

Abstract
Toxoplasma gondii, an intracellular protozoan parasite, is a major cause of opportunistic infectious disease affecting the brain and has been linked to an increased incidence of schizophrenia. In murine hosts, infection with T. gondii stimulates tryptophan degradation along the kynurenine pathway (KP), which contains several neuroactive metabolites, including 3-hydroxykynurenine (3-HK), quinolinic acid (QUIN) and kynurenic acid (KYNA). As these endogenous compounds may provide a mechanistic connection between T. gondii and the pathophysiology of schizophrenia, we measured KP metabolites in both brain and periphery of T. gondii-treated C57BL/6 mice 8 and 28 days post-infection. Infected mice showed early decreases in the levels of tryptophan in brain and serum, but not in the liver. These reductions were associated with elevated levels of kynurenine, KYNA, 3-HK and QUIN in the brain. In quantitative terms, the most significant increases in these KP metabolites were observed in the brain at 28 days post-infection. Notably, the anti-parasitic drugs pyrimethamine and sulfadiazine, a standard treatment of toxoplasmosis, significantly reduced 3-HK and KYNA levels in the brain of infected mice when applied between 28 and 56 days post-infection. In summary, T. gondii infection, probably by activating microglia and astrocytes, enhances the production of KP metabolites in the brain. However, during the first two months after infection, the KP changes in these mice do not reliably duplicate abnormalities seen in the brain of individuals with schizophrenia.
Keywords: Astrocytes, 3-Hydroxykynurenine, Kynurenic acid, Microglia, Quinolinic acid
1. Introduction
Toxoplasma gondii, an obligate intracellular protozoan parasite, infects approximately one-third of the human population worldwide (Desmonts and Couvreur, 1974; Hofhuis et al., 2011). Initial transmission occurs orally, and the parasite then starts its cycle of replication and dissemination with the tachyzoite stage (Dubey et al., 1998). During acute infection, the parasite is mainly present in peripheral tissues and blood, but also has access to the brain via circulating cells (Courret et al., 2006; Da Gama et al., 2004). One early feature of T. gondii-induced brain infection is the activation of glial cells, in particular astrocytes (Wilson and Hunter, 2004) and microglia (Hermes et al., 2008). The next stage is defined by the presence of bradyzoite tissue cysts in skeletal muscle and, most notably, in the brain. These dormant cysts are characteristic of chronic, latent infection (Dubey et al., 1998). In individuals suffering from acquired immune deficiencies, e.g. AIDS, or undergoing prolonged immune suppressive treatments, reactivation of the infection can lead to lethal toxoplasmic encephalitis (Dubey et al., 1998).
Latent toxoplasmosis, although frequently dismissed as asymptomatic and clinically unimportant, alters host behavior in both humans and rodents (Burkinshaw et al., 1953; Flegr, 2007; Kannan and Pletnikov, 2012; Webster et al., 2006; Wilson et al., 1980). In addition and possibly related, a growing number of epidemiological studies associate persistent T. gondii infection with an increased likelihood of developing schizophrenia (Dickerson et al., 2007; Torrey et al., 2012). Notably, elevated anti-T. gondii IgG antibody levels have been reported in patients with first-onset schizophrenia, suggesting an involvement of the parasite in the etiology of the disease (Torrey et al., 2007; Wang et al., 2006).
In immunocompetent hosts, infection with T. gondii leads to the production of interferon-γ (IFN-γ) and, consequently, the induction of indoleamine 2,3-dioxygenase (IDO), which converts the essential amino acid tryptophan to kynurenine and inhibits T. gondii growth in vitro (Dai et al., 1994; Däubener and MacKenzie, 1999) and in vivo (Silva et al., 2002). Kynurenine, in turn, is further degraded via a metabolic cascade - the kynurenine pathway (KP) -, which contains several neuroactive metabolites (“kynurenines”), such as 3-hydroxykynurenine (3-HK), a free radical generator, quinolinic acid (QUIN), an agonist of N-methyl-D-aspartate (NMDA) receptor, and kynurenic acid (KYNA), an endogenous antagonist of α7 nicotinic acetylcholine and NMDA receptors (Fig. 1). These compounds play distinct roles in brain physiology and have also been linked to the etiology of schizophrenia, as well as other major brain diseases (see Schwarcz et al., 2012, for review).
Fig. 1.

The kynurenine pathway of tryptophan degradation.
As KP metabolites in the brain are synthesized primarily in microglial cells and astrocytes (Espey et al., 1997; Guillemin et al., 2001; Heyes et al., 1996), glial activation that occurs during T. gondii infection may affect KP metabolism and thus provide a mechanistic link to the pathophysiology of schizophrenia (Schwarcz and Hunter, 2007). In susceptible mice, increased IDO mRNA expression after T. gondii infection is accompanied by elevations in the concentration of kynurenine in plasma, peripheral organs and brain (Engin et al., 2012; Fujigaki et al., 2002; Silva et al., 2002), and the presence of the parasite, IDO expression and kynurenine content in the brain were shown to peak approximately one month after the infection (Fujigaki et al., 2002; Silva et al., 2002). In these studies, no information was provided with regard to the fate of neuroactive downstream kynurenines, namely 3-HK, QUIN and KYNA, in either periphery or brain. We therefore designed experiments to fill this void and report here that substantial changes in KP metabolism take place in T. gondii-infected C57BL/6 mice over time. These changes turned out to be qualitatively and quantitatively distinct, and did not occur simultaneously, in the periphery and the brain. In addition, we tested the effect of a standard treatment of toxoplasmosis, the combination of pyrimethamine and sulfadiazine, on KP metabolism.
2. Materials and methods
2.1 Chemicals
L-Tryptophan, DL-3-hydroxykynurenine (3-HK), kynurenic acid (KYNA), quinolinic acid (QUIN), pentafluoropropionic anhydride, 2,2,3,3,3-pentafluoro-1-propanol, [2H6]L-kynurenine were purchased from Sigma-Aldrich (St. Louis, MO, USA). L-Kynurenine sulfate (“kynurenine”; purity: 99.4%) was obtained from Sai Advantium (Hyderabad, India). [2H3]Quinolinic acid was purchased from Synfine Research (Richmond Hill, Ontario, Canada), and [2H5]L-tryptophan was obtained from CDN Isotopes (Pointe-Claire, Quebec, Canada). Sulfadiazine and pyrimethamine were obtained from the Harvard Park Pharmacy.
All other chemicals were obtained from various commercial suppliers and were of the highest available purity.
2.2 Mice and infection
Female C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME, USA) were used in all experiments. All procedures were performed in accordance with the guidelines of the University of Pennsylvania Institutional Animal Care and Use Committee.
Mice (n=4-5 per group) were infected i.p. with 20 cysts of Me49 strain T. gondii at 8 weeks of age. The animals were euthanized by CO2 asphyxiation at various time points post-infection. Brain (minus cerebellum), liver and blood were rapidly harvested and placed on ice. Blood was allowed to clot at 4°C, and the supernatant serum was collected. All samples were then frozen and stored at −80°C until analysis.
2.3 Drug treatment
Twenty-eight days after T. gondii infection, one group of infected mice was treated orally with a combination of pyrimethamine (4 mg/kg) and sulfadiazine (100 mg/kg) daily for one month. A control group of infected mice received no drug treatment. A separate group of uninfected, naive mice was subjected to the drug treatment to evaluate possible effects on KP metabolism. All animals were euthanized 56 days after the infection, and their tissues was removed and stored as described above.
2.4 Histology
For histological studies, mice were euthanized, and the dissected brains were placed into 4% formalin. Saggital sections were H&E stained. For immunohistochemical studies, frozen sections were stained with rabbit anti-GFAP (Dako, Carpinteria, CA, USA) and anti-MHC Class II (eBioscience, Inc., San Diego, CA, USA) antibodies. Goat anti-rabbit Alexa 488 (Life Technologies, Grand Island, NY, USA) and goat-anti rat Cy3 (Jackson Immunoresearch, West Grove, PA, USA) were used as secondary antibodies. DAPI (Life Technologies) staining was used to highlight nuclei.
2.5 Real-time PCR
To measure parasite burden, real-time PCR was used as previously described (Wilson et al., 2005). Briefly, DNA was purified from 300 μL of whole brain homogenate using a High-Pure PCR template preparation kit (Roche). Real-time PCR specific for T. gondii was performed with a 2X SYBR green master mix (Applied Biosystems, Warrington UK) on an ABI 7500 Fast Real-time System using 500 ng of purified DNA per sample. The amount of T. gondii DNA in each sample was determined using a standard curve.
2.6 KYNA measurement
Tissues were weighed while frozen and then homogenized (1:5, w/v for brain, 1:50, w/v for liver) by sonication in ultrapure water. Serum was diluted 1:10 (v/v) in ultrapure water. KYNA was measured by HPLC as previously described (Pocivavsek et al., 2012).
2.7 Tryptophan, kynurenine, 3-HK and QUIN determination
Tryptophan, kynurenine, 3-HK and QUIN were quantified by GC/MS/MS. Briefly, the brain and liver homogenates used for the determination of KYNA were further diluted (1:4, v/v) in 0.1 % ascorbic acid in water (w/v), and serum was diluted 1:10 (v/v). Fifty μL of a solution containing internal standards ([2H ]quinolinic acid, [2 3 H6]L-kynurenine and [2H5]L-tryptophan) were added to 50 μL of the respective sample, and proteins were precipitated with 50 μL of acetone. After centrifugation (13,700 × g, 5 min), 50 μL of methanol:chloroform (20:50) were added to the supernatant, and the samples were centrifuged (13 700 × g, 10 min). The upper layer was added to a glass tube and evaporated to dryness (90 min). The samples were then derivatized with 120 μL of 2,2,3,3,3-pentafluoro-1-propanol and 130 μL of pentafluoropropionic anhydride at 75°C for 30 min, dried down again and reconstituted in 50 μL of ethyl acetate. One μL was injected into the gas chromatograph. GC/MS analysis was carried out with a 7890A GC coupled to a 7000B MS/MS (Agilent Technologies, Santa Clara, CA, USA), using electron capture negative chemical ionization (Notarangelo et al., 2012).
For kynurenine measurement in plasma, 50 μL of 6% perchloric acid were thoroughly mixed with 100 μL of plasma, and the precipitated proteins were removed by centrifugation (16 000 × g, 15 min). Kynurenine was measured by HPLC as described (Pocivavsek et al., 2012).
2.8 Statistical analysis
Results are expressed as the mean ± SEM. Student’s t-test was used to determine significance (p<0.05) in the drug treatment experiments. One-way ANOVA followed by Bonferroni’s post-hoc test was used in all other experiments.
3. Results
3.1 Tryptophan and KP metabolite levels in the periphery
Following i.p. injection, T. gondii infection is characterized by an acute phase, affecting mainly the periphery, and a chronic phase, which is specific to muscle and brain (Dubey et al., 1998). To investigate the effect of T. gondii on KP metabolism over time, we first determined the levels of KP metabolites 8 (acute phase) and 28 days (chronic phase) post-infection in the serum (Fig. 2) and liver (Table 1).
Fig. 2.

Total tryptophan and KP metabolite levels in the serum of T. gondii-infected mice. Analyses were performed as described in Materials and methods. Data are the mean ± SEM of 4-5 animals per experimental group. *p<0.05 and ***p<0.001 vs. uninfected controls (“Day 0”) (one-way Anova with Bonferroni’s post-hoc analysis).
Table 1.
KP metabolite levels in the liver of T. gondii-treated mice 8 and 28 days post-infection.
| Metabolite/Day | 0 | 8 | 28 |
|---|---|---|---|
| Tryptophan (pmoles/mg tissue) |
88.2 ± 3.1 | 82.6 ± 1.5 | 95.5 ± 4.9 |
|
Kynurenine (pmoles/mg tissue) |
4.0 ± 0.2 | 5.8 ± 0.3* | 3.7 ± 0.4 |
|
KYNA (fmoles/mg tissue) |
178.8 ± 44.0 | 93.0 ± 9.8 | 91.6 ± 3.9 |
|
3-HK (fmoles/mg tissue) |
100.2 ± 6.7 | 80.5 ± 4.6 | 101.4 ± 14.1 |
|
QUIN (pmoles/mg tissue) |
4.2 ± 0.1 | 3.4 ± 0.1 | 2.9 ± 0.2* |
Analyses were performed as described in Materials and methods. Data are the mean ± SEM of 4-5 animals per experimental group.
p<0.05 vs. uninfected controls (“Day 0”) (one-way Anova with Bonferroni’s post-hoc analysis).
Compared to non-infected controls, significant reductions in serum tryptophan levels were observed at 8 days (p<0.001) and 28 days (p<0.05) post-infection. Serum kynurenine and QUIN levels were elevated at 8 days (both p<0.001 vs. controls), but not at 28 days (both p>0.05 vs. controls). Notably, no changes in serum KYNA and 3-HK levels were measured in infected animals at any time interval assessed in the study.
With the exception of a 45% increase in kynurenine levels after 8 days (p<0.05 vs. controls), no noteworthy changes were observed in the liver at 8 or 28 days following an infection with T. gondii. Thus, the effects on KP metabolites in the periphery were mostly resolved by 28 days post-infection.
3.2 Tryptophan and kynurenines in the brain
We next determined the effect of parasite and phase of the infection on KP metabolism in the brain (Fig. 3). Eight days after a systemic T. gondii injection, at a time point when parasites are just starting to invade the CNS, tryptophan levels in the brain were decreased by 40% compared to non-infected controls (p<0.01). In contrast, we observed a significant elevation in brain kynurenine levels (p<0.05) and a tendency towards increased KYNA, 3-HK and QUIN levels in the same tissues. At 28 days post-infection, when mice have active toxoplasmic encephalitis, brain levels of tryptophan had returned to control levels (p>0.05), whereas the brain content of kynurenine, KYNA, 3-HK and QUIN was dramatically increased (25-, 50-, 50- and 50-fold, respectively, compared to control levels; all p<0.001).
Fig. 3.

Increased levels of KP metabolites in the brain of T. gondii-infected mice. Analyses were performed as described in Materials and methods. Data are the mean ± SEM of 4-5 animals per experimental group. *p<0.05, **p<0.01 and ***p<0.001 vs. uninfected controls (“Day 0”) (one-way Anova with Bonferroni’s post-hoc analysis).
3.3 Microscopic analysis
In the brain, KP metabolites are synthesized in microglia and astrocytes (Schwarcz et al., 2012). We therefore evaluated glial activation by immunocytochemistry at 28 days post-infection, i.e. at a time when major glial changes could be expected to parallel the remarkable elevation in KP metabolite levels. Antibodies directed against GFAP and MHC class II were used to visualize of astrocytes and microglia, respectively (different cell types express MHC II but microglial cells were identified by morphology). In line with previous studies (Hermes et al., 2008; Wilson and Hunter, 2004), mice showed pronounced increases in the numbers of both astrocytes and microglia during this phase of T. gondii infection (Fig. 4).
Fig. 4.
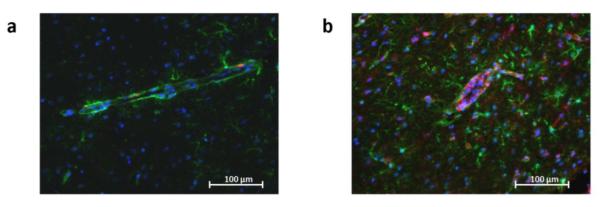
T. gondii infection increases the number of reactive astrocytes and microglial cells in the brain. Representative brain sections from uninfected (a) and infected (b) mice 28 days post-infection were stained with antibodies against GFAP (green) and class II MHC (red). Nuclei were stained with DAPI (blue). Sections are from the frontal cortex (n=5 per group).
3.4 Effects of drug treatment
In a separate group of animals, we evaluated the effect of pyrimethamine/sulfadiazine, a drug combination frequently used to treat toxoplasmosis (Antinori et al., 1992; Fung and Kirschenbaum, 1996; Leport et al., 1988), on the brain of infected mice. To this end, one group of T. gondii-infected mice received a 4 week-drug treatment beginning at 28 days post-infection (i.e. at a timepoint that showed very high cerebral KP metabolite levels and glial activation). All animals were killed 56 days post-infection. As structural and chemical changes in this phase of the infection are by far most prominent in cerebral tissue (see above), we confined our analyses to the brain of these animals.
Drug treatment caused a trend toward reduction in parasite burden (Fig. 5a). Histological analysis revealed reduced inflammatory infiltrates and astrocyte activation in the treated group compared to the untreated group of infected mice (Fig. 5b). Drug treatment significantly affected KP metabolite levels in the brain of chronically infected mice (Fig. 5c). KYNA and 3-HK levels were significantly decreased in the treated group compared to untreated infected mice (both p<0.001). No effects on KP metabolite levels were seen in age-matched, uninfected mice that were treated with the drugs for 4 weeks (data not shown).
Fig. 5.

Treatment of T. gondii-infected mice with pyrimethamine/sulfadiazine reduces KP metabolite levels, inflammatory infiltrates and parasite burden. Infected mice were treated for 4 weeks with a combination of pyrimethamine/sulfadiazine, starting at 28 days post-infection (see Materials and methods for details). a) Real-time PCR specific for T. gondii DNA as measure of parasite burden in the brain; b) H & E staining (top panel) and GFAP staining (bottom panel) of brain sections of treated and untreated T. gondii-infected mice; c) KYNA and 3-HK brain levels in T. gondii-infected mice with (treated group) or without (untreated group) treatment. ***p<0.001 vs. untreated group (Student’s t-test).
4. Discussion
Prompted by our long-standing interest in the possible role of neuroactive kynurenines in the pathophysiology of schizophrenia (Wonodi and Schwarcz, 2010), and the proposed role of T. gondii in the etiology of the disease (Torrey et al., 2012), the present study was designed to investigate the effects of T. gondii on KP metabolism in both brain and periphery at various times post-infection. To this end, we infected T. gondii-susceptible mice with the low virulence Me-49 strain of the parasite and measured the levels of tryptophan and several of its catabolic products in brain, liver and serum 8 and 28 days later. Infected mice showed early decreases in the levels of tryptophan in brain and serum, but not in the liver. These reductions were associated with elevated levels of kynurenine, KYNA, 3-HK and QUIN. In quantitative terms, by far the greatest increases in the levels of these metabolites were observed in the brain at 28 days post-infection. Finally, we demonstrated that a drug regimen used for the treatment of toxoplasmosis significantly affected parasite burden, inflammation and KP metabolite levels in the brain.
Our results confirmed the previously reported rapid reduction in serum tryptophan levels in mice within a few days after a T. gondii infection (Engin et al., 2012; Fujigaki et al., 2002; Silva et al., 2002). Here, we also observed a significant decrease in brain tryptophan levels after 8 days, though this effect was only reported as a trend in an earlier study (Fujigaki et al., 2002). Consistently, however, we and other investigators showed a substantial elevation in serum kynurenine levels during the first 1-2 weeks following infection but, notably, not after 4 weeks (Fujigaki et al., 2002; Silva et al., 2002). This time course was in stark contrast to the T. gondii-induced elevation of kynurenine seen in the brain.
Although an increase in IDO may explain most of the infection-related changes in tryptophan and kynurenine levels (Fujigaki et al., 2002; Taylor and Feng, 1991; see Silva et al., 2002, for discussion), analysis of downstream metabolic events suggest a more complex picture. Thus, serum levels of 3-HK and KYNA in infected mice remained consistently at control values, even when the level of their common precursor, kynurenine, was elevated. This, as well as the increase in serum QUIN levels 8 days after the infection, indicates that kynurenine is preferentially degraded to 3-HK and further to its downstream metabolite QUIN in the acute phase of the infection when the parasite is mainly present in peripheral tissues and blood (Chiarugi et al., 2001; Dubey et al., 1997).
In marked contrast to the periphery, the brain content of KYNA, 3-HK and QUIN was dramatically increased 4 weeks after infection with T. gondii, i.e. at a time when the parasite is largely controlled in the periphery (Vyas et al., 2007). This chronic phase is characterized by the presence of numerous cysts and inflammation in the brain, with activation of astrocytes and microglia (Hermes et al., 2008; Wilson and Hunter, 2004), as confirmed here by histological means. As these glial cells produce IDO and the other enzymes that are responsible for the cerebral biosynthesis of neuroactive kynurenines, and as the formation of these metabolites in the brain is known to be enhanced after immune stimulation (see Schwarcz et al., 2012 for review), it is likely that synthesis within the brain accounted for the observed changes in brain KP metabolism at this stage after infection.
Interestingly, 3-HK and KYNA levels were still elevated in the brain 56 days post-infection. However, the increases were less pronounced than at 28 days, suggesting that metabolite levels may gradually return to normal values over time. Administration of pyrimethamine and sulfadiazine, a drug combination used to treat toxoplasmosis (Antinori et al., 1992; Fung and Kirschenbaum, 1996; Leport et al., 1988), decreased the brain content of the KP metabolites further, to levels close to endogenous concentrations. This is likely a result of direct drug effects on the parasite and an associated reduction in inflammatory infiltrates and glial activation. Experiments currently in progress are designed to evaluate the status of KP metabolism in infected mice beyond the two-month period and to investigate the consequences of drug intervention during various phases of infection.
Stimulation of KP metabolism following T. gondii infection may be part of a biological defense strategy against T. gondii infections (Montoya and Liesenfeld, 2004). Activation of IDO (and possibly also of tryptophan 2,3-dioxygenase; Schmidt et al., 2009) has antimicrobial and anti-proliferative effects (MacKenzie et al., 2007) and suppresses adverse immune reactivity (Bauer et al., 2005; Frumento et al., 2002). These effects could be mediated by activation of the aryl hydrocarbon receptor, which is a target of both kynurenine and KYNA (DiNatale et al., 2010; Mezrich et al., 2010), or a variety of other receptors of KYNA, 3-HK and QUIN (Moroni et al., 2012; Stone et al., 2013).
Inadvertently, T. gondii-induced impairment of the KP may also be involved in the etiology of schizophrenia. Thus, epidemiological and serological studies have demonstrated an association between toxoplasmosis and an increased incidence of this disease (Nascimento et al., 2012; Torrey et al., 2007; Torrey et al., 2012; Wang et al., 2006). Of interest in this regard, antipsychotic drugs inhibit parasite replication in vitro (Jones-Brando et al., 2003) and reverse some behavioral abnormalities in T. gondii-infected rats (Webster et al., 2006). As KP abnormalities, including increases in kynurenine and KYNA levels, are seen in brain and cerebrospinal fluid of individuals with schizophrenia (see Erhardt et al., 2009 and Wonodi and Schwarcz, 2010, for reviews), we had previously proposed that an in-depth investigation of KP metabolism in mice infected with T. gondii may uncover relevant molecular clues regarding the pathophysiology of the disease (Schwarcz and Hunter, 2007).
Although the underlying concept connecting T. gondii infection, abnormal KP metabolism and schizophrenia remains conceptually intriguing, the present study indicates that the KP changes observed in the brain of infected C57BL/6 mice during a relatively short period (i.e. 28 or 56 days post-infection) do not accurately duplicate the abnormalities seen in the brain of individuals with schizophrenia (Sathyasaikumar et al., 2011; Schwarcz et al., 2001). However, longer chronic infections in mouse strains more resistant to the parasite, for example BALB/c mice (Johnson, 1984; Lu et al., 2005), may better mimic the situation in humans, where a T. gondii infection is mostly asymptomatic (Montoya and Liesenfeld, 2004). Other experimental strategies could involve the infection of mutant mice with defined KP abnormalities reminiscent of schizophrenia (Thomas et al., 2012) or the use of other immune stimulants that have been linked to schizophrenia (Brown and Derkits, 2010). Together with more detailed analyses of the effects of T. gondii infection on KP metabolism in humans, we expect these studies to eventually allow a rigorous evaluation of the hypothetical link between T. gondii infection, KP metabolism and schizophrenia.
Acknowledgments
Role of funding source This work was in part supported by USPHS grant MH083729.
Abbreviations
- IDO
indoleamine 2,3-dioxygenase
- KYNA
Kynurenic acid
- 3-HK
3-hydroxykynurenine
- QUIN
quinolinic acid
- T. gondii
Toxoplasma gondii
Footnotes
Conflict of interest The authors declare that they have no conflict of interest
Contributors Conception of study – FMN, EHW, THH, CAH, RS
Data collection – FMN, KJH, MART, THH, QF
Interpretation of results and manuscript drafting – FMN, EHW, THH
Review of manuscript – CAH, RS
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Antinori A, Ammassari A, Maiuro G, Camilli G, Damiano F, Federico G, Pizzigallo E, Tamburrini E. Comparison of two medications in central nervous system toxoplasmosis in patients with AIDS. Ital. J. Neurol. Sci. 1992;13(6):475–479. doi: 10.1007/BF02230867. [DOI] [PubMed] [Google Scholar]
- Bauer TM, Jiga LP, Chuang JJ, Randazzo M, Opelz G, Terness P. Studying the immunosuppressive role of indoleamine 2,3-dioxygenase: tryptophan metabolites suppress rat allogeneic T-cell responses in vitro and in vivo. Transpl. Int. 2005;18(1):95–100. doi: 10.1111/j.1432-2277.2004.00031.x. [DOI] [PubMed] [Google Scholar]
- Brown AS, Derkits EJ. Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am J Psychiatry. 2010;167(3):261–280. doi: 10.1176/appi.ajp.2009.09030361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkinshaw J, Kirman BH, Sorsby A. Toxoplasmosis in relation to mental deficiency. Br. Med. J. 1953;1(4812):702–704. doi: 10.1136/bmj.1.4812.702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarugi A, Calvani M, Meli E, Traggiai E, Moroni F. Synthesis and release of neurotoxic kynurenine metabolites by human monocyte-derived macrophages. J. Neuroimmunol. 2001;120(1-2):190–198. doi: 10.1016/s0165-5728(01)00418-0. [DOI] [PubMed] [Google Scholar]
- Courret N, Darche S, Sonigo P, Milon G, Buzoni-Gatel D, Tardieux I. CD11c- and CD11b-expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain. Blood. 2006;107(1):309–316. doi: 10.1182/blood-2005-02-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Gama LM, Ribeiro-Gomes FL, Guimaraes U, Jr., Arnholdt AC. Reduction in adhesiveness to extracellular matrix components, modulation of adhesion molecules and in vivo migration of murine macrophages infected with Toxoplasma gondii. Microbes Infect. 2004;6(14):1287–1296. doi: 10.1016/j.micinf.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Dai W, Pan H, Kwok O, Dubey JP. Human indoleamine 2,3-dioxygenase inhibits Toxoplasma gondii growth in fibroblast cells. J. Interferon Res. 1994;14(6):313–317. doi: 10.1089/jir.1994.14.313. [DOI] [PubMed] [Google Scholar]
- Däubener W, MacKenzie CR. IFN-gamma activated indoleamine 2,3-dioxygenase activity in human cells is an antiparasitic and an antibacterial effector mechanism. Adv. Exp. Med. Biol. 1999;467:517–524. doi: 10.1007/978-1-4615-4709-9_64. [DOI] [PubMed] [Google Scholar]
- Desmonts G, Couvreur J. Congenital toxoplasmosis. A prospective study of 378 pregnancies. N. Engl. J. Med. 1974;290(20):1110–1116. doi: 10.1056/NEJM197405162902003. [DOI] [PubMed] [Google Scholar]
- Dickerson F, Boronow J, Stallings C, Origoni A, Yolken R. Toxoplasma gondii in individuals with schizophrenia: association with clinical and demographic factors and with mortality. Schizophr. Bull. 2007;33(3):737–740. doi: 10.1093/schbul/sbm005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNatale BC, Murray IA, Schroeder JC, Flaveny CA, Lahoti TS, Laurenzana EM, Omiecinski CJ, Perdew GH. Kynurenic acid is a potent endogenous aryl hydrocarbon receptor ligand that synergistically induces interleukin-6 in the presence of inflammatory signaling. Toxicol. Sci. 2010;115(1):89–97. doi: 10.1093/toxsci/kfq024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey JP, Lindsay DS, Speer CA. Structures of Toxoplasma gondii tachyzoites, bradyzoites, and sporozoites and biology and development of tissue cysts. Clin. Microbiol. Rev. 1998;11(2):267–299. doi: 10.1128/cmr.11.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey JP, Speer CA, Shen SK, Kwok OC, Blixt JA. Oocyst-induced murine toxoplasmosis: life cycle, pathogenicity, and stage conversion in mice fed Toxoplasma gondii oocysts. J. Parasitol. 1997;83(5):870–882. [PubMed] [Google Scholar]
- Engin AB, Dogruman-Al F, Ercin U, Celebi B, Babur C, Bukan N. Oxidative stress and tryptophan degradation pattern of acute Toxoplasma gondii infection in mice. Parasitol. Res. 2012;111(4):1725–1730. doi: 10.1007/s00436-012-3015-6. [DOI] [PubMed] [Google Scholar]
- Erhardt S, Olsson SK, Engberg G. Pharmacological manipulation of kynurenic acid: potential in the treatment of psychiatric disorders. CNS Drugs. 2009;23(2):91–101. doi: 10.2165/00023210-200923020-00001. [DOI] [PubMed] [Google Scholar]
- Espey MG, Chernyshev ON, Reinhard JF, Jr., Namboodiri MA, Colton CA. Activated human microglia produce the excitotoxin quinolinic acid. Neuroreport. 1997;8(2):431–434. doi: 10.1097/00001756-199701200-00011. [DOI] [PubMed] [Google Scholar]
- Flegr J. Effects of toxoplasma on human behavior. Schizophr. Bull. 2007;33(3):757–760. doi: 10.1093/schbul/sbl074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frumento G, Rotondo R, Tonetti M, Damonte G, Benatti U, Ferrara GB. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J. Exp. Med. 2002;196(4):459–468. doi: 10.1084/jem.20020121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujigaki S, Saito K, Takemura M, Maekawa N, Yamada Y, Wada H, Seishima M. L-tryptophan-L-kynurenine pathway metabolism accelerated by Toxoplasma gondii infection is abolished in gamma interferon-gene-deficient mice: cross-regulation between inducible nitric oxide synthase and indoleamine-2,3-dioxygenase. Infect. Immun. 2002;70(2):779–786. doi: 10.1128/iai.70.2.779-786.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung HB, Kirschenbaum HL. Treatment regimens for patients with toxoplasmic encephalitis. Clin. Ther. 1996;18(6):1037–1056. doi: 10.1016/s0149-2918(96)80059-2. discussion 1036. [DOI] [PubMed] [Google Scholar]
- Guillemin GJ, Kerr SJ, Smythe GA, Smith DG, Kapoor V, Armati PJ, Croitoru J, Brew BJ. Kynurenine pathway metabolism in human astrocytes: a paradox for neuronal protection. J. Neurochem. 2001;78(4):842–853. doi: 10.1046/j.1471-4159.2001.00498.x. [DOI] [PubMed] [Google Scholar]
- Hermes G, Ajioka JW, Kelly KA, Mui E, Roberts F, Kasza K, Mayr T, Kirisits MJ, Wollmann R, Ferguson DJ, Roberts CW, Hwang JH, Trendler T, Kennan RP, Suzuki Y, Reardon C, Hickey WF, Chen L, McLeod R. Neurological and behavioral abnormalities, ventricular dilatation, altered cellular functions, inflammation, and neuronal injury in brains of mice due to common, persistent, parasitic infection. J. Neuroinflammation. 2008;5:48. doi: 10.1186/1742-2094-5-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyes MP, Achim CL, Wiley CA, Major EO, Saito K, Markey SP. Human microglia convert l-tryptophan into the neurotoxin quinolinic acid. Biochem. J. 1996;320(Pt 2):595–597. doi: 10.1042/bj3200595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofhuis A, van Pelt W, van Duynhoven YT, Nijhuis CD, Mollema L, van der Klis FR, Havelaar AH, Kortbeek LM. Decreased prevalence and age-specific risk factors for Toxoplasma gondii IgG antibodies in The Netherlands between 1995/1996 and 2006/2007. Epidemiol. Infect. 2011;139(4):530–538. doi: 10.1017/S0950268810001044. [DOI] [PubMed] [Google Scholar]
- Johnson AM. Strain-dependent, route of challenge-dependent, murine susceptibility to toxoplasmosis. Z. Parasitenkd. 1984;70(3):303–309. doi: 10.1007/BF00927816. [DOI] [PubMed] [Google Scholar]
- Jones-Brando L, Torrey EF, Yolken R. Drugs used in the treatment of schizophrenia and bipolar disorder inhibit the replication of Toxoplasma gondii. Schizophr. Res. 2003;62(3):237–244. doi: 10.1016/s0920-9964(02)00357-2. [DOI] [PubMed] [Google Scholar]
- Kannan G, Pletnikov MV. Toxoplasma gondii and cognitive deficits in schizophrenia: an animal model perspective. Schizophr. Bull. 2012;38(6):1155–1161. doi: 10.1093/schbul/sbs079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leport C, Raffi F, Matheron S, Katlama C, Regnier B, Saimot AG, Marche C, Vedrenne C, Vilde JL. Treatment of central nervous system toxoplasmosis with pyrimethamine/sulfadiazine combination in 35 patients with the acquired immunodeficiency syndrome. Efficacy of long-term continuous therapy. Am. J. Med. 1988;84(1):94–100. doi: 10.1016/0002-9343(88)90014-9. [DOI] [PubMed] [Google Scholar]
- Lu F, Huang S, Hu MS, Kasper LH. Experimental ocular toxoplasmosis in genetically susceptible and resistant mice. Infect. Immun. 2005;73(8):5160–5165. doi: 10.1128/IAI.73.8.5160-5165.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie CR, Heseler K, Muller A, Däubener W. Role of indoleamine 2,3-dioxygenase in antimicrobial defence and immuno-regulation: tryptophan depletion versus production of toxic kynurenines. Curr. Drug Metab. 2007;8(3):237–244. doi: 10.2174/138920007780362518. [DOI] [PubMed] [Google Scholar]
- Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 2010;185(6):3190–3198. doi: 10.4049/jimmunol.0903670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montoya JG, Liesenfeld O. Toxoplasmosis. Lancet. 2004;363(9425):1965–1976. doi: 10.1016/S0140-6736(04)16412-X. [DOI] [PubMed] [Google Scholar]
- Moroni F, Cozzi A, Sili M, Mannaioni G. Kynurenic acid: a metabolite with multiple actions and multiple targets in brain and periphery. J. Neural Transm. 2012;119(2):133–139. doi: 10.1007/s00702-011-0763-x. [DOI] [PubMed] [Google Scholar]
- Nascimento FS, de Rosalmeida Dantas C, Netto MP, Mella LF, Suzuki LA, Banzato CE, Rossi CL. Prevalence of antibodies to Toxoplasma gondii in patients with schizophrenia and mood disorders. Schizophr. Res. 2012;142(1-3):244–245. doi: 10.1016/j.schres.2012.08.036. [DOI] [PubMed] [Google Scholar]
- Pocivavsek A, Wu QH, Elmer GI, Bruno JP, Schwarcz R. Pre- and postnatal exposure to kynurenine causes cognitive deficits in adulthood. Eur. J. Neurosci. 2012;35(10):1605–1612. doi: 10.1111/j.1460-9568.2012.08064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathyasaikumar KV, Stachowski EK, Wonodi I, Roberts RC, Rassoulpour A, McMahon RP, Schwarcz R. Impaired kynurenine pathway metabolism in the prefrontal cortex of individuals with schizophrenia. Schizophr. Bull. 2011;37(6):1147–1156. doi: 10.1093/schbul/sbq112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt SK, Muller A, Heseler K, Woite C, Spekker K, MacKenzie CR, Daubener W. Antimicrobial and immunoregulatory properties of human tryptophan 2,3-dioxygenase. Eur. J. Immunol. 2009;39(10):2755–2764. doi: 10.1002/eji.200939535. [DOI] [PubMed] [Google Scholar]
- Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ. Kynurenines in the mammalian brain: when physiology meets pathology. Nat. Rev. Neurosci. 2012;13(7):465–477. doi: 10.1038/nrn3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarcz R, Hunter CA. Toxoplasma gondii and schizophrenia: linkage through astrocyte-derived kynurenic acid? Schizophr. Bull. 2007;33(3):652–653. doi: 10.1093/schbul/sbm030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarcz R, Rassoulpour A, Wu HQ, Medoff D, Tamminga CA, Roberts RC. Increased cortical kynurenate content in schizophrenia. Biol. Psychiatry. 2001;50(7):521–530. doi: 10.1016/s0006-3223(01)01078-2. [DOI] [PubMed] [Google Scholar]
- Silva NM, Rodrigues CV, Santoro MM, Reis LF, Alvarez-Leite JI, Gazzinelli RT. Expression of indoleamine 2,3-dioxygenase, tryptophan degradation, and kynurenine formation during in vivo infection with Toxoplasma gondii: induction by endogenous gamma interferon and requirement of interferon regulatory factor 1. Infect. Immun. 2002;70(2):859–868. doi: 10.1128/iai.70.2.859-868.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone TW, Stoy N, Darlington LG. An expanding range of targets for kynurenine metabolites of tryptophan. Trends Pharmacol. Sci. 2013;34(2):136–143. doi: 10.1016/j.tips.2012.09.006. [DOI] [PubMed] [Google Scholar]
- Taylor MW, Feng GS. Relationship between interferon-gamma, indoleamine 2,3-dioxygenase, and tryptophan catabolism. FASEB J. 1991;5(11):2516–2522. [PubMed] [Google Scholar]
- Thomas M, Pocivavsek A, Sathyasaikumar KV, Elmer GI, Giorgini F, Muchowski PJ, Schwarcz R. Behavioral abnormalities in mice deficient in kynurenine 3-monooxygenase: Relevance to schizophrenia. Soc. Neurosci. Abstr. 2012;27:663.29. [Google Scholar]
- Torrey EF, Bartko JJ, Lun ZR, Yolken RH. Antibodies to Toxoplasma gondii in patients with schizophrenia: a meta-analysis. Schizophr. Bull. 2007;33(3):729–736. doi: 10.1093/schbul/sbl050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrey EF, Bartko JJ, Yolken RH. Toxoplasma gondii and other risk factors for schizophrenia: an update. Schizophr. Bull. 2012;38(3):642–647. doi: 10.1093/schbul/sbs043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas A, Kim SK, Giacomini N, Boothroyd JC, Sapolsky RM. Behavioral changes induced by Toxoplasma infection of rodents are highly specific to aversion of cat odors. Proc. Natl. Acad. Sci. U S A. 2007;104(15):6442–6447. doi: 10.1073/pnas.0608310104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HL, Wang GH, Li QY, Shu C, Jiang MS, Guo Y. Prevalence of Toxoplasma infection in first-episode schizophrenia and comparison between Toxoplasmaseropositive and Toxoplasma-seronegative schizophrenia. Acta Psychiatr. Scand. 2006;114(1):40–48. doi: 10.1111/j.1600-0447.2006.00780.x. [DOI] [PubMed] [Google Scholar]
- Webster JP, Lamberton PH, Donnelly CA, Torrey EF. Parasites as causative agents of human affective disorders? The impact of anti-psychotic, mood-stabilizer and anti-parasite medication on Toxoplasma gondii’s ability to alter host behaviour. Proc. Biol. Sci. 2006;273(1589):1023–1030. doi: 10.1098/rspb.2005.3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CB, Remington JS, Stagno S, Reynolds DW. Development of adverse sequelae in children born with subclinical congenital Toxoplasma infection. Pediatrics. 1980;66(5):767–774. [PubMed] [Google Scholar]
- Wilson EH, Hunter CA. The role of astrocytes in the immunopathogenesis of toxoplasmic encephalitis. Int. J. Parasitol. 2004;34(5):543–548. doi: 10.1016/j.ijpara.2003.12.010. [DOI] [PubMed] [Google Scholar]
- Wilson EH, Wille-Reece U, Dzierszinski F, Hunter CA. A critical role for IL-10 in limiting inflammation during toxoplasmic encephalitis. J. Neuroimmunol. 2005;165(1-2):63–74. doi: 10.1016/j.jneuroim.2005.04.018. [DOI] [PubMed] [Google Scholar]
- Wonodi I, Schwarcz R. Cortical kynurenine pathway metabolism: a novel target for cognitive enhancement in Schizophrenia. Schizophr. Bull. 2010;36(2):211–218. doi: 10.1093/schbul/sbq002. [DOI] [PMC free article] [PubMed] [Google Scholar]