

Abstract
Positron emission tomography (PET) is an increasingly important biomedical imaging technique that relies on the development of radiotracers labeled with positron-emitters to achieve biochemical specificity. Fluorine-18 (t1/2 = 109.7 min) is an attractive positron-emitting radiolabel for organic radiotracers, primarily because of its longer half-life and greater availability relative to those for the main alternative, carbon-11 (t1/2 = 20.4 min). Rapid simple methods are sought for labeling prospective PET radiotracers with fluorine-18 from cyclotron-produced aqueous [18F]fluoride ion, which must often be converted first into a suitably reactive labeling synthon for use in a subsequent labelling reaction. Use of 18F-labeled synthons in ‘click chemistry’ attracts increasing attention for labeling PE Tradiotracers. Here we describe rapid single-step radiosyntheses of azido- or azidomethyl-bearing [18F]fluoroarenes from the reactions of diaryliodonium salts with no-carrier-added [18F]fluoride ion within a microfluidic apparatus to provide previously poorly accessible 18F-labeled click synthons in radiochemical yields of 15% for [18F]4-fluorophenyl azide and about 40% for each of the [18F](azidomethyl)-fluorobenzenes. The radiosyntheses of the latter synthons was possible under ‘wet conditions’, so obviating the need to dry the cyclotron-produced [18F]fluoride ion and greatly enhancing the practicality of the method.
Keywords: Click chemistry, Hypervalent compounds, Imaging agents, Isotopic labeling, Radiochemistry
Introduction
Efficient and rapid methods for labeling organic compounds with the short lived positron-emitter fluorine-18 (t1/2 = 109.7 min) are sought for producing novel radiotracers for biomedical imaging with positron emission tomography (PET).[1] Fluorine-18 can be obtained in high yields and in high specific radioactivity from a cyclotron by the use of the 18O(p,n)18F reaction on 18O-enriched water.[2] This process provides the fluorine-18 as aqueous [18F]fluoride ion. Methods for producing 18F-labeled radiotracers from this source must either use [18F]fluoride ion directly or proceed through conversion of the [18F]fluoride ion into a labeling synthon. 18F-Labeled organoazides are potentially useful synthons in ‘click radiochemistry’, based on the well-known mild, rapid and efficient copper(I)-catalyzed azide-alkyne 1,3-dipolar cycloaddition reaction[3], for attaching fluorine-18 to biomolecules, especially macromolecules.[4] Despite the attractive features of the ‘click reaction’ itself for radiolabeling, the prior preparation of a suitable radioactive synthon is still necessary and radiosynthetically demanding.
[18F]Fluorolkyl azides[5] and [18F]azidobenzyl fluorides[6] have been explored as labeling synthons in click radiochemistry. Generally, these labeling synthons may be prepared in a single efficient step from [18F]fluoride ion. However, potential radiotracers prepared from these synthons may be prone to radiodefluorination in vivo.[7] Such radiodefluorination results in uptake of [18F]fluoride ion into bone which may compromise the performance of the radiotracer. Because aryl C-18F bonds are usually more stable than alkyl C-18F bonds in vivo, azide synthons with fluorine-18 attached to an aryl ring have been sought. Hashizume et al.[8] reported the synthesis of several [18F]fluorophenyl azides ([18F]1a–f; Figure 1) through isotopic exchange of [18F]fluoride ion with fluorophenyl azides, and they used these synthons to label a protein, cytochrome C, photochemically. The decay-corrected radiochemical yields (RCYs) of the [18F]fluorophenyl azides were very low (0–12%). Moreover, a major disadvantage of such exchange labeling is that the resultant specific radioactivity of the labeled synthon and of any derived radioactive product becomes greatly decreased from that of the no-carrier-added (NCA) [18F]fluoride ion source. [18F]1-(Azidomethyl)-4-fluorobenzene ([18F]2a; Figure 1) has been used as a labeling synthon to prepare 18F-labeled peptides,[9,10] sometimes in excellent RCYs (19–90%). Potential to use this synthon to label oligonucleotides quantitatively has also been demonstrated.[11] Nonetheless, the reported radiosynthesis of [18F]2a required four radiochemical steps from 4-formyl N,N,N-trimethylanilinium triflate performed over 75 min and gave a low overall RCY (34%).[9] This type of procedure was recently automated to give [18F]2a in RCYs up to 60% and the ortho isomer [18F]2b in 30% RCY.[12]
Figure 1.

Previously reported [18F]fluoroaryl azides.
Herein, we describe rapid one-step radiosyntheses of various [18F]fluoroarenes bearing azide functionality from azide-functionalized diaryliodonium salts as accessible ‘18F-labeled click synthons’, including [18F]2a and [18F]2b.
Results and Discussion
Diaryliodonium salts have served as uniquely effective precursors for introducing [18F]fluoride ion into electron-rich as well as electron-deficient aryl rings (Figure 2).[13-15] Such diaryliodonium salts are gradually gaining application in the preparation of PET radiotracers, as improved methods for the synthesis of elaborate diaryliodonium salts become available.[16]
Figure 2.
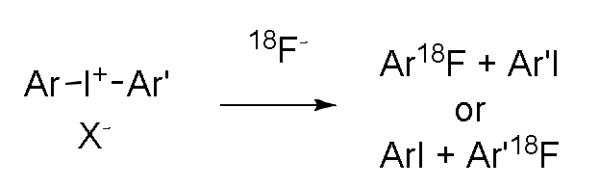
Radiosynthesis of [18F]fluoroarenes from diaryliodonium salts.
We considered that the use of diaryliodonium salt precursors might permit advantageous single-step radiosyntheses of click synthons, such as [18F]1f and [18F]2a, because the azide group is regarded as electronically neutral, and like other electronically neutral groups might be expected to be well-tolerated in the radiofluorination of diaryliodiodonium salts. An electron-rich aryl ring, such as a 4-methoxyphenyl or 2-thienyl ring, is generally known to direct [18F]fluoride into a relatively electron-deficient ring in a diaryliodonium salt.[13-15] Accordingly, we sought a convenient method for preparing diaryliodonium salts having an azide group directly or indirectly attached to one aryl ring, with the other ring being relatively electron-rich. Moreover, we wished to avoid highly nucleophilic or fluorine-containing anions to prevent competing reaction or dilution of specific radioactivity, respectively, in the radiofluorination process. Recently, we have reported success in preparing functionalized diaryliodonium tosylates by generating reactive [hydroxy(tosyl)iodo]arenes in situ for reaction with activated arenes or arylstannanes.[17] Yamamoto and Togo[18] have reported the syntheses of [hydroxy(sulfonyloxy)iodo]arenes from iodoarenes and mCPBA (m-chloroperbenzoic acid) in the presence of various sulfonic acids. Because this method uses a mild oxidizing agent under ambient temperature, we were attracted to its use for preparing azide-bearing [hydroxy(tosyloxy)iodo]arenes that might then be converted in situ into the target diaryliodonium tosylates by treatment with anisole or thiophene (Figure 3).
Figure 3.
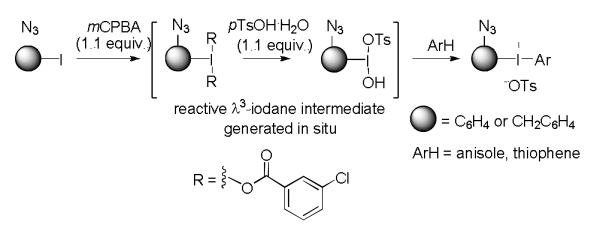
mCPBA-mediated one-pot syntheses of azide–bearing diaryliodonium tosylates.
With this one-pot approach, we were successful in converting iodoaryl azides into azide-bearing diaryliodonium tosylates in moderate to good yields (53–74%, Scheme 1). Many of the prepared diaryliodonium tosylates were also conveniently converted into diaryliodonium halides in excellent yields (76–92%) by treatment with inorganic halides in aqueous acetonitrile (Scheme 1).
Scheme 1.

One-pot syntheses of diaryliodonium tosylates from iodoaryl azides and subsequent conversion into their corresponding halides. General reaction conditions: i) iodoaryl azide (1 equiv.), mCPBA (1.1 equiv.); ii) pTsOH·H2O (1.1 equiv.), iii) CHCl3 and ArH (excess, 5–10 fold); iv) MX = NH4Cl (9–11, 14), KBr (12) or KI (13).
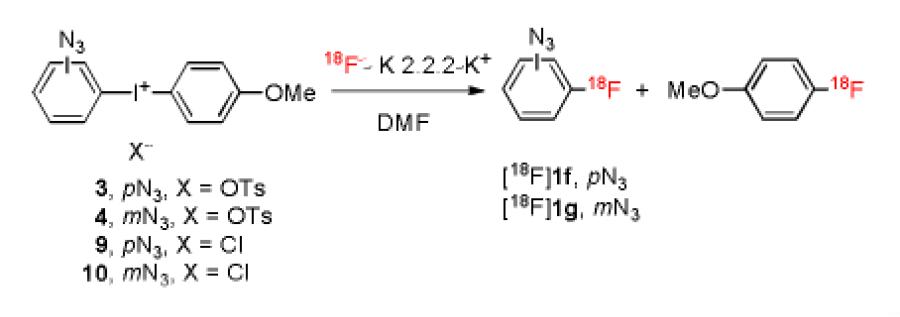
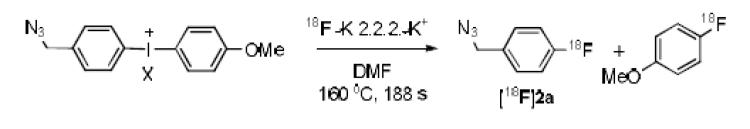
The prepared azide-bearing diaryliodonium salts (3–14) were of two types, those in which the azide group was attached directly to the aryl ring (3, 4, 9 and 10) and those in which the azide group was linked to the aryl ring by a methylene group (5–8 and 11–14). Each salt was subjected to labeling with NCA [18F]fluoride ion in the commercial microfluidic apparatus that we have described previously.[14] Thus, dried [18F]fluoride ion-kryptofix 2.2.2 complex (18F−-K 2.2.2- K+) was dissolved in reaction solvent, either DMF or MeCN. A solution of the diaryliodonium salt was prepared in matching solvent. Each reagent solution was then loaded into a storage loop. Reaction was implemented by infusing each reagent solution into the micro-reactor at equal set rates (4–10 μL/min) and at a fixed temperature. Effluents were quenched with aq. MeCN and analyzed with reverse phase HPLC equipped with a radioactivity detector. Reaction temperature and time were varied to obtain optimal RCYs. Radioactivity adsorption is a well-known phenomenon in radiofluorination with dry NCA [18F]fluoride ion.[19] In this study, adsorption of radioactivity within the microfluidic apparatus was overall generally moderate but somewhat variable (25 ± 9%; mean ± SD, n = 22).
For substrates having an azide group attached directly to the aromatic ring, low RCYs of [18F]1-azido-4-fluorobenzene ([18F]1f) or [18F]1-azido-3-fluorobenzene ([18F]1g) were obtained in DMF from tosylates (Table 1, entries 1 and 5). Changing solvent to acetonitrile (entry 3) or the anion to chloride (entries 4 and 7) gave similar or lower RCYs. Accompanying formation of [18F]4-fluoroanisole was relatively low. Hashizume et al.[8] failed to produce [18F]1f by exchange of 1f with [18F]fluoride ion, and hence these results show some benefit of using an iodonium salt precursor. Nonetheless, the low RCYs are surprising given that analogous diaryliodonium salts, only differing by having a non electron-withdrawing substituent (e.g., Me, MeO) other than azide, give high radiochemical yields of the substituted radiofluorinated arenes under comparable conditions.[14,15] Inclusion of the free radical scavenger, TEMPO (2,2,6,6-tetramethylpiperidine 1-oxyl) in some fluoridation reactions of diaryliodonium salts has been reported to give improved yields.[20] We found that inclusion of TEMPO improved the RCY of [18F]1f appreciably (c.f., entries 1 and 2) but had less effect on the RCY of [18F]1g (c.f., entries 5 and 6). In view of the still modest RCYs for [18F]1f and [18F]1g, we decided to explore the radiofluorination of diaryliodonium salts having the azido group indirectly linked to an aryl ring.
Table 1.
RCYs of radioactive products from the radiofluorination of azide-functionalized diaryliodonium salts in DMF.
| |||||
|---|---|---|---|---|---|
| Entry | Substrate | Conditions][a] |
RCY [%][b] |
||
| Temp. [°C] |
Time [s] |
[18F]Click synthon |
[18F]4- Fluoro- anisole |
||
| 1 | 3 | 160 | 188 | [18F]1f, 10 | 0.7 |
| 2[c] | 3 | 160 | 188 | [18F]1f, 15 | 1.7 |
| 3[d] | 3 | 160 | 126 | [18F]1f, 12 | 0 |
| 4 | 9 | 160 | 188 | [18F]1f, 9 | 0.6 |
| 5 | 4 | 160 | 188 | [18F]1g,10 | 1 |
| 6[c] | 4 | 160 | 188 | [18F]1g,12 | 0.7 |
| 7 | 10 | 180 | 118 | [18F]1g, 4 | 0 |
Conditions are those giving optimal RCY from ~ 8 runs under various conditions.
RCYs were determined chromatographically and are corrected for radioactivity adsorption in the apparatus. Adsorption was 29 ± 12% (mean ± SD, n = 7).
Reaction mixture included 1 equiv. of TEMPO with respect to the iodonium salt.
Reaction performed in MeCN.
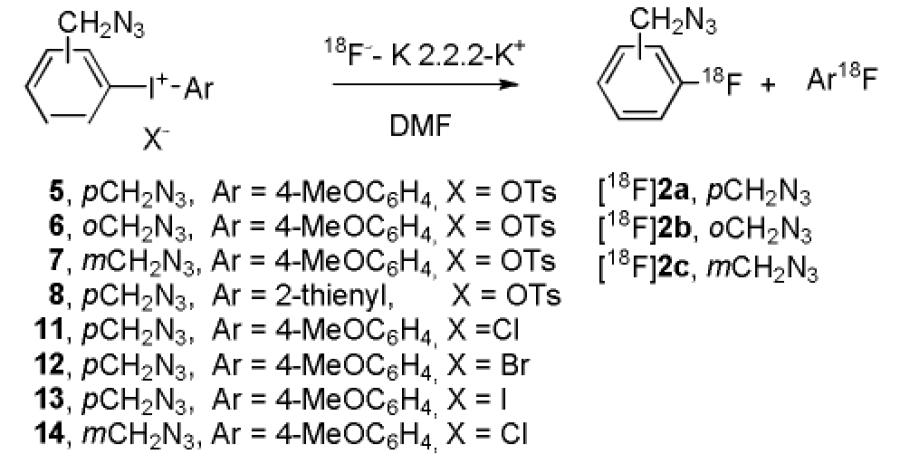
Gratifyingly, the radiofluorination of the aryl(4-methoxyphenyl)iodonium toyslates bearing azidomethyl groups generally proceeded rapidly in DMF to give the desired 18F-labeled fluorobenzyl azides in useful RCYs (~ 40%), irrespective of the position of the azidomethyl group on the aryl ring (Table 2, entries 1–3). Replacement of the 4-methoxy aryl ring in the salt with a 2-thienyl ring reduced the attainable RCY to 14% (Table 2, c.f., entries 4 and 1). Variation in optimal RCYs with salt counterion was small (Table 2, entries 1–3 and 5–8). When the radiofluorinations of the four 4-azidomethylphenyl(4-methoxyphenyl)iodonium salts (5, 11–13) were compared under conditions of identical temperature (160 °C) and reaction time (188 s), the chloride salt gave the highest RCY (Table 3).
Table 2.
Single-step NCA radiofluorination of diaryliodonium salts bearing azidomethyl groups in DMF.
| |||||
|---|---|---|---|---|---|
| Entry | Substrate | Conditions[a] |
RCY [%][b] |
||
| Temp [°C] |
Time [s] |
[18F]Click synthon |
Ar18F | ||
| 1 | 5 | 160 | 188 | [18F]2a, 37 | 1.8 |
| 2 | 6 | 180 | 188 | [18F]2b, 41 | <0.7 |
| 3 | 7 | 180 | 188 | [18F]2c, 39 | 0.9 |
| 4 | 8 | 160 | 188 | [18F]2a, 14 | 0 |
| 5 | 11 | 200 | 94 | [18F]2a, 45 | 2.2 |
| 6 | 12 | 200 | 94 | [18F]2a, 40 | 2.4 |
| 7 | 13 | 200 | 188 | [18F]2a, 38 | 1.7 |
| 8 | 14 | 180 | 157 | [18F]2c, 35 | 0.8 |
Conditions giving optima l RCYs from ~ 8 runs under various conditions.
RCYs we re determined chromatographically and are corrected for radioactivity adsorption; adsorption was 19 ± 7% (mean ± SD, n = 8).
Table 3.
Effect of counterions on the radiofluorination of 4- azidomethylphenyl(4-methoxyphenyl)iodonium salts in DMF.
| ||||
|---|---|---|---|---|
| Entry | Substrate[a] | X | RCY [%][b] |
|
| [18F]2a | [18]4-Fluoro- anisole |
|||
| 1 | 5 | OTs | 37 | 1.8 |
| 2 | 11 | CI | 51 | 2.7 |
| 3 | 12 | Br | 40 | 1.6 |
| 4 | 13 | I | 36 | 1.7 |
5 mM substrate concentration in DMF.
RCYs are those determined chromatographically with correction for adsorption. Radioactivity recovery from the apparatus was 82 ± 7% (mean ± SD, n = 4).
When radiofluorinations were performed at high temperature (> 180 °C), a small amount of [18F]fluorobenzaldehyde (< 3% RCY) was also produced, as detected by radio-HPLC (Figure 4). This radioactive product presumably arises through aerobic oxidation of the [18F]fluorobenzyl azide. Otherwise, all the radiofluorinations proceeded cleanly and selectively to the desired [18F]fluorobenzyl azides, with only [18F]4-fluoroanisole appearing as a minor byproduct.
Figure 4.
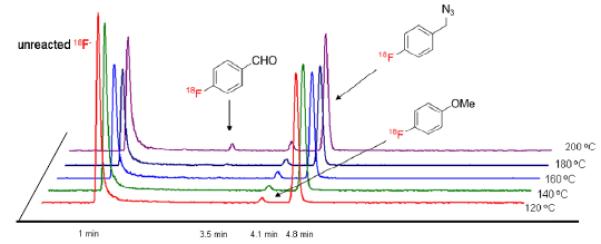
Radio-HPLC chromatograms for the analysis of radioactive products from the radiofluorination of 11 in the temperature range 120–200 °C.
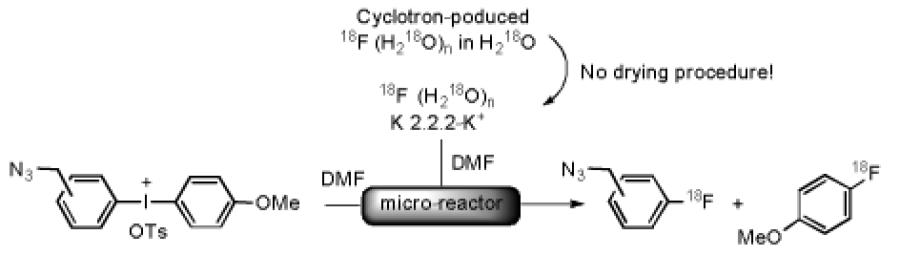
Because [18F]fluoride ion is generally produced by proton irradiation of 18O-enriched water, the first stage in radiotracer synthesis is virtually always removal of the water to generate highly nucleophilic ‘naked’ [18F]fluoride ion.[1] This can be a tedious and lengthy process, that wastes useful radioactivity. We have previously shown that radiofluorinations of diaryliodonium salts in DMF proceed effectively even in the presence of low concentrations of water (0.25 vol.-% in DMF).[14] With a view to developing a more streamlined procedure for preparing the click synthons, [18F]2a–c, we investigated the radiofluorination of azide-functionalized diaryliodonium salts with ‘wet’ cyclotron-produced NCA [18F]fluoride ion. Thus, one of the storage loops of the microfluidic apparatus was loaded with a mixture of cyclotron-produced [18F]fluoride ion in [18O]water and a solution of potassium carbonate and K 2.2.2 in DMF. The concentration of water in this storage loop was ~ 70 vol.-%. The radiofluorination of the p-azidomethyl salt 5 in aqueous DMF in this format produced a low but appreciable RCY (17%) of [18F]2a, while remarkably the radiofluorination of the o-azidomethyl salt 7 produced the synthon [18F]2b in very useful RCY (46%) (Table 4). Radiofluorination of 7 in aqueous MeCN also gave [18F]2b in even higher RCY (53%) in a shorter reaction time and at lower temperature. This RCY even exceeds that obtained under dry conditions (41%; Table 2, entry 2). These results show a tolerance of the radiofluorination of iodonium salts for the presence of high concentrations of water, in this case 35 vol-.%. Again they show the tolerance of these reactions to bulky non-electron withdrawing substituents in ortho position to the hypervalent iodine. These are distinct advantages over traditional radiofluorination through aromatic nucleophilic substitution reactions.
Table 4.
Radiofluorination of diaryliodonium salts under ‘wet’ conditions.
| |||||
|---|---|---|---|---|---|
| Entry | substrate[a] | conditions | RCY [%][b] | ||
|
|
|||||
| Temp. [°C] |
Time [S] |
[18F]Fluoro- benzyl azide |
[18F]4- Fluoro anisole |
||
| 1 | 3 | 200 | 188 | [18F]2a, 17 | 1 |
| 2 | 8 | 200 | 188 | [18F]2c, 46 | 1 |
| 3[c] | 8 | 180 | 94 | [18F]2c, 53 | 2 |
5 mM in DMF.
Optimal RCY from 6-8 different runs under different conditions, determined chromatographically with correction for radioactivity adsorption; adsorption in the microfluidic apparatus was 29 ± 2% (n = 3, mean ± SD).
Reaction performed in MeCN.
The very rapid production of [18F]2b in moderate radiochemical yield under ‘wet’ radiofluorination conditions, avoiding the need to dry the cyclotron-produced [18F]fluoride ion, is highly attractive for application of this synthon to the click labeling of macromolecules or even small molecules. Aqueous DMF and MeCN are often used a solvents for click chemistry, and hence conceivably the synthons [18F]2a–c may be used in the solvents in which they are prepared. The radiosyntheses and use of these labelling synthons should be highly amenable to simple automation for radiation safety, and therefore to operation with very high amounts of radioactivity.
Conclusions
In summary, azide-bearing diaryliodonium salts were readily prepared. Those bearing an azidomethyl group on one aryl ring, and having a partner 4-methoxyphenyl ring, were readily radiofluorinated with cyclotron-produced NCA [18F]fluoride ion, even in the presence of a high proportion of water, to give the useful click labeling synthons [18F]2a–c rapidly in useful RCYs. This represents a major advance on the previous multi-step radiosyntheses[9,12] of [18F]2a and [18F]2b, which required a drying stage and four radiochemical steps. The synthons [18F]2a–c are now readily accessible for application in click radiochemistry.
Experimental Section
Materials
Anhydrous chloroform and acetonitrile were purchased in Sure/SealTM bottles from Sigma-Aldrich (Milwaukee, WI) and used without further treatment. Iodobenzyl bromide, fluorobenzyl bromide, mCPBA, and pTsOH.H2O were purchased from Sigma-Aldich and used as received. Iodobenzyl azides used were prepared according to a known procedure.[21] Fluorobenzyl azides were prepared similarly from fluorobenzyl bromides. All other reagents were purchased from either Sigma-Aldrich or Alfa Aesar (Ward Hill, MA) and used without purification. Acetonitrile (high purity solvent, Burdick & Jackson; Morristown, NJ) for HPLC was also used without further treatment. QMA anionic resin cartridges were supplied by ORTG (Oakdale, TN). NCA [18F]fluoride ion was obtained through the 18O(p,n)18F nuclear reaction by irradiating [18O]water (95 atom %) for 90–120 min with a proton beam (17 MeV; 20 μA) produced with a PETrace cyclotron (GE Medical Systems; Milwaukee, MI).
General
1H (400 MHz) and 13C (100 MHz) NMR spectra were recorded at room temperature on Avance-400 spectrometer (Bruker; Billerica, MA). 1H and 13C chemical shifts are reported in δ units (ppm) downfield relative to the signal for tetramethylsilane. Abbreviations br, s, d, t, and m denote broad, singlet, doublet, triplet, and multiplet, respectively. High resolution mass spectra (HRMS) were acquired from the Mass Spectrometry Laboratory, University of Illinois at Urbana-Champaign (Urbana, IL, USA) under electron ionization conditions using a double-focusing high-resolution mass spectrometer (Autospec; Micromass Inc., USA). Melting points were measured with a Mel-Temp manual melting point apparatus (Electrothermal; Fisher Scientific, Pitsburg, PA).
Note
Many organoazides with high azido content are known to be explosive.[22] Great care should be exercised during the heating needed for diaryliodonium salt synthesis (mostly ~ 40 °C, with up to 7 mmol of organic azide in this study). Due to possible friction and impact-sensitive character, the isolation of the final iodonium salts should be conducted in a properly shielded hood with safety precautions. Also, the test of complete consumption of mCPBA should be performed with either aqueous potassium iodide or KI starch paper to ensure absence of organic peracids after the reaction.[23]
General procedure for diaryliodonium tosylate synthesis
Azide-functionalized diaryliodonium tosylates were readily obtained in a single-step from the corresponding iodoarenes with mCPBA/pTsOH.H2O and an electron-rich arene such as anisole and thiophene. The procedure is exemplified by the synthesis of 3.
4-Azidophenyl(4’-methoxyphenyl)iodonium tosylate (3)
mCPBA (0.49 g, 2.2 mmol, 77% max. content) was added to a stirred solution of 1-azido-4-iodobenzene (0.49 g, 2.0 mmol) in CHCl3 (20 mL) at r.t. The mixture was stirred at r.t. until it became a pale yellow solution (ca. 15 min). pTsOH.H2O (0.42 g, 2.2 mmol) was added, followed by anisole (1.08 g, 10 mmol). The reaction mixture was gradually heated to 40 °C and held at this temperature for about 2 h while the mixture became a yellow solution. Consumption of 1-azido-4-iodobenzene was monitored by TLC (Rf = 0.53, hexane) and formation of intermediate [hydroxy(tosyl)iodo]arene was verified with KI starch paper. Reaction solvent was then removed in vacuo. The residual yellow oil was triturated with Et2O, giving a solid that was washed with Et2O and recrystallized from MeOH/Et2O to give 3 (white solid; 0.58 g, 56%). mp = 150–151 °C; 1H-NMR (MeOD-d4) δ 8.11–8.05 (m, 4H), 7.69 (d, J = 8 Hz, 2H), 7.23–7.17 (m, 4H), 7.05 (d, J = 8.8 Hz, 2H), 3.84 (s, 3H), 2.36 (s, 3H); 13C-NMR (MeOD-d4) δ 164.7, 146.6, 143.8, 141.8, 138.6, 138.0, 130.0, 127.1, 123.6, 119.0, 110.4, 105.1, 56.5, 21.4; HRMS [M–OTs].+ Calc’d. for C13H11N3OI: 351.9947, Found: 351.9947.
The following diaryliodonium tosylates were prepared similarly.
3-Azidophenyl(4’-methoxyphenyl)iodonium tosylate (4)
White solid (0.71 g, 68%). mp = 143–145 °C; 1H-NMR (MeOD-d4) δ 8.12–8.08 (m, 2H), 7.87–7.83 (m, 2H), 7.68 (d, J = 8.4 Hz, 2H), 7.49 (t, J = 8 Hz, 1H), 7.36-7.33 (m, 1H), 7.21 (d, J = 7.6 Hz, 2H), 7.08-7.05 (m, 2H), 3.84 (s, 3H), 2.36 (s, 3H); 13C-NMR (MeOD-d4) δ 164.8, 144.8, 143.8, 141.8, 138.9, 134.1, 132.0, 129.9, 126.3, 124.1, 119.1, 116.9, 104.7, 56.5, 21.5; HRMS [M–OTs].+ Calc’d. for C13H11N3OI: 351.9947, Found: 351.9951.
[4-(Azidomethyl)phenyl](4’-methoxyphenyl)iodonium tosylate (5)
White solid (0.72 g, 74%). mp = 149–151 °C; 1H-NMR (MeOD-d4) δ 8.12–8.06 (m, 4H), 7.67 (d, J = 8 Hz, 2H), 7.48 (d, J = 8.8 Hz, 2H), 7.21 (d, J = 8 Hz, 2H), 7.05 (dd, J = 2, 9.2 Hz, 2H), 4.78 (s, 2H), 3.84 (s, 3H), 2.36 (s, 3H); 13C-NMR (MeOD-d4) δ 164.7, 143.8, 142.6, 141.8, 138.7, 136.5, 132.6, 130.0, 127.1, 119.0, 115.7, 104.7, 56.5, 54.6, 21.5; HRMS [M–OTs].+ Calc’d. for C14H13N3OI: 366.0103, Found: 366.0107.
[2-(Azidomethyl)phenyl](4’-methoxyphenyl)iodonium tosylate (6)
White solid (0.78 g, 73%). mp = 167–169 °C; 1H-NMR (MeOD-d4) δ 8.30 (d, J = 8.4 Hz, 1H), 8.09 (dd, J = 2, 9.2 Hz, 2H), 7.70–7.67 (m, 4H), 7.49–7.45 (m, 1H), 7.21 (d, J = 8 Hz, 2H), 7.06 (dd, J = 2, 8.8 Hz, 2H), 4.74 (s, 2H), 3.84 (s, 3H), 2.36 (s, 3H); 13C-NMR (MeOD-d4) δ 164.5, 143.7, 141.8, 139.3, 138.8, 138.6, 134.6, 133.3, 133.1, 129.9, 127.1, 119.8, 119.0, 104.5, 57.4, 56.5, 21.4; HRMS [M–OTs].+ Calc’d. for C14H13N3OI: 366.0103, Found: 366.0096.
[3-(Azidomethyl)phenyl](4’-methoxyphenyl)iodonium tosylate (7)
White solid (0.82 g, 69%). mp = 136–138 °C; 1H-NMR (MeOD-d4) δ 8.15 (s, 1H), 8.11–8.07 (m, 3H), 7.71–7.64 (m, 3H), 7.53 (t, J = 8 Hz, 1H), 7.22 (d, J = 7.6 Hz, 2H), 7.06 (dd, J = 2, 6.8 Hz, 2H), 4.47 (s, 2H), 3.84 (s, 3H), 2.37 (s, 3H); 13C-NMR (MeOD-d4) δ 164.8, 143.8, 142.1, 141.8, 138.8, 135.6, 135.4, 133.4, 133.2, 130.0, 127.1, 119.1, 116.8, 104.7, 56.5, 54.5, 21.5; HRMS [M–OTs].+ Calc’d. for C14H13N3OI: 366.0103, Found: 366.0105.
[4-(Azidomethyl)phenyl](2’-thienyl)iodonium tosylate (8)
White solid (0.38 g, 53%). mp = 163–165 °C; 1H-NMR (MeOD-d4) δ 8.17 (d, J = 8.4 Hz, 2H), 8.01 (dd, J = 1.2, 4 Hz, 1H), 7.88 (dd, J = 1.2, 5.6 Hz, 1H), 7.69 (d, J = 8 Hz, 2H), 7.50 (d, J = 8.4 Hz, 2H), 7.22 (d, J = 8 Hz, 2H), 7.17 (dd, J = 5.6, 9.2 Hz, 1H), 4.49 (s, 2H), 2.36 (s, 3H); 13C-NMR (MeOD-d4) δ 143.7, 142.9, 142.4, 141.8, 138.8, 136.3, 132.7, 131.0, 130.0, 127.1, 118.1, 99.3, 54.6, 21.5; HRMS [M–OTs].+ Calc’d. for C11H9N3SI: 341.9562, Found: 341.9559.
Syntheses of diaryliodonium halides
Diaryliodonium chlorides, bromides and iodides were obtained from the corresponding diaryliodonium tosylates by anion metathesis with sat. NH4Cl (aq), sat. KBr (aq), and sat. KI (aq), respectively, as explified for 9.
4-Azidophenyl(4’-methoxyphenyl)iodonium chloride (9)
4-Azidophenyl(4’-methoxyphenyl)iodonium tosylate (3, 0.20 g, 0.38 mmol) was dissolved in MeCN/H2O (H2O 10 vol.- %, 2 mL) at r.t. and heated to 50 °C. The mixture became a clear solution and was stirred for a further 5 min at 50 °C. Sat. NH4Cl (aq) was added dropwise. The reaction mixture was slowly cooled to r.t. over 30 min. A white solid precipitated out, and this was filtered off, washed with ice-cold water (2 mL) and Et2O (20 mL), and further dried in vacuo for 3 h to give 9 (0.13 g, 84%). mp = 180–182 °C; 1H-NMR (DMF-d7) δ 8.21 (d, J = 8.8 Hz, 2H), 8.14 (d, J = 9.2 Hz, 2H), 7.21 (d, J = 8.8 Hz, 2H), 7.05 (d, J = 9.2 Hz, 2H), 3.86 (s, 3H); 13C-NMR (DMF-d7) δ 162.6, 143.1, 137.0, 136.6, 122.0, 117.3, 117.2, 111.7, 55.7; HRMS [M–Cl].+ Calc’d. for C13H11N3OI: 351.9947, Found: 351.9949.
The following diaryliodonium halides were prepared similarly.
3-Azidophenyl(4’-methoxyphenyl)iodonium chloride (10)
White solid (0.11 g, 91%). mp = 182–184 °C; 1H-NMR (DMSO-d6) δ 8.12 (dd, J = 2.4, 7.2 Hz, 2H), 7.98 (t, J = 2 Hz, 1H), 7.02 (dt, J = 0.8, 5.6 Hz, 1H), 7.47 (t, J = 8.4 Hz, 1H), 7.33–7.30 (m, 1H), 7.03 (dd, J = 2, 6.8 Hz, 2H), 3.79 (s, 3H); 13C-NMR (DMSO-d6) δ 161.5, 141.6, 136.9, 132.3, 130.7, 124.9, 121.8, 120.6, 117.1, 108.8, 55.6; HRMS [M–Cl].+ Calc’d. for C14H13N3OI: 351.9947, Found: 351.9957.
[4-(Azidomethyl)phenyl](4’-methoxyphenyl)iodonium chloride (11)
White solid (89 mg, 76%). mp = 163–165 °C; 1H-NMR (DMSO-d6) δ 8.11 (dd, J = 7.6, 15.2 Hz, 4H), 7.44 (d, J = 7.6 Hz, 2H), 7.02 (d, J = 8 Hz, 2H), 4.52 (s, 2H), 3.78 (s, 3H); 13C-NMR (DMSO-d6) δ 161.5, 139.2, 136.8, 134.9, 130.8, 119.0, 117.1, 108.5, 55.6, 52.7; HRMS [M–Cl].+ Calc’d. for C14H13N3OI: 366.0103, Found: 366.0106.
[4-(Azidomethyl)phenyl](4’-methoxyphenyl)iodonium bromide (12)
White solid (0.10 g, 77%). mp = 176–177 °C; 1H-NMR (DMSO-d6) δ 8.15 (dd, J = 8, 12.4 Hz, 4H), 7.46 (d, J = 8 Hz, 2H), 7.04 (d, J = 8.4 Hz, 2H), 4.53 (s, 2H), 3.79 (s, 3H); 13C-NMR (DMSO-d6) δ 161.7, 139.5, 137.0, 135.0, 131.0, 117.9, 117.2, 107.3, 55.6, 52.3; HRMS [M–I].+ Calc’d. for C14H13N3OI: 366.0103, Found: 366.0104.
[4-(Azidomethyl)phenyl](4’-methoxyphenyl)iodonium iodide (13)
White solid (0.15 g, 92%). mp = 152–153 °C; 1H-NMR (DMSO-d6) δ 8.17 (dd, J = 8, 10.8 Hz, 4H), 7.48 (d, J = 8 Hz, 2H), 7.06 (d, J = 8.8 Hz, 2H), 4.54 (s, 2H), 3.79 (s, 3H); 13C-NMR (DMSO-d6) δ 161.8, 139.7, 137.1, 135.1, 131.1, 117.3, 117.0, 106.3, 55.7, 52.6; HRMS [M–I].+ Calc’d. for C14H13N3OI: 366.0103, Found: 366.0105.
[3-(Azidomethyl)phenyl](4’-methoxyphenyl)iodonium chloride (14)
White solid (0.13 g, 83%). mp = 177–179 °C; 1H-NMR (DMF-d7) δ 8.27 (s, 1 H), 8.19-8.15 (m, 3H), 7.63 (d, J = 7.6 Hz, 1H), 8.14 (t, J = 8 Hz, 1H), 7.05 (d, J = 9.2 Hz, 2H), 4.60 (s, 2H), 3.85 (s, 3H); 13C-NMR (DMF-d7) δ 162.6, 139.8, 137.1, 134.4, 134.3, 131.7, 131.0, 122.4, 117.3, 110.9, 55.7, 53.5; HRMS [M–Cl].+ Calc’d. for C14H13N3IO: 366.0103, Found: 366.0107.
Radiochemistry
Dry radiofluorination
Cyclotron-produced NCA [18F]fluoride ion (3.7–7.4 GBq) in [18O]water (250–400 μL) was adsorbed onto a QMA anionic resin cartridge within a CE module of a NanoTek apparatus (Advion; Louisville, TN), and then released with a solution of K2CO3 (0.8 mg; 5 nmol) plus K 2.2.2 (4.5 mg; 11 nmol) in MeCN-H2O (9: 1 v/v; 450 μL) into a 2-mL V-vial. The solution was dried by two successive cycles of azeotropic evaporation with acetonitrile (0.6 mL) under nitrogen flow at 95 °C. Dried 18F−-K 2.2.2-K+ complex (3.7–5.5 GBq) was dissolved in reaction solvent (typically, DMF or MeCN; 260 μL). A solution of diaryliodonium salt (10 mM) was prepared in matching solvent. Each solution (260 μL) was loaded into a separate storage loop of the microfluidic apparatus. (The detailed configuration of the microfluidic system has been described in our earlier publication).[14] For radiofluorination reaction, each solution (10–20 μL) was infused simultaneously into the micro-reactor (4-m coiled glass silica tube; internal diameter, 100 μm; internal volume, 31.4 μL) of the apparatus at a set flow rate in the range 8–20 μL/min and at a pre-set temperature. The micro-reactor output was directly quenched with MeCN-H2O (1: 1 v/v; 3 mL). The RCYs of products were measured by reverse phase radio-HPLC on a Luna C18 column (250 × 4.6 mm i.d., 10 μm; Phenomenex, Torrance, CA) eluted at 1.75 mL/min with a gradient of MeCN-H2O (60: 40, v/v) with the percentage of MeCN increased linearly from 60 to 90% over 7 min. Radioactive products were identified by their comobility with reference fluoro compounds. These chromatographically determined RCYs were corrected for adsorption of radioactivity in the apparatus, to give RCYs from the total [18F]fluoride ion originally introduced into the micro-reactor. Amounts of precursor, temperature and flow rates were varied to obtain high RCYs.
Wet radiofluorination
Cyclotron-produced no-carrier-added [18F]fluoride ion (3.7–7.4 MBq) in [18O]water (200 μL) from the cyclotron target was mixed with DMF (200 μL) and K 2.2.2 (3 mg)-K2CO3 (0.5 mg) solution in aq. MeCN (5% vol.-%, 100 μL). The solution was left to stand for 5 min at r.t., and loaded into a storage loop of the microfluidic apparatus. Reactions were then implemented and analyzed as for dry radiofluorination reactions.
Supplementary Material
Acknowledgments
We thank the Intramural Research Program of the National Institutes of Health (NIMH) for the support of this research and are grateful to NIH Clinical PET department (Chief, Dr. P. Herscovitch) for production of fluorine-18.
Footnotes
Supporting Information (see footnote on the first page of this article): Details of the syntheses of reference azido compounds. Copies of the 1H, and 13C NMR spectroscopic data for all prepared compounds and sample radiochromatograms.
References
- 1.Cai L, Lu S, Pike VW. Eur. J. Org. Chem. 2008;178:2853–2873. [Google Scholar]
- 2.a) Ruth TJ, Wolf AP. Radiochim. Acta. 1979;26:21–24. [Google Scholar]; b) Guillaume M, Luxen A, Nebeling B, Argentini M, Clark JC, Pike VW. Appl. Radiat. Isot. 1991;42:749–762. [Google Scholar]; c) Qaim SM, Clark JC, Crouzel C, Guillaume M, Helmeke HJ, Nebeling B, Pike VW, Stöcklin G. In: Radiopharmaceuticals for Positron Emission Tomography. Stöcklin G, Pike VW, editors. Kluwer Academic Publishers; Dordrecht, Netherlands: 1993. pp. 1–43. [Google Scholar]
- 3.a) Huisgen R. In: 1,3-Dipolar Cycloaddition Chemistry. Padwa A, editor. Wiley; New York: 1984. [Google Scholar]; b) Kolb HC, Finn MG, Sharpless KB. Angew. Chem. 2001;113:2056–2075. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; c) Kolb HC, Sharpless KB. Drug Discovery Today. 2003;8:1128–1137. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]; d) Mamidyala SK, Finn MG. Chem. Soc. Rev. 2010;39:1252–1261. doi: 10.1039/b901969n. [DOI] [PubMed] [Google Scholar]
- 4.a) Miller PM, Long NJ, Vilar R, Gee AD. Angew. Chem. 2008;120:9136–9172. doi: 10.1002/anie.200800222. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2008;47:8998–9033. doi: 10.1002/anie.200800222. [DOI] [PubMed] [Google Scholar]; b) Glaser M, Robbins EG, Label J. Compds. Radiopharm. 2009;52:407–414. [Google Scholar]; c) Mamat C, Ramenda T, Wuest FR. Mini-Rev. Org. Chem. 2009;6:21–34. [Google Scholar]; d) Wängler C, Schirrmacher R, Bartenstein P, Wängler B. Curr. Med. Chem. 2010;17:1092–1116. doi: 10.2174/092986710790820615. [DOI] [PubMed] [Google Scholar]; e) Nwe K, Brechbiel MW. Cancer Biotherapy Radiopharmaceut. 2009;24:289–302. doi: 10.1089/cbr.2008.0626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Glaser M, Arstadt E. Bioconj. Chem. 2007;18:989–993. doi: 10.1021/bc060301j. [DOI] [PubMed] [Google Scholar]; b) Sirion U, Kim HJ, Lee JH, Seo JW, Lee BS, Lee SJ, Oh SJ, Chi DY. Tetrahedron Lett. 2007;48:3953–3957. [Google Scholar]
- 6.Kuboyama T, Nakahara M, Yoshimo M, Cui Y, Sako T, Wada Y, Imanishi T, Obika S, Watanabe Y, Suzuki M, Doi H. Bioorg. Med. Chem. 2011;19:249–255. doi: 10.1016/j.bmc.2010.11.033. [DOI] [PubMed] [Google Scholar]
- 7.Magata Y, Lang L, Kiesewetter DO, Jagoda EM, Channing MA, Eckelman WC. Nucl. Med. Biol. 2000;27:163–168. doi: 10.1016/s0969-8051(99)00108-0. [DOI] [PubMed] [Google Scholar]
- 8.Hashizume K, Hashimoto N, Miyake Y. J. Org. Chem. 1995;60:6681–6681. [Google Scholar]
- 9.Thonon D, Kech C, Paris J, Lemaire C, Luxen A. Bioconjugate Chem. 2009;20:817–823. doi: 10.1021/bc800544p. [DOI] [PubMed] [Google Scholar]
- 10.Campbell-Verdyn LS, Mirfeizi L, Schoonen AK, Dierckx RA, Elsinga PH, Feringa BL. Angew. Chem. 2011;123:11313–11316. doi: 10.1002/anie.201105547. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2011;50:11117–11120. doi: 10.1002/anie.201105547. [DOI] [PubMed] [Google Scholar]
- 11.Shiraishi T, Kitamura Y, Ueno Y, Kitade Y. Chem. Commun. 2011;47:2691–2693. doi: 10.1039/c0cc04979d. [DOI] [PubMed] [Google Scholar]
- 12.Lemaire C, Libert L, Plenevaux A, Aerts J, Franci X, Luxen A. J. Fluorine Chem. 2012;138:48–55. [Google Scholar]
- 13.a) W. Pike V, I. Aigbirhio F. J. Chem. Soc. Chem. Commun. 1995:2215–2216. [Google Scholar]; b) Shah A, Pike VW, Widdowson DA. J. Chem. Soc., Perkin. Trans 1. 1998:2043–2046. [Google Scholar]; c) Ross TL, Ermert T, Hocke C, Coenen HH. J. Am. Chem. Soc. 2007;129:8018–8025. doi: 10.1021/ja066850h. [DOI] [PubMed] [Google Scholar]
- 14.Chun J-H, Lu SY, Lee Y-S, Pike VW. J. Org. Chem. 2010;75:3332–3338. doi: 10.1021/jo100361d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chun J-H, Lu SY, Pike VW. Eur. J. Org. Chem. 2011;23:4439–4447. doi: 10.1002/ejoc.201100382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a) Zhang MR, Kumata K, Suzuki K. Tetrahedron. Lett. 2007;48:8632–8635. [Google Scholar]; b) Telu S, Chun J-H, Siméon FG, Lu S, Pike VW. Org. Biomol. Chem. 2011;9:6629–6638. doi: 10.1039/c1ob05555k. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lee BC, Kim JS, Kim BS, Son JY, Hong SK, Park HS, Moon BS, Jung JH, Jeong JM, Kim SE. Bioorg. Med. Chem. 2011;19:2980–2990. doi: 10.1016/j.bmc.2011.03.029. [DOI] [PubMed] [Google Scholar]
- 17.Chun J-H, Pike VW. J. Org. Chem. 2012;77:1931–1938. doi: 10.1021/jo202517v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamamoto Y, Togo H. Synlett. 2005:2486–2488. [Google Scholar]
- 19.Brodack JW, Kilbourn MR, Welch MJ, Katzenellenbogen JA. Appl. Radiat. Isot. 1986;37:217–221. doi: 10.1016/0883-2889(86)90174-7. [DOI] [PubMed] [Google Scholar]
- 20.Carroll MA, Nairne J, Smith G, Widdowson DA. J. Fluorine Chem. 2007;128:127–132. [Google Scholar]
- 21.Qu W, Kung M-P, Hou C, Oya S, Kung HF. J. Med. Chem. 2007;50:3380–3387. doi: 10.1021/jm070467l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keicher T, Löbbecke S, Banert K. In: Organic Azides: Syntheses and Applications. Bräse S, editor. Wiley; Chichester: 2010. pp. 3–27. Ch.1. [Google Scholar]
- 23.Mosher HS, Turner L, Carlsmith A. Org. Synth. Coll. 1963;4:828. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.