

Abstract
Effective treatment of the cognitive symptoms of schizophrenia has remained an elusive goal. Despite the intense focus on treatments acting at or via cholinergic mechanisms, little remains known about the dynamic cholinergic abnormalities that contribute to the manifestation of the cognitive symptoms in patients. Evidence from basic neuroscientific and psychopharmacological investigations assists in proposing detailed cholinergic mechanisms and treatment targets for enhancement of attentional performance. Dynamic, cognitive performance-dependent abnormalities in cholinergic activity have been observed in animal models of the disorder and serve to further refine such proposals. Finally, the potential usefulness of individual groups of cholinergic drugs and important issues concerning the interactions between pro-cholinergic and antipsychotic treatments are addressed. The limited evidence available from patient studies and animal models indicates pressing research needs in order to guide the development of cholinergic treatments of the cognitive symptoms of schizophrenia.
Keywords: Acetylcholine, Cognition, Schizophrenia, Attention, Cortex, Translational research
1. Introduction
There is wide agreement in psychiatric practice and the literature that treatment with dopamine D2 receptor antagonists benefits the positive symptoms of a majority of patients with schizophrenia (Agid et al., 2006; Kapur et al., 2005; Kapur and Mamo, 2003; Kinon et al., 2010a, 2010b). However, the cognitive impairments, which are present in all patients, albeit at highly variable degrees of severity (Fioravanti et al., 2005; Heinrichs and Zakzanis, 1998), and which greatly limit the patient's ability to interact with the world and engage in a productive life (e.g., Green, 1996), are only minimally or not at all improved by antipsychotic drug treatment (for review see van Os and Kapur, 2009).
Treatments targeted at the cholinergic system are currently intensely studied candidates for addressing the cognitive deficits that remain after antipsychotic treatment and in remission. We will argue in this review that much of the current reasoning about cholinergic neuropsychopharmacology is based on a back-translation of the hypothesized mechanisms of acetylcholinesterase inhibitor treatments. Perhaps even more importantly, current drug development and theoretical work often treats the cholinergic system in a rather generic and static way, and does not consider the situational or temporal dynamics of its involvement in cognitive function. As will be discussed below, it is very likely that acetylcholine plays a role in the attention deficits of schizophrenia. However, we believe that a real and detailed understanding of what that role might be is still at the earliest stages. If drug development is to be effective, there is a strong need for truly mechanistic research that takes the complexities and interactions of the cholinergic system's functions in attention into account, and details which aspects are dysfunctional in both the active and remitted states of the disease (Sarter et al., 2009a).
For more than two decades, basic neuroscience research in animals as well as psychopharmacological studies in humans, including studies employing pharmaco-fMRI approaches, have confirmed that forebrain cholinergic systems, specifically the cortical cholinergic input system, are a major and indeed necessary modulator of the brain's fronto-parietal attention system (e.g., Deco and Thiele, 2009; Giocomo and Hasselmo, 2007; Hahn et al., 2007; Hasselmo and Sarter, 2010; Sarter et al., 2005). Cholinergic treatment approaches for the cognitive symptoms of schizophrenia have often started from a static hypothesis predicting a role for both abnormally low and high levels of cholinergic neurotransmission in the expression of cognitive symptoms (e.g., Money et al., 2010; Stip et al., 2005; Tandon and Greden, 1989). Recent research has begun to describe the multiple time scales that characterize the cholinergic mediation of attentional processes and capacities (Parikh et al., 2007; Parikh and Sarter, 2008) and thereby indicate new challenges and opportunities for the development of cholinergic therapies of attentional impairments. We begin with a review of the current literature, before giving a more detailed description of these new findings and their potential implications.
2. Attentional symptoms in schizophrenia and role of cholinergic systems
Among the domains of cognitive functions impaired in schizophrenia, considerable research has focused on the ability to sustain attention, specifically in situations which require that internal representations are employed in order to maintain attentional orienting toward the target location, target type, and/or the timing of targets (e.g., Gold et al., 2007; Grillon et al., 1990; Jazbec et al., 2007; Nuechterlein et al., 2009). Several reasons justify such a focus. Chronic impairments in the ability to detect and select relevant targets interfere with the ability to learn about the world and to update memory with realistic relationships between cues, actions and outcomes. Moreover, impairments in the management of attentional resources further weaken the patient's ability to employ experience, expectations and outcome to guide attention toward selected targets. These impairments are not attenuated in a clinically remitted state and thus are not secondary to other symptom clusters or acute clinical illness. Attentional impairments therefore have been postulated to serve as “neurocognitive rate-limiting factors” (Green, 1996) and consequently have remained a primary target for the development of therapies aiming at improving the cognitive status of schizophrenic patients.
The attentional performance of schizophrenic patients has been consistently demonstrated to be vulnerable to the detrimental effects of distractors (Grillon et al., 1990; Oltmanns, 1978; Oltmanns and Neale, 1975; Rappaport, 1967; Spring et al., 1989). Our own studies, using the human version of the sustained attention task (SAT) and its distractor condition (dSAT; see Demeter et al., 2008, 2011 for validation and neuroimaging data), found that exposure to the distractor condition produced robustly larger performance decline in schizophrenic patients than in age-matched, healthy controls. Importantly, this effect was not due to distractor-evoked impairments in perceptual mechanisms (Demeter et al., 2010). Our findings are consistent with the general hypothesis that patients are specifically impaired in employing cognitive control mechanisms to guide attention (Gold et al., 2007; Jazbec et al., 2007; Luck and Gold, 2008).
Given the evidence indicating an essential role of the cholinergic system for attention (Hasselmo and Sarter, 2010; Sarter et al., 2005), the cognitive symptoms of schizophrenia have long been hypothesized to be mediated, at least in part, by a dysregulated forebrain cholinergic system (e.g., Davis et al., 1975). Such dysregulation may reflect primary abnormalities in the organization or regulation of cholinergic neurons, as suggested by evidence indicating abnormalities in the expression of muscarinic and nicotinic acetylcholine receptors (m/nAChRs; Adler et al., 1998; Crook et al., 2000, 2001; De Luca et al., 2006; Freedman et al., 1997), and/or may be secondary to structural abnormalities that affect the functions of telencephalic-limbic circuits that converge on cholinergic neurons in the basal forebrain and/or cholinergic terminals in cortical and hippocampal target regions (Alexander et al., 2009; Floresco et al., 2009).
A potential role of cholinergic abnormalities in the manifestation of the symptoms of schizophrenia was first deduced from observations indicating that accidental or experimental exposure to irreversible acetylcholine esterase (AChE) inhibitors can induce or activate symptoms of psychosis in healthy humans and schizophrenic patients, respectively (Gershon and Shaw, 1961; Rowntree et al., 1950). However, direct evidence indicating abnormally enhanced, lowered, or abnormally responsive levels of neurotransmission in patients has remained extremely difficult to obtain. Postmortem neurochemical assessment of cholinergic neurotransmission levels continues to be a challenging objective, in part because the abundance of soluble acetylcholinesterase (AChE) rapidly hydrolyzes acetylcholine (ACh) in extracted tissues (hence the need for rapid microwave irradiation of the head in older neurochemical studies in animals; Bluth et al.,1980). Neither one of the two key enzymes of the cholinergic system, choline acetyltransferase (ChAT) or AChE, limit the rate of synthesis or metabolism, respectively. Therefore, decreases in the activity or density of ChAT or AChE indicate the loss of cholinergic neurons or terminals but are less likely to reveal more dynamic dysregulation in cholinergic neurotransmission. Likewise, the ex vivo assessment of the density of the choline transporter (CHT) density in synaptic plasma membrane, which is a rate-limiting factor in the synthesis of ACh, requires substantial amounts of fresh, unfrozen tissue and thus has been attempted extremely rarely (Bissette et al., 1996; Ferguson et al., 2003). Thus, it is perhaps unsurprising that alterations in the density of cholinergic neurons or of the morphological characteristics of cholinergic neurons in brains of schizophrenic patients have rarely been reported and are widely considered not to be present (Haroutunian et al., 1994; Powchik et al., 1998).
While the status of the presynaptic component of cholinergic neurotransmission in patients has remained unsettled, research focusing on cholinergic receptors has been informative. Several studies, including one SPECT study conducted in unmedicated patients and controlling for effects of smoking (Raedler et al., 2003) demonstrated that the density and expression of muscarinic M1 and M4 receptors are reduced in telencephalic regions of schizophrenic patients (Crook et al., 2000, 2001; Deng and Huang, 2005; Mancama et al., 2003; for review see Raedler et al., 2007). The presence of a subgroup of schizophrenic patients, characterized by a particularly marked decrease in cortical M1 receptor density, has been proposed (Scarr et al., 2009). The cognitive phenotype of this subgroup remains unclear, as does the impact of a potentially compensatory upregulation of M1 second-messenger signaling (Salah-Uddin et al., 2009). Finally, nAChR polymorphisms and decreased expression of the α7 and α3/α5 subunit-containing nAChRs were demonstrated (De Luca et al., 2006; Petrovsky et al., 2010).
Studies aiming at demonstrating the displacement of labeled ligands for nicotinic or muscarinic acetylcholine receptors (n/mAChRs) are potentially capable of revealing putative alterations in the synthetic capacity of cholinergic neurons, or even more dynamic, cognitive activity-dependent cholinergic dysregulation. Although several ligands for these receptors and other markers of cholinergic terminal function have been developed for use in positron emission tomography (PET) or single photon computerized emission tomography (SPECT; e.g. Bohnen and Frey, 2007; Frey et al., 1992; Horti and Villemagne, 2006; Mach et al., 1997; Mulholland et al., 1998; Nobuhara et al., 2000, 2001; Tsukada et al., 2001), to our knowledge, there are no studies that employed these ligands to determine potentially abnormal displacement rates in schizophrenic patients performing cognitive tasks (for a demonstration of the feasibility of such studies, in the context of aging research, see Cohen et al., 2006).
Collectively, and with the exception of a reduced availability of m/nAChRs in telencephalic regions, the status of cholinergic neurotransmission in schizophrenic patients, specifically the potential dysregulatory characteristics that are hypothesized to contribute to cognitive symptoms, is not well understood. As was the case for the “dopaminergic hypothesis,” where the empirical demonstration of abnormally regulated dopaminergic neurotransmission during acute disease periods came many decades after the original proposal (Laruelle et al., 1999), PET studies to demonstrate altered cholinergic neurotransmission during cognitive performance in schizophrenia appear technically feasible, but to our knowledge have not yet occurred.
As we will discuss next, evidence from basic research, including research in animal models, may provide clues as to the potential cholinergic regulatory abnormalities in schizophrenia. This includes evidence indicating a tight link between mesolimbic dopaminergic and basal forebrain cholinergic activity (Neigh et al., 2001, 2004) and suggests that potential cholinergic dysregulation in schizophrenia involves complex interactions between multiple modes of neurotransmission (tonic/phasic) and the timing, rise time and/or decay rates of the phasic (or transient) increases in cholinergic activity (on the scale of seconds). These transients mediate essential attentional operations (below) but for now may remain below the temporal and spatial resolution of current human neuroimaging methods.
3. Cholino-cognitive dynamics: what could go wrong and why?
3.1. Basic methodological and conceptual issues
In this section we will focus on evidence indicating how the forebrain cholinergic system, specifically the cortical cholinergic input system, mediates attentional performance and describe the implications of evidence from animal models of schizophrenia for the generation of hypotheses about the role of the cholinergic systems in schizophrenia's cognitive symptoms. An initial clarification of two important methodological and conceptual issues will assist in illuminating a complex array of evidence from neurochemical studies.
Different neurochemical methods employed to monitor ACh release in task-performing animals generate data at vastly different temporal resolutions. Depending on the individual method, data points indicating extracellular concentrations of ACh may be based on collections over several minutes and even tens of minutes (microdialysis), to seconds and milliseconds (amperometric measures using enzyme-coated microelectrodes).
Importantly, this issue is not merely a technical one. Current evidence and circuitry models suggest that cholinergic inputs to prefrontal regions consist of two populations of neurons, each with distinct transmission modes, tonic versus phasic, which associate with different aspects of the cholinergic regulation of attention (Hasselmo and Sarter, 2010). One population of cholinergic neurons tonically influences the gain of glutamatergic inputs from the thalamus (see dark red neuron in Fig. 1). In these neurons, cholinergic activity levels fluctuate over minutes. In functional terms, these cholinergic inputs enhance the general responsiveness of cortical circuitry for thalamic input processing.
Fig. 1.

Schematic illustration of the main elements of prefrontal (PFC) circuitry mediating the detection of cues and of the tonic (dark red cholinergic neuron) and phasic (bright red cholinergic neurons) components of cholinergic neurotransmission (a). In the cortex, cholinergic transients are generated in part by the stimulation of ionotropic glutamate receptors and by glutamate released from mediodorsal thalamic (MD) afferents (Parikh et al., 2008, 2010). Mediodorsal inputs and thus glutamatergic transients are modulated by tonic (changes occurring on the scale of minutes) cholinergic stimulation of α4β2* nicotinic acetylcholine receptors (nAChRs). This allows levels of motivation and the subjects's readiness for utilizing external cues to influence the probability and efficacy of cue detection. An example of such tonic cholinergic activity measured by using microdialysis is shown in b, illustrating tonic prefrontal ACh before, during, and after four blocks of 8-min trials of sustained attention performance (data adopted from Paolone et al., 2010). In the PFC, glutamatergic and cholinergic transients interact to recruit efferent circuitry for the generation of the behavioral response. Figure c shows prefrontal glutamatergic and cholinergic transients recorded during cued trials yielding hits (the onset of the cue and the times of lever extension and correct lever presses are indicated below the abscissa). Note that the timing of detection-indicating behavior is related to the timing of the initial rise in transient cholinergic activity, as opposed to transient peak time (Howe and Sarter, 2010; Parikh et al., 2007).
In contrast, the terminals of separate cholinergic neurons are the target of these glutamateric inputs and generate the short, second-based cholinergic transients (bright red cholinergic neuron in Fig. 1a) that mediate the actual cue detection process. These transients are indirectly modulated by the tonic cholinergic component, which as described above enhances glutamatergic inputs from thalamus. These glutamatergic inputs in turn influence the amplitudes of the cholinergic transients (Parikh et al., 2008, 2010). Electrochemical recordings outside the thalamic input layers have failed to indicate the presence of detection-mediating cholinergic transients (Howe and Sarter, 2010; unpublished results).
Measurements of ACh release using microdialysis studies do not differentiate between the release from these two populations. However, neuropharmacological studies indicate that drug treatments can lead to opposite effects on release measured by microdialysis versus the amplitudes of transients measured amperometrically (Howe et al., 2010; Paolone et al., 2010), suggesting that measures generated by microdialysis selectively indicate the tonic component of cholinergic neurotransmission. Conversely, the amperometric method is optimized for measuring transients. Furthermore, changes in tonic levels of cholinergic neurotransmission may occur simultaneously across large regions of the cortex; in contrast, cue detection-mediating transients appear to be restricted to prefrontal regions (Parikh et al., 2007). Thus, schizophrenia-related abnormalities in cholinergic neurotransmission could occur on either or both time scales of cholinergic neurotransmission.
The issue of cognitive context, and in particular the demands placed on the cognitive system, is important when assessing cholinergic neurotransmission and potential disease-related abnormalities in the system (Sarter et al., 2007). Tonic levels of performance-associated cortical ACh release covary with demands on attention, rather than levels of attentional performance (Kozak et al., 2006; Passetti et al., 2000; Sarter et al., 2006). Increasing the demands on top-down control of attention, for example by presenting a distractor, impairs attentional performance but at the same time further increases cortical levels of cholinergic neurotransmission (see release levels of control subjects in Fig. 2a and b). Moreover, results from a recent study indicated a highly significant relationship between distractor-evoked augmentation of performance-associated cholinergic activity and the severity of the performance decline (E. Demeter et al., unpublished results). This relationship suggests that higher levels of cholinergic activity more effectively protect against distractor-induced decline in attentional performance, consistent with the hypothesis that tonic levels of cortical cholinergic indicate the degree of top-down control deployed to support attentional performance.
Fig. 2.

Illustration of hypotheses about attentional performance-associated abnormalities in the regulation of tonic cholinergic activity in controls and schizophrenia. Although reflecting the evidence described in Kozak et al. (2007), the data shown represent a paradigmatic scenario. In the absence of a challenge on top-down control (a), performance-associated increases in cholinergic activity remain relatively stable during the performance period in control subjects as well as the disease model. However, while unchallenged performance does not differ significantly between the animal model and healthy controls, robustly higher levels of cholinergic activity mediate the performance in the animal model. This finding has been discussed in terms of reflecting inordinate requirements on cognitive control in order to support performance even in the absence of challenges, in part because levels of cholinergic activity in the animal model and the absence of challenges were found to correspond with the elevated levels observed after a top-down challenges in healthy subjects (b). In control subjects, manipulations that evoke top-down control of attentional performance, including the presentation of a distractor, moderately impair attentional performance, followed by (partial) performance recovery toward the end of the session. In these subjects, cortical cholinergic activity levels during the distractor period exceed the levels seen during standard task performance. In contrast, the more severe disruption of performance in the animal model is associated with, and perhaps caused by (for evidence supporting a causal relationship see Kozak et al., 2007), a rapid loss and return to pre-task levels of task-associated cholinergic activity.
As our experiments on the regulation of performance-associated tonic cholinergic activity in animal models of schizophrenia consistently found that baseline (or pre-task) ACh release levels remained normal and did not predict the abnormalities in performance-associated cortical ACh release, this review we will not address studies that were limited to measures of basal ACh release. Instead, we will describe hypotheses that address how different demands on attentional performance, specifically on top-down control, may reveal complex and diverging abnormalities in cholinergic neurotransmission in schizophrenia.
3.2. Attentional performance-associated tonic and phasic cholinergic neurotrans-mission in intact animals
Measures of tonic ACh release in the cortex of attentional task-performing animals revealed increases reaching 120–160% over baseline (e.g., Fig. 1b). Such increases are not seen in the cortex of animals performing behavioral control procedures devoid of explicit demands on attention (Arnold et al., 2002; Dalley et al., 2001; Himmelheber et al., 1997, 2001; Kozak et al., 2006, 2007). While more evidence is needed, it seems that such increases can be observed not just in prefrontal regions but also in parietal and posterior parietal regions (R. Kozak and M. Sarter, unpublished data). Fronto-parietal increases in tonic cholinergic neurotransmission may support overlapping mechanisms involved in cue detection and distractor filtering in prefrontal and parietal regions (Broussard et al., 2006, 2009; Gill et al., 2000).
Attentional performance can be challenged by manipulations of task parameters, specifically the presentation of distractors or by pharmacological challenges. In motivated subjects, these manipulations recruit (top-down) mechanisms designed to limit the impact of these challenges and to enhance the detection of cues (for a review of neuronal mechanisms mediating such effects see Sarter et al., 2006). As a result of such challenges, and while performance remains below baseline and recovers only slowly, ACh release levels are substantially higher than during standard task performance (Fig. 2; Kozak et al., 2006; Passetti et al., 2000). These augmented increases of tonic cholinergic activity are hypothesized to result from activation of prefrontal-mesolimbic circuitry, specifically involving the nucleus accumbens, which projects to basal forebrain neurons (Zmarowski et al., 2005, 2007). Such distractor-induced activation of prefrontal-mesolimbic circuitry may be evoked by prediction errors, possibly involving activation of dopaminergic inputs to prefrontal, mesolimbic as well as basal forebrain regions (Pessiglione et al., 2006; Pezze et al., 2007, 2009; Valentin and O'Doherty, 2009).
As already mentioned, increases in cholinergic neurotransmission over seconds (“transients”) in prefrontal regions mediate, and are necessary for, the incorporation of cues into ongoing cognitive activity. The rise time and peak time of these transients are precisely orchestrated and sensitive to variations of cue-reward intervals, but they are not evoked by reward-related behavior (Parikh et al., 2007; see Fig. 1a).
3.3. Altered cholino-cognitive dynamics in animal models of schizophrenia
Evidence addressing this issue was generated by studying an animal model that focused on the long-term attentional consequences of prior administration of an escalating-dose amphetamine regimen to attentional task-performing animals (Kozak et al., 2007; for discussion of the face and construct validity of this model see Sarter et al., 2009a). Furthermore, as low-dose psychostimulant exposure can evoke acute disease periods in schizophrenic patients (Davis, 1974; Yui et al., 1999), the model exhibits construct validity in that the disrupted behavior observed during acute disease periods was provoked by low-dose challenges of amphetamine. Performance under normal conditions, without the addition of the additional amphetamine challenge, has been considered a model of the remitted state, whereas performance after such challenge is thought to model the acute disease state.
3.3.1. Tonic cholinergic activity
When in the “remitted” state, these animals' attentional performance under standard, non-distracting conditions does not differ from that of control subjects. However, prefrontal ACh release is robustly higher than in control animals, reaching levels seen in control animals only in response to performance challenges (Fig. 2a; Kozak et al., 2007). This suggests that standard attentional performance in these animals may require an abnormal degree of top-down control. In intact animals, highly practiced attentional performance may largely be based on a combination of default responses in the absence of signals (correct rejections) and signalevoked – bottom-up switches out of this default behavior to report a hit. In contrast, maintaining high performance by the animals modeling the disease may require control mechanisms akin to those evoked by distractors in controls, including the filtering of noise as implicit distractor, enhanced processing of signals, and perhaps mechanisms enhancing the animals' ability to constrain behaviors to those that are task-relevant and sustain monitoring of the signal state. Abnormal regulation in larger, specifically prefrontal-mesolimbic circuits that converge on the cholinergic basal forebrain (Alexander et al., 2009) likely is involved in the cholinergic and cognitive mediation of attentional performance in the animal model.
When these animals were given a low-dose amphetamine challenge, conceptualized as a model for triggering an acute disease period, performance was characterized by near random lever selection. Correspondingly, ACh release levels did not rise from pre-task, baseline levels (Fig. 2b). Additional data, described in Kozak et al. (2007) suggests that the attenuation of performance-associated ACh release was not merely a correlate of, or secondary to, the disruption of performance. Rather, the “freezing” of ACh release at baseline levels may have prevented above-chance performance.
The results from these experiments indicate an abnormal regulation of the tonic component of cholinergic neurotransmission as a function of the demands on top-down regulation. Abnormally high levels of cholinergic neurotransmission support standard task performance in the animal model. These high levels may limit an effective response to additional demands on attentional control mechanisms.
3.3.2. Phasic cholinergic activity
There are not yet data on cue detection-mediating cholinergic transients in this animal model. We may speculate that such transients are less precisely orchestrated than in intact controls and that they exhibit highly variable rise and peak times and decay rates (see Fig. 3). Furthermore, it seems likely that such transients are generated spontaneously or in response to task events other than target signal presentation, such as to a cue or extending levers indicating the onset of a response period. Such false transients would begin to explain the propensity of patients and animal models to generate false alarms (Fig. 3). Clearly, the neuronal mechanisms, locally in the PFC and involving larger circuits, that generate and modulate these transients in intact animals and animal models of schizophrenia, remain poorly understood.
Fig. 3.
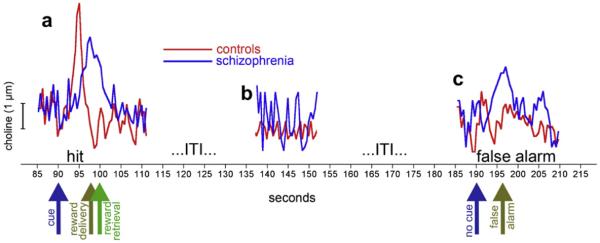
Attentional performance-associated abnormalities in the regulation of transient cholinergic activity in controls and schizophrenia. Cholinergic transients are shown during hit (a) and miss trials (c) and during intertrial intervals (b), in control subjects and, speculatively, in schizophrenia. The hit-related cholinergic transient as well as the absence of such transients is based on evidence from intact animals (Howe and Sarter, 2010; Parikh et al., 2007). As discussed in the main text, cues that are detected by patients, albeit likely with lower probability and longer response latencies than in healthy controls. Such performance may be mediated via transients that rise later and exhibit slower and less linear rise rates, peak at lower levels and more variable time points, and decay less rapidly than in healthy control subjects (a). Indeed, slower decay rates, indicative of ongoing and slowly diminishing release, were demonstrated to be associated with less effective attentional performance (Howe et al., 2010). The attentional performance of animal models and patients is characterized in part by an increased rate of false alarms, specifically in response to performance challenges. As cholinergic transients are required for detection, we can speculate that such transients, although relatively poorly organized, occur sporadically during blank trials (c). Finally, in the absence of performance, transient cholinergic activity is speculated to fluctuate more markedly (b), indicative of a less effective and stable state of the prefrontal detection network. These speculations serve to illustrate that normalization of cholinergic neurotransmission are less a subject of simple corrections of levels of neurotransmitter activity but presumably require restoring the timing and rise and decay dynamics of such transients.
4. Specific cholinergic treatment strategies
In this section, we consider the implications of the dynamic and demand-dependent aspects of the cholinergic system's involvement in attention for the major classes of cholinergically-targeted treatments that are either in use or under active development. For each, we describe the demonstrated or likely effectiveness in terms of the circuitry model described above, and indicate what evidence may further establish and refine their clinical utility.
4.1. Acetylcholinesterase inhibitors
Cholinergic treatment strategies in schizophrenia have focused on cholinesterase inhibitors, largely because of the availability of donepezil and galantamine for safe, clinical trials and the hypothesis that there are commonalities between the status of cholinergic neurotransmission in patients with schizophrenia and patients with dementia (e.g., Barak, 2009). The potential efficacy of galantamine has been of particular interest as this compound, in addition to blocking the AChE, also modulates nAChRs (Samochocki et al., 2003), although the clinical relevance of such modulation remains unclear. Beneficial cognitive, including attentional effects of AChE inhibitors have occasionally been documented in clinical studies (e.g., Schubert et al., 2006). However, the collective evidence does not convincingly suggest that AChE inhibitors are useful adjunct treatment for improving the patients' cognitive abilities (Buchanan et al., 2008; Chouinard et al., 2007; Dyer et al., 2008; Erickson et al., 2005; Friedman et al., 2002; Keefe et al., 2008; Kohler et al., 2007; Lindenmayer and Khan, 2010).
It is conceivable that these compounds' broad effects on extracellular ACh concentrations, including the ACh release-inhibiting effects of stimulation of presynaptic mAChRs, do not assist in optimizing the tonic component of cholinergic activity and even interfere with the generation of transients. Furthermore, if it is the case that cognitive performance-associated levels of tonic cholinergic activity are already elevated in patients (see also Tandon and Greden, 1989), it would not be clear how further increasing cholinergic activity would benefit the modulation of cortical target circuitry.
With respect to effect on phasic cholinergic neurotransmission, the effects of AChE inhibitors uncouple postsynaptic activity from presynaptic mechanisms and thus may not benefit the precisely orchestrated generation of cholinergic transients. In particular, inhibition of presynaptic ACh release as a result of mostly M2 receptor stimulation is expected to prevent the generation of precisely timed transients. Furthermore, the amplitudes of cholinergic transients are relatively small and a dampening of those release events as a result of presynaptic autoreceptor stimulation would not be consistent with a prediction of enhancement of attentional performance.
However, it is important to acknowledge that our understanding of the compartmentalized regulation of the proteins capable of hydrolyzing ACh remains extremely poor (Darvesh et al., 2003; Meerson et al., 2010; Meshorer and Soreq, 2006), that the pharmacological profiles of individual AChE-inhibitors differ considerably, and that therefore the potential usefulness of these compounds, specifically for patients with defined pharmacogenetic profiles (e.g., O'Brien et al., 2003) needs to be further studied (see also Ribeiz et al.). Finally, as will be further discussed below, smoking in patients may mask potentially more efficacious beneficial as well as detrimental effects of AChE inhibitors.
4.2. α4β2* nAChR agonists
Agonists at α4β2* nAChRs are thought to mimic the tonic effects of ACh at glutamatergic thalamic inputs, thereby amplifying the cortical representation of cues (Fig. 1a). In functional terms, the effects of such compounds may be described in relatively traditional terms as enhancing the general readiness for input processing or cortical arousal. Irrespective of such conceptualizations, these compounds enhance attentional performance in animals, selectively increasing the animals' detection rate and in interaction with performance challenges or poor performance levels (Grottick et al., 2003; Howe et al., 2010; McGaughy et al., 1999; Mohler et al., 2010). The circuitry model we have described for the effects of such compounds (Hasselmo and Sarter, 2010; Sarter et al., 2009b) correctly predicts that enhanced attentional performance as a result of treatment with α4β2* nAChR agonists is ultimately due to augmented amplitudes of cholinergic transients.
α4β2* nAChR agonists appear to represent a promising class of drugs for treating specifically the persistent cognitive impairments that are revealed by demands on top-down control and that are likely associated with attenuated levels of tonic cholinergic activity (above). While pilot studies on the cognitive effects of such compounds in patients with ADHD reported promising results (Wilens et al., 1999, 2006) we are not aware of clinical trials of such compounds in schizophrenic patients, except for a press release indicating that in a Phase IIb study, ispronicline (AZD3480; TC-1734) “did not meet the trial's criteria for statistical significance on the primary outcome endpoints, improvement on various cognitive domains measured by the IntegNeuro computerized test battery” (Press Release by AstraZeneca and Targacept Inc., Dec 8, 2008; taken from www.astrazeneca.com/pressrelease). This Press Release further indicates that patients “were active smokers”; it is therefore possible that high levels of nicotine competed directly with the efficacy of this compound at α4β2* nAChRs and, perhaps more importantly, that the effects of nicotine at other nAChRs limited the efficacy of the selective agonist (Howe et al., 2010). High smoking levels of schizophrenic patients may represent a variable that generally limits the efficacy of nAChR agonists (10 cigarettes or more sustain >5 ng nicotine per ml serum; Lawson et al., 1998). On the other hand, treatment with α4β2* nAChR agonists may allow a concomitant reduction in smoking in patients (Evins and Goff, 2008; Smith et al., 2009).
4.3. α7 nAChR agonists
Inhibitory sensory gating deficits exhibited by schizophrenic patients have been linked to mutations in the gene for the α7 nAChR. Furthermore, nicotine was shown to attenuate gating deficits (Adler et al., 1998; Freedman et al., 1997). Additional research indicated decreased α7 nAChR densities in thalamic and telencephalic regions of schizophrenic patients (Freedman et al., 1995; Marutle et al., 2001). Collectively, this evidence elevated the α7 nAChR to a primary target for treatment of the cognitive, specifically attentional impairments of schizophrenia (see also Olincy and Stevens, 2007). However, evidence from animal studies did not consistently support the hypothesis that stimulation of this receptor benefit attention (Grottick and Higgins, 2000; Hahn et al., 2003). Furthermore, the partial α7 nAChR agonist GTS-21 did not enhance cognition in a Phase II trial (Freedman et al., 2008). Furthermore, animal studies have shown that the beneficial attentional effects of nicotine are unrelated to effects at this receptor (Blondel et al., 2000). In our experiments, the beneficial attentional effects of nicotine were statistically robust when its effects at the α7 nAChR were blocked (Howe et al., 2010). Such blockade allowed nicotine to evoke cholinergic transients that rose much more rapidly and were cleared much more rapidly when compared with transients evoked by nicotine in the absence of α7 nAChR blockade. These findings are consistent with the hypothesis that cue detection is more effectively mediated by “sharper” transients than by transients that are less precisely locked to the cue (Howe et al., 2010; Parikh et al., 2007). It would be of interest to determine whether pharmacological blockade of the α7 nAChR augments the pro-attentional effects of nicotine in healthy (Hahn et al., 2009, 2007) and schizophrenic patients (Barr et al., 2008; Hong et al., 2009).
Owing to its profound calcium influx and thus effects on a wide array of cellular pathways (e.g., Bitner et al., 2007), stimulation of α7 nAChR would be expected to produce lasting structural neuronal and behavioral effects. It is more difficult to see how such a mechanism may normalize the abnormal processing of information in forebrain circuits that support trial-to-trial based performance. It seems more likely that such compounds are of potential interest for treating broader and more severe cognitive impairments resulting from more severe structural decline of neuronal circuitry.
4.4. mAChR agonists
We know surprisingly little about the specific roles of mAChRs in attentional performance, in part because of the past lack of selective ligands for these receptors and our extremely limited understanding of the distribution and functions of mAChR subtypes (e.g., Cea-del Rio et al., 2010; Jeon et al., 2010). Even the detrimental effects of non-selective mAChR antagonists such as scopolamine or atropine are difficult to interpret as the substantial increases in ACh release that result from blocking presynaptic mAChRs would excessively stimulate nAChRs (Hasselmo and Sarter, 2010). Generally, stimulation of postsynaptic mAChRs is assumed to contribute to the recruitment of postsynaptic circuitry that mediates the execution of the detection process (Nelson et al., 2005). Various mAChR agonists have been proposed for treatment of the cognitive symptoms of schizophrenia (Bradley et al., 2010; Brady et al., 2008; Leach et al., 2010; Shekhar et al., 2008).
4.5. Interactions with antipsychotic drugs
The preclinical and clinical development of compounds for the treatment of the cognitive symptoms of schizophrenia is burdened by the complexities that arise from the presumably compulsive co-treatment with first- or second-generation antipsychotic drugs. First, the selection of antipsychotic compounds for such studies involves an enormous array of issues, ranging from the receptor profile of individual compounds (e.g., concerns about interfering effects of muscarinic receptor antagonism; Raedler et al., 2000) to clinical antipsychotic and pro-cognitive efficacy (Riedel et al., 2007, 2010). Second, antipsychotic drug dosing issues need to be clarified in view of evidence indicating that low-dose treatment with anti-psychotic drugs may lead to beneficial cognitive effects (Green et al., 2002; Keefe et al., 2006; Mishara and Goldberg, 2004) while higher doses may exacerbate cognitive impairments (Castner et al., 2000; Uchida et al., 2009). In this context, antipsychotic drug doses and the administration regimen used in preclinical studies designed to characterize treatment combinations will need to be carefully selected and justified (Kapur et al., 2003).
Third, and most relevant in the present context, the cholinergic mechanisms of putative cognition enhancers will need to be studied in the presence of D2 receptor blockade (Kapur and Mamo, 2003). For example, how would concurrent D2 receptor blockade modulate ACh release in naive animals (Ichikawa et al., 2002), assuming that such effects generalize to animal models of the disorder and contexts in which demands on attention actually recruit cholinergic activity? How would D2 blockade modulate the enhancement of cue detection-mediating cholinergic transients by potential treatments and what might be the predictive validity of such data? Such evidence appears important for proposing compounds for clinical development and to begin conducting translational psychopharmacological research on treatments for cognitive symptoms.
5. Conclusions
Efforts to find and develop treatments for the cognitive symptoms of schizophrenia remain intensely focused on drugs acting at cholinergic mechanisms (e.g., Conn et al., 2009; Leach et al., 2010; Lieberman et al., 2008; Money et al., 2010). As discussed above, such a focus seems well-justified based on basic neuroscientific and, specifically with respect to nAChR agonists and psychopharmacological research in animal models and healthy humans. However, the nature of cholinergic dysregulation in patients and its impact on cognitive, including attentional, functioning remains poorly understood. In order to define drug-development targets, cognitive-psychopharmacological and pharmaco-fMRI studies in patients should make close contact with the available evidence from neuroscientific and psychopharmacological studies in animals and healthy humans. To this end, the use of a task for the measurement of attentional functions that exhibit face and construct validity in laboratory animals and humans, including schizophrenic patients (Demeter et al., 2008, 2010, 2011; Nuechterlein et al., 2009) represents an important necessity as it assures that all these levels of analysis address similar cognitive operations. For example, as we learn more about the pharmacological manipulation of cholinergic transients by nicotinic receptor ligands, including the impact of altered transients for on attentional performance (above), the use of a task with cross-species validity will enable arguably more useful predictions about how different compounds will modify performance in healthy humans. The need for these cross-species task and the general challenges of investigations that cross disciplines and species boundaries are not unique to the discovery and evaluation of cholinergic treatments (e.g., Barnett et al., 2010).
Our discussion of the nature of the abnormalities of tonic and phasic cholinergic neurotransmission in schizophrenia is necessarily hypothetical. These speculations were derived in part from available evidence from an animal model. Considering that levels of, or demands on, cognitive performance yield different predictions about the nature of cholinergic dysregulation, and that these predictions may entail variation in the timing and slope of rise and decay rates of transients, one may conclude that these complexities render a rational development of cholinergic cognition enhancers to be a rather challenging goal. However, this is not the point of this discussion. These complexities indicate that potential treatments will need to be characterized across the range of measures of tonic and phasic cholinergic neurotransmission, and that cognitive activity will need to be treated as an independent variable, not just an outcome measure. These complexities do not, however, inform about the usefulness, or lack thereof, of a particular pharmacological target mechanisms. For example, it is plausible that a rather simple mechanism, such as manipulation of a receptor subtype of the cholinergic or GABAergic systems, would normalize the timing of the transient onset or enhance a rapid attenuation of such a transient. We hope to enhance awareness of the complex nature of cholinergic neurotransmission in order to advance the predictive validity of preclinical research on cognition enhancers.
Acknowledgements
The research described in this manuscript has been supported by NIH grants MH080332, MH080426, MH086530 (MS), MH086701 and MH086701 (SFT) and NSF grant 0726285 (CL).
References
- Adler L, Olincy A, Waldo M, Harris J, Griffith J, Stevens K, Flach K, Nagamoto H, Bickford P, Leonard S, Freedman R. Schizophrenia, sensory gating, and nicotinic receptors. Schizophr. Bull. 1998;24:189–202. doi: 10.1093/oxfordjournals.schbul.a033320. [DOI] [PubMed] [Google Scholar]
- Agid O, Seeman P, Kapur S. The “delayed onset” of antipsychotic action–an idea whose time has come and gone. J. Psychiatry Neurosci. 2006;31:93–100. [PMC free article] [PubMed] [Google Scholar]
- Alexander K, Brooks J, Sarter M, Bruno JP. Disruption of mesolimbic regulation of prefrontal cholinergic transmission in an animal model of schizophrenia and normalization by chronic clozapine treatment. Neuropsychopharmacology. 2009;34:2710–2720. doi: 10.1038/npp.2009.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold H, Burk J, Hodgson E, Sarter M, Bruno J. Differential cortical acetylcholine release in rats performing a sustained attention task versus behavioral control tasks that do not explicitly tax attention. Neuroscience. 2002;114:451–460. doi: 10.1016/s0306-4522(02)00292-0. [DOI] [PubMed] [Google Scholar]
- Barak S. Modeling cholinergic aspects of schizophrenia: focus on the anti-muscarinic syndrome. Behav. Brain Res. 2009;204:335–351. doi: 10.1016/j.bbr.2009.04.006. [DOI] [PubMed] [Google Scholar]
- Barnett JH, Robbins TW, Leeson VC, Sahakian BJ, Joyce EM, Blackwell AD. Assessing cognitive function in clinical trials of schizophrenia. Neurosci. Biobehav Rev. 2010;34:1161–1177. doi: 10.1016/j.neubiorev.2010.01.012. [DOI] [PubMed] [Google Scholar]
- Barr R, Culhane M, Jubelt L, Mufti R, Dyer M, Weiss A, Deckersbach T, Kelly J, Freudenreich O, Goff D, Evins A. The effects of transdermal nicotine on cognition in nonsmokers with schizophrenia and nonpsychiatric controls. Neuropsychopharmacology. 2008;33:480–490. doi: 10.1038/sj.npp.1301423. [DOI] [PubMed] [Google Scholar]
- Bissette G, Seidler F, Nemeroff C, Slotkin T. High affinity choline transporter status in Alzheimer's disease tissue from rapid autopsy. Ann. N. Y Acad. Sci. 1996;777:197–204. doi: 10.1111/j.1749-6632.1996.tb34419.x. [DOI] [PubMed] [Google Scholar]
- Bitner R, Bunnelle W, Anderson D, Briggs C, Buccafusco J, Curzon P, Decker M, Frost J, Gronlien J, Gubbins E, Li J, Malysz J, Markosyan S, Marsh K, Meyer M, Nikkel A, Radek R, Robb H, Timmermann D, Sullivan J, Gopalakrishnan M. Broad-spectrum efficacy across cognitive domains by alpha7 nicotinic acetylcholine receptor agonism correlates with activation of ERK1/2 and CREB phosphorylation pathways. J. Neurosci. 2007;27:10578–10587. doi: 10.1523/JNEUROSCI.2444-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondel A, Sanger D, Moser P. Characterisation of the effects of nicotine in the five-choice serial reaction time task in rats: antagonist studies. Psychopharmacology (Berl.) 2000;149:293–305. doi: 10.1007/s002130000378. [DOI] [PubMed] [Google Scholar]
- Bluth R, Langnickel R, Raubach K, Fink H. A simplified enzymatic assay for the determination of acetylcholine and choline in discrete structures of rat brain. Acta Biol. Med. Ger. 1980;39:881–887. [PubMed] [Google Scholar]
- Bohnen N, Frey K. Imaging of cholinergic and monoaminergic neurochemical changes in neurodegenerative disorders. Mol. Imaging Biol. 2007;9:243–257. doi: 10.1007/s11307-007-0083-6. [DOI] [PubMed] [Google Scholar]
- Bradley S, Lameh J, Ohrmund L, Son T, Bajpai A, Nguyen D, Friberg M, Burstein E, Spalding T, Ott T, Schiffer H, Tabatabaei A, McFarland K, Davis R, Bonhaus D. AC-260584, an orally bioavailable M(1) muscarinic receptor allosteric agonist, improves cognitive performance in an animal model. Neuropharmacology. 2010;58:365–373. doi: 10.1016/j.neuropharm.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Brady A, Jones C, Bridges T, Kennedy J, Thompson A, Heiman J, Breininger M, Gentry P, Yin H, Jadhav S, Shirey J, Conn P, Lindsley C. Centrally active allosteric potentiators of the M4 muscarinic acetylcholine receptor reverse amphetamine-induced hyperlocomotor activity in rats. J. Pharmacol. Exp. Ther. 2008;327:941–953. doi: 10.1124/jpet.108.140350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broussard J, Sarter M, Givens B. Neuronal correlates of signal detection in the posterior parietal cortex of rats performing a sustained attention task. Neuroscience. 2006;143:407–417. doi: 10.1016/j.neuroscience.2006.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broussard JI, Karelina K, Sarter M, Givens B. Cholinergic optimization of cue-evoked parietal activity during challenged attentional performance. Eur. J. Neurosci. 2009;29:1711–1722. doi: 10.1111/j.1460-9568.2009.06713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan RW, Conley RR, Dickinson D, Ball MP, Feldman S, Gold JM, McMahon RP. Galantamine for the treatment of cognitive impairments in people with schizophrenia. Am. J. Psychiatry. 2008;165:82–89. doi: 10.1176/appi.ajp.2007.07050724. [DOI] [PubMed] [Google Scholar]
- Castner S, Williams G, Goldman-Rakic P. Reversal of antipsychotic-induced working memory deficits by short-term dopamine D1 receptor stimulation. Science. 2000;287:2020–2022. doi: 10.1126/science.287.5460.2020. [DOI] [PubMed] [Google Scholar]
- Cea-del Rio C, Lawrence J, Tricoire L, Erdelyi F, Szabo G, McBain C. M3 muscarinic acetylcholine receptor expression confers differential cholinergic modulation to neurochemically distinct hippocampal basket cell subtypes. J. Neurosci. 2010;30:6011–6024. doi: 10.1523/JNEUROSCI.5040-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouinard S, Stip E, Poulin J, Melun J, Godbout R, Guillem F, Cohen H. Rivastigmine treatment as an add-on to antipsychotics in patients with schizophrenia and cognitive deficits. Curr. Med. Res. Opin. 2007;23:575–583. doi: 10.1185/030079906X167372. [DOI] [PubMed] [Google Scholar]
- Cohen R, Carson R, Filbey F, Szczepanik J, Sunderland T. Age and APOE-epsilon4 genotype influence the effect of physostigmine infusion on the in-vivo distribution volume of the muscarinic-2-receptor dependent tracer [18F] FP-TZTP. Synapse. 2006;60:86–92. doi: 10.1002/syn.20276. [DOI] [PubMed] [Google Scholar]
- Conn P, Jones C, Lindsley C. Subtype-selective allosteric modulators of muscarinic receptors for the treatment of CNS disorders. Trends Pharmacol. Sci. 2009;30:148–155. doi: 10.1016/j.tips.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crook J, Tomaskovic-Crook E, Copolov D, Dean B. Decreased muscarinic receptor binding in subjects with schizophrenia: a study of the human hippocampal formation. Biol. Psychiatry. 2000;48:381–388. doi: 10.1016/s0006-3223(00)00918-5. [DOI] [PubMed] [Google Scholar]
- Crook J, Tomaskovic-Crook E, Copolov D, Dean B. Low muscarinic receptor binding in prefrontal cortex from subjects with schizophrenia: a study of Brodmann's areas 8, 9, 10, and 46 and the effects of neuroleptic drug treatment. Am. J. Psychiatry. 2001;158:918–925. doi: 10.1176/appi.ajp.158.6.918. [DOI] [PubMed] [Google Scholar]
- Dalley J, McGaughy J, O'Connell M, Cardinal R, Levita L, Robbins T. Distinct changes in cortical acetylcholine and noradrenaline efflux during contingent and noncontingent performance of a visual attentional task. J. Neurosci. 2001;21:4908–4914. doi: 10.1523/JNEUROSCI.21-13-04908.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darvesh S, Hopkins D, Geula C. Neurobiology of butyrylcholinesterase. Nat. Rev. Neurosci. 2003;4:131–138. doi: 10.1038/nrn1035. [DOI] [PubMed] [Google Scholar]
- Davis JM. A two factor theory of schizophrenia. J. Psychiatr. Res. 1974;11:25–29. doi: 10.1016/0022-3956(74)90065-x. [DOI] [PubMed] [Google Scholar]
- Davis KL, Hollister LE, Berger PA, Barchas JD. Cholinergic imbalance hypotheses of psychoses and movement disorders: strategies for evaluation. Psychopharmacol. Commun. 1975;1:533–543. [PubMed] [Google Scholar]
- De Luca V, Voineskos S, Wong G, Kennedy J. Genetic interaction between alpha4 and beta2 subunits of high affinity nicotinic receptor: analysis in schizophrenia. Exp. Brain Res. 2006;174:292–296. doi: 10.1007/s00221-006-0458-y. [DOI] [PubMed] [Google Scholar]
- Deco G, Thiele A. Attention: oscillations and neuropharmacology. Eur. J. Neurosci. 2009;30:347–354. doi: 10.1111/j.1460-9568.2009.06833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demeter E, Sarter M, Lustig C. Rats and humans paying attention: cross-species task development for translational research. Neuropsychology. 2008;22:787–799. doi: 10.1037/a0013712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demeter E, Hernandez-Garcia L, Sarter M, Lustig C. Challenges to attention: a continuous arterial spin labeling (ASL) study of the effects of distraction on sustained attention. Neuroimage. 2011;54:1518–1529. doi: 10.1016/j.neuroimage.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demeter E, St. Peters M, Lustig C, M S. Society for Neuroscience Annual Meeting. San Diego: 2010. The Distractor Condition Sustained Attention Task: A Translational Tool for Attentional Control in Mice, Rats, Healthy Humans and Schizophrenic Patients. [Google Scholar]
- Deng C, Huang X. Decreased density of muscarinic receptors in the superior temporal gyrusin schizophrenia. J. Neurosci. Res. 2005;81:883–890. doi: 10.1002/jnr.20600. [DOI] [PubMed] [Google Scholar]
- Dyer MA, Freudenreich O, Culhane MA, Pachas GN, Deckersbach T, Murphy E, Goff DC, Evins AE. High-dose galantamine augmentation inferior to placebo on attention, inhibitory control and working memory performance in nonsmokers with schizophrenia. Schizophr Res. 2008;102:88–95. doi: 10.1016/j.schres.2007.12.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson S, Schwarzkopf S, Palumbo D, Badgley-Fleeman J, Smirnow A, Light G. Efficacy and tolerability of low-dose donepezil in schizophrenia. Clin. Neuropharmacol. 2005;28:179–184. doi: 10.1097/01.wnf.0000173714.61744.e6. [DOI] [PubMed] [Google Scholar]
- Evins AE, Goff DC. Varenicline treatment for smokers with schizophrenia: a case series. J. Clin. Psychiatry. 2008;69:1016. doi: 10.4088/jcp.v69n0620a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson S, Savchenko V, Apparsundaram S, Zwick M, Wright J, Heilman C, Yi H, Levey A, Blakely R. Vesicular localization and activity-dependent trafficking of presynaptic choline transporters. J. Neurosci. 2003;23:9697–9709. doi: 10.1523/JNEUROSCI.23-30-09697.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioravanti M, Carlone O, Vitale B, Cinti M, Clare L. A meta-analysis of cognitive deficits in adults with a diagnosis of schizophrenia. Neuropsychol. Rev. 2005;15:73–95. doi: 10.1007/s11065-005-6254-9. [DOI] [PubMed] [Google Scholar]
- Floresco S, Zhang Y, Enomoto T. Neural circuits subserving behavioral flexibility and their relevance to schizophrenia. Behav. Brain Res. 2009;204:396–409. doi: 10.1016/j.bbr.2008.12.001. [DOI] [PubMed] [Google Scholar]
- Freedman R, Hall M, Adler L, Leonard S. Evidence in postmortem brain tissue for decreased numbers of hippocampal nicotinic receptors in schizophrenia. Biol. Psychiatry. 1995;38:22–33. doi: 10.1016/0006-3223(94)00252-X. [DOI] [PubMed] [Google Scholar]
- Freedman R, Coon H, Myles-Worsley M, Orr-Urtreger A, Olincy A, Davis A, Polymeropoulos M, Holik J, Hopkins J, Hoff M, Rosenthal J, Waldo M, Reimherr F, Wender P, Yaw J, Young D, Breese C, Adams C, Patterson D, Adler L, Kruglyak L, Leonard S, Byerley W. Linkage of a neurophysiological deficit in schizophrenia to a chromosome 15 locus. Proc. Natl. Acad. Sci. U S A. 1997;94:587–592. doi: 10.1073/pnas.94.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman R, Olincy A, Buchanan R, Harris J, Gold J, Johnson L, Allensworth D, Guzman-Bonilla A, Clement B, Ball M, Kutnick J, Pender V, Martin L, Stevens K, Wagner B, Zerbe G, Soti F, Kem W. Initial phase 2 trial of a nicotinic agonist in schizophrenia. Am. J. Psychiatry. 2008;165:1040–1047. doi: 10.1176/appi.ajp.2008.07071135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey K, Koeppe R, Mulholland G, Jewett D, Hichwa R, Ehrenkaufer R, Carey J, Wieland D, Kuhl D, Agranoff B. In vivo muscarinic cholinergic receptor imaging in human brain with [11C]scopolamine and positron emission tomography. J. Cereb. Blood Flow Metab. 1992;12:147–154. doi: 10.1038/jcbfm.1992.18. [DOI] [PubMed] [Google Scholar]
- Friedman J, Adler D, Howanitz E, Harvey P, Brenner G, Temporini H, White L, Parrella M, Davis K. A double blind placebo controlled trial of donepezil adjunctive treatment to risperidone for the cognitive impairment of schizophrenia. Biol. Psychiatry. 2002;51:349–357. doi: 10.1016/s0006-3223(01)01342-7. [DOI] [PubMed] [Google Scholar]
- Gershon S, Shaw F. Psychiatric sequelae of chronic exposure to organo-phosphorus insecticides. Lancet. 1961;1:1371–1374. doi: 10.1016/s0140-6736(61)92004-9. [DOI] [PubMed] [Google Scholar]
- Gill TM, Sarter M, Givens B. Sustained visual attention performance-associated prefrontal neuronal activity: evidence for cholinergic modulation. J. Neurosci. 2000;20:4745–4757. doi: 10.1523/JNEUROSCI.20-12-04745.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giocomo L, Hasselmo M. Neuromodulation by glutamate and acetylcholine can change circuit dynamics by regulating the relative influence of afferent input and excitatory feedback. Mol. Neurobiol. 2007;36:184–200. doi: 10.1007/s12035-007-0032-z. [DOI] [PubMed] [Google Scholar]
- Gold J, Fuller R, Robinson B, Braun E, Luck S. Impaired top-down control of visual search in schizophrenia. Schizophr Res. 2007;94:148–155. doi: 10.1016/j.schres.2007.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green M. What are the functional consequences of neurocognitive deficits in schizophrenia? Am. J. Psychiatry. 1996;153:321–330. doi: 10.1176/ajp.153.3.321. [DOI] [PubMed] [Google Scholar]
- Green M, Marder S, Glynn S, McGurk S, Wirshing W, Wirshing D, Liberman R, Mintz J. The neurocognitive effects of low-dose haloperidol: a two-year comparison with risperidone. Biol. Psychiatry. 2002;51:972–978. doi: 10.1016/s0006-3223(02)01370-7. [DOI] [PubMed] [Google Scholar]
- Grillon C, Courchesne E, Ameli R, Geyer M, Braff D. Increased distractibility in schizophrenic patients. Electrophysiologic and behavioral evidence. Arch. Gen. Psychiatry. 1990;47:171–179. doi: 10.1001/archpsyc.1990.01810140071010. [DOI] [PubMed] [Google Scholar]
- Grottick A, Higgins G. Effect of subtype selective nicotinic compounds on attention as assessed by the five-choice serial reaction time task. Behav. Brain Res. 2000;117:197–208. doi: 10.1016/s0166-4328(00)00305-3. [DOI] [PubMed] [Google Scholar]
- Grottick A, Haman M, Wyler R, Higgins G. Reversal of a vigilance decrement in the aged rat by subtype-selective nicotinic ligands. Neuropsychopharmacology. 2003;28:880–887. doi: 10.1038/sj.npp.1300102. [DOI] [PubMed] [Google Scholar]
- Hahn B, Sharples C, Wonnacott S, Shoaib M, Stolerman I. Attentional effects of nicotinic agonists in rats. Neuropharmacology. 2003;44:1054–1067. doi: 10.1016/s0028-3908(03)00099-6. [DOI] [PubMed] [Google Scholar]
- Hahn B, Ross T, Yang Y, Kim I, Huestis M, Stein E. Nicotine enhances visuospatial attention by deactivating areas of the resting brain default network. J. Neurosci. 2007;27:3477–3489. doi: 10.1523/JNEUROSCI.5129-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn B, Ross T, Wolkenberg F, Shakleya D, Huestis M, Stein E. Performance effects of nicotine during selective attention, divided attention, and simple stimulus detection: an fMRI study. Cereb. Cortex. 2009;19:1990–2000. doi: 10.1093/cercor/bhn226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haroutunian V, Davidson M, Kanof P, Perl D, Powchik P, Losonczy M, McCrystal J, Purohit D, Bierer L, Davis K. Cortical cholinergic markers in schizophrenia. Schizophr Res. 1994;12:137–144. doi: 10.1016/0920-9964(94)90071-x. [DOI] [PubMed] [Google Scholar]
- Hasselmo M, Sarter M. Modes and models of forebrain cholinergic neuromodulation of cognition. Neuropsychopharmacology. 2010 doi: 10.1038/npp.2010.104. Epub 7/28/2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrichs R, Zakzanis K. Neurocognitive deficit in schizophrenia: a quantitative review of the evidence. Neuropsychology. 1998;12:426–445. doi: 10.1037//0894-4105.12.3.426. [DOI] [PubMed] [Google Scholar]
- Himmelheber A, Sarter M, Bruno J. Operant performance and cortical acetylcholine release: role of response rate, reward density, and non-contingent stimuli. Cogn. Brain Res. 1997;6:23–36. doi: 10.1016/s0926-6410(97)00014-1. [DOI] [PubMed] [Google Scholar]
- Himmelheber A, Sarter M, Bruno J. The effects of manipulations of attentional demand on cortical acetylcholine release. Cogn. Brain Res. 2001;12:353–370. doi: 10.1016/s0926-6410(01)00064-7. [DOI] [PubMed] [Google Scholar]
- Hong L, Schroeder M, Ross T, Buchholz B, Salmeron B, Wonodi I, Thaker G, Stein E. Nicotine enhances but does not normalize visual sustained attention and the associated brain network in schizophrenia. Schizophr Bull. 2009 doi: 10.1093/schbul/sbp089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horti A, Villemagne V. The quest for Eldorado: development of radioligands for in vivo imaging of nicotinic acetylcholine receptors in human brain. Curr. Pharm. Des. 2006;12:3877–3900. doi: 10.2174/138161206778559605. [DOI] [PubMed] [Google Scholar]
- Howe W, Sarter M. Prefrontal glutamatergic-cholinergic interactions for attention: glutamatergic coding of signal salience as a function of performance levels. In: Westerink B, Clinckers R, Smolders I, Sarre S, Michotte Y, editors. Monitoring Molecules in Neuroscience. Vrije Universiteit Brussel; Brussels, Belgium: 2010. pp. 57–59. [Google Scholar]
- Howe W, Ji J, Parikh V, Williams S, Mocaer E, Trocme-Thibierge C, Sarter M. Enhancement of attentional performance by selective stimulation of alpha4beta2* nAChRs: underlying cholinergic mechanisms. Neuropsychopharmacology. 2010;35:1391–1401. doi: 10.1038/npp.2010.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa J, Dai J, O'Laughlin I, Fowler W, Meltzer H. Atypical, but not typical, antipsychotic drugs increase cortical acetylcholine release without an effect in the nucleus accumbens or striatum. Neuropsychopharmacology. 2002;26:325–339. doi: 10.1016/S0893-133X(01)00312-8. [DOI] [PubMed] [Google Scholar]
- Jazbec S, Pantelis C, Robbins T, Weickert T, Weinberger D, Goldberg T. Intra-dimensional/extra-dimensional set-shifting performance in schizophrenia: impact of distractors. Schizophr Res. 2007;89:339–349. doi: 10.1016/j.schres.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Jeon J, Dencker D, Wortwein G, Woldbye D, Cui Y, Davis A, Levey A, Schutz G, Sager T, Mork A, Li C, Deng C, Fink-Jensen A, Wess J. A subpopulation of neuronal M4 muscarinic acetylcholine receptors plays a critical role in modulating dopamine-dependent behaviors. J. Neurosci. 2010;30:2396–2405. doi: 10.1523/JNEUROSCI.3843-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur S, Mamo D. Half a century of antipsychotics and still a central role for dopamine D2 receptors. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2003;27:1081–1090. doi: 10.1016/j.pnpbp.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Kapur S, VanderSpek S, Brownlee B, Nobrega J. Antipsychotic dosing in preclinical models is often unrepresentative of the clinical condition: a suggested solution based on in vivo occupancy. J. Pharmacol. Exp. Ther. 2003;305:625–631. doi: 10.1124/jpet.102.046987. [DOI] [PubMed] [Google Scholar]
- Kapur S, Arenovich T, Agid O, Zipursky R, Lindborg S, Jones B. Evidence for onset of antipsychotic effects within the first 24 hours of treatment. Am. J. Psychiatry. 2005;162:939–946. doi: 10.1176/appi.ajp.162.5.939. [DOI] [PubMed] [Google Scholar]
- Keefe R, Seidman L, Christensen B, Hamer R, Sharma T, Sitskoorn M, Rock S, Woolson S, Tohen M, Tollefson G, Sanger T, Lieberman J. Long-term neurocognitive effects of olanzapine or low-dose haloperidol in first-episode psychosis. Biol. Psychiatry. 2006;59:97–105. doi: 10.1016/j.biopsych.2005.06.022. [DOI] [PubMed] [Google Scholar]
- Keefe R, Malhotra A, Meltzer H, Kane J, Buchanan R, Murthy A, Sovel M, Li C, Goldman R. Efficacy and safety of donepezil in patients with schizophrenia or schizoaffective disorder: significant placebo/practice effects in a 12-week, randomized, double-blind, placebo-controlled trial. Neuropsychopharmacology. 2008;33:1217–1228. doi: 10.1038/sj.npp.1301499. [DOI] [PubMed] [Google Scholar]
- Kinon B, Chen L, Ascher-Svanum H, Stauffer V, Kollack-Walker S, Zhou W, Kapur S, Kane J. Early response to antipsychotic drug therapy as a clinical marker of subsequent response in the treatment of schizophrenia. Neuropsychopharmacology. 2010a;35:581–590. doi: 10.1038/npp.2009.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinon B, Chen L, Ascher-Svanum H, Stauffer V, Kollack-Walker S, Zhou W, Kapur S, Kane J, Naber D. Challenging the assumption that improvement in functional outcomes is delayed relative to improvement in symptoms in the treatment of schizophrenia. Schizophr Res. 2010b doi: 10.1016/j.schres.2009.12.013. [DOI] [PubMed] [Google Scholar]
- Kohler C, Martin E, Kujawski E, Bilker W, Gur R, Gur R. No effect of donepezil on neurocognition and social cognition in young persons with stable schizophrenia. Cogn. Neuropsychiatry. 2007;12:412–421. doi: 10.1080/13546800701307263. [DOI] [PubMed] [Google Scholar]
- Kozak R, Bruno J, Sarter M. Augmented prefrontal acetylcholine release during challenged attentional performance. Cereb. Cortex. 2006;16:9–17. doi: 10.1093/cercor/bhi079. [DOI] [PubMed] [Google Scholar]
- Kozak R, Martinez V, Young D, Brown H, Bruno J, Sarter M. Toward a neurocognitive animal model of the cognitive symptoms of schizophrenia: disruption of cortical cholinergic neurotransmission following repeated amphetamine exposure in attentional task-performing, but not non-performing, rats. Neuropsychopharmacology. 2007;32:2074–2086. doi: 10.1038/sj.npp.1301352. [DOI] [PubMed] [Google Scholar]
- Laruelle M, Abi-Dargham A, Gil R, Kegeles L, Innis R. Increased dopamine transmission in schizophrenia: relationship to illness phases. Biol. Psychiatry. 1999;46:56–72. doi: 10.1016/s0006-3223(99)00067-0. [DOI] [PubMed] [Google Scholar]
- Lawson G, Hurt R, Dale L, Offord K, Croghan I, Schroeder D, Jiang N. Application of serum nicotine and plasma cotinine concentrations to assessment of nicotine replacement in light, moderate, and heavy smokers undergoing transdermal therapy. J. Clin. Pharmacol. 1998;38:502–509. doi: 10.1002/j.1552-4604.1998.tb05787.x. [DOI] [PubMed] [Google Scholar]
- Leach K, Loiacono R, Felder C, McKinzie D, Mogg A, Shaw D, Sexton P, Christopoulos A. Molecular mechanisms of action and in vivo validation of an M4 muscarinic acetylcholine receptor allosteric modulator with potential antipsychotic properties. Neuropsychopharmacology. 2010;35:855–869. doi: 10.1038/npp.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman J, Javitch J, Moore H. Cholinergic agonists as novel treatments for schizophrenia: the promise of rational drug development for psychiatry. Am. J. Psychiatry. 2008;165:931–936. doi: 10.1176/appi.ajp.2008.08050769. [DOI] [PubMed] [Google Scholar]
- Lindenmayer JP, Khan A. Galantamine augmentation of long-acting injectable risperidone for cognitive impairments in chronic schizophrenia. Schizophr Res. 2010 doi: 10.1016/j.schres.2010.08.021. [DOI] [PubMed] [Google Scholar]
- Luck SJ, Gold JM. The construct of attention in schizophrenia. Biol. Psychiatry. 2008;64:34–39. doi: 10.1016/j.biopsych.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mach R, Voytko M, Ehrenkaufer R, Nader M, Tobin J, Efange S, Parsons S, Gage H, Smith C, Morton T. Imaging of cholinergic terminals using the radiotracer [18F](+)-4-fluorobenzyltrozamicol: in vitro binding studies and positron emission tomography studies in nonhuman primates. Synapse. 1997;25:368–380. doi: 10.1002/(SICI)1098-2396(199704)25:4<368::AID-SYN8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Mancama D, Arranz M, Landau S, Kerwin R. Reduced expression of the muscarinic 1 receptor cortical subtype in schizophrenia. Am. J. Med. Genet. B. Neuropsychiatr Genet. 2003;119B:2–6. doi: 10.1002/ajmg.b.20020. [DOI] [PubMed] [Google Scholar]
- Marutle A, Zhang X, Court J, Piggott M, Johnson M, Perry R, Perry E, Nordberg A. Laminar distribution of nicotinic receptor subtypes in cortical regions in schizophrenia. J. Chem. Neuroanat. 2001;22:115–126. doi: 10.1016/s0891-0618(01)00117-x. [DOI] [PubMed] [Google Scholar]
- McGaughy J, Decker M, Sarter M. Enhancement of sustained attention performance by the nicotinic acetylcholine receptor agonist ABT-418 in intact but not basal forebrain-lesioned rats. Psychopharmacology (Berl.) 1999;144:175–182. doi: 10.1007/s002130050991. [DOI] [PubMed] [Google Scholar]
- Meerson A, Cacheaux L, Goosens K, Sapolsky R, Soreq H, Kaufer D. Changes in brain MicroRNAs contribute to cholinergic stress reactions. J. Mol. Neurosci. 2010;40:47–55. doi: 10.1007/s12031-009-9252-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshorer E, Soreq H. Virtues and woes of AChE alternative splicing in stress-related neuropathologies. Trends Neurosci. 2006;29:216–224. doi: 10.1016/j.tins.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Mishara A, Goldberg T. A meta-analysis and critical review of the effects of conventional neuroleptic treatment on cognition in schizophrenia: opening a closed book. Biol. Psychiatry. 2004;55:1013–1022. doi: 10.1016/j.biopsych.2004.01.027. [DOI] [PubMed] [Google Scholar]
- Mohler E, Franklin S, Rueter L, Fox G, Decker M, Browman K. ABT-594 improves performance in the 5-choice serial reaction time task under conditions of increased difficulty, sub-chronic dosing, and in poorly-performing subjects. Pharmacol. Biochem. Behav. 2010;95:146–157. doi: 10.1016/j.pbb.2009.12.019. [DOI] [PubMed] [Google Scholar]
- Money T, Scarr E, Udawela M, Gibbons A, Jeon W, Seo M, Dean B. Treating schizophrenia: novel targets for the cholinergic system. CNS Neurol. Disord. Drug Targets. 2010;9:241–256. doi: 10.2174/187152710791012062. [DOI] [PubMed] [Google Scholar]
- Mulholland G, Wieland D, Kilbourn M, Frey K, Sherman P, Carey J, Kuhl D. [18F]fluoroethoxy-benzovesamicol, a PET radiotracer for the vesicular acetylcholine transporter and cholinergic synapses. Synapse. 1998;30:263–274. doi: 10.1002/(SICI)1098-2396(199811)30:3<263::AID-SYN4>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Neigh G, Arnold H, Sarter M, Bruno J. Dissociations between the effects of intra-accumbens administration of amphetamine and exposure to a novel environment on accumbens dopamine and cortical acetylcholine release. Brain Res. 2001;894:354–358. doi: 10.1016/s0006-8993(01)02059-5. [DOI] [PubMed] [Google Scholar]
- Neigh G, Arnold H, Rabenstein R, Sarter M, Bruno J. Neuronal activity in the nucleus accumbens is necessary for performance-related increases in cortical acetylcholine release. Neuroscience. 2004;123:635–645. doi: 10.1016/j.neuroscience.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Nelson C, Sarter M, Bruno J. Prefrontal cortical modulation of acetylcholine release in posterior parietal cortex. Neuroscience. 2005;132:347–359. doi: 10.1016/j.neuroscience.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Nobuhara K, Halldin C, Hall H, Karlsson P, Farde L, Hiltunen J, McPherson D, Savonen A, Bergstrom K, Pauli S, Swahn C, Larsson S, Schnell P, Sedvall G. Z-IQNP: a potential radioligand for SPECT imaging of muscarinic acetylcholine receptors in Alzheimer's disease. Psychopharmacology (Berl.) 2000;149:45–55. doi: 10.1007/s002139900356. [DOI] [PubMed] [Google Scholar]
- Nobuhara K, Farde L, Halldin C, Karlsson P, Swahn C, Olsson H, Bergstrom K, Larsson S, Schnell P, McPherson D, Savonen A, Hiltunen J, Sedvall G. SPET imaging of central muscarinic acetylcholine receptors with iodine-123 labelled E-IQNP and Z-IQNP. Eur. J. Nucl. Med. 2001;28:13–24. doi: 10.1007/s002590000390. [DOI] [PubMed] [Google Scholar]
- Nuechterlein K, Luck S, Lustig C, Sarter M. CNTRICS final task selection: control of attention. Schizophr Bull. 2009;35:182–196. doi: 10.1093/schbul/sbn158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien K, Saxby B, Ballard C, Grace J, Harrington F, Ford G, O'Brien J, Swan A, Fairbairn A, Wesnes K, del Ser T, Edwardson J, Morris C, McKeith I. Regulation of attention and response to therapy in dementia by butyrylcholinesterase. Pharmacogenetics. 2003;13:231–239. doi: 10.1097/00008571-200304000-00008. [DOI] [PubMed] [Google Scholar]
- Olincy A, Stevens K. Treating schizophrenia symptoms with an alpha7 nicotinic agonist, from mice to men. Biochem. Pharmacol. 2007;74:1192–1201. doi: 10.1016/j.bcp.2007.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltmanns TF. Selective attention in schizophrenic and manic psychoses: the effect of distraction on information processing. J. Abnorm. Psychol. 1978;87:212–225. doi: 10.1037//0021-843x.87.2.212. [DOI] [PubMed] [Google Scholar]
- Oltmanns TF, Neale JM. Schizophrenic performance when distractors are present: attentional deficit or differential task difficulty? J. Abnorm. Psychol. 1975;84:205–209. doi: 10.1037/h0076721. [DOI] [PubMed] [Google Scholar]
- Paolone G, Howe W, Gopalakrishnan M, Decker M, Sarter M. Regulation and function of the tonic component of cortical acetylcholine release. In: Westerink B, Clinckers R, Smolders I, Sarre S, Michotte Y, editors. Monitoring Molecules in Neuroscience. Vrije Universiteit Brussel; Brussels, Belgium: 2010. pp. 363–365. [Google Scholar]
- Parikh V, Sarter M. Cholinergic mediation of attention: contributions of phasic and tonic increases in prefrontal cholinergic activity. Ann. N. Y. Acad. Sci. 2008;1129:225–235. doi: 10.1196/annals.1417.021. [DOI] [PubMed] [Google Scholar]
- Parikh V, Kozak R, Martinez V, Sarter M. Prefrontal acetylcholine release controls cue detection on multiple timescales. Neuron. 2007;56:141–154. doi: 10.1016/j.neuron.2007.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh V, Man K, Decker M, Sarter M. Glutamatergic contributions to nicotinic acetylcholine receptor agonist-evoked cholinergic transients in the prefrontal cortex. J. Neurosci. 2008;28:3769–3780. doi: 10.1523/JNEUROSCI.5251-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh V, Ji J, Decker M, Sarter M. Prefrontal beta2 subunit-containing and alpha7 nicotinic acetylcholine receptors differentially control glutamatergic and cholinergic signaling. J. Neurosci. 2010;30:3518–3530. doi: 10.1523/JNEUROSCI.5712-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passetti F, Dalley J, O'Connell M, Everitt B, Robbins T. Increased acetylcholine release in the rat medial prefrontal cortex during performance of a visual attentional task. Eur. J. Neurosci. 2000;12:3051–3058. doi: 10.1046/j.1460-9568.2000.00183.x. [DOI] [PubMed] [Google Scholar]
- Pessiglione M, Seymour B, Flandin G, Dolan R, Frith C. Dopamine-dependent prediction errors underpin reward-seeking behaviour in humans. Nature. 2006;442:1042–1045. doi: 10.1038/nature05051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovsky N, Quednow B, Ettinger U, Schmechtig A, Mossner R, Collier D, Kuhn K, Maier W, Wagner M, Kumari V. Sensorimotor gating is associated with CHRNA3 polymorphisms in schizophrenia and healthy volunteers. Neuropsychopharmacology. 2010;35:1429–1439. doi: 10.1038/npp.2010.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezze M, Dalley J, Robbins T. Differential roles of dopamine D1 and D2 receptors in the nucleus accumbens in attentional performance on the five-choice serial reaction time task. Neuropsychopharmacology. 2007;32:273–283. doi: 10.1038/sj.npp.1301073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezze M, Dalley J, Robbins T. Remediation of attentional dysfunction in rats with lesions of the medial prefrontal cortex by intra-accumbens administration of the dopamine D(2/3) receptor antagonist sulpiride. Psychopharmacology (Berl.) 2009;202:307–313. doi: 10.1007/s00213-008-1384-4. [DOI] [PubMed] [Google Scholar]
- Powchik P, Davidson M, Haroutunian V, Gabriel S, Purohit D, Perl D, Harvey P, Davis K. Postmortem studies in schizophrenia. Schizophr Bull. 1998;24:325–341. doi: 10.1093/oxfordjournals.schbul.a033330. [DOI] [PubMed] [Google Scholar]
- Raedler T, Knable M, Jones D, Lafargue T, Urbina R, Egan M, Pickar D, Weinberger D. In vivo olanzapine occupancy of muscarinic acetylcholine receptors in patients with schizophrenia. Neuropsychopharmacology. 2000;23:56–68. doi: 10.1016/S0893-133X(99)00162-1. [DOI] [PubMed] [Google Scholar]
- Raedler T, Knable M, Jones D, Urbina R, Gorey J, Lee K, Egan M, Coppola R, Weinberger D. In vivo determination of muscarinic acetylcholine receptor availability in schizophrenia. Am. J. Psychiatry. 2003;160:118–127. doi: 10.1176/appi.ajp.160.1.118. [DOI] [PubMed] [Google Scholar]
- Raedler T, Bymaster F, Tandon R, Copolov D, Dean B. Towards a muscarinic hypothesis of schizophrenia. Mol. Psychiatry. 2007;12:232–246. doi: 10.1038/sj.mp.4001924. [DOI] [PubMed] [Google Scholar]
- Rappaport M. Competing voice messages. Effects of message load and drugs on the ability of acute schizophrenics to attend. Arch. Gen. Psychiatry. 1967;17:97–103. doi: 10.1001/archpsyc.1967.01730250099014. [DOI] [PubMed] [Google Scholar]
- Ribeiz SR, Bassitt DP, Arrais JA, Avila R, Steffens DC, Bottino CM. Cholinesterase inhibitors as adjunctive therapy in patients with schizophrenia and schizoaffective disorder: a review and meta-analysis of the literature. CNS Drugs. 24:303–317. doi: 10.2165/11530260-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Riedel M, Muller N, Spellmann I, Engel R, Musil R, Valdevit R, Dehning S, Douhet A, Cerovecki A, Strassnig M, Moller H. Efficacy of olanzapine versus quetiapine on cognitive dysfunctions in patients with an acute episode of schizophrenia. Eur. Arch. Psychiatry Clin. Neurosci. 2007;257:402–412. doi: 10.1007/s00406-007-0748-9. [DOI] [PubMed] [Google Scholar]
- Riedel M, Schennach-Wolff R, Musil R, Dehning S, Cerovecki A, Opgen-Rhein M, Matz J, Seemuller F, Obermeier M, Engel R, Muller N, Moller H, Spellmann I. Neurocognition and its influencing factors in the treatment of schizophrenia-effects of aripiprazole, olanzapine, quetiapine and risperidone. Hum. Psychopharmacol. 2010;25:116–125. doi: 10.1002/hup.1101. [DOI] [PubMed] [Google Scholar]
- Rowntree D, Nevin S, A W. The effects of diisopropylfluorophosphonate in schizophrenia and manic depressive psychosis. J. Neurol. Neurosurg. Psychiatry. 1950;13:47–62. doi: 10.1136/jnnp.13.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salah-Uddin H, Scarr E, Pavey G, Harris K, Hagan J, Dean B, Challiss R, Watson J. Altered M(1) muscarinic acetylcholine receptor (CHRM1)-Galpha(q/11) coupling in a schizophrenia endophenotype. Neuropsychopharmacology. 2009;34:2156–2166. doi: 10.1038/npp.2009.41. [DOI] [PubMed] [Google Scholar]
- Samochocki M, Hoffle A, Fehrenbacher A, Jostock R, Ludwig J, Christner C, Radina M, Zerlin M, Ullmer C, Pereira EF, Lubbert H, Albuquerque EX, Maelicke A. Galantamine is an allosterically potentiating ligand of neuronal nicotinic but not of muscarinic acetylcholine receptors. J. Pharmacol. Exp. Ther. 2003;305:1024–1036. doi: 10.1124/jpet.102.045773. [DOI] [PubMed] [Google Scholar]
- Sarter M, Hasselmo M, Bruno J, Givens B. Unraveling the attentional functions of cortical cholinergic inputs: interactions between signal-driven and cognitive modulation of signal detection. Brain Res. Brain Res. Rev. 2005;48:98–111. doi: 10.1016/j.brainresrev.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Sarter M, Gehring W, Kozak R. More attention must be paid: the neurobiology of attentional effort. Brain Res. Rev. 2006;51:145–160. doi: 10.1016/j.brainresrev.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Sarter M, Bruno J, Parikh V. Abnormal neurotransmitter release underlying behavioral and cognitive disorders: toward concepts of dynamic and function-specific dysregulation. Neuropsychopharmacology. 2007;32:1452–1461. doi: 10.1038/sj.npp.1301285. [DOI] [PubMed] [Google Scholar]
- Sarter M, Martinez V, Kozak R. A neurocognitive animal model dissociating between acute illness and remission periods of schizophrenia. Psychopharmacology (Berl.) 2009a;202:237–258. doi: 10.1007/s00213-008-1216-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarter M, Parikh V, Howe W. nAChR agonist-induced cognition enhancement: integration of cognitive and neuronal mechanisms. Biochem. Pharmacol. 2009b;78:658–667. doi: 10.1016/j.bcp.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarr E, Cowie T, Kanellakis S, Sundram S, Pantelis C, Dean B. Decreased cortical muscarinic receptors define a subgroup of subjects with schizophrenia. Mol. Psychiatry. 2009;14:1017–1023. doi: 10.1038/mp.2008.28. [DOI] [PubMed] [Google Scholar]
- Schubert MH, Young KA, Hicks PB. Galantamine improves cognition in schizophrenic patients stabilized on risperidone. Biol. Psychiatry. 2006;60:530–533. doi: 10.1016/j.biopsych.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Shekhar A, Potter W, Lightfoot J, Lienemann J, Dube S, Mallinckrodt C, Bymaster F, McKinzie D, Felder C. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am. J. Psychiatry. 2008;165:1033–1039. doi: 10.1176/appi.ajp.2008.06091591. [DOI] [PubMed] [Google Scholar]
- Smith RC, Lindenmayer JP, Davis JM, Cornwell J, Noth K, Gupta S, Sershen H, Lajtha A. Cognitive and antismoking effects of varenicline in patients with schizophrenia or schizoaffective disorder. Schizophr Res. 2009;110:149–155. doi: 10.1016/j.schres.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Spring B, Lemon M, Weinstein L, Haskell A. Distractibility in schizophrenia: state and trait aspects. Br. J. Psychiatry Suppl. 1989:63–68. [PubMed] [Google Scholar]
- Stip E, Chouinard S, Boulay L. On the trail of a cognitive enhancer for the treatment of schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2005;29:219–232. doi: 10.1016/j.pnpbp.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Tandon R, Greden J. Cholinergic hyperactivity and negative schizophrenic symptoms. A model of cholinergic/dopaminergic interactions in schizophrenia. Arch. Gen. Psychiatry. 1989;46:745–753. doi: 10.1001/archpsyc.1989.01810080075010. [DOI] [PubMed] [Google Scholar]
- Tsukada H, Takahashi K, Miura S, Nishiyama S, Kakiuchi T, Ohba H, Sato K, Hatazawa J, Okudera T. Evaluation of novel PET ligands (+)N-[11C] methyl-3-piperidyl benzilate ([11C](+)3-MPB) and its stereoisomer [11C](−)3-MPB for muscarinic cholinergic receptors in the conscious monkey brain: a PET study in comparison with. Synapse. 2001;39:182–192. doi: 10.1002/1098-2396(200102)39:2<182::AID-SYN10>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Uchida H, Rajji T, Mulsant B, Kapur S, Pollock B, Graff-Guerrero A, Menon M, Mamo D. D2 receptor blockade by risperidone correlates with attention deficits in late-life schizophrenia. J. Clin. Psychopharmacol. 2009;29:571–575. doi: 10.1097/JCP.0b013e3181bf4ea3. [DOI] [PubMed] [Google Scholar]
- Valentin V, O'Doherty J. Overlapping prediction errors in dorsal striatum during instrumental learning with juice and money reward in the human brain. J. Neurophysiol. 2009;102:3384–3391. doi: 10.1152/jn.91195.2008. [DOI] [PubMed] [Google Scholar]
- van Os J, Kapur S. Schizophrenia. Lancet. 2009;374:635–645. doi: 10.1016/S0140-6736(09)60995-8. [DOI] [PubMed] [Google Scholar]
- Wilens T, Biederman J, Spencer T, Bostic J, Prince J, Monuteaux M, Soriano J, Fine C, Abrams A, Rater M, Polisner D. A pilot controlled clinical trial of ABT-418, a cholinergic agonist, in the treatment of adults with attention deficit hyperactivity disorder. Am. J. Psychiatry. 1999;156:1931–1937. doi: 10.1176/ajp.156.12.1931. [DOI] [PubMed] [Google Scholar]
- Wilens T, Verlinden M, Adler L, Wozniak P, West S. ABT-089, a neuronal nicotinic receptor partial agonist, for the treatment of attention-deficit/hyperactivity disorder in adults: results of a pilot study. Biol. Psychiatry. 2006;59:1065–1070. doi: 10.1016/j.biopsych.2005.10.029. [DOI] [PubMed] [Google Scholar]
- Yui K, Goto K, Ikemoto S, Ishiguro T, Angrist B, Duncan G, Sheitman B, Lieberman J, Bracha S, Ali S. Neurobiological basis of relapse prediction in stimulant-induced psychosis and schizophrenia: the role of sensitization. Mol. Psychiatry. 1999;4:512–523. doi: 10.1038/sj.mp.4000575. [DOI] [PubMed] [Google Scholar]
- Zmarowski A, Sarter M, Bruno J. NMDA and dopamine interactions in the nucleus accumbens modulate cortical acetylcholine release. Eur. J. Neurosci. 2005;22:1731–1740. doi: 10.1111/j.1460-9568.2005.04333.x. [DOI] [PubMed] [Google Scholar]
- Zmarowski A, Sarter M, Bruno J. Glutamate receptors in nucleus accumbens mediate regionally selective increases in cortical acetylcholine release. Synapse. 2007;61:115–123. doi: 10.1002/syn.20354. [DOI] [PubMed] [Google Scholar]