

Abstract
Objective
We previously showed that endothelial epsin deficiency causes elevated VEGFR2 and enhanced VEGF signaling, resulting in aberrant tumor angiogenesis and tumor growth in adult mice. However, direct evidence demonstrating that endothelial epsins regulate angiogenesis specifically through VEGFR2 downregulation is still lacking. In addition, whether the lack of epsins causes abnormal angiogenesis during embryonic development remains unclear.
Approach and Results
A novel strain of endothelial epsin-deleted mice that are heterozygous for VEGFR2 (Epn1fl/fl; Epn2−/−; Flkfl/+; iCDH5 Cre mice) was created. Analysis of embryos at different developmental stages shows that deletion of epsins causes defective embryonic angiogenesis and retards embryo development. In vitro angiogenesis assays using isolated primary endothelial cells (EC) from Epn1fl/fl; Epn2−/−; iCDH5 Cre (EC-iDKO) and Epn1fl/fl; Epn2−/−; Flkfl/+; iCDH5 Cre (EC-iDKO-Flkfl/+) mice demonstrated that VEGFR2 reduction in epsin depleted cells is sufficient to restore normal VEGF signaling, EC proliferation, EC migration and EC network formation. These findings were complemented by in vivo wound healing, inflammatory angiogenesis, and tumor angiogenesis assays in which reduction of VEGFR2 was sufficient to rescue abnormal angiogenesis in endothelial epsin-deleted mice.
Conclusions
Our results provide the first genetic demonstration that epsins function specifically to downregulate VEGFR2 by mediating activated VEGFR2 internalization and degradation and that genetic reduction of VEGFR2 level protects against excessive angiogenesis caused by epsin loss. Our findings indicate epsins may be a potential therapeutic target in conditions where tightly regulated angiogenesis is crucial, such as in diabetic wound healing and tumors.
Keywords: Epsin, VEGFR2, Angiogenesis
Introduction
Vascular endothelial growth factor (VEGF) signaling is essential for angiogenesis during development and postnatal organ growth, as well as in pathological conditions such as tumor.1-8 VEGF binds to its receptor, VEGFR2 (Flk), on endothelial cells (EC), inducing VEGFR2 homodimerization and autophosphorylation at several tyrosine residues to initiate signaling cascades required for EC proliferation and migration.9-11 The amount of VEGFR2 on the cell surface controls the magnitude of signal transmission.12 Its abundance is negatively regulated by internalization and degradation.13-16 These tightly regulated processes are vital for the development and maintenance of a normal vascular system. Prolonged VEGF signaling causes vascular leakage and inflammatory responses.15
Epsins are a family of endocytic adaptor proteins required for internalization and degradation of ubiquitinated receptors.17-22 Mammals express three distinct epsin genes (Epn1, Epn2, and Epn3).23 While epsin 3 expression is localized primarily to the stomach and epidermis, epsins 1 and 2 are widely expressed and have redundant functions.18, 19, 23-25 Specifically, global deletion of epsins 1 and 2 result in embryonic lethality, largely due to epsins role in promoting Notch signaling in all cell types.23 In addition to the endocytic adaptor function, the multivalent properties of epsins facilitate additional functions such as the regulation of GTPases involved in actin remodeling.26, 27 However, the specific role epsins play in different cell types, or their implications in normal physiology and disease, remains elusive.
Endothelial-specific deletion of epsins 1 and 2 specifically increases VEGF signaling, but not signaling of other angiogenic factors including FGF, PDGF, EGF and TGFβ.28 Furthermore, mice lacking endothelial epsins 1 and 2 exhibited excessive tumor angiogenesis with highly disorganized vessels and significantly increased vascular permeability. Epsin interacts with VEGFR2 through its ubiquitin interacting motifs (UIMs), leading to the VEGF-dependent downregulation of VEGFR2, thus acting as a negative regulator of VEGF signaling.13, 28, 29 However, the role of epsins in regulating tumor angiogenesis, where surrounding tumor cells secrete exaggerated amounts of VEGF, may not represent the role of epsins during embryonic/developmental angiogenic conditions.4 Furthermore, while our previous data established a correlation between epsin loss and augmented VEGF signaling, presumably owing to increased VEGFR2 levels in the absence of epsin-mediated internalization and degradation, direct evidence that epsin loss causes augmented VEGFR2 levels is missing. In other words, it remains to be determined whether epsins directly and specifically downregulate VEGFR2 to control developmental and pathological angiogenesis in vivo.
Here we report that endothelial epsin deletion during development caused defective embryonic angiogenesis and lethality. Using a novel strain of endothelial epsin-deleted mice that are also heterozygous for VEGFR2, we found that reducing VEGFR2 expression in vivo significantly improved the defective wound healing and pathological angiogenesis produced by the loss of endothelial epsins. Furthermore, reducing VEGFR2 expression in primary mouse EC (MEC) suppressed heightened VEGF signaling and angiogenic responses including EC proliferation and migration. Our findings provide the first direct evidence that endothelial epsins function to control angiogenesis by specifically downregulating VEGFR2 to modulate the VEGF signaling fundamental for developmental or pathologic angiogenesis.
Materials and Methods
Materials and methods are available in the online-only Data Supplement.
Results
Endothelial epsins are essential for embryonic angiogenesis
To determine the role of endothelial epsins in regulating angiogenesis, we first engineered mice constitutively lacking endothelial epsins 1 and 2 (EC-DKO) by crossing Epn1fl/fl; Epn2−/− mice with the EC-specific Cre recombinase expressing Tie2 Cre deleter mice (Supplemental Figure IB).30 To rule out any undesirable effects of the Cre expression, we also crossed WT and Epn1fl/fl; Epn2+/+ mice with the Tie2 Cre deleter mice. These mice exhibited similar phenotypes to WT (data not shown). After several litters in which no EC-DKO pups were born, we used timed mating of the Epn1fl/fl; Epn2−/− and Tie2 Cre deleter mice to determine if loss of endothelial epsins resulted in embryonic lethality. Similar to previously reported global DKO embryos, E11 EC-DKO embryos were significantly smaller than WT with striking vascular defects (Figure 1A), suggesting that loss of endothelial epsins is a cause of the defective angiogenesis resulting in embryonic lethality.23 Immunostaining with CD31, a vascular endothelial marker, revealed major vascular developmental defects, including increased vascular density and disorganized vascular networks, in E10 EC-DKO embryos (Figure 1B).31 Further immunofluorescent staining analyses of cross sections from isolated embryonic midbrain, hindbrain, skin, and intestine revealed much denser and highly disorganized vascular networks in the EC-DKO embryonic tissues compared to WT (Figure 1C-J; Supplemental Figure II). In addition, detailed analysis of hindbrain cross sections revealed that loss of endothelial epsins promoted formation of a more elaborate subventricular vascular plexus (Figure 1G). Collectively, our findings demonstrate that endothelial epsins are critical for the regulation of embryonic angiogenesis.
Figure 1. Aberrant embryonic angiogenesis caused by endothelial epsin deletion.

A, Whole mount E10 WT or EC-DKO embryos. B, Telencephalic region of E10 embryos immunostained by CD31. Arrows indicate regions of disorganized vasculature. C, E, I, CD31 immunostaining of midbrain (C), hindbrain (E), and skin (I) of E10 WT or EC-DKO embryos. G, CD31 immunostaining of hindbrain cross sections of E10 WT or EC-DKO embryos showing the subventricular vascular plexus. D, F, H, J, CD31-positive surface area in C, E, G, and I were quantified by SlideBook software. Error bars indicate the mean ± s.e.m. n > 5, *p < 0.05. Scale bars: A, 500 μm; B, 225 μm; C and G; 50 μm; E, I,100 μm.
Epsins negatively regulate VEGF-induced angiogenic responses in endothelial cells
To determine if epsins regulate embryonic angiogenesis through modulating EC proliferation, migration, or network formation, we employed in vitro angiogenesis assays using isolated primary mouse endothelial cells (MECs).28, 32 We measured EC proliferation by culturing WT or DKO MECs with or without VEGF stimulation in the presence of 5-ethynl-2′-deoxyuridine (EdU), which labels cells actively undergoing S-phase DNA replication.33 The small VEGF-dependent increase in WT MEC EdU incorporation (Figure 2A) is consistent with previously established roles for VEGF signaling in EC proliferation.7, 9, 10, 28, 34, 35 Depletion of epsins exaggerated VEGF-dependent proliferation in the DKO MECs (Figure 2A). We next used an in vitro scratch assay and matrigel tube formation assay to measure EC migration and network formation, respectively.32 In the scratch assay, confluent WT or DKO MEC monolayers were evenly scarred then stimulated with VEGF to induce migration. MEC migration was determined by measuring scratch width reduction. Unlike the proliferation assay, VEGF stimulation did not affect WT MEC migration but did significantly increase DKO MEC migration (Figure 2B, C). Similarly, the in vitro tube formation assay revealed a more robust VEGF-dependent endothelial tube network in DKO MECs (Figure 2D, E). In summary, our in vitro functional assays support an anti-angiogenic function for epsins in the regulation of angiogenesis through limiting VEGF-dependent EC proliferation, migration, and tube formation.
Figure 2. Epsin deficiency promotes VEGF-dependent in vitro angiogenesis.

A, Quantification of EdU incorporation into WT or DKO MECs after labeling in the absence or presence of VEGF-A (50 ng/mL) for 24 h. B, WT or DKO MEC monolayers were subjected to a scratch assay in the absence or presence of VEGF-A (50 ng/ml) for 12 h. C, Wound distance in B at 12 h was quantified using NIH ImageJ software. D, WT or DKO MECs were subjected to a tube formation assay by culturing on matrigel for 16 h in the absence or presence of VEGF-A (50 ng/ml). E, Tube formation in D at 16 h was quantified as in C. F, Plasma membrane fractions from WT or DKO MECs stimulated with 50 ng/mL VEGF-A for 0, 5 and 15 min were immunoblotted using the indicated antibodies. A, C and E, error bars indicate the mean ± s.e.m. n > 5, *p < 0.05. Scale bars: B and D, 50 μm.
We also examined whether VEGF can stimulate the phosphorylation of accumulated cell surface VEGFR2 in DKO MECs. Five minutes of VEGF stimulation was sufficient to induce phosphorylation of VEGFR2 in both WT and DKO MECs with heightened VEGFR2 phosphorylation in DKO MECs (Figure 2F). Prolonged stimulation of WT MECs resulted in the loss of both phosphorylated and cell surface VEGFR2, indicating receptor degradation. In contrast, phosphorylated and cell surface VEGFR2 remained elevated in the DKO MECs (Figure 2F). To directly visualize the VEGF-induced phosphorylation of VEGFR2, WT or DKO MECs were immunostained using total and phospho-VEGFR2 antibodies. Consistently, VEGF stimulation rapidly induced the phosphorylation of VEGFR2 at the plasma membrane of both WT and DKO MECs (Supplemental Figure III). In WT MECs, ten minutes of VEGF stimulation resulted in the internalization and colocalization of phosphorylated VEGFR2 with EEA1 (Supplemental Figure III). In contrast, a significant portion of the phosphorylated VEGFR2 in DKO MECs remained localized at the plasma membrane and failed to colocalize with EEA1 (Supplemental Figure III). In summary, our biochemical and cell biology approaches revealed that endothelial epsin depletion significantly impairs VEGFR2 internalization and degradation, resulting in prolonged VEGFR2 phosphorylation. It is important, however, to note that the cell surface level of a house keeping protein, transferrin receptor, did not change in response to epsin depletion, indicating the specificity of epsins for regulating VEGFR2 (Figure 2F).
Genetic reduction of VEGFR2 expression rescues in vitro angiogenesis in the absence of epsins
To directly test whether the defective angiogenesis caused by endothelial epsin deletion is a result of heightened VEGFR2 and enhanced VEGF signaling, we crossed Epn1fl/fl; Epn2−/− mice with iCDH5 Cre deleter and Flkfl/+ mice (Supplemental Figure IC-E).36 The iCDH5 Cre deleter mice specifically express Cre in ECs after tamoxifen treatment, resulting in a VEGFR2 heterozygote on an inducible EC-specific epsins 1 and 2 deletion background (EC-iDKOFlkfl/+).37, 38 Loss of a single VEGFR allele was sufficient to reduce VEGFR2 expression by approximately fifty-percent in the liver, lungs, and heart extracted from EC-iDKO-FLKfl/+ mice (Figure 3A). It is important to note that postnatal deletion of epsins did not affect quiescent postnatal angiogenesis.28 Furthermore, to ensure the tamoxifen treatment, the Flkfl/+ genotype, or the Cre expression did not produce undesirable effects, we also created WT; iCDH5 Cre deleter, WT; Flkfl/+, Epn1fl/fl; Epn2+/+; Flkfl/+ and Epn1fl/fl; Epn2+/+; iCDH5 Cre deleter mice; all of which produce phenotypes consistent with WT (data not shown). We also isolated and treated MECs with tamoxifen ex vivo, resulting in sufficient epsin 1 deletion and an approximate 50% reduction of the VEGFR2 in DKO-Flkfl/+ MECs (Figure 3B). We measured EC proliferation via both in vitro EdU incorporation and in vivo BrdU (5-bromo-2-deoxyuridine) incorporation. In the in vitro study, WT, DKO or DKO-Flkfl/+ MECs were treated with VEGF in the presence of EdU as described above. Immunofluorescent detection revealed reduced EdU incorporation in the DKOFlkfl/+ MECs to levels comparable to WT (Figure 3C, D). To confirm these findings in vivo, we injected BrdU into WT, EC-iDKO and EC-iDKO-Flkfl/+ P6 pups then isolated the intestine, a tissue that continuously undergoes postnatal angiogenesis. Consistently, reducing VEGFR2 expression in the EC-iDKO-Flkfl/+ pups reduced the BrdU incorporation within the intestinal vasculature (Figure 3E; Supplemental Figure IV). The findings strongly suggest that reducing VEGFR2 is sufficient to rescue the enhanced proliferative phenotype resulting from loss of epsins.
Figure 3. Genetic reduction of VEGFR2 expression rescues proliferation caused by epsin deletion.

A, Liver, lung and heart tissue isolated from WT, EC-iDKO, or EC-iDKO-Flkfl/+ mice analyzed by immunoblot using the specified antibodies. B, MECs isolated from WT, EC-iDKO, or EC-iDKO-Flkfl/+ mice were treated ex vivo with 4-hydroxytamoxifen for 2 d then whole cell lysates were analyzed by immunoblot using the specified antibodies. C, MEC monolayers isolated from WT, EC-iDKO, or EC-iDKO-Flkfl/+ mice were labeled with EdU for 16 h in the presence of 50 ng/mL VEGF-A then fixed and immunostained for CD31 and EdU. D, Quantification of in vitro EdU incorporation in C. Error bars indicate the mean ± s.e.m. n > 5, *p < 0.05. E, Intestines isolated from BrdU injected WT, EC-iDKO, or EC-iDKO-Flkfl/+ mice were processed for in vivo CD31 and BrdU immunostaining. Scale bars: C and E, 50 μm.
Next, we determined whether reducing VEGFR2 also recovers normal levels of VEGFR2 phosphorylation, signaling, and cell migration in DKO MECs. First, using immunoblotting, we determined that reducing the VEGFR2 expression in DKO-Flkfl/+ MECs significantly reduced the VEGF-dependent phosphorylation of VEGFR2, as well as the downstream activation of PLCγ, Akt and ERK (Figure 4A; Supplemental Figure V). More importantly, these reductions corresponded to levels similar to WT MECs. Next, we compared the migration of DKO-Flkfl/+ MECs to WT and DKO MECs using the scratch assay. Reducing VEGFR2 levels significantly hindered DKO MEC migration such that the wound distance in the DKO-Flkfl/+ MECs was significantly larger than DKO MECs, but comparable to WT MECs (Figure 4B, C). In summary, our findings strongly support our hypothesis that the pro-angiogenic phenotype of the epsin-depleted ECs is caused specifically by impaired downregulation of VEGFR2 activation.
Figure 4. Genetic reduction of VEGFR2 expression rescues in vitro angiogenesis caused by epsin deletion.

A, WT, DKO or DKO-Flkfl/+ MECs were stimulated with 50 ng/mL VEGF-A then whole cell lysates were immunoblotted using the indicated antibodies. B, WT, DKO or DKO-Flkfl/+ MEC monolayers were subjected to a scratch assay in the absence or presence of VEGF-A (50 ng/ml) for 12 h. C, Wound distance in B at 12 h was quantified using NIH ImageJ software. Error bars indicate the mean ± s.e.m. n > 5, *p < 0.05. Scale bars: B, 50 μm.
Genetic reduction of VEGFR2 levels rescues in vivo angiogenesis during wound healing
To test whether genetic reduction of VEGFR2 is sufficient to rescue abnormal angiogenesis in epsin deficiency in vivo, we utilized a wound healing assay that measures angiogenesis after postnatal dorsal wounds are generated by biopsy punches.28 We found that EC-iDKO mice exhibited disorganized wound vasculature and a significant delay in wound healing, suggesting defective angiogenesis (Figure 5A-C). Importantly, the EC-iDKO-Flkfl/+ mice exhibited a wound healing curve and wound vasculature comparable to that of WT mice (Figure 5B, C). In addition, we employed a STZ-induced diabetic mouse model, where angiogenesis is greatly compromised, to investigate the effects of epsin deficiency and modulating VEGFR2 expression in wound healing. Compared to STZ-WT mice, STZ-EC-iDKO mice exhibited more rapid wound healing (Figure 5D), suggesting that boosting angiogenesis by depleting epsins plays a beneficial role in promoting diabetic wound healing. These results demonstrate the importance of epsins for balancing VEGFR2 protein levels during angiogenesis and suggest that epsins are a potential novel therapeutic target in treating diabetic ulcer.
Figure 5. Genetic reduction of VEGFR2 expression rescues aberrant angiogenesis in epsin deleted mice.

A, Representative wound injuries at 0 and 9 days in WT, EC-iDKO or EC-iDKO-Flkfl/+ mice. B, Wound areas from A were calculated and reported as a wound healing curve. C, CD31 immunostaining of wounds from A isolated at day 9. D, WT or EC-iDKO mice were injected with STZ, followed by wound injury as described in A. E, Representative matrigel plugs (supplemented with or without 200 ng/mL VEGF-A) isolated 5 days post s.c. injection into WT, EC-iDKO or EC-iDKO-Flkfl/+ mice. F, CD31 immunostaining of matrigel plugs from E. G, Representative WT, EC-iDKO or EC-iDKO-Flkfl/+ LLC tumors harvested at 15 days. H, Quantification of tumor volumes in G. I, CD31 immunostaining of tumors from G. J, Quantification of CD31immunostaining in I using SlideBook software. B, D, H and J, error bars indicate the mean ± s.e.m. n > 5, *p < 0.05. Scale bars: A, 1 mm; C, F, and I, 50 μm.
Genetic reduction of VEGFR2 levels rescues defective angiogenesis under pathological conditions in vivo
To determine whether VEGFR2 reduction rescues abnormal pathological angiogenesis in the absence of epsins, we first subcutaneously implanted VEGF-containing matrigel plugs in WT, EC-iDKO, or EC-iDKO-Flkfl/+ mice.39 VEGF-dependent angiogenesis, revealed by both gross appearance and CD31 immunofluorescent staining of the matrigel plugs, was dramatically increased in the EC-iDKO mice, compared to WT (Figure 5E, F). Reducing VEGFR2 expression normalized the excessive angiogenic phenotype in EC-iDKO-Flkfl/+ mice to levels comparable to WT (Figure 5F). Aberrant angiogenesis in the EC-iDKO matrigel plugs is reminiscent of our previously established phenotype in which endothelial epsin deletion significantly enhanced the formation of dysfunctional, leaky vasculature in tumors.28 However, it is unknown whether reducing the VEGFR2 expression in the tumor is sufficient to rescue this epsin-deletion phenotype. To test this, we used a Lewis Lung Carcinoma (LLC) xenograft model in the WT, EC-iDKO, and EC-iDKO-Flkfl/+ mice. Consistent with our previous findings, epsin deletion significantly reduced tumor growth (Figure 5G, H) as a result of the enhanced formation of non-productive vasculature within the tumor (Figure 5I, J). Interestingly, reducing VEGFR2 expression promoted tumor growth in the EC-iDKO-Flkfl/+ mice to sizes comparable to WT (Figure 5G, H). Furthermore, the increased tumor growth correlated with normalized vasculature in the tumor (Figure 5I, J). Cumulatively, these findings support our model that endothelial epsin deletion results in aberrant embryonic and pathologic angiogenesis by prolonging activation of VEGFR2. These results indicate that endothelial epsins specifically downregulate VEGFR2 as a negative feedback regulator for VEGFR2 activation and angiogenesis.
Discussion
In this study, we demonstrate a role for endothelial epsins as negative regulators to limit unwanted embryonic angiogenesis. Most importantly, by crossing endothelial epsin-deleted mice with mice heterozygous for VEGFR2, we provide the first direct evidence that endothelial epsin deficiency promotes angiogenesis by specifically increasing the total amount of cell surface VEGFR2 during angiogenesis (Figure 6A).
Figure 6. Schematics of endothelial epsin deletion and reduced VEGFR2 expression effects on angiogenesis.
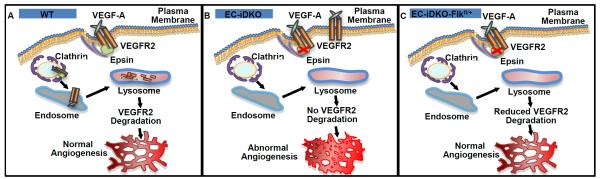
A, Epsins bind to and facilitate the internalization and degradation of activated VEGFR2 to modulate normal angiogenesis. B, Epsin deletion impairs VEGFR2 internalization and degradation resulting in elevated cell surface VEGFR2 and aberrant angiogenesis. C, Reduced VEGFR2 expression normalizes the amount of VEGFR2 on the cell surface resulting in normalized angiogenesis in EC-iDKO-Flkfl/+ mice.
We utilized complementary constitutive and inducible endothelial epsin deletion models to address the specific and temporal role of endothelial epsins. First, utilizing the constitutive Tie2 Cre deleter mice allowed us to identify, for the first time, an important role for endothelial epsins in embryonic angiogenesis. Importantly, the Tie2 Cre deleter mice exhibited delayed lethality and reduced phenotype severity than the global epsins 1 and 2 deletion, consistent with the hypothesis that EC-specific epsin deficiency specifically altered VEGFR2 signaling and developmental angiogenesis. Although the Tie2 Cre deleter mice also express Cre in hematopoietic cells,30 we did not observe any gross defects in blood cells, suggesting epsin deletion in hematopoietic cells does not contribute to defective embryonic angiogenesis and lethality. Secondly, the inducible iCDH5 Cre deleter mice allowed us to study a specific role of endothelial epsins in regulating VEGFR2 in adult mice. Importantly, postnatal epsin deletion does not affect quiescent angiogenesis.28
Our findings support a model in which inhibiting activated VEGFR2 internalization and degradation through loss of epsins accumulates receptors on cell surface, prolongs VEGF signaling and promotes angiogenesis. Reduction of VEGFR2 by genetic means suppresses the enhanced VEGF signaling and restores normal angiogenesis. Although receptor internalization is a recognized mechanism of signal modification, it is still unclear whether VEGFR2 internalization promotes or hinders VEGF signaling to modulate angiogenesis. Impaired internalization or intracellular trafficking of phosphorylated VEGFR2 reportedly inhibits certain intracellular signals, such as RhoA activation, but enhance others, such as Akt activation.38, 42, 43 For example, loss of the endocytic adaptor protein, Dab2, impairs VEGFR2 internalization resulting in reduced downstream signaling to ERK and impaired neovascular angiogenesis.42 Similarly, depletion of the class II PI3K, C2α, impairs VEGFR2 intracellular trafficking resulting in impaired RhoA activation and formation of endothelial focal adhesions. Given the implicated roles of class II PI3K and Dab2 in receptor endocytosis and trafficking to the endosomes, it is tempting to speculate that impaired internalization of phosphorylated VEGFR2 prevents VEGFR2 traveling to or affects the formation of signaling endosomes responsible for signal enhancement. In contrast, epsins interact with ubiquitinated VEGFR2, presumably after prolonged activation, and, as suggested by our findings, targets ubiquitinated VEGFR2 for lysosomal degradation thus effectively diminishing VEGF signaling.28 Loss of epsins prevents targeted VEGFR2 degradation, which may, in turn, promote VEGFR2 recycling and prolonged signaling, resulting in enhanced EC proliferation, migration and network formation responsible for aberrant angiogenesis. However, we cannot discount that, through impaired internalization of phosphorylated VEGFR2, epsins may also be inhibiting RhoA activation resulting in impaired focal adhesion formation in vivo thus contributing to our leaky vascular phenotype. Our findings, in combination with those from Drs. Takuwa group, strongly suggest that tight modulation of VEGF signaling through VEGFR2 internalization and differential downstream signal regulation is necessary to ensure proper angiogenesis.
Our previous findings support epsins as potential anti-cancer therapeutic targets that promote aberrant tumor angiogenesis and retarded tumor growth.28 We speculated that altering epsins may prove favorable in angiogenic-dependent conditions such as wound healing. Although we demonstrated that endothelial epsin deletion impairs, rather than promotes, wound healing under normal conditions, interestingly, in conditions where angiogenesis is compromised, such as diabetic ulcers, we found that epsin deletion promoted wound healing in STZ-induced diabetic models, suggesting that modulating endothelial epsins may be beneficial. Cumulatively, these findings establish a role for epsins in developmental and pathological angiogenesis and further fortify that epsins modulate VEGF signaling by specifically downregulating VEGFR2 levels.
Supplementary Material
SIGNIFICANCE.
Vascular endothelial growth factor (VEGF) signaling is essential for physiological and pathological angiogenesis. VEGF activates its receptor, VEGFR2, on endothelial cells to initiate angiogenic signaling cascades. Cell surface VEGFR2 levels control the magnitude of signal transmission. VEGFR2 abundance is negatively regulated by endocytosis and degradation. We found that epsins, a family of endocytic adaptor proteins, function in the regulation of tumor angiogenesis by downregulating VEGF signaling. However, whether epsins regulate embryonic angiogenesis is unclear. Here, we report for the first time an important role for endothelial epsins in embryonic angiogenesis. Furthermore, by engineering a novel genetic model with epsins deletion and reduced VEGFR2 expression in endothelial cells, we provide direct evidence that epsins regulate angiogenesis by specifically modulating total VEGFR2. Our findings suggest that modulating endothelial epsins may be a useful therapeutic strategy in diabetic ulcers and tumors.
Acknowledgments
We thank the OMRF Imaging Core for help with the histology studies.
Sources of Funding Work was supported by NIH Grants R01HL-093242, P20 RR018758, Oklahoma Center for Advanced Science Technology (OCAST) Grant HR09-116, and Department of Defense Grant W81XWH-11-1-00226 to H. Chen; OCAST Grant AR11-043 to Y. Dong; AHA postdoctoral fellowships 13POST16940008 to K.L. Tessneer and 13POST17270006 to S. Pasula.
Abbreviations
- EC-DKO
constitutive endothelial epsins deletion mice
- EC-iDKO
tamoxifen inducible endothelial epsins deletion mice
- EC-iDKO-Flkfl/+
tamoxifen inducible epsins deletion mice with reduced VEGFR2
- EdU
5-ethynl-2′-deoxyuridine
Footnotes
Disclosures None.
References
- 1.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Folkman J, Merler E, Abernathy C, Williams G. Isolation of a tumor factor responsible for angiogenesis. The Journal of experimental medicine. 1971;133:275–288. doi: 10.1084/jem.133.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jain RK. Molecular regulation of vessel maturation. Nature medicine. 2003;9:685–693. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 4.Kerbel RS. Tumor angiogenesis. The New England journal of medicine. 2008;358:2039–2049. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 6.Rossant J, Howard L. Signaling pathways in vascular development. Annual review of cell and developmental biology. 2002;18:541–573. doi: 10.1146/annurev.cellbio.18.012502.105825. [DOI] [PubMed] [Google Scholar]
- 7.Nagy JA, Feng D, Vasile E, Wong WH, Shih SC, Dvorak AM, Dvorak HF. Permeability properties of tumor surrogate blood vessels induced by vegf-a. Laboratory investigation; a journal of technical methods and pathology. 2006;86:767–780. doi: 10.1038/labinvest.3700436. [DOI] [PubMed] [Google Scholar]
- 8.Yancopoulos GD, Davis S, Gale NW, Rudge JS, Wiegand SJ, Holash J. Vascular-specific growth factors and blood vessel formation. Nature. 2000;407:242–248. doi: 10.1038/35025215. [DOI] [PubMed] [Google Scholar]
- 9.Ferrara N, Gerber HP, LeCouter J. The biology of vegf and its receptors. Nature medicine. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 10.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. Vegf receptor signalling - in control of vascular function. Nature reviews. Molecular cell biology. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 11.Weinstein BM, Lawson ND. Arteries, veins, notch, and vegf. Cold Spring Harbor symposia on quantitative biology. 2002;67:155–162. doi: 10.1101/sqb.2002.67.155. [DOI] [PubMed] [Google Scholar]
- 12.Mukherjee S, Tessema M, Wandinger-Ness A. Vesicular trafficking of tyrosine kinase receptors and associated proteins in the regulation of signaling and vascular function. Circulation research. 2006;98:743–756. doi: 10.1161/01.RES.0000214545.99387.e3. [DOI] [PubMed] [Google Scholar]
- 13.Duval M, Bedard-Goulet S, Delisle C, Gratton JP. Vascular endothelial growth factor-dependent down-regulation of flk-1/kdr involves cbl-mediated ubiquitination. Consequences on nitric oxide production from endothelial cells. The Journal of biological chemistry. 2003;278:20091–20097. doi: 10.1074/jbc.M301410200. [DOI] [PubMed] [Google Scholar]
- 14.Lanahan AA, Hermans K, Claes F, Kerley-Hamilton JS, Zhuang ZW, Giordano FJ, Carmeliet P, Simons M. Vegf receptor 2 endocytic trafficking regulates arterial morphogenesis. Developmental cell. 2010;18:713–724. doi: 10.1016/j.devcel.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murdaca J, Treins C, Monthouel-Kartmann MN, Pontier-Bres R, Kumar S, Van Obberghen E, Giorgetti-Peraldi S. Grb10 prevents nedd4-mediated vascular endothelial growth factor receptor-2 degradation. The Journal of biological chemistry. 2004;279:26754–26761. doi: 10.1074/jbc.M311802200. [DOI] [PubMed] [Google Scholar]
- 16.Hicke L. Ubiquitin-dependent internalization and down-regulation of plasma membrane proteins. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 1997;11:1215–1226. doi: 10.1096/fasebj.11.14.9409540. [DOI] [PubMed] [Google Scholar]
- 17.Chen H, De Camilli P. The association of epsin with ubiquitinated cargo along the endocytic pathway is negatively regulated by its interaction with clathrin. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:2766–2771. doi: 10.1073/pnas.0409719102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen H, Fre S, Slepnev VI, Capua MR, Takei K, Butler MH, Di Fiore PP, De Camilli P. Epsin is an eh-domain-binding protein implicated in clathrin-mediated endocytosis. Nature. 1998;394:793–797. doi: 10.1038/29555. [DOI] [PubMed] [Google Scholar]
- 19.Rosenthal JA, Chen H, Slepnev VI, Pellegrini L, Salcini AE, Di Fiore PP, De Camilli P. The epsins define a family of proteins that interact with components of the clathrin coat and contain a new protein module. The Journal of biological chemistry. 1999;274:33959–33965. doi: 10.1074/jbc.274.48.33959. [DOI] [PubMed] [Google Scholar]
- 20.Shih SC, Katzmann DJ, Schnell JD, Sutanto M, Emr SD, Hicke L. Epsins and vps27p/hrs contain ubiquitin-binding domains that function in receptor endocytosis. Nature cell biology. 2002;4:389–393. doi: 10.1038/ncb790. [DOI] [PubMed] [Google Scholar]
- 21.Wendland B. Epsins: Adaptors in endocytosis? Nature reviews. Molecular cell biology. 2002;3:971–977. doi: 10.1038/nrm970. [DOI] [PubMed] [Google Scholar]
- 22.Tessneer KL, Cai X, Pasula S, Dong Y, Liu X, Chang B, McManus J, Hahn S, Yu L, Chen H. Epsin family of endocytic adaptor proteins as oncogenic regulators of cancer progression. Journal of Cancer Research Updates. 2013;2:144–150. doi: 10.6000/1929-2279.2013.02.03.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen H, Ko G, Zatti A, Di Giacomo G, Liu L, Raiteri E, Perucco E, Collesi C, Min W, Zeiss C, De Camilli P, Cremona O. Embryonic arrest at midgestation and disruption of notch signaling produced by the absence of both epsin 1 and epsin 2 in mice. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:13838–13843. doi: 10.1073/pnas.0907008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ko G, Paradise S, Chen H, Graham M, Vecchi M, Bianchi F, Cremona O, Di Fiore PP, De Camilli P. Selective high-level expression of epsin 3 in gastric parietal cells, where it is localized at endocytic sites of apical canaliculi. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:21511–21516. doi: 10.1073/pnas.1016390107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spradling KD, McDaniel AE, Lohi J, Pilcher BK. Epsin 3 is a novel extracellular matrix-induced transcript specific to wounded epithelia. The Journal of biological chemistry. 2001;276:29257–29267. doi: 10.1074/jbc.M101663200. [DOI] [PubMed] [Google Scholar]
- 26.Coon BG, Burgner J, Camonis JH, Aguilar RC. The epsin family of endocytic adaptors promotes fibrosarcoma migration and invasion. The Journal of biological chemistry. 2010;285:33073–33081. doi: 10.1074/jbc.M110.124123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coon BG, Direnzo DM, Konieczny SF, Aguilar RC. Epsins’ novel role in cancer cell invasion. Communicative & integrative biology. 2011;4:95–97. doi: 10.4161/cib.4.1.14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pasula S, Cai X, Dong Y, Messa M, et al. Endothelial epsin deficiency decreases tumor growth by enhancing vegf signaling. The Journal of clinical investigation. 2012;122:4424–4438. doi: 10.1172/JCI64537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ewan LC, Jopling HM, Jia H, Mittar S, Bagherzadeh A, Howell GJ, Walker JH, Zachary IC, Ponnambalam S. Intrinsic tyrosine kinase activity is required for vascular endothelial growth factor receptor 2 ubiquitination, sorting and degradation in endothelial cells. Traffic. 2006;7:1270–1282. doi: 10.1111/j.1600-0854.2006.00462.x. [DOI] [PubMed] [Google Scholar]
- 30.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-cre transgenic mice: A new model for endothelial cell-lineage analysis in vivo. Developmental biology. 2001;230:230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 31.Fu J, Gerhardt H, McDaniel JM, et al. Endothelial cell o-glycan deficiency causes blood/lymphatic misconnections and consequent fatty liver disease in mice. The Journal of clinical investigation. 2008;118:3725–3737. doi: 10.1172/JCI36077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang H, He Y, Dai S, Xu Z, Luo Y, Wan T, Luo D, Jones D, Tang S, Chen H, Sessa WC, Min W. Aip1 functions as an endogenous inhibitor of vegfr2-mediated signaling and inflammatory angiogenesis in mice. The Journal of clinical investigation. 2008;118:3904–3916. doi: 10.1172/JCI36168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Salic A, Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:2415–2420. doi: 10.1073/pnas.0712168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lobov IB, Renard RA, Papadopoulos N, Gale NW, Thurston G, Yancopoulos GD, Wiegand SJ. Delta-like ligand 4 (dll4) is induced by vegf as a negative regulator of angiogenic sprouting. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:3219–3224. doi: 10.1073/pnas.0611206104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patel-Hett S, D’Amore PA. Signal transduction in vasculogenesis and developmental angiogenesis. The International journal of developmental biology. 2011;55:353–363. doi: 10.1387/ijdb.103213sp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hooper AT, Butler JM, Nolan DJ, et al. Engraftment and reconstitution of hematopoiesis is dependent on vegfr2-mediated regeneration of sinusoidal endothelial cells. Cell stem cell. 2009;4:263–274. doi: 10.1016/j.stem.2009.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sorensen I, Adams RH, Gossler A. Dll1-mediated notch activation regulates endothelial identity in mouse fetal arteries. Blood. 2009;113:5680–5688. doi: 10.1182/blood-2008-08-174508. [DOI] [PubMed] [Google Scholar]
- 38.Wang Y, Nakayama M, Pitulescu ME, et al. Ephrin-b2 controls vegf-induced angiogenesis and lymphangiogenesis. Nature. 2010;465:483–486. doi: 10.1038/nature09002. [DOI] [PubMed] [Google Scholar]
- 39.Valapala M, Thamake SI, Vishwanatha JK. A competitive hexapeptide inhibitor of annexin a2 prevents hypoxia-induced angiogenic events. Journal of cell science. 2011;124:1453–1464. doi: 10.1242/jcs.079236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kerbel R, Folkman J. Clinical translation of angiogenesis inhibitors. Nature reviews. Cancer. 2002;2:727–739. doi: 10.1038/nrc905. [DOI] [PubMed] [Google Scholar]
- 41.Kim BS, Chen J, Weinstein T, Noiri E, Goligorsky MS. Vegf expression in hypoxia and hyperglycemia: Reciprocal effect on branching angiogenesis in epithelial-endothelial co-cultures. Journal of the American Society of Nephrology : JASN. 2002;13:2027–2036. doi: 10.1097/01.asn.0000024436.00520.d8. [DOI] [PubMed] [Google Scholar]
- 42.Nakayama M, Nakayama A, van Lessen M, Yamamoto H, Hoffmann S, Drexler HC, Itoh N, Hirose T, Breier G, Vestweber D, Cooper JA, Ohno S, Kaibuchi K, Adams RH. Spatial regulation of vegf receptor endocytosis in angiogenesis. Nature cell biology. 2013;15:249–260. doi: 10.1038/ncb2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoshioka K, Yoshida K, Cui H, et al. Endothelial pi3k-c2alpha, a class ii pi3k, has an essential role in angiogenesis and vascular barrier function. Nature medicine. 2012;18:1560–1569. doi: 10.1038/nm.2928. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.