

Abstract
Diffuse alveolar hemorrhage is characterized by the presence of red blood cells and free hemoglobin in the alveoli and complicates a number of serious medical and surgical lung conditions including the pulmonary vasculitides and acute respiratory distress syndrome. In this study we investigated the hypothesis that exposure of human alveolar epithelial cells to hemoglobin and its breakdown products regulates chemokine release via iron- and oxidant-mediated activation of the transcription factor NF-κB. Methemoglobin alone stimulated the release of IL-8 and MCP-1 from A549 cells via activation of the NF-κB pathway; additionally, IL-8 required ERK activation and MCP-1 required JNK activation. Neither antioxidants nor iron chelators and knockdown of ferritin heavy and light chains affected these responses, indicating that iron and reactive oxygen species are not involved in the response of alveolar epithelial cells to methemoglobin. Incubation of primary cultures of human alveolar type 2 cells with methemoglobin resulted in a similar pattern of chemokine release and signaling pathway activation. In summary, we have shown for the first time that methemoglobin induced chemokine release from human lung epithelial cells independent of iron- and redox-mediated signaling involving the activation of the NF-κB and MAPK pathways. Decompartmentalization of hemoglobin may be a significant proinflammatory stimulus in a variety of lung diseases.
Keywords: alveolar epithelial cells, cytokines, methemoglobin
diffuse alveolar hemorrhage (DAH) may result from trauma (10), coagulation disorders, vasculitis, and the acute respiratory distress syndrome (ARDS) (34). Hemolysis that inevitably follows DAH leads to the release of free hemoglobin (Hb), which may affect both prooxidant and proinflammatory processes. Indeed, both free Hb and red blood cells induced lung injury in experimental animal models (19). In patients with sepsis, the plasma concentration of free Hb was an independent predictor of mortality (1, 25), and serum concentrations of the soluble Hb scavenger receptor CD163 have been proposed as a prognostic biomarker in sepsis (17).
Within red blood cells the Hb tetramer (α2β2) is maintained predominantly as oxyhemoglobin, in which iron is coordinated within heme in the reduced or ferrous (Fe2+) state. Oxyhemoglobin (HbFe2+) is the predominant carrier of oxygen in the red blood cell, where only a small proportion (1–2%) occurs as oxidized methemoglobin (HbFe3+). During hemolytic events the release of tetrameric oxyhemoglobin is followed by dissociation of the parent tetramer into αβ dimers, the majority of which are comprised of oxyhemoglobin with some (<10%) present in the methemoglobin form (4, 42, 49). Methemoglobin formation is more pronounced in the presence of inflammation owing to the relative abundance of oxidizing species such as hydrogen peroxide (H2O2), hypochlorous acid (HOCl), nitric oxide (NȮ), and peroxynitrite (ONOO−) produced by activated macrophages and neutrophils (20). Further degradation of methemoglobin can occur, leading to the release of heme and iron (4). The catalytic iron present in Hb can induce the formation of reactive oxygen species (ROS) leading to oxidative damage in lung tissues (54). The resulting disrupted iron metabolism and oxidative stress may contribute to systemic and lung-specific oxidative damage, altered signaling, and inflammatory responses (28).
Plasma contains haptoglobin, hemopexin, transferrin, and lactoferrin, which bind Hb, heme, and free iron, respectively, thereby providing antioxidant protection and a mechanism for removal of the products of hemolysis from the extracellular compartment. These strata can become saturated when bleeding is significant, allowing free Hb and associated breakdown products to accumulate. Extracellular heme (either free or as part of Hb) can exhibit peroxidase-like activity generating oxidants capable of initiating lipid peroxidation (41). Iron released from heme through interaction with H2O2 can catalyze the formation of aggressive oxidants from innocuous species (Fenton reaction) leading to oxidative damage and tissue injury (23). Such changes in oxidant balance also impact on an array of redox-mediated inflammatory responses (40). Cellular uptake of heme or Hb can itself augment proinflammatory responses; neutrophil lifespan is extended by exposure to these agents owing to antiapoptotic processes related to the induction of inducible heme oxygenase (HO-1). The heme oxygenase enzymes catalyze the breakdown of heme to bilirubin, carbon monoxide, and ferrous iron (Fe2+). This prooxidant iron is subsequently sequestered for storage in an unreactive form by ferritin, an iron storage protein that consists of a hollow protein shell composed of 24 subunits of both heavy (FTH) and light (FTL) chain origin. Deficits in ferritin expression or exhaustion of iron storage capacity have implications for cellular iron-catalyzed oxidative stress and signaling responses, including that derived from heme catabolism.
Although some literature supports a role for Hb-stimulated proinflammatory responses in several cell types (31, 35), as yet no exploration has been undertaken in human alveolar epithelial cells. Given the potential for Hb and its derivatives to modulate inflammation, we hypothesized that exposure of human lung epithelial cells to Hb and its breakdown products regulates chemokine release via iron- and oxidant-mediated activation of the transcription factor NF-κB. Using A549 human lung epithelial cells, we aimed to identify the forms of Hb that best induce release of the chemokines IL-8 and MCP-1, which have established roles in lung inflammation (15, 36, 45), and to characterize the signaling pathways involved. Finally, we aimed to determine the role of intracellular iron and oxidative stress in mediating the effects of free Hb using a combination of pharmacological and gene silencing techniques.
MATERIALS AND METHODS
Cells and reagents.
General reagents were from Sigma (Dorset, UK), Invitrogen (Paisley, UK), VWR (Lutterworth, UK), or Fisher Scientific (Loughborough, UK) and were of the highest purity available or of cell culture grade.
A549 cells were purchased from the European Collection of Cell Cultures (Porton Down, UK) and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum, 2 mM glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin.
Primary cell isolation and culture.
Human alveolar epithelial (hAT2) cells were isolated from samples of normal lung parenchyma from patients undergoing thoracic surgery at the Royal Brompton Hospital, by use of trypsin digest followed by differential adherence (14). Culture medium (DCCM-1) was replaced after 24 h and the cell monolayer was washed with HBSS 48 h after isolation to remove nonadherent cells. The cells formed a visually confluent monolayer ready for experimentations 3–4 days after isolation.
Precision-cut human lung slices.
Macroscopically normal human lung tissue (∼6 g) was washed twice with ice-cold HBSS containing penicillin (100 μg/ml) and streptomycin (100 μg/ml). Tissue was immersed in ice-cold HBSS containing penicillin and streptomycin and inflated by injection of 3% (wt/vol) low-melting-point agarose using a total volume of ∼3–5 ml/g wet weight of tissue. The tissue was cooled and cut with a microtome blade into slices ∼0.5 cm thick. With use of an 8-mm coring tool, cylindrical cores of the inflated tissue were prepared and cut into ∼400-μm-thick slices by a Krumdieck Tissue Slicer. Lung slices were transferred to a 12-well tissue culture plate containing 1 ml of RPMI supplemented with penicillin, streptomycin, l-glutamine, and gentamicin and incubated at 37°C in an atmosphere of 95% air-5% CO2. Slices were washed six times in medium with a 30-min incubation period between each wash to remove endogenous factors released from the slices. Slices were then incubated overnight and medium was removed and replaced with 1 ml of fresh medium ∼30 min prior to treatment protocols.
Adenovirus transfection.
The dominant negative (dn) IKK-2 adenovirus was titrated by end point dilution and plaque assay to determine plaque-forming units. A multiplicity of infection (MOI) dose curve was constructed (MOI 0–500) to determine optimum conditions. The appropriate amount of virus (MOI 10) was diluted in DMEM and added to cells. After 24 h the medium was replaced and the cells were ready for experiments. An empty vector served as a transfection control.
Knockdown.
A549 cells were treated with either 10 nM FTL, 25 nM FTH siRNA (Smartpool, Dharmacon, Epsom, UK), or corresponding amounts of scrambled siRNA, as per the manufacturers' instructions. The cells were then treated with methemoglobin (10 μM) for 2 or 24 h, after 40 h for FTL and 18 h for FTH knockdowns.
Treatment protocols.
Spectrophotometric analysis was performed on commercially sourced human hemoglobins (oxy- and methemoglobin; Sigma-Aldrich, Dorset, UK) to confirm the oxidation state (data not shown). Human methemoglobin and oxyhemoglobin were prepared in culture medium at a stock of 200 μM. The solutions were sterile filtered and further diluted to the desired concentrations for experimental protocols. Cells were incubated with Hb for 24 h for IL-8 and MCP-1 protein determination and for 2 h for mRNA experiments. Cells were incubated with hemin (hemin chloride, Frontier Scientific Europe, Lancashire, UK) or iron salts for 24 h.
The IKK-2 inhibitors [5-(p-fluorophenyl)-2-ureido]thiophene-3-carboxamide (TPCA-1)] and SC-514 (Cambridge Biosciences, Cambridge, UK) were added 1 and 2 h, respectively, before the addition of methemoglobin. The MAP kinase pathway inhibitor U0126 (Cambridge Biosciences) was added 2 h prior to methemoglobin and SP600125 (R&D Systems Europe, Oxford, UK) 1 h before methemoglobin. The iron chelators desferrioxamine, phenanthroline, and 2,2′-bipyridyl (Sigma); the hydroxyl radical scavenger N,N′-di(2-hydroxybenzyl)ethylenediamine-N,N′-diacetic acid monohydrochloride hydrate (HBED; Insight Biotechnologies, Wembley, UK), and the antioxidants N-acetyl-cysteine (NAC) and α-tocopherol (Sigma) were all preincubated with the cells for 1 h prior to the addition of methemoglobin.
Contaminating LPS in the methemoglobin was determined by the Limulus amoebocyte lysate assay (Rapid Endotest, Cambrex, Verviers, Belgium).
Cell proliferation.
The CyQUANT cell proliferation assay kit (Invitrogen) was used to measure cell proliferation, following the manufacturer's instructions.
Cell viability.
Epithelial cell respiration and hence cell viability was quantified by the mitochondrial reduction of yellow MTT [3-(4,5-dimethyl-2;thiazolyl)-2–5-diphnyltetrazloium bromide] to formazan (purple, which is measured spectrophotometrically at 550 nm).
Preparation of cell lysates and nuclear extracts.
Cell lysates were prepared by adding cell lysis buffer (New England Biolabs, Hitchin, UK) containing 20 mM Tris·HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin, and 1 mM PMSF. Cells were scraped from the culture plates and centrifuged to remove debris.
Nuclear extracts were prepared by a kit method following the manufacturer's instructions (Active Motif).
Total protein in cell lysates and nuclear extracts was determined by use of the Bradford protein assay (Bio-Rad Laboratories, Hemel Hempstead, UK).
ELISA.
IL-8 and MCP-1 were quantified by ELISA (R&D Systems Europe) according to the manufacturer's protocol. Ferritin light and heavy chain ELISA kits were from USCN Life Science (Wuhan, China).
RT-PCR.
Total RNA was extracted by using the RNeasy Mini preparation kit (Qiagen, Crawley, UK) following the manufacturer's instructions. RNA purity and concentration were determined by use of a Nano-drop spectrophotometer, and 0.6 μg of total RNA was used for reverse transcription to produce cDNA. Real-time PCR was performed with SYBR green and the following primers on a Rotor-Gene 6000 PCR machine (Qiagen): IL-8 forward TCAGGGAGGCTACCACTTC, reverse TACTCCAAACCTTTCCACCC; MCP-1 forward GATCTCAGTGCAGAGGCTCG, reverse TGCTTGTCCAGGTGGTCCAT; GAPDH: forward GAAGGTGAAGGTCGGAGT, reverse GAAGATGGTGATGGGATTTC.
Western blot.
Proteins were resolved on 10% SDS-PAGE gels and transferred to nitrocellulose membranes. After blocking with 5% milk, membranes were probed with antibodies directed against either human IκBα, phospho-IκBα, NF-κB-p65, phospho-p65-ser536, ERK and phospho-ERK, JNK and phospho-JNK, p38 and phospho-p38, all from New England Biolabs. Human β-actin (New England Biolabs) was used as a loading control. Primary antibodies were detected with horseradish peroxidase-conjugated anti-rabbit (New England Biolabs) secondary antibody. Peroxidase activity was detected via ECL reagent (GE Healthcare, Buckinghamshire, UK).
Statistical analysis.
Data are presented as means ± SE of the stated number of experiments. Statistical analyses were performed with GraphPad Prism version 4 (GraphPad Software, San Diego, CA). Data normality was assessed by the Kolmogorov-Smirnov test. The statistical tests used for individual experiments are described; significance was defined at the 95% level.
Ethics declaration.
The use of normal lung tissue has been approved by the Royal Brompton and Harefield NHS Trust Research Ethics Committee (ethics number 01-165). All patients gave written, informed consent prior to the use of their lung tissue.
RESULTS
Methemoglobin caused the release of IL-8 and MCP-1 from human lung epithelial cells A549.
Only methemoglobin (3 and 10 μM), and not oxyhemoglobin (3 and 10 μM), azide-bound methemoglobin (10 μM), which is redox inactivated, hemin (10, 30 and 50 μM), nor the iron salts ammonium iron sulfate (Fe2+) and ferric citrate (Fe3+; 100 μM; Fig. 1) resulted in a significant (P < 0.01) time- and dose-dependent increase in both IL-8 and MCP-1 release (Fig. 2, A and B) and mRNA expression (Fig. 2, C and D) in A549 cells. At higher nonphysiological concentrations of 50 μM methemoglobin the same response was apparent with greater levels of IL-8 and MCP-1 release; at 100 μM release was even greater but with significant cell death (data not shown). There was minimal spontaneous conversion of oxyhemoglobin to the methemoglobin form after 24-h incubation under experimental conditions, consistent with the lack of IL-8 and MCP-1 release seen after treatment with oxyhemoglobin (data not shown).
Fig. 1.

Effect of oxyhemoglobin, hemoglobin degradation products, and azide-bound methemoglobin on A549 cells. A549 cells were incubated for 24 h with the indicated concentrations of oxyhemoglobin (oxyhb; A and B), hemin (C and D), ammonium iron sulfate (Fe2+; E and F), ferric citrate (Fe3+; G and H), or redox-inactivated azide-bound methemoglobin (methb; I). Methemoglobin (10 μM) was incubated overnight at 4°C with increasing amounts of azide prior to the addition to A549 cells. Levels of IL-8 (left) and MCP-1 (right) were determined in the supernatants by ELISA. Data are expressed as means ± SE from 4 separate experiments. Comparison between conditions was made by 1-way ANOVA with Bonferroni posttest, *P < 0.05, **P < 0.01, and ***P < 0.001 compared with control.
Fig. 2.

Time course of IL-8 and MCP-1 expression and release after methemoglobin challenge. A and B: A549 cells were incubated with 0 (open bars), 3 (shaded bars), or 10 μM (solid bars) methemoglobin for the times indicated. Levels of IL-8 (A) and MCP-1 (B) were determined in the supernatants by ELISA. C and D: A549 cells were incubated with 0 or 10 μM methemoglobin for the indicated times and RNA was extracted. Reverse-transcribed cDNA was subject to RT-PCR. Levels of IL-8 (C) and MCP-1 (D) mRNA are expressed as a ratio relative to the housekeeping gene. Data are expressed as means ± SE from 4 independent experiments. Comparison between conditions was made by 1-way ANOVA with Bonferroni posttest, *P < 0.05, **P < 0.01, and ***P < 0.001 compared with control at the indicated time points.
To exclude the possibility that the methemoglobin used was contaminated by LPS, the endotoxin content of the methemoglobin was measured at 9 ng/ml. This concentration failed to cause significant release of IL-8 and MCP-1 from A549 cells. Secondly, polymyxin B, a molecule that binds to and inactivates endotoxin, did not suppress the methemoglobin-mediated release of IL-8 and MCP-1 but did so for LPS-stimulated chemokine release. Finally, we prepared methemoglobin using Pierce High-Capacity endotoxin removal resin columns (ThermoScientific, Loughborough, Leicestershire, UK), which remove 99% of LPS contamination from solutions. Methemoglobin prepared by this method still resulted in the release of IL-8 from A549 cells and primary human lung slices (data not shown). Similarly, cell proliferation was excluded as the cause of the increase in IL-8 and MCP-1 from A549, since there was no significant increase in cell proliferation in response to methemoglobin (3 and 10 μM, as determined by Cyquant assay) over the time course of the relevant experiments.
Methemoglobin induced IL-8 and MCP-1 through activation of NF-κB.
Incubation of A549 cells with methemoglobin (10 μM) led to phosphorylation and partial degradation of the NF-κB inhibitor IκBα (Fig. 3, A–C) but had no effect on IκBβ (data not shown). Methemoglobin significantly (P < 0.001) induced the nuclear translocation and phosphorylation of NF-κB p65 serine-536 after 15 min (Fig. 3, D and E), but not NF-κB p65 serine-276 (data not shown). The IKK-2 inhibitors TPCA-1 (1 μM) and SC-514 (20 μM) both significantly (P < 0.001) inhibited methemoglobin-mediated phosphorylation of NF-κB p65 serine-536 (Fig. 4, A and B), the increase in IL-8 and MCP-1 protein release (Fig. 4, C–F), and mRNA expression (Fig. 4, G–J) in A549 cells.
Fig. 3.
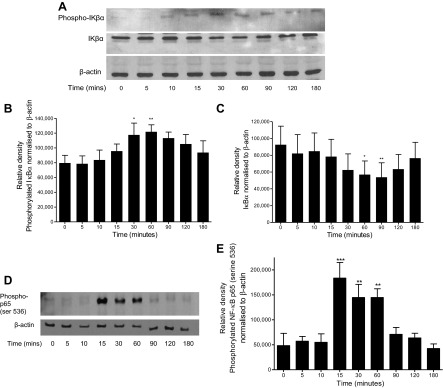
Time course of NF-κB activation after methemoglobin challenge. A549 cells were incubated with 10 μM methemoglobin for 0–180 min, and the cells were harvested in lysis buffer (A–C) or nuclear extracts were prepared (D and E). Western blots were performed to detect the presence of phosphorylated IκBα, IκBα, phosphorylated NF-κB p65 (ser536), and β-actin as a loading control; representative blots are shown (A). Western blots were quantified by use of Image J software. The values for phosphorylated IκBα and IκBα are presented, respectively, in B and C, and phosphorylated NF-κB p65 (ser536) in E. Data are presented as means ± SE of 4 independent experiments. Comparison between conditions was made by 1-way ANOVA with Bonferroni posttest: *P < 0.05, **P < 0.01, and ***P < 0.001 compared with control (time 0).
Fig. 4.

Effect of IKK-2 inhibitors on NF-κB activation and IL-8 and MCP-1 expression and release. A549 cells were pretreated with 1 μM [5-(p-fluorophenyl)-2-ureido]thiophene-3-carboxamide (TPCA-1) for 1 h or 20 μM SC-514 for 2 h prior to incubation with 10 μM methemoglobin. Cells were collected at the indicated times and nuclear extracts were prepared. Western blot analysis was performed to detect the presence of phosphorylated NF-κB p65 (ser536) and β-actin (loading control); representative blots are shown (A). Western blots were quantified by use of Image J software. The values for phosphorylated NF-κB p65 (ser536) normalized to β-actin are presented in Fig. 3B. A549 cells were pretreated with either TPCA-1 (left) for 1 h or SC-514 (right) for 2 h prior to the addition of methemoglobin. IL-8 and MCP-1 levels were determined in the cell supernatants after 24 h by ELISA (Fig. 3, C–F). IL-8 and MCP-1 mRNA levels relative to the housekeeping gene were determined after 2 h (G–J). In all panels data are presented as means ± SE of 4 independent experiments. Comparison between conditions was made by 1-way ANOVA with Bonferroni posttest, *P < 0.05, **P < 0.01, and ***P < 0.001. Asterisks on top of columns refer to comparison with the control (medium only); asterisks above lines refer to comparison between indicated conditions.
When A549 cells were transfected with the dominant negative form of IKK-2, phosphorylation of NF-κB p65 serine-536 was prevented (Fig. 5, A and B) and the expression of IL-8 (Fig. 5, C and D) and MCP-1 (Fig. 5, E and F) protein and mRNA levels were significantly (P < 0.001) suppressed.
Fig. 5.

Effect of dnIKK-2 transfection on NF-κB activation and IL-8 and MCP-1 expression and release. A and B: transfected cells (dnIKK-2; shaded bars) or normal nontransfected A549 cells (solid bars) were incubated with 10 μM methemoglobin for 0–60 min. Western blot analysis was performed to detect the presence of phosphorylated NF-κB p65 (ser536) and β-actin as loading control (blot shown in Fig. 4A). Western blots were quantified by use of Image J software and the values for phosphorylated NF-κB p65 (ser536) normalized to β-actin are presented in B as means ± SE of 3 independent experiments. C–F: A549 cells were transfected with dnIKK-2 (shaded bars) or an empty vector (open bars) for 24 h, transfection medium was removed, and cells incubated with methemoglobin for 2 or 24 h. IL-8 and MCP-1 were determined in the cell supernatants after 24 h by ELISA (C and E). Cells were harvested after 2 h for the detection of IL-8 and MCP-1 mRNA expression (D and F). IL-8 and MCP-1 mRNA levels were quantified by RT-PCR compared with housekeeping gene. Data are presented as means ± SE of 4 independent experiments. Comparison between conditions was made by 1-way ANOVA with Bonferroni posttest, *P < 0.05, **P < 0.01, and ***P < 0.001. Asterisks on top of column refer to comparison with the appropriate control; asterisks above lines refer to comparison between indicated conditions.
Methemoglobin induced IL-8 and MCP-1 via activation of the ERK1/2 and JNK MAPK.
Treatment of A549 cells with methemoglobin (10 μM) activated the ERK1/2 and JNK but not the p38 MAPK pathways. Methemoglobin caused a significant (P < 0.01) increase in the phosphorylation of p44/42 between 10 and 30 min, which decreased to basal levels after 3 h (Fig. 6, A–C). Preincubation with the MEK1/2 inhibitor U0126 (10 μM) suppressed ERK1/2 phosphorylation- and methemoglobin-mediated increase in IL-8 mRNA and protein (P < 0.05; Fig. 6, D and E) but had no effect on the increase in MCP-1 (Fig. 6, F and G).
Fig. 6.

Methemoglobin stimulated ERK activation in A549 cells. A–C: A549 cells were incubated with 10 μM methemoglobin for 0–180 min and cells were harvested in lysis buffer. Western blot analysis was performed (A). Data from 4 independent experiments were analyzed by use of Image J software to quantify the density of the bands, and the values for phospho-ERK42 and phospho-ERK44 against total ERK42 and ERK44 after normalization to β-actin are presented, respectively, in B and C. D–F: A549 cells were preincubated with 10 μM U0126 for 2 h prior to the addition of 3 μM (shaded bars) or 10 μM (solid bars) methemoglobin. IL-8 (D) and MCP-1 (F) were determined in the cell supernatants after 24 h by ELISA. Cells were treated with 10 μM methemoglobin and harvested after 2 h for the detection of IL-8 (E) and MCP-1 (G) mRNA expression. Data are presented as means ± SE of 4 independent experiments. Comparison between conditions was made by 1-way ANOVA with Bonferroni posttest, *P < 0.05, **P < 0.01, and ***P < 0.001. Asterisks on top of bars refer to comparison with the control (medium only); asterisks above lines refer to comparison between indicated conditions.
Incubation of A549 cells with methemoglobin (10 μM) resulted in a significant (P < 0.001) increase in the phosphorylation of JNK between 15 and 60 min. Levels returned to baseline after 2 h (Fig. 7, A–C). Preincubation of A549 with the JNK pathway inhibitor SP600125 (1–10 μM) resulted in a significant (P < 0.001) decrease in the methemoglobin-mediated increase in MCP-1 mRNA and protein (Fig. 7, D and E) but had no effect on methemoglobin-mediated IL-8 release (data not shown), suggesting that ERK1/2 pathway activation leads to the upregulation of IL-8 (but not MCP-1) and that activation of the JNK pathway is required for upregulation of MCP-1 (but not IL-8) in lung epithelial cells.
Fig. 7.

Methemoglobin stimulated JNK activation in A549 cells. A–C: A549 cells were incubated with 10 μM methemoglobin for 0–180 min and cells were harvested in lysis buffer. Western blot analysis was performed to detect the presence of phosphorylated JNK, JNK, and β-actin in 20 μg of cell lysate protein; representative blots are shown in A. Data from 4 independent Western blot experiments were analyzed by use of Image J software to quantify the density of the bands and the values for phospho-JNK46 and phospho-JNK54 against JNK46 and JNK54 after normalization to β-actin are presented, respectively, in B and C. D and E: A549 cells were preincubated with SP600125 for 1 h prior to the addition of methemoglobin for 2 or 24 h. MCP-1 (D) was determined in the cell supernatants after 24 h by ELISA. Cells were harvested after 2 h and total RNA was extracted. MCP-1 mRNA levels were quantified by real-time PCR compared with a housekeeping gene (E). Data are presented as means ± SE of 4 independent experiments. Comparison between conditions was made by 1-way ANOVA with Bonferroni posttest, *P < 0.05, **P < 0.01, and ***P < 0.001. Asterisks on top of bar refer to comparison with the control (medium only); asterisks above lines refer to comparison between indicated conditions.
Effect of antioxidants and manipulating iron levels on the response of epithelial cells to methemoglobin.
The iron chelators phenanthroline (500 μM), desferrioxamine (100 μM), and 2,2′-bipyridyl (10 μM) had no effect on methemoglobin-mediated release of IL-8 and MCP-1 from A549 cells; additionally, HBED (10 μM), an iron chelator and hydroxyl radical scavenger, was also ineffective; however, responses to hydrogen peroxide (100 μM) were suppressed (data not shown). Similarly, neither water nor lipid-soluble antioxidants, NAC (1 mM) and α-tocopherol (vitamin E, 1 mM), respectively, inhibited methemoglobin-mediated release of IL-8 and MCP-1 from A549 cells (data not shown).
In addition, the role of the iron storage protein ferritin was investigated. Knockdown of neither the FTL (Fig. 8A) nor FTH (data not shown) chains affected methemoglobin-mediated increase in IL-8 and MCP-1 mRNA expression and protein release (Fig. 7, B–E). Taken together these results suggest that the proinflammatory response of human epithelial cells to methemoglobin does not involve an iron- or ROS-driven mechanism.
Fig. 8.

Effect of ferritin light chain (FTL) knockdown on methemoglobin-mediated IL-8 and MCP-1 expression and release. A: A549 cells were treated with 10 nM control (Ctl) or FTL siRNA for 40 h. Cells were harvested and levels of FTL were measured by ELISA relative to total protein. Comparison between conditions was made by Student's t-test, **P < 0.01. B–E: A549 cells were treated with siRNA as above, followed by 10 μM methemoglobin (shaded bars) or medium (open bars). After 24 h IL-8 and MCP-1 levels (B and C) were measured in the cell supernatant by ELISA. Total RNA was harvested after treatment with methemoglobin for 2 h. Levels of IL-8 and MCP-1 mRNA relative to the housekeeping gene were quantified by real-time PCR (D and E).
Methemoglobin caused the release of IL-8 and MCP-1 from human lung alveolar type II cells.
Incubation of primary cultures of hAT2 cells with methemoglobin (3 and 10 μM) resulted in a significant (P < 0.01) increase in IL-8 and MCP-1, a result similar to that obtained in the A549 cell line (Fig. 9, A and B). Pharmacological inhibition of IKK-2 suppressed methemoglobin-mediated release of both IL-8 and MCP-1 (Fig. 9, C–F). Inhibition of ERK prevented the methemoglobin-mediated increase in IL-8 (Fig. 9G) but not MCP-1 (Fig. 9H) in AT2 cells. The JNK pathway inhibitor (SP600125, 10 μM) significantly inhibited methemoglobin-mediated MCP-1 release but had no effect on IL-8 release (Fig. 9, J and I), results identical to those obtained with A549 cells.
Fig. 9.

Methemoglobin stimulated release of IL-8 and MCP-1 from human AT2 cells. A and B: hAT2 cells were incubated with 0 (open bars), 3 (shaded bars), or 10 μM (solid bars) methemoglobin for 24 h. IL-8 (A) and MCP-1 (B) levels were determined in the supernatant by ELISA. Data are presented as means ± SE of 5 independent experiments. C–J: hAT2 cells were preincubated with 1 μM TPCA-1 (C and D) for 1 h, 20 μM SC-514 (E and F) for 2 h, 10 μM U0126 (G and H) for 2 h, or 10 μM SP600125 (I and J) for 1 h, prior to the addition of methemoglobin for 24 h. IL-8 and MCP-1 levels were determined in the cell supernatants by ELISA. Data are presented as means ± SE of 4 independent experiments. Comparison between conditions was made by Kruskal-Wallis test with Dunn's posttest, *P < 0.05, **P < 0.01, and ***P < 0.001. Asterisks on top of bar refer to comparison with the control (medium only); asterisks above the line refer to comparison between indicated conditions.
Cell viability.
For all the experiments carried out the MTT assay for cellular respiration was utilized to ensure that stimuli and inhibitors did not affect cell viability; no significant change in cell viability was evident with any of the conditions studied, unless stated in the figure legend.
DISCUSSION
In this study we have shown for the first time that human methemoglobin, but not oxyhemoglobin nor the Hb degradation products hemin and iron (Fe2+ or Fe3+) nor redox-inactivated methemoglobin (azide-bound methemoglobin) stimulated the release of IL-8 and MCP-1 from lung-derived alveolar epithelial cells. Via activation of the NF-κB pathway, methemoglobin caused maximum IL-8 and MCP-1 mRNA upregulation at 2 h. IL-8 expression also required ERK activation and MCP-1 required JNK activation; both MAPK pathways were activated after incubation of A549 cells with methemoglobin. Neither iron chelators nor antioxidants prevented methemoglobin-stimulated release of IL-8 and MCP-1 from A549 cells. Furthermore, knockdown of FTH or FTL, the protein complex responsible for intracellular iron storage, had no effect on methemoglobin-induced release of IL-8 and MCP-1. Finally, incubation of primary cultures of hAT2 cells with methemoglobin (10 μM for 24 h) resulted in a significant (P < 0.01) release of both IL-8 and MCP-1 protein. The IKK-2 (TPCA-1 and SC-514) and MAPK pathway (U0126 and SP600125) inhibitors prevented methemoglobin-stimulated IL-8 and MCP-1 release from hAT2 cells, yielding similar data to that obtained with A549 cells.
These results imply that methemoglobin activates the NF-κB and MAPK pathways leading to the release of IL-8 and MCP-1 via a mechanism that is independent of iron and ROS. Methemoglobin, oxyhemoglobin, and hemolysate induced the expression of inflammatory mediators and adhesion molecules in a variety of other cell types and experimental systems through the activation of NF-κB (31, 32, 47). In a rodent model of blast injury, which results in hemorrhagic acute lung injury, NF-κB activation was associated with whole lung levels of MCP-1 mRNA five to six times higher than in controls (10). We have shown for the first time that the ERK1/2 but not the p38 or JNK MAP kinase pathways mediate methemoglobin-stimulated induction of IL-8 in alveolar epithelial cells. Methemoglobin can activate numerous signaling pathways, including JNK, p38, and MAP kinase signal transduction pathways, which along with the NF-κB pathway were required for adhesion molecule expression by vascular endothelial cells (47). In neuronal cells, Hb treatment released lactate dehydrogenase and ROS via the activation of the ERK pathway (11, 24).
Methemoglobin induced IL-8 production in endothelial cells in a similar (31) time- and dose-dependent fashion. Others have reported that oxyhemoglobin and methemoglobin did not cause a proinflammatory response, but ferryl-hemoglobin, an oxidation product, stimulated endothelial cells to express adhesion molecules (47). Methemoglobin and oxyhemoglobin stimulated the release of IL-8 and TNF-α from mononuclear leukocytes (35) although nonphysiological doses were employed (10 mg/ml, which equates to ∼155 μM). Additionally, Hb caused the release of TNF-α (9), IL-6, and IL-8 (33) by monocytes and IL-1β, TNF-α, IL-6, and IL-8 by macrophages (7). Finally, hemolysate stimulated the expression of ICAM-1 and MCP-1 in brain microvascular endothelial cells, but again a very high concentration of Hb (1 mM) was used (32).
Some studies have indicated that heme or iron and not Hb instigates chemokine release after hemolysis (4, 5, 52). For example, hemin caused the release of IL-8 and upregulation of ICAM-1 in endothelial cells (39, 46), and IL-8 from neutrophils (21). Moreover, hemozoin, a polymer of heme, produced as a result of the malarial parasite's digestion of host Hb, stimulated the release of MCP-1 by murine macrophages and, when applied to human endothelial cells, induced IL-6, IL-8, and MCP-1 release (22). Furthermore, iron present in coal fly ash induced IL-8 release from epithelial cells (48). However, in the studies presented here, hemin and iron salts (Fe2+ or Fe3+) did not cause the release of IL-8 and MCP-1 from A549 cells, suggesting that the cells were responding to methemoglobin and not to its degradation products.
Outside the protective environment of the red blood cell, Hb predominantly remains in the ferrous state but in the presence of oxidants can undergo oxidation resulting in the transition of iron from the ferrous (Fe2+) to the ferric (Fe3+) state within the heme moiety. During this process ROS are generated that can further sustain a cycle of oxidative reactions. Indeed, the autooxidation of Hb itself leads to the production of superoxide and methemoglobin (37). Furthermore, Hb scavenges NȮ and in so doing can disrupt the balance between biological peroxides and redox regulators: superoxide, peroxynitrite, and hydrogen peroxide, and NO itself (2, 50). The molecules with which these species react to either stimulate or repress a given signaling pathway have not been identified, but downstream targets include MAPK and NF-κB (38). Additionally, NF-κB is activated by iron-dependent mechanisms (53), for example, in hepatic macrophages (29).
Patients with ARDS have elevated free Hb in the air space compared with patients with hydrostatic edema, and the quantity of blood was associated with markers of lipid peroxidation (6). In the present study, the antioxidants NAC and α-tocopherol and iron chelators phenanthroline, desferrioxamine, bipyridyl, and HBED (also a hydroxyl radical scavenger) failed to suppress methemoglobin-stimulated IL-8 and MCP-1 release from A549 cells, and knockdown of FTH and FTL chains had no effect on its proinflammatory effects. This implies that ROS and intracellular free iron are not essential for this response in alveolar epithelial cells. In endothelial cells, NAC but not the iron chelator desferrioxamine inhibited methemoglobin-stimulated production of IL-6 and IL-8, and membrane expression of E-selectin, although a high concentration of NAC (50 mM) was used (31). By contrast, the proinflammatory effects of ferryl-hemoglobin on vascular endothelial cells could not be inhibited by the antioxidants NAC and catalase (47).
Questions remain relating to how the signaling response is initiated by methemoglobin. In monocytes and macrophages, CD163 acts as an uptake receptor for Hb and haptoglobin/Hb complexes. We investigated the possibility that a similar uptake mechanism might be operational within epithelial cells by Western blotting, flow cytometry, and RT-PCR techniques, but we were unable to demonstrate the presence or expression of CD163 (data not shown). We cannot exclude the involvement of Toll-like receptors (TLRs) and pattern recognition receptors (PRR) (rather than a mechanism requiring cellular uptake) in that heme is a known activator of TLR4, and hemozoin activates TLR9 in macrophages (12, 18). Additionally proteases within the lungs of patients with cystic fibrosis cleave Hb (from microbleeds), and the resultant heme stimulates bronchial epithelial cells to release IL-8 via a novel TLR4 and EGF receptor pathway (13). TLR4 signaling requires a number of coreceptors (CD14 and MD-2 in particular), which are diminished or lost in A549 cells; hence this cell type fails to elicit a TLR4 response (44, 51). However, a recent study has shown hemoglobin to be proinflammatory in endothelial cells via activation of the myeloid differentiation primary response gene-88 (MyD88)-dependent pathway that activates NF-κB but is independent of TLR4 (30). Given our findings it is noteworthy that the authors suggested that it is the HbFe3+ form of hemoglobin that is responsible for the activation of MyD88 and that Hb could be classified as an endogenous danger-associated molecular pattern (DAMP) molecule. The actual TLR or PRR that Hb activates is of great interest but is as yet still unknown.
Finally, we investigated the possibility that iron released from the cellular processing of heme directly or via alteration of oxidant composition/fluxes modulates the signaling responses characterized in our study. We used iron chelators and antioxidants to exclude this possibility; although such approaches have been criticized experimentally because of lack of effective uptake for some chelators and the selective nature of some antioxidants, by using several chelators, both aqueous and lipid-phase antioxidants, and appropriate positive controls, we feel these criticisms have been largely answered.
Methemoglobin stimulated IL-6 and -8 release from endothelial cells (31) has been attributed to contamination with LPS (47). Although the methemoglobin used in our experiments contained 9 ng/ml LPS, incubation of A549 cells with 10 ng/ml LPS did not result in a significant increase in IL-8 or MCP-1, suggesting that LPS was unlikely to be solely responsible for the proinflammatory effect. Additionally, when methemoglobin was preincubated with the LPS chelator polymyxin B, which binds LPS with greater affinity than hemoglobin (8) or passed through an endotoxin-removal column, and then exposed to A549 cells and precision cut human lung slices, no decrease in IL-8 or MCP-1 release was observed. The lack of effect of removing LPS from a solution containing methemoglobin makes it much less likely that the stimulatory effect was dependent on LPS synergy (8, 26, 27, 43) under these circumstances.
Thus decompartmentalization of Hb within the lung, which complicates many causes of lung injury, results in the release of chemokines that have the potential to attract and activate neutrophils and monocytes, leading to an amplification of the original injury: in effect, a “second hit.” IL-8 and MCP-1 were used as end point markers to investigate the inflammatory effects of methemoglobin on the alveolar epithelium; these chemokines have been suggested as mediators and prognostic biomarkers of ARDS (15, 36) and other inflammatory lung conditions (3, 16, 45).
In summary, we have shown that in human lung epithelial cells methemoglobin activates the NF-κB and MAPK pathways, leading to the release of IL-8 and MCP-1, via as yet unidentified mechanisms that do not involve iron and ROS signaling. Free Hb within the lung potentially results in chemokine release, which attracts and activates inflammatory cells to the alveoli, thus potentiating a cycle of proinflammatory mediator release and cell activation. Decompartmentalization of Hb is therefore likely to provide a significant proinflammatory stimulus in the context of DAH. Limiting levels of lung free Hb and its downstream consequences may prove a potential therapeutic target worthy of further exploration.
GRANTS
This work was funded by grants from The Henry Smith Charity, The J P Moulton Charitable Foundation, The David Boston Trust, and The Royal Brompton Hospital Charitable Fund.
This work was also supported by the NIHR Respiratory Disease Biomedical Research Unit at the Royal Brompton and Harefield NHS Foundation Trust and Imperial College London. The views expressed in this publication are those of the authors and not necessarily those of the NHS, the National Institute for Health Research, or the Department of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
S.M., M.J.D.G., and G.J.Q. conception and design of research; S.M. and L.R. performed experiments; S.M. and L.R. analyzed data; S.M., L.R., T.W.E., M.J.D.G., and G.J.Q. interpreted results of experiments; S.M. and L.R. prepared figures; S.M. drafted manuscript; S.M., L.R., T.W.E., M.J.D.G., and G.J.Q. edited and revised manuscript; S.M., L.R., T.W.E., M.J.D.G., and G.J.Q. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors would like to thank Dr. Duncan Rogers and Natasha Madge for preparation of the precision-cut human lung slices.
REFERENCES
- 1.Adamzik M, Hamburger T, Petrat F, Peters J, de Groot H, Hartmann M. Free hemoglobin concentration in severe sepsis: methods of measurement and prediction of outcome. Crit Care 16: R125, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alayash AI, Patel RP, Cashon RE. Redox reactions of hemoglobin and myoglobin: biological and toxicological implications. Antioxid Redox Signal 3: 313–327, 2001 [DOI] [PubMed] [Google Scholar]
- 3.Antoniades HN, Neville-Golden J, Galanopoulos T, Kradin RL, Valente AJ, Graves DT. Expression of monocyte chemoattractant protein 1 mRNA in human idiopathic pulmonary fibrosis. Proc Natl Acad Sci USA 89: 5371–5375, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balla J, Jacob HS, Balla G, Nath K, Eaton JW, Vercellotti GM. Endothelial-cell heme uptake from heme proteins: induction of sensitization and desensitization to oxidant damage. Proc Natl Acad Sci USA 90: 9285–9289, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balla J, Vercellotti GM, Jeney V, Yachie A, Varga Z, Jacob HS, Eaton JW, Balla G. Heme, heme oxygenase, and ferritin: how the vascular endothelium survives (and dies) in an iron-rich environment. Antioxid Redox Signal 9: 2119–2137, 2007 [DOI] [PubMed] [Google Scholar]
- 6.Bastarache JA, Sebag SC, Clune JK, Grove BS, Lawson WE, Janz DR, Roberts LJ, 2nd, Dworski R, Mackman N, Ware LB. Low levels of tissue factor lead to alveolar haemorrhage, potentiating murine acute lung injury and oxidative stress. Thorax 67: 1032–1039, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bodet C, Chandad F, Grenier D. Hemoglobin and LPS act in synergy to amplify the inflammatory response. J Dent Res 86: 878–882, 2007 [DOI] [PubMed] [Google Scholar]
- 8.Brandenburg K, Garidel P, Andra J, Jurgens G, Muller M, Blume A, Koch MH, Levin J. Cross-linked hemoglobin converts endotoxically inactive pentaacyl endotoxins into a physiologically active conformation. J Biol Chem 278: 47660–47669, 2003 [DOI] [PubMed] [Google Scholar]
- 9.Carrillo EH, Gordon LE, Richardson JD, Polk HC., Jr. Free hemoglobin enhances tumor necrosis factor-alpha production in isolated human monocytes. J Trauma 52: 449–452, 2002 [DOI] [PubMed] [Google Scholar]
- 10.Chavko M, Adeeb S, Ahlers ST, McCarron RM. Attenuation of pulmonary inflammation after exposure to blast overpressure by N-acetylcysteine amide. Shock 32: 325–331, 2009 [DOI] [PubMed] [Google Scholar]
- 11.Chen-Roetling J, Li Z, Chen M, Awe OO, Regan RF. Heme oxygenase activity and hemoglobin neurotoxicity are attenuated by inhibitors of the MEK/ERK pathway. Neuropharmacology 56: 922–928, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coban C, Ishii KJ, Kawai T, Hemmi H, Sato S, Uematsu S, Yamamoto M, Takeuchi O, Itagaki S, Kumar N, Horii T, Akira S. Toll-like receptor 9 mediates innate immune activation by the malaria pigment hemozoin. J Exp Med 201: 19–25, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cosgrove S, Chotirmall SH, Greene CM, McElvaney NG. Pulmonary proteases in the cystic fibrosis lung induce interleukin 8 expression from bronchial epithelial cells via a heme/meprin/epidermal growth factor receptor/Toll-like receptor pathway. J Biol Chem 286: 7692–7704, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dobbs LG. Isolation and culture of alveolar type II cells. Am J Physiol Lung Cell Mol Physiol 258: L134–L147, 1990 [DOI] [PubMed] [Google Scholar]
- 15.Donnelly SC, Strieter RM, Kunkel SL, Walz A, Robertson CR, Carter DC, Grant IS, Pollok AJ, Haslett C. Interleukin-8 and development of adult respiratory distress syndrome in at-risk patient groups. Lancet 341: 643–647, 1993 [DOI] [PubMed] [Google Scholar]
- 16.Elssner A, Jaumann F, Dobmann S, Behr J, Schwaiblmair M, Reichenspurner H, Furst H, Briegel J, Vogelmeier C. Elevated levels of interleukin-8 and transforming growth factor-beta in bronchoalveolar lavage fluid from patients with bronchiolitis obliterans syndrome: proinflammatory role of bronchial epithelial cells. Munich Lung Transplant Group. Transplantation 70: 362–367, 2000 [DOI] [PubMed] [Google Scholar]
- 17.Feng L, Zhou X, Su LX, Feng D, Jia YH, Xie LX. Clinical significance of soluble hemoglobin scavenger receptor CD163 (sCD163) in sepsis, a prospective study. PLoS One 7: e38400, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, Oliveira MF, Oliveira PL, Graca-Souza AV, Bozza MT. Characterization of heme as activator of Toll-like receptor 4. J Biol Chem 282: 20221–20229, 2007 [DOI] [PubMed] [Google Scholar]
- 19.Ghio AJ, Richards JH, Crissman KM, Carter JD. Iron disequilibrium in the rat lung after instilled blood. Chest 118: 814–823, 2000 [DOI] [PubMed] [Google Scholar]
- 20.Gow AJ, Luchsinger BP, Pawloski JR, Singel DJ, Stamler JS. The oxyhemoglobin reaction of nitric oxide. Proc Natl Acad Sci USA 96: 9027–9032, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Graca-Souza AV, Arruda MA, de Freitas MS, Barja-Fidalgo C, Oliveira PL. Neutrophil activation by heme: implications for inflammatory processes. Blood 99: 4160–4165, 2002 [DOI] [PubMed] [Google Scholar]
- 22.Griffith JW, Sun T, McIntosh MT, Bucala R. Pure Hemozoin is inflammatory in vivo and activates the NALP3 inflammasome via release of uric acid. J Immunol 183: 5208–5220, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Halliwell B, Gutteridge J. Free Radicals in Biology and Medicine. New York: Oxford University Press, 2007 [Google Scholar]
- 24.Huang FP, Xi G, Keep RF, Hua Y, Nemoianu A, Hoff JT. Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J Neurosurg 96: 287–293, 2002 [DOI] [PubMed] [Google Scholar]
- 25.Janz DR, Bastarache JA, Peterson JF, Sills G, Wickersham N, May AK, Roberts LJ, 2nd, Ware LB. Association between cell-free hemoglobin, acetaminophen, and mortality in patients with sepsis: an observational study. Crit Care Med 41: 784–790, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jurgens G, Muller M, Koch MH, Brandenburg K. Interaction of hemoglobin with enterobacterial lipopolysaccharide and lipid A. Physicochemical characterization and biological activity. Eur J Biochem 268: 4233–4242, 2001 [DOI] [PubMed] [Google Scholar]
- 27.Kaca W, Roth RI, Levin J. Hemoglobin, a newly recognized lipopolysaccharide (LPS)-binding protein that enhances LPS biological activity. J Biol Chem 269: 25078–25084, 1994 [PubMed] [Google Scholar]
- 28.Lagan AL, Melley DD, Evans TW, Quinlan GJ. Pathogenesis of the systemic inflammatory syndrome and acute lung injury: role of iron mobilization and decompartmentalization. Am J Physiol Lung Cell Mol Physiol 294: L161–L174, 2008 [DOI] [PubMed] [Google Scholar]
- 29.Lin M, Rippe RA, Niemela O, Brittenham G, Tsukamoto H. Role of iron in NF-κB activation and cytokine gene expression by rat hepatic macrophages. Am J Physiol Gastrointest Liver Physiol 272: G1355–G1364, 1997 [DOI] [PubMed] [Google Scholar]
- 30.Lisk C, Kominsky D, Ehrentraut S, Bonaventura J, Nuss R, Hassell K, Nozik-Grayck E, Irwin DC. Hemoglobin-induced endothelial cell permeability is controlled, in part, via a myeloid differentiation primary response gene-88-dependent signaling mechanism. Am J Respir Cell Mol Biol 49: 619–626, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu X, Spolarics Z. Methemoglobin is a potent activator of endothelial cells by stimulating IL-6 and IL-8 production and E-selectin membrane expression. Am J Physiol Cell Physiol 285: C1036–C1046, 2003 [DOI] [PubMed] [Google Scholar]
- 32.Lu H, Shi JX, Zhang DM, Shen J, Lin YX, Hang CH, Yin HX. Hemolysate-induced expression of intercellular adhesion molecule-1 and monocyte chemoattractant protein-1 expression in cultured brain microvascular endothelial cells via through ROS-dependent NF-κB pathways. Cell Mol Neurobiol 29: 87–95, 2009 [DOI] [PubMed] [Google Scholar]
- 33.Lumadue JA, Lanzkron SM, Kennedy SD, Kuhl DT, Kickler TS. Cytokine induction of platelet activation. Am J Clin Pathol 106: 795–798, 1996 [DOI] [PubMed] [Google Scholar]
- 34.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest 122: 2731–2740, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McFaul SJ, Bowman PD, Villa VM, Gutierrez-Ibanez MJ, Johnson M, Smith D. Hemoglobin stimulates mononuclear leukocytes to release interleukin-8 and tumor necrosis factor alpha. Blood 84: 3175–3181, 1994 [PubMed] [Google Scholar]
- 36.Miller EJ, Cohen AB, Nagao S, Griffith D, Maunder RJ, Martin TR, Weiner-Kronish JP, Sticherling M, Christophers E, Matthay MA. Elevated levels of NAP-1/interleukin-8 are present in the airspaces of patients with the adult respiratory distress syndrome and are associated with increased mortality. Am Rev Respir Dis 146: 427–432, 1992 [DOI] [PubMed] [Google Scholar]
- 37.Misra HP, Fridovich I. The generation of superoxide radical during the autoxidation of hemoglobin. J Biol Chem 247: 6960–6962, 1972 [PubMed] [Google Scholar]
- 38.Muller JM, Rupec RA, Baeuerle PA. Study of gene regulation by NF-κB and AP-1 in response to reactive oxygen intermediates. Methods 11: 301–312, 1997 [DOI] [PubMed] [Google Scholar]
- 39.Natarajan R, Fisher BJ, Fowler AA., 3rd. Hypoxia inducible factor-1 modulates hemin-induced IL-8 secretion in microvascular endothelium. Microvasc Res 73: 163–172, 2007 [DOI] [PubMed] [Google Scholar]
- 40.Quinlan GJ, Evans TW, Gutteridge JM. Iron and the redox status of the lungs. Free Radic Biol Med 33: 1306–1313, 2002 [DOI] [PubMed] [Google Scholar]
- 41.Reeder BJ, Wilson MT. Hemoglobin and myoglobin associated oxidative stress: from molecular mechanisms to disease states. Curr Med Chem 12: 2741–2751, 2005 [DOI] [PubMed] [Google Scholar]
- 42.Reiter CD, Wang X, Tanus-Santos JE, Hogg N, Cannon RO, 3rd, Schechter AN, Gladwin MT. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat Med 8: 1383–1389, 2002 [DOI] [PubMed] [Google Scholar]
- 43.Roth RI. Hemoglobin enhances the production of tissue factor by endothelial cells in response to bacterial endotoxin. Blood 83: 2860–2865, 1994 [PubMed] [Google Scholar]
- 44.Schulz C, Farkas L, Wolf K, Kratzel K, Eissner G, Pfeifer M. Differences in LPS-induced activation of bronchial epithelial cells (BEAS-2B) and type II-like pneumocytes (A-549). Scand J Immunol 56: 294–302, 2002 [DOI] [PubMed] [Google Scholar]
- 45.Sekine Y, Yasufuku K, Heidler KM, Cummings OW, Van Rooijen N, Fujisawa T, Brown J, Wilkes DS. Monocyte chemoattractant protein-1 and RANTES are chemotactic for graft infiltrating lymphocytes during acute lung allograft rejection. Am J Respir Cell Mol Biol 23: 719–726, 2000 [DOI] [PubMed] [Google Scholar]
- 46.Shiu YT, Udden MM, McIntire LV. Perfusion with sickle erythrocytes up-regulates ICAM-1 and VCAM-1 gene expression in cultured human endothelial cells. Blood 95: 3232–3241, 2000 [PubMed] [Google Scholar]
- 47.Silva G, Jeney V, Chora A, Larsen R, Balla J, Soares MP. Oxidized hemoglobin is an endogenous proinflammatory agonist that targets vascular endothelial cells. J Biol Chem 284: 29582–29595, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith KR, Veranth JM, Hu AA, Lighty JS, Aust AE. Interleukin-8 levels in human lung epithelial cells are increased in response to coal fly ash and vary with the bioavailability of iron, as a function of particle size and source of coal. Chem Res Toxicol 13: 118–125, 2000 [DOI] [PubMed] [Google Scholar]
- 49.Solomon SB, Wang D, Sun J, Kanias T, Feng J, Helms CC, Solomon MA, Alimchandani M, Quezado M, Gladwin MT, Kim-Shapiro DB, Klein HG, Natanson C. Mortality increases after massive exchange transfusion with older stored blood in canines with experimental pneumonia. Blood 121: 1663–1672, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Suzuki YJ, Forman HJ, Sevanian A. Oxidants as stimulators of signal transduction. Free Radic Biol Med 22: 269–285, 1997 [DOI] [PubMed] [Google Scholar]
- 51.Thorley AJ, Grandolfo D, Lim E, Goldstraw P, Young A, Tetley TD. Innate immune responses to bacterial ligands in the peripheral human lung—role of alveolar epithelial TLR expression and signalling. PLoS One 6: e21827, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tsemakhovich VA, Bamm VV, Shaklai M, Shaklai N. Vascular damage by unstable hemoglobins: the role of heme-depleted globin. Arch Biochem Biophys 436: 307–315, 2005 [DOI] [PubMed] [Google Scholar]
- 53.Xiong S, She H, Takeuchi H, Han B, Engelhardt JF, Barton CH, Zandi E, Giulivi C, Tsukamoto H. Signaling role of intracellular iron in NF-κB activation. J Biol Chem 278: 17646–17654, 2003 [DOI] [PubMed] [Google Scholar]
- 54.Yang F, Haile DJ, Berger FG, Herbert DC, Van Beveren E, Ghio AJ. Haptoglobin reduces lung injury associated with exposure to blood. Am J Physiol Lung Cell Mol Physiol 284: L402–L409, 2003 [DOI] [PubMed] [Google Scholar]