

Abstract
The reactivity of three cytotoxic trans-PtII complexes bearing aliphatic amine ligands, with transferrin and single-stranded oligonucleotides as DNA models, was investigated by ESI-MS and the results obtained are discussed in comparison with cisplatin. Tandem MS studies provided additional information on the preferential Pt binding sites. To determine whether trans-PtII complexes can migrate from a peptide to an oligonucleotide, transfer experiments were also performed using ESI-MS, and competitive binding of the trans-PtII complexes toward a model peptide and different oligonucleotides was also investigated. Significant differences in the reactivity of the trans complexes with respect to cisplatin were observed. In general, adduct formation with the selected peptide is favored for the trans compounds, whereas cisplatin shows a preference for oligonucleotides, especially if adjacent G–G residues are present. The results are discussed in relation to the possible mechanism of action of the trans-PtII complexes.
Keywords: cancer, mass spectrometry, oligonucleotides, peptides, platinum
Introduction
The search for new anticancer platinum drugs with improved pharmacological properties over those of cisplatin has been focused on cis geometric compounds for a long time. This bias originates from the fact that transplatin, the trans isomer of cisplatin, is not an active anticancer agent.[1,2] However, since the first reports on trans-Pt complexes with relevant in vitro cytotoxicity, and in some cases endowed with significant in vivo activity,[3–6]
trans-platinum complexes have been widely investigated as “rule breaker” anticancer drugs. Some of these compounds, with aliphatic amines as ligands (non-leaving groups), of general formula trans-[PtLL′Cl2] (L and L′=aliphatic amines), were developed by some of us.[7] The prototypical compound of this family is trans-[PtCl2(dma)(ipa)] (1) (dma=dimethylamine, ipa=isopropylamine);[8] notably, this compound was able to overcome cisplatin resistance in tumor cells overexpressing the ras oncogene and exhibited promising effects in tumor xenografts.[7,9] Compound 1 has also been shown to readily react with DNA, the cellular target for cisplatin,[10] forming interstrand cross-links.[8,11] Subsequently, trans-[PtCl2(dma)(ma)] (2) (ma=methylamine) was found to be more cytotoxic than 1.[12]

As the mechanism of action of this promising family of Pt complexes is not fully known, and might involve interactions with biomolecules other than DNA, adduct formation with proteins must also be considered. Notably, the exact mechanism of platinum drug delivery into cells is still not elucidated, although it appears that cisplatin enters cells by both passive diffusion and facilitated transporter-mediated uptake.[13–15] In the cytoplasm, which has a chloride concentration of ~4 mm compared with > 100 mm in the bloodstream, cisplatin undergoes aquation and hydrolysis reactions to generate various reactive platinum species that can bind to several nucleophiles, including glutathione, metallothionein and DNA nucleobases.[16]
There are currently only a few published studies that describe the interactions of trans-platinum(II) complexes with proteins.[17–24] The reactivity of 1 and 2 with the model protein cytochrome c was studied by electrospray ionization mass spectrometry (ESI-MS) and the nature of protein-bound metal-containing molecular fragments was identified.[25] In contrast with the results obtained for cisplatin and carboplatin,[26,27] the platinum-bound aliphatic amine ligands were not lost upon protein binding. In addition, inductively coupled plasma optical emission spectroscopy (ICP-OES) was used to evaluate the degree of platination of serum proteins by 1 and 2, showing that platinum binding was not only relevant, but similar to cisplatin. Met65 was also identified as the coordination site for these complexes by 2D [1H15N] and [1H13C] HSQC NMR and 2D [1H1H] NOESY NMR experiments.[25]
Among the proteins that are involved in metallodrug transport, delivery, and storage, serum transferrin (Tf) has been widely investigated.[28] Tfs are a family of large non-heme iron-binding glycoproteins present in the blood and other mammalian fluids including bile, amniotic fluid, cerebrospinal fluid, lymph, colostrum, and milk, and play a major role in maintaining iron homeostasis by sequestering free iron in the extracellular milieu to deliver it to cells and tissues. The concentration of Tf in blood is ~2.5 gL−1 (35 μm), in which ~30% is loaded with two Fe3+ ions. When Tf is saturated with iron it binds strongly to its receptor, whereupon it is internalized by cells, the iron is released, and the protein is re-circulated. Human serum Tf is a single-chain peptide with a molecular mass of 79570 Da, containing 679 amino acids and comprising two structurally related lobes referred to as the N and C lobes.[29] Each lobe is further divided into two subdomains by a cleft, with each cleft able to coordinate an Fe3+ ion via two tyrosine residues, an aspartate and a histidine residue, as well as an exogenous bidentate anion (a so-called “synergistic anion”), usually carbonate,[30] forming a pseudo-octahedral geometry.
Because the role of Tf in the mechanism of action of trans-PtII complexes has yet to be explored, and in view of the fact that protein-bound metallofragments could represent active anti-cancer species, we studied the reactivity of 1 and 2 and their analogue trans-[PtCl2(ipa)(ma)][31] (3) with Tf using ESI-MS. The reactivity of the complexes toward single-stranded oligonucleotides was also studied and compared with cisplatin. Moreover, to elucidate whether the PtII ion can bind selectively or could be transferred from a peptide or protein such as Tf to another biomolecular target such as DNA, competitive binding toward a model peptide, mimicking one of the iron-binding pockets of Tf, and different oligonucleotides was also investigated by ESI-MS. The outcome of these studies is described herein.
Experimental Section
Reagents
Human serum transferrin, cisplatin, ammonium bicarbonate and ammonium acetate were purchased from Sigma–Aldrich and used as received. The trans-platinum complexes were prepared using published protocols.[7] HPLC-purified synthetic oligonucleotides were purchased from Microsynth (Balgach, Switzerland) and DNA Technology A/S (Risskov, Denmark); the HPLC-purified model peptide (KDCHLAQVPSHTV) was obtained from PSL (Heidelberg, Germany). HPLC-grade solvents (water, n-propanol, and methanol) were obtained from Acros (Geel, Belgium).
ESI-MS
ESI-MS data of Tf adducts were recorded on an Ultima II q-TOF mass spectrometer (Waters, Manchester, UK) operated in positive ion mode. Data acquisition and analysis were carried out using the MassLynx software package (Waters). The instrument was calibrated daily using a 0.01% phosphoric acid solution in 50% acetonitrile. Samples were prepared with a metallodrug/protein ratio of 3:1 in 20 mm carbonate buffer (pH 7.4) and incubated for up to 24 h at 37°C. Prior to analysis, samples were ultracentrifuged using 30 kDa cutoff filters (VWR, Switzerland) to remove unbound metallodrugs and injected into the ESI-MS system after dilution with acetonitrile and formic acid (final concentrations 25% and 0.05%, respectively).
For ESI-MS analysis of the oligonucleotides, samples contained a complex/oligonucleotide ratio of 3:1 (75:25 mm) in 10 mm ammonium acetate (pH 7.4) and were incubated for 24 h at 37°C. ESI-MS data were recorded using the above-mentioned q-TOF instrument in negative ion mode by diluting the samples 1:1 with methanol.
For ESI-MS experiments with the model peptide, samples were prepared with a metallodrug/peptide ratio of 3:1 (75:25 μm) in 20 mm ammonium hydrogen carbonate (pH 7.4) and incubated for 24 h at 37°C prior to analysis. Samples were then mixed with acetonitrile and formic acid (final concentration 25% and 0.05%, respectively) and placed into a 96-well plate in an Advion TriVersa robot (Advion Biosciences, Ithaca, NY, USA) equipped with a 5.5 μm nozzle chip. The ESI robot was controlled with ChipSoft v. 7.2.0 software with a gas pressure of 0.45 psi and voltage set at 1.7 kV. The samples were analyzed in positive ion mode using an LTQ XL mass spectrometer (ThermoFisher Scientific, Bremen, Germany). The same instrument was also used for fragmentation studies for the oligonucleotides; in this case, samples were diluted 10-fold with a solution containing methanol/water/n-propanol in a ratio of 65:20:5 immediately prior to analysis. For all measurements, the mass spectrometer was operated in enhanced resolution mode, and for fragmentation experiments an isolation width of 3.0 m/z and a relative collision energy of 35% were used. The Xcalibur software package was used for data acquisition and data analysis. For identification of unmodified fragments, the web-based Mongo Oligo Mass calculator v. 2.06 was used:[32] http://library.med.utah.edu/masspec/mongo.htm (accessed May 17, 2010).
For transfer experiments, the peptide was incubated for 24 h at 37°C with the corresponding metallodrug in a 1:1 ratio (50 μm) in 20 mm ammonium hydrogen carbonate (pH 7.4). Before adding an equimolar amount of an oligonucleotide, binding of the metallodrugs to the peptide was checked by ESI-MS. Monitoring of the free Pt complexes as well as drug–peptide adducts assured that only a very small amount of platinum compounds, if any, was still present free in solution before adding the oligonucleotide. Subsequently, the mixture was incubated for 72 h, with samples taken after 24 and 48 h. Immediately prior to analysis, methanol and formic acid were added (final respective concentrations of 50 and 0.05%). For analysis of the metallodrug–peptide adducts, the q-TOF instrument was operated in positive ion mode and metallodrug–oligonucelotide adducts were analyzed in negative ion mode. Instrument settings in positive ion mode: capillary voltage 4.0 kV, cone voltage 70 V, source temperature 100°C, desolvation temperature 200°C. Instrument settings in negative ion mode: capillary voltage −4.5 kV, cone voltage −55 V, source temperature 80°C, desolvation temperature 100°C.
In the case of direct competition experiments, samples containing a metallodrug/peptide/oligonucleotide ratio of 1:1:1 (25 μm) and incubated for up to 72 h (aliquots taken after 24 and 48 h) at 37°C were analyzed as described for the transfer experiments (Table 1).
Table 1.
Sequences and monoisotopic masses of the oligonucleotides and model peptide.
| Sequence | Mass [Da] | |
|---|---|---|
| SS1 | 5′-d(TACACACCGGTAC)-3′ | 3901.7 |
| SS2 | 5′-d(TAATTAAGCATAATAT)-3′ | 4885.9 |
| Tf-Peptide | H-KDCHLAQVPSHTV-OH | 1433.7 |
Results and Discussion
Transferrin and peptide binding studies
The reactivity of 1–3 with human apo-Tf under simulated physiological conditions (pH 7.4; 37°C) was studied at a drug/protein molar ratio of 3:1. As can be seen in Figure 1 for compound 3, binding toward Tf is observed after 24 h incubation, but with the major peaks corresponding to the unmodified protein. Deconvolution of the spectrum yields 79550 Da as the mass of the protein, which is in good agreement with published data; other peaks of much lower relative intensity between 79250 and 79 700 Da probably correspond to post-transitionally modified isoforms of the glycoprotein.[33] Additionally, an intense peak at 79834 Da, corresponding to a Tf–[Pt-(ipa)(ma)] adduct (calculated mass increase: 283 Da), is observed in the spectrum. Similar spectra were obtained for compounds 1 and 2; the formed adducts correspond to the complexes after loss of the chlorido ligands. After an incubation period of 24 h the adducts reach a maximum relative intensity of ~19% in the case of 2 and 3, but only 9% for 1. This percentage is relatively low in comparison with cisplatin, for which only a minor peak corresponding to the unmodified protein is detected after 24 h, as observed in previous ESI-MS, capillary electrophoresis–ICP-MS and LC–ICP-MS studies.[34–36]
Figure 1.

ESI mass spectra of Tf incubated with 3 for 24 h under simulated physiological conditions: spectrum at left shows the charge envelope of both the pure and modified (★) protein; at right is the deconvoluted spectrum.
Only mono-adducts were detected in which the metallofragments retain both the amine ligands. This was to be expected, as chlorido ligands are displaced more readily by aqua intermediates to finally afford protein binding. Although caution must be taken in quantification of the formed adducts, it seems that protein binding is significantly less pronounced for complex 1 than it is for 2 and 3. These results are in good agreement with previously reported studies on cytochrome c,[25] and with a study concerning transplatin–protein interactions.[26] A possible explanation for the decreased reactivity of 1 relative to 2 and 3 might be the presence of bulkier amine ligands (dma and ipa), which impede coordination of the PtII center to the protein. The differences in the binding rate of 1–3 may be related to differences in their aquation behavior. [1H15N] HSQC NMR spectroscopic studies of the aquation of trans-[PtCl2(15N-amine)(15N-amine)] complexes 1–3 showed that the mono-aquation step is rapid (t½ : 1.3–3.5 h at 298 K) and faster than that observed for cisplatin (t½: 8.1 h), [37] but aquation occurs only to a limited extent, with the equilibrium favoring the dichlorido species, and formation of the diaqua complexes is scarcely observed.[12] Aquation is significantly slower for 1 and 3 than it is for 2, which presumably contributes to the slower reactivity of 1 with Tf. Although aquation occurs more rapidly for 2, it is the least aquated of the three complexes at equilibrium. Notably, 2 is more cytotoxic than 1 and 3, suggesting an important role for hydrolysis in the mechanism of antitumor activity.
Tf peptide mimic
To identify which amino acids might be responsible for adduct formation between the Pt compounds and the protein, 1–3 were incubated for up to 24 h with a peptide resembling one of the binding pockets of Tf, and the adducts were subsequently analyzed by tandem MS. Collision-induced dissociation (CID) is a widely employed technique for the characterization of peptides in proteomics and can also be used to identify modifications of single amino acid residues. The technique has only been used recently to determine metallodrug binding sites on proteins,[21] with bottom-up approaches previously used.[38]
The metallodrugs form bifunctional adducts with the peptide, i.e., [Pep+Pt(amine)(amine′)], following loss of the two chlorido ligands. Intense peaks exhibiting isotopic patterns characteristic for platinum at m/z: 578.2, 568.8, and 573.5 (most intense isotope, 3+ charge) are observed for compounds 1, 2, and 3, respectively, which were subsequently subjected to CID. As expected, a similar pattern was identified in the fragment ion spectra for all three trans-Pt complexes; the two major peaks in the spectra correspond to the intact peptide with platinum and one of the amine ligands, resulting in signals at m/z: 558.4 and 563.1 assigned to [Pep+Pt(dma)]3+ and [Pep+Pt(ipa)]3+ in the case of 1, as depicted in Figure 2.
Figure 2.

CID spectrum of the Tf iron binding site model peptide after incubation with 1 in the range from 200 to 1100 m/z indicating cysteine as the major binding partner. The fragmented parent ion corresponds to [Pep+Pt(dma)(ipa)+H]3+. The insert shows a close-up of the platinated peaks at 722.2 and 736.1 m/z corresponding to [b4+Pt(dma)]+ and [b4+Pt(ipa)]+ 4 , respectively, with the distinctive Pt isotope pattern.
The exact binding site on the peptide is elucidated by combining information of the complementary b (containing an intact N terminus) and y (containing an intact C terminus) fragment ion series (see Figure 3 for nomenclature). Analysis of the bn-type fragments formed starting from the N terminus of the peptide reveals that b2+ is present in its unmodified form (m/z: 244.1), whereas b4 (containing both the cysteine and histidine residues) is platinated. This modification results in four peaks at m/z: 361.6 and 368.6 for the doubly charged species [b4+Pt(dma)]2+ and [b4+Pt(ipa)]2+ (note that either of the amine ligands is lost during fragmentation), and at m/z: 722.2 and 736.1 for the corresponding singly charged product ions. Whereas complete sequence coverage is obtained for the unmodified peptide upon CID, the only further ion attributable to the bn series in the CID spectrum of the platinated peptide corresponds to [b8+Pt(dma)]2+ at m/z: 567.1. This indicates that dramatic structural changes are induced upon binding of a platinum moiety, leading to decreased fragmentation efficiency of the platinated species relative to the unmodified peptide. Complementary yn ions starting from the C terminus of the peptide remain unmodified up to the y9 ion (m/z: 476.2 and 951.4 for the doubly and singly charged species, respectively), and also a minor peak can be assigned to unmodified y10+ (m/z: 1088.3). In contrast, y112+ and y132+ (both containing the histidine and cysteine residue) are platinated, with [y11+Pt(dma)]2+ assigned to the peak at m/z: 715.2, [y13+Pt(dma)2+] at 837.2, and [y13+Pt(ipa)]2+ at 843.7 (the peak corresponding to [y11+Pt(ipa)]2+ overlaps with [b4+Pt(dma)]+). The obtained results suggest that the cysteine residue is the major binding partner (as it is included in all platinated fragment ions), although its vicinal histidine is probably also involved in the bidentate adduct formation by providing the second coordination site. In a previous study, the same residues were also found to interact with cisplatin.[36] All four platinum compounds bind more rapidly to the peptide than to Tf itself. This difference underscores the strong influence of the tertiary structure of the protein on metal binding, compared with the essentially linear peptide that has much more flexibility, and therefore facilitates adduct formation without steric restrains.
Figure 3.
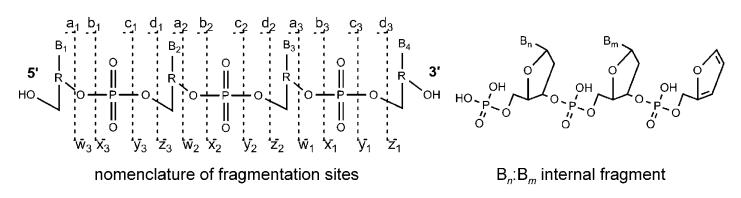
Nomenclature of oligonucleotide fragments observed by tandem MS; a-d correspond to fragments with an intact 5′ terminus and w-z-type fragments have an intact 3′ terminus. Internal fragments resulting from double fragmentation usually occur at the a/w site.
Oligonucleotide binding studies
The applicability of a tandem top-down MS approach for determining the binding sites of cisplatin on oligonucleotides has already been demonstrated.[39–41] Due to the characteristic isotopic pattern of platinum upon coordination to the oligonucleotide, modified fragments can easily be distinguished from those that remain unplatinated. Consequently, this approach was used to elucidate the binding sites of the trans-Pt complexes 1–3 on two different DNA models: SS1 (5′-d(TAC ACA CCG GTA C)-3′) and SS2 (5′-d(TAA TTA AGC ATA ATA T)-3′), which essentially differ in the number of guanine residues present.
The commonly applied nomenclature for oligonucleotide fragments is summarized in Figure 3. For n-type fragments, Bn (B=one of the nucleobases A, C, G, or T) is usually lost by an elimination reaction prior to strand breaking due to a second elimination reaction, leading to a furan ring system.[42] Internal fragments, resulting from two strand breaks at the a/w site, possess a phosphate group at their 5′ terminus, whereas the 3′ terminus carries a furan system.
The full-scan mass spectra of oligonucleotides SS1 and SS2 incubated with 1–3 and cisplatin showed the formation of similar adducts corresponding to [SS+Pt(amine)(amine′)] moieties (data not shown). In general, the reactivity of the trans compounds was lower than that of cisplatin.
As can be seen in figure S1 (Supporting Information) for the SS1 oligonucleotide that contains a G–G sequence (see Table 1) incubated with 2, fragmentation of the modified oligonucleotides ([SS1+Pt(dma)(ma)]3− at m/z: 1390 in the full-scan MS data in this case) leads to a spectrum in which a number of Pt-modified species are observed. Starting from the 5′ end, no modification of the nucleobases up to G9 are observed, as evidenced, for example, by the peaks at m/z: 1103.0 and 1248.0, which were assigned to [a8–C]2− and [a9–G]2−. From there on, (an–Bn) fragments exhibit an isotopic pattern characteristic of platinum and a mass increase corresponding to the addition of [Pt(dma)(ma)]2+ (269 Da for the neutral species). Consequently, the peaks at m/z: 1547.0 and 1863.4 were assigned to [a10−G+Pt(dma)(ma)]2− and [a12−A+Pt(dma)(ma)]2− fragments. Starting from the 3′ end of the oligonucleotide, all fragments up to w4 (m/z: 1252.0), which includes G10 but not G9, remain unmodified, whereas all further identified fragments, which include G9, are platinated. This is evidenced, for example, by peaks at m/z: 1214.0 and 1515.0, which may be assigned to [w7+Pt(dma)(ma)]2− and [w9+Pt(dma)(ma)]2−, respectively.
Combining the information from both ends of the strand, G9 is the major binding partner, as it is contained in all platinated fragments. As the binding is bidentate, G10 could serve as the second binding partner, as observed for cisplatin, oxaliplatin, and carboplatin.[41]
For 1 and 2, fragmentation spectra showed similar binding characteristics. In general, lower reactivity toward SS2 than toward SS1 was observed for all compounds, also indicating a strong influence of the number of guanine residues.
To evaluate if the binding is still selective toward guanine residues if adenine and thymine are present in large excess, tandem MS experiments were carried out using SS2 (containing only one G residue, see Table 1). Supporting Information figure S2 shows a CID spectrum of the parent ion [SS2+Pt-(dma)(ma)]4− at m/z: 1287.8. Similar to the experiments with SS1, the adducts are formed with the [Pt(ipa)(ma)]2+ moiety, resulting in peaks such as [a9−C+Pt(dma)(ma)]2− and [a10−A+Pt(dma)(ma)]2− at m/z: 1429.0 and 1574.0, respectively, both of which contain the guanine residue. Furthermore, the peak at m/z: 1264.9 may be assigned to [a8−G+Pt-(dma)(ma)]2−, indicating that adenine residue at position 8 may also serve as a coordination site for the platinum moiety. More generally, (an–Bn) fragments with n<8 (none of which contain guanine) remain unmodified, whereas all product ions of this series with n>8 are platinated. When analyzing fragments with an intact 3′ terminus (w ion series), the first modification is observed for the w82− ion, which is present in both native and platinated forms (peaks at m/z: 1234.4 and 1369.4, respectively). This is rather surprising, as w8 includes the cytosine, but not its vicinal guanine residue. All wn-type fragments with n>8 are detected only in their platinated forms, as evidenced, for example, by [w9+Pt-(dma)(ma)]2− and [w10+Pt(dma)(ma)]2− at m/z: 1533.9 and 1690.3, respectively.
Combining the assignments for both (an–Bn)- and wn-type ions, G9 and its neighboring A8 and C10 residues correspond to the major binding sites. Additionally, as SS2 contains six more bases than SS1, an increased number of internal (Bn:Bm) fragments such as [A3:A7]− at m/z: 1723.9 are also detected, which also indicated that G9 is a binding partner. As the binding is bidentate, it may be assumed that adduct formation occurs by intrastrand bridging of two nucleobases. Similar observations were made for 2 and 3.
Transfer experiments
To determine whether Pt complexes can transfer from a peptide to an oligonucleotide, transfer experiments were performed in which the Tf model peptide was pre-incubated with one of the platinum compounds under physiological-type conditions for 24 h, after which the oligonucleotide (SS1 or SS2) was added (see Experimental Section above for details). The sample was analyzed by ESI-MS at various times over 72 h. Such an experiment imitates, to some extent, transporter-mediated delivery to DNA nucleobases. Figures 4 and 5 show representative spectra, recorded in both positive and negative modes, for 3 and cisplatin, respectively. Table 2 lists the peaks’ full assignment. Note that although ionization conditions were optimized, small peaks corresponding to the oligonucleotide can be seen in positive ion mode (especially following Pt binding, which introduces additional positive charges) as well as peptide peaks in the negative ion mode. For the non-platinated peptide, peaks of low relative intensity corresponding to dimers (labeled [2Pep] in the spectra) are observed after 72 h of incubation, indicating oxidation of the cysteine residue with the concomitant formation of a disulfide bond. The fact that no platinum adducts were detected for the dimer further substantiates the results from the binding site determination studies, that cysteine is involved in adduct formation, as this residue is not accessible after dimerization.
Figure 4.

Spectra obtained following incubation of 3 with the model peptide for 24 h, followed by addition of SS1 and further incubation for 72 h: A) positive ion spectrum (peptide), B) negative ion spectrum (oligonucleotide); see Table 2 for the assignment of platinated peaks.
Figure 5.

Spectra obtained following incubation of cisplatin with the model peptide for 24 h, followed by addition of SS2 and further incubation for 72 h: A) positive ion spectrum (peptide), B) negative ion spectrum (oligonucleotide); see Table 2 for the assignment of platinated peaks.
Table 2.
Calculated and experimental m/z values (most abundant isotope) for platinated species in the transfer and competition experiments (see Figures 4–7).
| Ion | m/z | Transfer | Competition | |
|---|---|---|---|---|
| Calcd | Exptl | |||
| Cisplatin: | ||||
| [Pep+2Pt(NH3)]3+ | 618.9 | 619.1 | + | − |
| [Pep+2Pt(NH3)2]3+ | 630.2 | 630.3 | + | − |
| [Pep+Pt(NH3)]2+ | 823.3 | 823.2 | + | − |
| [Pep+Pt(NH3)2]2+ | 831.9 | 831.9 | + | − |
| [Pep+2Pt(NH3)]2+ | 927.8 | 927.9 | + | − |
| [Pep+Pt(NH3)+Pt(NH3)2]2+ | 936.4 | 936.5 | + | − |
| [Pep+2Pt(NH3)2]2+ | 944.9 | 944.7 | + | − |
| [SS2+Pt(NH3)2]7− | 729.6 | 729.5 | + | + |
| [SS2+2Pt(NH3)2]7− | 762.1 | 762.3 | − | + |
| [SS2+Pt(NH3)2]6− | 851.3 | 851.5 | + | + |
| [SS2+2Pt(NH3)2]6− | 889.7 | 890.0 | − | + |
| [SS2+Pt(NH3)2]5− | 1021.9 | 1022.0 | + | + |
| [SS2+2Pt(NH3)2]5− | 1067.3 | 1068.0 | − | + |
| trans-[PtCl2(ipa)(ma)] (3): | ||||
| [Pep+Pt(ma)]3+ | 553.9 | 554.0 | + | + |
| [Pep+Pt(ipa)]3+ | 563.2 | 563.1 | + | + |
| [Pep+Pt(ipa)(ma)]3+ | 573.5 | 573.7 | + | + |
| [Pep+Pt(ma)]2+ | 830.4 | 830.3 | + | + |
| [Pep+Pt(ipa)]2+ | 844.4 | 844.3 | + | + |
| [Pep+Pt(ipa)(ma)]2+ | 859.5 | 859.4 | + | + |
| [SS1+Pt(ipa)(ma)]6− | 697.0 | 697.0 | + | + |
| [SS1+Pt(ipa)(ma)]5− | 836.5 | 836.6 | + | + |
| [SS1+Pt(ipa)(ma)]4− | 1045.7 | 1045.6 | + | + |
For samples of 3 incubated with the peptide and SS1, the peak of highest relative intensity in the positive ion spectrum corresponds to [Pep+Pt(ma)]3+ and [Pep+Pt(ipa)]3+ (Figure 4A) 72 h after addition of the oligonucleotide. The amount of the metallofragment transferred to the oligonucleotide is rather low and depends on its sequence, that is, the number of guanines, as expected from the oligonucleotide binding experiments. Even when using SS1 (two adjacent G residues), only peaks of low relative intensity such as [SS1+Pt(ipa)(ma)]5− at m/z: 836.5 were detected (Figure 4B), indicating a preference for the peptide under these conditions. Compounds 1 and 2 displayed a very similar behavior in these experiments, with only a low transfer of the platinum fragment from the peptide to the oligonucleotides observed.
When performing the same experiment with cisplatin, the influence of the number of guanines is more pronounced. In the case of addition of SS1 to the reaction mixture, only minor peaks can be assigned to peptide–Pt adducts in positive ion mode, whereas major peaks in the negative ion mode spectra correspond to [SS1+Pt(NH3)2] ions. As depicted in Figure 5, this is not the case with SS2; in positive ion mode, a multitude of peaks can be assigned to platinated peptide species (Figure 5A). The most significant peaks correspond to [Pep+Pt(NH3)2]2+ at m/z: 831.9, but also to bis-adducts such as [Pep+Pt(NH3)+Pt(NH3)2]2+ at m/z: 936.0, indicating even tridentate coordination with the peptide can occur. Minor peaks stem from adduct formation with sodium or potassium ions. Only a minor amount of the Pt is bound to the oligonucleotide, evidenced by low-intensity peaks for platinated species in the negative ion spectrum (Figure 5B).
For all of the trans complexes, only limited transfer of the metallodrug fragments from the peptide was observed, with the majority of the Pt adducts still bound to the peptide 72 h after addition of either oligonucleotide. For cisplatin, the distribution depended strongly on the sequence of the DNA model; for SS1 (adjacent G–G residues), most of the metallodrug adduct was transferred to the oligonucleotide, whereas relatively little transfer was observed for SS2 (one G residue).
Competition experiments
Direct competition experiments, in which the peptide and oligonucleotide were added simultaneously to the metallodrug solution in equimolar amounts, were undertaken. This setup simulates, to some extent, interactions of the free drug in the cytoplasm where a multitude of binding partners such as peptides and proteins, but also single nucleobases, oligonucleotides, and RNA molecules are present. Figure 6 shows representative spectra for the interaction of 3 with the model peptide and SS1. Although caution must be taken in quantifying the extent of adduct formation using ESI-MS, it is clear that the amount of platinated oligonucleotide is higher under competitive conditions relative to the case in which the Pt is initially bound to the peptide. The peak of highest relative intensity in the positive ion spectrum corresponds to [Pep+Pt(ipa)]3+ and [Pep+Pt(ma)]3+ (Figure 6A), whereas the most prominent peak in the negative ion spectrum may be assigned to [SS1+Pt(ipa)(ma)]5− (Figure 6B). Using SS2 (only one G residue), only minor peaks can be assigned to platinated oligonucleotides, with the vast majority bound to the peptide. The situation with 1 and 2 is very similar for both mixtures containing the peptide and SS1 or SS2.
Figure 6.

Spectra obtained following incubation of 3 with the model peptide and SS1 for 72 h: A) positive ion spectrum (peptide), B) negative ion spectrum (oligonucleotide); see Table 2 for the assignment of platinated peaks.
For cisplatin, the distribution of the drug between the binding partners is altered more dramatically. As depicted in Figure 7 for the incubation with the model peptide and SS2 under competitive conditions, no platinated peptide species could be detected in positive ion mode, whereas significant adduct formation with the oligonucleotide takes place. Even with only one guanine present in the strand, the formation of bis-adducts is observed (peaks at m/z: 851.5 and 890.0 for [SS2+Pt(NH3)2]6− and [SS2+2Pt(NH3)2]6−, respectively). When exchanging SS2 for SS1, which provides the preferential G–G binding site, similar spectra are obtained, with no platinated peptide peaks detected.
Figure 7.

Spectra obtained following incubation of cisplatin with the model peptide and SS2 for 72 h: A) positive ion spectrum (peptide), B) negative ion spectrum (oligonucleotide); see Table 2 for the assignment of platinated peaks.
In general, in direct competition experiments, no platination of the peptide could be detected for cisplatin, independent of the oligonucleotide used. In contrast, the trans complexes are distributed about equally between the peptide and the oligonucleotide when using SS1, whereas with SS2 the complexes are almost exclusively bound to the peptide.
Notably, published studies have already highlighted the differences between cis- and trans-PtII complexes in their preferential binding to sulfur-containing amino acids and nucleobases.[43] For example, studies of the reactivity of the model cis compound [PtCl(dien)]+ with sulfur donor nucleophiles showed that the Pt–thioether bond could be displaced by N7 of guanine.[44,45] Other studies on various trans-PtII complex ternary systems with sulfur-containing nucleophiles and DNA model compounds have evidenced different reactivity patterns depending on the Pt spectator ligands and the kind of model biomolecules used.[46–48] Recently, the affinity of trans-PtII complexes for cysteine residues has been described in a comparative study on the reactivity of the cysteine-rich protein metallothionein-2 with cisplatin– and transplatin–(5′-GMP) adducts (5′-GMP=5′-guanosine monophosphate).[49] The analysis of the reaction products over time by NMR spectroscopy showed that while the formation of the ternary GMP–trans-Pt–MT adduct is accompanied by 5′-GMP release, a stable ternary GMP–cis-Pt–MT complex is formed.
Conclusions
ESI-MS was used to study the reactivity of 1–3 with apo-Tf, establishing the nature of the protein-bound metal fragments and the stoichiometry of the adducts. The compounds retain both aliphatic amine ligands upon protein binding, in agreement with previous studies involving model proteins.[25] Differences in the reactivity of these compounds may be correlated to differences in aquation kinetics, but could also partly be ascribed to steric/hydrophobic effects, as adduct formation is decreased for the complexes with bulkier ligands. Relative to cisplatin, the extent of adduct formation is lower, highlighting the influence of the cis geometry on reactivity. Using a Tf model peptide in combination with tandem MS, a preference of 1–3 for the sulfur-containing amino acid cysteine was observed. This preference is in accordance with data obtained for cisplatin,[36] indicating that the PtII compounds exhibit similar binding preferences toward peptides independent of their geometry. However, it must be taken into account that a short peptide possesses much more flexibility than a protein, making all residues equally accessible.
The binding of 1–3 toward single-stranded oligonucleotides containing different numbers of guanine residues was also investigated, and [Pt(amine)(amine′)] moieties were identified as the bound species. In all cases, cisplatin was found to be more reactive than the trans complexes. The transfer of the metallodrug adducts from the Tf model peptide to the oligonucleotides was also studied, as well as the competitive binding of the complexes toward peptide–oligonucleotide mixtures. In general, significant differences between the trans-PtII complexes and cisplatin were observed, indicating the preferential binding of the trans compounds toward the peptide, whereas cisplatin shows a preference for binding to oligonucleotides (especially those containing adjacent G–G residues). This selectivity could be relevant to the mode of action of the trans compounds. Should adduct formation with DNA (either directly or by transfer from another biomolecule) prove to be inefficient to damage DNA, proteinaceous targets could be involved in inhibiting cellular proliferation, especially for compounds of similar cytotoxicity. Further biochemical and biological studies will be conducted to substantiate this hypothesis.
Supplementary Material
Acknowledgements
The authors thank the COST D39 Action for financial support, and in particular WG1 and WG6 for fruitful scientific discussions. C.N.R., A.G.Q., and L.C. thank the Spanish Comisión Interministerial de Ciencia y Tecnología (SAF-2009-09431) for funding. A.C. gratefully acknowledges the Swiss National Science Foundation (AMBIZIONE project no. PZ00P2_121933), the Swiss Confederation (Action COST D39 – Accord de recherche – SER project no. C09.0027). M.G. thanks the Austrian Science Fund for financial support (Schrödinger Fellowship J2882-N19). The authors also thank Prof. Yury Tsybin for assistance in the MS measurements.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cmdc.201000104: CID mass spectra showing the preferential binding site of compound 2 on the oligonucleotides SS1 and SS2.
References
- [1].Lippert B. Met. Ions Biol. Syst. 1996;33:105–141. [PubMed] [Google Scholar]
- [2].Reedijk J. Eur. J. Inorg. Chem. 2009;10:1303–1312. [Google Scholar]
- [3].Coluccia M, Natile G. Anti-Cancer Agents Med. Chem. 2007;7:111–123. doi: 10.2174/187152007779314080. [DOI] [PubMed] [Google Scholar]
- [4].Farrell N, Kelland LR, Roberts JD, Van Beusichem M. Cancer Res. 1992;52:5065–5072. [PubMed] [Google Scholar]
- [5].Zorbas-Seifried S, Jakupec MA, Kukushkin NV, Groessl M, Hartinger CG, Semenova O, Zorbas H, Kukushkin VY, Keppler BK. Mol. Pharmacol. 2006;71:357–365. doi: 10.1124/mol.106.030726. [DOI] [PubMed] [Google Scholar]
- [6].Pérez JM, Kelland LR, Montero EI, Boxall FE, Fuertes MA, Alonso C, Navarro-Ranninger C. Mol. Pharmacol. 2003;63:933–944. doi: 10.1124/mol.63.4.933. [DOI] [PubMed] [Google Scholar]
- [7].Montero EI, Diaz S, Gonzalez-Vadillo AM, Pérez JM, Alonso C, Navarro-Ranninger C. J. Med. Chem. 1999;42:4264–4268. doi: 10.1021/jm991015e. [DOI] [PubMed] [Google Scholar]
- [8].Pérez JM, Montero EI, Gonzalez AM, Solans X, Font-Bardia M, Fuertes MA, Alonso C, Navarro-Ranninger C. J. Med. Chem. 2000;43:2411–2418. doi: 10.1021/jm000925p. [DOI] [PubMed] [Google Scholar]
- [9].Pérez JM, Fuertes MA, Alonso C, Navarro-Ranninger C. Crit. Rev. Oncol. Hematol. 2000;35:109–120. doi: 10.1016/s1040-8428(00)00053-6. [DOI] [PubMed] [Google Scholar]
- [10].Barnes KR, Lippard SJ. Met. Ions Biol. Syst. 2004;42:143–177. [PubMed] [Google Scholar]
- [11].Montero EI, Pérez JM, Schwartz A, Fuertes MA, Malinge JM, Alonso C, Leng M, Navarro-Ranninger C. ChemBioChem. 2002;3:61–67. doi: 10.1002/1439-7633(20020104)3:1<61::AID-CBIC61>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- [12].Cubo L, Quiroga AG, Zhang JY, Thomas DS, Carnero A, Navarro-Ranninger C, Berners-Price SJ. Dalton Trans. 2009;18:3457–3466. doi: 10.1039/b819301k. [DOI] [PubMed] [Google Scholar]
- [13].Hall MD, Okabe M, Shen DW, Liang XJ, Gottesman MM. Annu. Rev. Pharmacol. Toxicol. 2008;48:495–535. doi: 10.1146/annurev.pharmtox.48.080907.180426. [DOI] [PubMed] [Google Scholar]
- [14].Klein AV, Hambley TW. Chem. Rev. 2009;109:4911–4920. doi: 10.1021/cr9001066. [DOI] [PubMed] [Google Scholar]
- [15].Arnesano F, Scintilla S, Natile G. Angew. Chem. 2007;119:9220–9222. doi: 10.1002/anie.200703271. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2007;46:9062–9064. doi: 10.1002/anie.200703271. [DOI] [PubMed] [Google Scholar]
- [16].Gibson D. Dalton Trans. 2009;48:10681–10689. doi: 10.1039/b918871c. [DOI] [PubMed] [Google Scholar]
- [17].Peleg-Shulman T, Najajreh Y, Gibson D. J. Inorg. Biochem. 2002;91:306–311. doi: 10.1016/s0162-0134(02)00362-8. [DOI] [PubMed] [Google Scholar]
- [18].Anzellotti AI, Liu Q, Bloemink MJ, Scarsdale JN, Farrell N. Chem. Biol. 2006;13:539–548. doi: 10.1016/j.chembiol.2006.04.004. [DOI] [PubMed] [Google Scholar]
- [19].Casini A, Gabbiani C, Mastrobuoni G, Pellicani RZ, Intini FP, Arnesano F, Natile G, Moneti G, Francese S, Messori L. Biochemistry. 2007;46:12220–12230. doi: 10.1021/bi701516q. [DOI] [PubMed] [Google Scholar]
- [20].Knipp M, Karotki AV, Chesnov S, Natile G, Sadler PJ, Brabec V, Vasak M. J. Med. Chem. 2007;50:4075–4086. doi: 10.1021/jm070271l. [DOI] [PubMed] [Google Scholar]
- [21].Hartinger CG, Tsybin YO, Fuchser J, Dyson PJ. Inorg. Chem. 2008;47:17–19. doi: 10.1021/ic702236m. [DOI] [PubMed] [Google Scholar]
- [22].Mandal R, Teixeira C, Li XF. Analyst. 2003;128:629–634. doi: 10.1039/b212797k. [DOI] [PubMed] [Google Scholar]
- [23].Mandal R, Kalke R, Li XF. Chem. Res. Toxicol. 2004;17:1391–1397. doi: 10.1021/tx049868j. [DOI] [PubMed] [Google Scholar]
- [24].Mandal R, Sawyer MB, Li XF. Rapid Commun. Mass Spectrom. 2006;20:2533–2538. doi: 10.1002/rcm.2622. [DOI] [PubMed] [Google Scholar]
- [25].Cubo L, Casini A, Gabbiani C, Mastrobuoni G, Messori L, Jimenez-Barbero J, Navarro-Ranninger C, Quiroga AG. Chem. Eur. J. 2009;15:9139–9146. doi: 10.1002/chem.200901090. [DOI] [PubMed] [Google Scholar]
- [26].Casini A, Gabbiani C, Mastrobuoni G, Messori L, Moneti G, Pieraccini G. ChemMedChem. 2006;1:413–417. doi: 10.1002/cmdc.200500079. [DOI] [PubMed] [Google Scholar]
- [27].Gabbiani C, Casini A, Mastrobuoni G, Kirshenbaum N, Moshel O, Pieraccini G, Moneti G, Messori L, Gibson D. J. Biol. Inorg. Chem. 2008;13:755–764. doi: 10.1007/s00775-008-0361-z. [DOI] [PubMed] [Google Scholar]
- [28].Qian ZM, Li HY, Sun HZ, Ho K. Pharmacol. Rev. 2002;54:561–587. doi: 10.1124/pr.54.4.561. [DOI] [PubMed] [Google Scholar]
- [29].Wally J, Halbrooks PJ, Vonrhein C, Rould MA, Everse SJ, Mason AB, Buchanan SK. J. Biol. Chem. 2006;281:24934–24944. doi: 10.1074/jbc.M604592200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Schlabach MR, Bates GW. J. Biol. Chem. 1975;250:2182–2188. [PubMed] [Google Scholar]
- [31].Pérez JM, Montero EI, Gonzalez AM, Alvarez-Valdes A, Alonso C, Navarro-Ranninger C. J. Inorg. Biochem. 1999;77:37–42. doi: 10.1016/s0162-0134(99)00143-9. [DOI] [PubMed] [Google Scholar]
- [32].Ni JS, Pomerantz SC, Rozenski J, Zhang YH, McCloskey JA. Anal. Chem. 1996;68:1989–1999. doi: 10.1021/ac960270t. [DOI] [PubMed] [Google Scholar]
- [33].Thevis M, Loo RRO, Loo JA. J. Am. Soc. Mass Spectrom. 2003;14:635–647. doi: 10.1016/S1044-0305(03)00199-5. [DOI] [PubMed] [Google Scholar]
- [34].Timerbaev AR, Aleksenko KS, Polec-Pawlak K, Ruzik R, Semenova O, Hartinger CG, Oszwaldowski S, Galanski M, Jarosz M, Keppler BK. Electrophoresis. 2004;25:1988–1995. doi: 10.1002/elps.200305984. [DOI] [PubMed] [Google Scholar]
- [35].Khalaila I, Allardyce CS, Verma CS, Dyson PJ. ChemBioChem. 2005;6:1788–1795. doi: 10.1002/cbic.200500067. [DOI] [PubMed] [Google Scholar]
- [36].Groessl M, Terenghi M, Casini A, Elviri L, Lobinski R, Dyson PJ. J. Anal. At. Spectrom. 2010 [Google Scholar]
- [37].Davies MS, Berners-Price SJ, Hambley TW. J. Inorg. Biochem. 2000;79:167–172. doi: 10.1016/s0162-0134(99)00180-4. [DOI] [PubMed] [Google Scholar]
- [38].Allardyce CS, Dyson PJ, Coffey J, Johnson N. Rapid Commun. Mass Spectrom. 2002;16:933–935. doi: 10.1002/rcm.662. [DOI] [PubMed] [Google Scholar]
- [39].Egger AE, Hartinger CG, Ben Hamidane H, Tsybin YO, Keppler BK, Dyson PJ. Inorg. Chem. 2008;47:10626–10633. doi: 10.1021/ic801371r. [DOI] [PubMed] [Google Scholar]
- [40].Nyakas A, Eymann M, Schurch S. J. Am. Soc. Mass Spectrom. 2009;20:792–804. doi: 10.1016/j.jasms.2008.12.018. [DOI] [PubMed] [Google Scholar]
- [41].Groessl M, Tsybin YO, Hartinger C, Keppler BK, Dyson PJ. J. Biol. Inorg. Chem. 2010 doi: 10.1007/s00775-010-0635-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].McLuckey SA, Van Berkel GJ, Glish GL. J. Am. Soc. Mass Spectrom. 1992;3:60–70. doi: 10.1016/1044-0305(92)85019-G. [DOI] [PubMed] [Google Scholar]
- [43].Chen Y, Guo Z, Sadler PJ. In: Cisplatin. Lippert B, editor. Verlag Helvetica Chimica Acta; Zurich: 2006. pp. 293–318. [Google Scholar]
- [44].Van Boom SSGE, Reedijk J. J. Chem. Soc. Chem. Commun. 1993;18:1397–1398. [Google Scholar]
- [45].Reedijk J. Chem. Rev. 1999;99:2499–2510. doi: 10.1021/cr980422f. [DOI] [PubMed] [Google Scholar]
- [46].Bierbach U, Farrell N. J. Biol. Inorg. Chem. 1998;3:570–580. [Google Scholar]
- [47].Quintal SMO, Qu Y, Quiroga AG, Moniodis J, Nogueira HIS, Farrell N. Inorg. Chem. 2005;44:5247–5253. doi: 10.1021/ic050062w. [DOI] [PubMed] [Google Scholar]
- [48].Li C, Li ZY, Sletten E, Arnesano F, Losacco M, Natile G, Liu YZ. Angew. Chem. 2009;121:8649–8652. doi: 10.1002/anie.200902948. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2009;48:8497–8500. doi: 10.1002/anie.200902948. [DOI] [PubMed] [Google Scholar]
- [49].Karotki AV, Vasak M. Biochemistry. 2008;47:10961–10969. doi: 10.1021/bi801253x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.