stress damage leads to decreased mitochondrial fragmentation and lower expression of mitochondrial fission proteins. Increased mitochondrial fission events are normally associated with apoptosis (18). Several parameters of the apoptotic cascade were measured to determine whether apoptosis was decreased in relation to lower mitochondrial fragmentation accompanied by overexpression of mitochondrial Ogg1 (Fig. 8A). For instance, the ratio of mitochondrial cytochrome c/cytoplasmic cytochrome c was significantly higher in cells transduced with Adv-Ogg1 (Fig. 8B, P < 0.0001). The activated caspase-3 (Fig. 8C) and activated caspase-9 (Fig. 8D) protein levels were lower in the Adv-Ogg1 transduced cells than in the Adv-control cells (P < 0.05). This outcome suggests that increasing mitochondrial Ogg1 expression in oxidative stress conditions enhances cell survival.
Fig. 8.
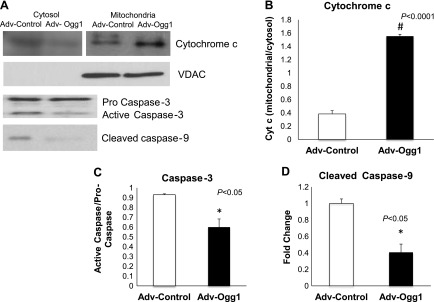
A: representative Western blots showing markers of apoptosis in H9C2 cells after menadione treatment. Ogg1 overexpression prevents cytochrome c release from the mitochondria and leads to decreased protein levels of activated caspases. The scans of cytochrome c levels in mitochondrial and cytosolic fractions are from two separate blots. B–D: graphs represent the fold change of protein levels relative to control. Bars indicated means ± SE. Experiments were repeated 3 times, and assays were done in triplicate.*P < 0.05, #P < 0.0001.
DISCUSSION
The mitochondrial DNA base modification 8-OHdG is the major DNA lesion induced by oxidative stress and is considered a marker of oxidative DNA damage (27, 28). Elevated levels of 8-OHdG have been associated with increased mtDNA deletions, mutation, and mtDNA loss (3, 6, 15, 21). For instance, the levels of 8-OHdG and the mtDNA content were 50% higher and 75% lower, respectively, in the left ventricular tissue from end-stage heart failure patients compared with non-heart failure patients (22). Tutsui et al. found that a decrease in mtDNA content and decline in mitochondrial function play major roles in the development of heart failure that occurs after myocardial infarction (37, 38). In addition, age-related progressive decline of ventricular performance and a correlative increase in 8-OHdG with mtDNA deletions has been demonstrated in normal subjects, as well as positive controls. Increased mtDNA deletions have been proposed as the molecular basis for progressive mitochondrial dysfunction and the decline of cellular activity (24, 28). In kidneys of diabetic rats, a correlation between the levels of mitochondrial 8-OHdG and a 4,834-bp mtDNA deletion was suggested to be involved in the pathogenesis of diabetic nephropathy (25). In this study, we found that H9C2 cardiac muscle cells with higher concentrations of 8-OHdG had elevated levels of a 4,834-bp mtDNA deletion and lower mtDNA content. However, we showed that decreasing the formation of 8-OHdG by elevated expression of mitochondrial Ogg1 was associated with decreased mtDNA deletions and higher mtDNA content. Importantly, we found that even when there was an increased expression of nuclear Ogg1 in Adv-Ogg1 cells, we failed to detect 8-OHdG in the nuclear DNA of both Adv-control and Adv-Ogg1 cells. Our results bring more evidence that mtDNA is more susceptible to oxidative damage and that the nucleus has more efficient DNA repair mechanisms, as other authors have reported (44). In accordance with our findings, Chouteau et al. (7) failed to detect glucose oxidase-induced nuclear DNA damage in rat lungs, but mtDNA lesion was prevented by OGG1 fusion protein accumulated in lung cell mitochondria within 30 min of addition to the perfusion medium. While in our study the mechanisms of action of OGG1 seem to be associated with increased mtDNA repair activity, in the work by Chouteau et al. the effects seem to be associated with other rapid actions of OGG1 such as stabilizing mtDNA-protein interactions, as speculated by these authors. Further studies exploring the short-and long-terms effects of increased mitochondrial Ogg1 expression under oxidative stress conditions will be useful to elucidate the different mechanisms of action of OGG1.
Elevated OGG1 activity may paradoxically be associated with increased mtDNA fragmentation. OGG1 carries out the recognition and removal of 8-OHdG, leading to the formation of AP sites, which are then processed by APE1. As such, excessive OGG1 activity could theoretically overcome the repair capacity of APE1 which could lead to excessive numbers of AP sites, triggering single- and double-strand breaks and large mtDNA deletions. In practice, we found that this did not occur and that cells with high levels of OGG1 had lower levels of AP sites. Interestingly, we also found that the mRNA levels of Ape1 and DNA poly γ, both downstream enzymes in the BER pathway, were increased in cells overexpressing mitochondrial Ogg1. These results suggest that elevated levels of mitochondrial OGG1 may upregulate the expression of Ape1 and DNA poly γ and stimulate the global BER pathway leading to an improvement in DNA repair activity. This outcome supports the mtDNA hypothesis of OGG1 as the rate-limiting enzyme in the BER pathway, as suggested by other investigators (34). Thus it is possible that any event that adversely affects mitochondrial Ogg1 expression and/or activity would trigger a global BER pathway dysfunction and would consequently increase mtDNA damage. Indeed, studies have found that decreased BER activity, mostly by a downregulation of mitochondrial Ogg1 activity, was associated with increased mtDNA deletions in a rat model of aging (46).
The mtDNA deletions resulting from excessive 8-OHdG comprise a region that encodes protein subunits of ETC complex I, complex IV, and complex V. As such, these mtDNA deletions can decrease the protein levels of the ETC subunits and adversely affect the activity of the ETC. We observed that the protein levels of ND4 and COXI, mtDNA-encoded subunits of complex I and IV, respectively, were elevated in cells with high levels of mitochondrial OGG1. These outcomes highlight the important role of OGG1 in maintaining the integrity of mtDNA and preventing the loss of the mitochondrial protein subunits of the ETC.
Oxidative stress increases fission and fragmentation of the mitochondria, promotes the mitochondrial permeability transition pore (mPTP) opening, collapses the mitochondrial membrane potential, decreases ATP synthesis, and induces release of cytochrome c from mitochondria which activates the caspase cascade and increases apoptosis (18, 39, 42, 43). Numerous studies have demonstrated that oxidative stress damage of mainly mtDNA may be the factor responsible for triggering those adverse effects (2, 19, 20, 22, 23, 33, 45). Chatterjee et al. (5) demonstrated that mitochondrially targeted mutant OGG1 resulted in more cell death than nuclear-targeted mutant OGG1 upon exposure of cells to oxidative damage. Those results suggested that deficiencies of mitochondrial OGG1 lead to reduced mitochondrial DNA integrity, resulting in decreased cell viability. In this context, in vitro studies using different cell lines have shown that increased expression of mitochondrial Ogg1 as a means to repair damaged mtDNA attenuates or prevents mitochondrial dysfunction and apoptosis. For instance, targeting human OGG1 to mitochondria enhanced mtDNA repair and cellular survival after oxidative stress in HeLa cells (11). In a similar fashion, overexpression of Ogg1 in mitochondria reduced the release of cytochrome c from the inner mitochondrial membrane and the activation of caspase 9 in oligodendrocytes after exposure to menadione (12). In pulmonary artery endothelial cells overexpression of mitochondrial Ogg1 protected the cells against xanthine oxidase (XO)-induced mtDNA damage and cell death (10). A similar study in the same cell line found that increased activity of mitochondrial OGG1 suppressed the XO-induced mtDNA damage and the XO-mediated loss of mitochondrial membrane potential (33). Interestingly, mitochondrial Ogg1 overexpression attenuated XO-induced apoptosis as determined by suppression of caspase-3 activation, by reduced DNA fragmentation, and by blunted appearance of condensed, fragmented nuclei (33). In INS-1 cells, Ogg1 overexpression in mitochondria decreased fatty acid-induced inhibition of ATP production and protected cells from apoptosis (32). A recent study using transgenic mice with cardiac-specific overexpression of active mitochondrial Ogg1 presented reduced cardiac fibrosis following transaortic constriction (41). Although the authors did not find any significant reduction in cardiac hypertrophy or improvement in cardiac function measured by echocardiography 13 wk after surgery, they did not evaluate any parameter directly associated with mitochondrial dysfunction, or apoptosis. We observed, however, that increased expression of mitochondrial Ogg1 in H9C2 cells had an impact on mitochondrial function. Increased levels of mitochondrial OGG1 in H9C2 cells attenuated the loss of mitochondrial membrane potential and decreased mitochondrial fragmentation induced by menadione treatment. Decreased mitochondrial fragmentation was associated with decreased expression of fission proteins (e.g., DRP1 and FIS1) and with increased expression of fusion proteins (e.g., MFN2 and OPA1). These results show for the first time that mtDNA integrity may play a role in the regulation of mitochondrial fission and fusion events. Analysis of apoptotic markers indicated that higher and lower levels of mitochondrial and cytosolic cytochrome c, respectively, reflected protection of mtDNA against apoptosis by Ogg1 overexpression. In addition, the levels of activated caspase 3 and activated caspase 9 were lower in cells overexpressing mitochondrial Ogg1. Our results provide evidence that efficient mtDNA repair activity in cardiac cells exposed to oxidative stress conditions is crucial to maintaining mitochondrial function and cell viability. Moreover, these findings suggest that mitochondrial OGG1 is the rate-limiting enzyme in the mtDNA-repair pathway.
In summary, mitochondrial overexpression of Ogg1 decreased the formation of 8-OHdG induced by oxidative stress. In addition, decreased levels of 8-OHdG were associated with increased mtDNA content, decreased extensive mtDNA deletions, as well as with improved mitochondrial function and cell survival.
GRANTS
This work was supported by National Institutes of Health Grants 5R01HL066917-12 (to W. H. Dillman) and 3T32DK007044-31S1 (to W. H. Dillman/M. Torres-Gonzalez), UCSD Neuroscience Microscopy Facility Grant P30 NS047101, and the Department of Veteran's Affairs Merit Review Award 5I01BX001121-02 (to W. H. Dillman). This work was also supported by a grant from the P. Robert Majumder Charitable Foundation.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M.T.-G. and W.H.D. conception and design of research; M.T.-G., T.G., and B.T.S. performed experiments; M.T.-G., T.G., H.K., and B.T.S. analyzed data; M.T.-G., T.G., H.K., B.T.S., and W.H.D. interpreted results of experiments; M.T.-G. prepared figures; M.T.-G. drafted manuscript; M.T.-G., B.T.S., and W.H.D. edited and revised manuscript; M.T.-G., T.G., H.K., B.T.S., and W.H.D. approved final version of manuscript.
REFERENCES
- 1.Boesch P, Weber LF, Ibrahim N, Tarasenko V, Cosset A, Paulus F, Lightowlers RN, Dietrich A. DNA repair in organelles: pathways, organization, regulation, relevance in disease and aging. Biochim Biophys Acta 1813: 186–200, 2011 [DOI] [PubMed] [Google Scholar]
- 2.Bravard A, Vacher M, Gouget B, Coutant A, de Boisferon FH, Marsin S, Chevillard S, Radicella JP. Redox regulation of human OGG1 activity in response to cellular oxidative stress. Mol Cell Biol 26: 7430–7436, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cassano P, Sciancalepore AG, Lezza AM, Leeuwenburgh C, Cantatore P, Gadaleta MN. Tissue-specific effect of age and caloric restriction diet on mitochondrial DNA content. Rejuvenation Res 9: 211–214, 2006 [DOI] [PubMed] [Google Scholar]
- 4.Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet 46: 265–287, 2012 [DOI] [PubMed] [Google Scholar]
- 5.Chatterjee A, Mambo E, Zhang Y, Deweese T, Sidransky D. Targeting of mutant hogg1 in mammalian mitochondria and nucleus: effect on cellular survival upon oxidative stress. BMC Cancer 6: 235, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chou YF, Yu CC, Huang RF. Changes in mitochondrial DNA deletion, content, and biogenesis in folate-deficient tissues of young rats depend on mitochondrial folate and oxidative DNA injuries. J Nutr 137: 2036–2042, 2007 [DOI] [PubMed] [Google Scholar]
- 7.Chouteau JM, Obiako B, Gorodnya OM, Pastukh VM, Ruchko MV, Wright AJ, Wilson GL, Gillespie MN. Mitochondrial DNA integrity may be a determinant of endothelial barrier properties in oxidant-challenged rat lungs. Am J Physiol Lung Cell Mol Physiol 301: L892–L898, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ciapaite J, Bakker SJ, Van Eikenhorst G, Wagner MJ, Teerlink T, Schalkwijk CG, Fodor M, Ouwens DM, Diamant M, Heine RJ, Westerhoff HV, Krab K. Functioning of oxidative phosphorylation in liver mitochondria of high-fat diet fed rats. Biochim Biophys Acta 1772: 307–316, 2007 [DOI] [PubMed] [Google Scholar]
- 9.Coenen MJ, van den Heuvel LP, Smeitink JA. Mitochondrial oxidative phosphorylation system assembly in man: recent achievements. Curr Opin Neurol 14: 777–781, 2001 [DOI] [PubMed] [Google Scholar]
- 10.Dobson AW, Grishko V, LeDoux SP, Kelley MR, Wilson GL, Gillespie MN. Enhanced mtDNA repair capacity protects pulmonary artery endothelial cells from oxidant-mediated death. Am J Physiol Lung Cell Mol Physiol 283: L205–L210, 2002 [DOI] [PubMed] [Google Scholar]
- 11.Dobson AW, Xu Y, Kelley MR, LeDoux SP, Wilson GL. Enhanced mitochondrial DNA repair and cellular survival after oxidative stress by targeting the human 8-oxoguanine glycosylase repair enzyme to mitochondria. J Biol Chem 275: 37518–37523, 2000 [DOI] [PubMed] [Google Scholar]
- 12.Druzhyna NM, Hollensworth SB, Kelley MR, Wilson GL, Ledoux SP. Targeting human 8-oxoguanine glycosylase to mitochondria of oligodendrocytes protects against menadione-induced oxidative stress. Glia 42: 370–378, 2003 [DOI] [PubMed] [Google Scholar]
- 13.Edris W, Burgett B, Stine OC, Filburn CR. Detection and quantitation by competitive PCR of an age-associated increase in a 4.8-kb deletion in rat mitochondrial DNA. Mutat Res 316: 69–78, 1994 [DOI] [PubMed] [Google Scholar]
- 14.Fernandez-Vizarra E, Tiranti V, Zeviani M. Assembly of the oxidative phosphorylation system in humans: what we have learned by studying its defects. Biochim Biophys Acta 1793: 200–211, 2009 [DOI] [PubMed] [Google Scholar]
- 15.Fukagawa NK, Li M, Liang P, Russell JC, Sobel BE, Absher PM. Aging and high concentrations of glucose potentiate injury to mitochondrial DNA. Free Radic Biol Med 27: 1437–1443, 1999 [DOI] [PubMed] [Google Scholar]
- 16.Gadaleta M, Rainaldi G, Lezza AM, Milella F, Fracasso F, Cantatore P. Mitochondrial DNA copy number and mitochondrial DNA deletion in adult and senescent rats. Mutat Res 275: 181–193, 1992 [DOI] [PubMed] [Google Scholar]
- 17.Gawlowski TSJ, Scott B, Torres-Gonzalez M, Wang H, Schwappacher R, Han X, Yates J, Hoshijima M, Dillmann WH. Modulation of dynamin-related protein 1 (DRP1) function by increased O-linked-β-N-acetylglucosamine modification (O-GlcNAc) in cardiac myocytes. J Biol Chem 287: 30024–30024, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13: 589–598, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gredilla R, Bohr VA, Stevnsner T. Mitochondrial DNA repair and association with aging—an update. Exp Gerontol 45: 478–488, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hayakawa MKK, Yoneda M, Tanaka M, Sugiyama S, Ozawa T. Age-related extensive fragmentation of mitochondrial DNA into minicircles. Biochem Biophys Res Commun 226: 369–377, 1996 [DOI] [PubMed] [Google Scholar]
- 21.Hiona A, Sanz A, Kujoth GC, Pamplona R, Seo AY, Hofer T, Someya S, Miyakawa T, Nakayama C, Samhan-Arias AK, Servais S, Barger JL, Portero-Otin M, Tanokura M, Prolla TA, Leeuwenburgh C. Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS One 5: e11468, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, Utsumi H, Hamasaki N, Takeshita A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res 88: 529–535, 2001 [DOI] [PubMed] [Google Scholar]
- 23.Jiang ZBQ, Sun L, Huang X, Wang T, Zhang S, Li H, Zhang L. Possible role of mtDNA depletion and respiratory chain defects in aristolochic acid I-induced acute nephrotoxicity. Toxicol Appl Pharmacol 266: 198–203, 2013 [DOI] [PubMed] [Google Scholar]
- 24.Kajander OAKP, Jacobs HT. The relationship between somatic mtDNA rearrangements, human heart disease and aging. Hum Mol Genet 3: 317–324, 2002 [DOI] [PubMed] [Google Scholar]
- 25.Kakimoto M, Inoguchi T, Sonta T, Yu HY, Imamura M, Etoh T, Hashimoto T, Nawata H. Accumulation of 8-hydroxy-2′-deoxyguanosine and mitochondrial DNA deletion in kidney of diabetic rats. Diabetes 51: 1588–1595, 2002 [DOI] [PubMed] [Google Scholar]
- 26.Karamanlidis G, Nascimben L, Couper GS, Shekar PS, del Monte F, Tian R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ Res 106: 1541–1548, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klungland A, Bjelland S. Oxidative damage to purines in DNA: role of mammalian Ogg1. DNA Repair (Amst) 6: 481–488, 2007 [DOI] [PubMed] [Google Scholar]
- 28.Lu AL, Li X, Gu Y, Wright PM, Chang DY. Repair of oxidative DNA damage: mechanisms and functions. Cell Biochem Biophys 35: 141–170, 2001 [DOI] [PubMed] [Google Scholar]
- 29.Marin-Garcia J, Goldenthal MJ. The mitochondrial organelle and the heart. Rev Esp Cardiol 55: 1293–1310, 2002 [DOI] [PubMed] [Google Scholar]
- 30.Marshall OJ. PerlPrimer: cross-platform, graphical primer design for standard, bisulphite and real-time PCR. Bioinformatics 20: 2471–2472, 2004 [DOI] [PubMed] [Google Scholar]
- 31.Qian W, Van Houten B. Alterations in bioenergetics due to changes in mitochondrial DNA copy number. Methods 51: 452–457, 2010 [DOI] [PubMed] [Google Scholar]
- 32.Rachek LI, Thornley NP, Grishko VI, LeDoux SP, Wilson GL. Protection of INS-1 cells from free fatty acid-induced apoptosis by targeting hOGG1 to mitochondria. Diabetes 55: 1022–1028, 2006 [DOI] [PubMed] [Google Scholar]
- 33.Ruchko M, Gorodnya O, LeDoux SP, Alexeyev MF, Al-Mehdi AB, Gillespie MN. Mitochondrial DNA damage triggers mitochondrial dysfunction and apoptosis in oxidant-challenged lung endothelial cells. Am J Physiol Lung Cell Mol Physiol 288: L530–L535, 2005 [DOI] [PubMed] [Google Scholar]
- 34.Ruchko MV, Gorodnya OM, Zuleta A, Pastukh VM, Gillespie MN. The DNA glycosylase Ogg1 defends against oxidant-induced mtDNA damage and apoptosis in pulmonary artery endothelial cells. Free Radic Biol Med 50: 1107–1113, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh KK, Sigala B, Sikder HA, Schwimmer C. Inactivation of Saccharomyces cerevisiae OGG1 DNA repair gene leads to an increased frequency of mitochondrial mutants. Nucleic Acids Res 29: 1381–1388, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsutsui H, Kinugawa S, Matsushima S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc Res 81: 449–456, 2009 [DOI] [PubMed] [Google Scholar]
- 38.Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and mitochondrial DNA damage in heart failure. Circ J 72, Suppl A: A31–A37, 2008 [DOI] [PubMed] [Google Scholar]
- 39.Turner NA, Xia F, Azhar G, Zhang X, Liu L, Wei JY. Oxidative stress induces DNA fragmentation and caspase activation via the c-Jun NH2-terminal kinase pathway in H9c2 cardiac muscle cells. J Mol Cell Cardiol 30: 1789–1801, 1998 [DOI] [PubMed] [Google Scholar]
- 40.Vongsamphanh R, Wagner JR, Ramotar D. Saccharomyces cerevisiae Ogg1 prevents poly(GT) tract instability in the mitochondrial genome. DNA Repair (Amst) 5: 235–242, 2006 [DOI] [PubMed] [Google Scholar]
- 41.Wang J, Wang Q, Watson LJ, Jones SP, Epstein PN. Cardiac overexpression of 8-oxoguanine DNA glycosylase 1 protects mitochondrial DNA and reduces cardiac fibrosis following transaortic constriction. Am J Physiol Heart Circ Physiol 301: H2073–H2080, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wei YH, Wu SB, Ma YS, Lee HC. Respiratory function decline and DNA mutation in mitochondria, oxidative stress and altered gene expression during aging. Chang Gung Med J 32: 113–132, 2009 [PubMed] [Google Scholar]
- 43.Wu S, Zhou F, Zhang Z, Xing D. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission-fusion proteins. FEBS J 278: 941–954, 2011 [DOI] [PubMed] [Google Scholar]
- 44.Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci USA 94: 514–519, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yowe DL, Ames BN. Quantitation of age-related mitochondrial DNA deletions in rat tissues shows that their pattern of accumulation differs from that of humans. Gene 209: 23–30, 1998 [DOI] [PubMed] [Google Scholar]
- 46.Zhong Y, Hu YJ, Chen B, Peng W, Sun Y, Yang Y, Zhao XY, Fan GR, Huang X, Kong WJ. Mitochondrial transcription factor A overexpression and base excision repair deficiency in the inner ear of rats with d-galactose-induced aging. FEBS J 278: 2500–2510, 2011 [DOI] [PubMed] [Google Scholar]