

Abstract
Technological development led to an increased interest in systems biological approaches to characterize disease mechanisms and candidate genes relevant to specific diseases. We suggested that the human peripheral blood mononuclear cells (PBMC) network can be delineated by cellular reconstruction to guide identification of candidate genes. Based on 285 microarrays (7370 genes) from 98 heart transplant patients enrolled in the Cardiac Allograft Rejection Gene Expression Observational study, we used an information-theoretic, reverse-engineering algorithm called ARACNe (algorithm for the reconstruction of accurate cellular networks) and chromatin immunoprecipitation assay to reconstruct and validate a putative gene PBMC interaction network. We focused our analysis on transcription factor (TF) genes and developed a priority score to incorporate aspects of network dynamics and information from published literature to supervise gene discovery. ARACNe generated a cellular network and predicted interactions for each TF during rejection and quiescence. Genes ranked highest by priority score included those related to apoptosis, humoural and cellular immune response such as GA binding protein transcription factor (GABP), nuclear factor of κ light polypeptide gene enhancer in B-cells (NFκB), Fas (TNFRSF6)-associated via death domain (FADD) and c-AMP response element binding protein. We used the TF CREB to validate our network. ARACNe predicted 29 putative first-neighbour genes of CREB. Eleven of these (37%) were previously reported. Out of the 18 unknown predicted interactions, 14 primers were identified and 11 could be immunoprecipitated (78.6%). Overall, 75% (n= 22) inferred CREB targets were validated, a significantly higher fraction than randomly expected (P < 0.001, Fisher’s exact test). Our results confirm the accuracy of ARACNe to reconstruct the PBMC transcriptional network and show the utility of systems biological approaches to identify possible molecular targets and biomarkers.
Keywords: systems biology, cellular networks, cardiac transplant, rejection, gene expression, candidate gene
Introduction
Heart transplantation is complicated by different forms of cardiac allograft rejection [1]. The immune response against a transplanted heart is a complex and heterogeneous process mediated by various cellular pathways that depend on factors related to the recipient, the donor, ischemia-reperfusion injury and to the violation of the natural human barriers by the surgical procedure and the donor–recipient interaction [2]. To deal with this immune response, transplant physicians use a combination of drugs to impair the immune system in recognizing, eliciting and sustaining a response [3]. Three major forms of rejection have been recognized [1]. It is common to identify more than one type of rejection in the same biopsy specimen [1, 4]. Moreover, there are forms of graft dysfunction, which are assumed to be episodes of rejection, but for which no specific mechanism can be identified [5]. Despite this, therapy of these episodes follows classic approaches to rejection; in some cases, the clinical response is favourable.
A systems biological approach to the acute rejection process involves a hypothesis that the response against the transplanted graft is the result of a constellation of events mediated by various cellular pathways. These may polarize into a manifestation at the organ and system levels with a complexity related to the processes that act together. We identify these processes as modules with a specific size and a specific relationship between the modules or joint configuration. We envision that the rejection process, usually referred to as a single entity (i.e. acute cellular rejection or antibody mediated rejection), is the result of combinations of modular structures with different sizes and spatial configurations.
This hypothesis proposes that there is not a specific form of rejection but a constellation of biological processes that promote the development of a specific phenotype, which emerges from the complexity of the underlying biology. Questions that need to be asked are: (1) Do those modules exist? (2) Can we characterize them? (3) Do they have a specific plasticity that emerges as a phenotypic condition?
The answers to these questions are most likely not going to be resolved in the short term, and certainly not by us, but at least these questions can be explored by taking advantage of evolving technologies. Such answers, albeit still partial, have already proven useful as guides for therapeutic intervention [6]. They are expected to become drivers in the development of new personalized, diagnostic and therapeutic strategies [7].
The publication of the human genome sequence opened an avenue to understanding, in an unprecedented way, the acute rejection process by the application of high-throughput technologies [8]. Microarray expression profiles provide the capacity to study the activity of thousands of functionally characterized and uncharacterized genes; their use has advanced the field of transplantation [9]. Many of the limitations, challenges and solutions of studies related to gene expression profiles have been addressed [10–12].
Approaches involving systems biology can be used to observe, through quantitative measures, multiple components simultaneously. By rigorous data integration with mathematical models, as opposed to the study of single aspects of the system under limited conditions, the technique can be used to identify ‘emergent properties’. A systems biological approach suggests that dysregulation of modules in the regulatory network of the cell is ultimately responsible for the differential expression patterns of individual gene transcripts and of their protein products associated with the clinical phenotype. The property on the organ level is ‘emergent’ from the pattern of regulation on the molecular and cellular levels. By examining the joint behaviour of a set of related genes, coherent changes can be detected, even in cases where the expression of individual genes is not significantly different [13]. Systems approaches have been used in life and social sciences [14–18]. By use of a variety of data, including microarray expression profiles, they are being increasingly applied to the dissection of molecular pathways that are active or deregulated in specific cellular contexts [19]. In this report, we describe a retrospective study, involving technologies currently available [10, 19, 20], that allowed us to identify and characterize specific genes and modules associated with the problem of rejection using a systems biological approach.
Materials and methods
This project involved retrospective analysis of gene expression data generated by a large multicenter study, entitled the ‘Cardiac Allograft Rejection Gene Expression Observational’ (CARGO) Study. This study used gene microarrays as one of the steps to develop a molecular classifier [9]. Results of the CARGO study have been published elsewhere. In this retrospective analysis, we used gene expression analysis and a cellular reconstruction method to identify and characterize genes and gene modules that are components of the acute cellular rejection process. A summary of the steps followed in this study is shown in Table 1 and Described below.
Table 1.
Sequence in the reconstruction and analysis of the network
| Step | Procedure |
|---|---|
| 1 | Build a phenotype independent network |
| 2 | Focus on biologically relevant genes |
| 3 | Build exploratory phenotype specific networks |
| 4 | Compute priority score |
| 5 | Generate gene list & choose candidate TF |
| 6 | Validate results |
Patients
Patients in this study were selected from the CARGO study database. After heart transplantation, all patients were routinely evaluated with endomyocardial biopsies (EMBs) at specific time intervals or at the time of an adverse event. Samples of peripheral blood mononuclear cells (PBMC) were obtained each time a patient had a biopsy for microarray analysis. Results of PBMC microarrays were used to correlate with pathologic findings and for purposes of gene discovery. A total of 98 patients provided 285 PBMC samples that were used for microarray analysis. For the study population, donor and recipient characteristics were equivalent to those reported by the United Network for Organ Sharing for 2003 and have been described elsewhere [9].
Endomyocardial biopsy
EMBs performed by standard techniques were graded by local pathologists and by three independent ‘central’ pathologists, blinded to clinical information, following the 1990 classification of the cardiac allograft EMB [21]. This classification assigns different degrees of rejection depending of the severity of an inflammatory infiltrate and other factors. For the present analysis, we divided the EMBs into two major groups, ‘quiescent’[International Society of Heart and Lung Transplantation (ISHLT) grade 0] or ‘rejection’ (ISHLT grades 1A or higher).
Blood samples
PBMCs were isolated from 8 ml of venous blood by use of density gradient centrifugation (Cell Preparation Tube, Becton-Dickinson, Franklin Lakes, NJ, USA). Samples were frozen in lysis buffer (RLT) (Qiagen, Valencia, CA, USA) within 2 hrs of phlebotomy. The integrity of the purified RNA was tested by a bioanalyser; only the samples demonstrating an adequate 28S-to-18S ratio were used for microarray hybridization.
Microarray expression profiling
We used a custom leucocyte-focused, 7370 gene microarray (TeleChem International, Inc., Sunnyvale, CA, USA) that was utilized in the initial phase of the multicenter CARGO study [9]. This microarray was developed by use of RNA sequences expressed in stimulated and resting human leucocytes (PCR Select, Clontech Laboratories, Inc., Mountain View, CA, USA) and from publicly available sequence databases. Any gene arrays showing significant systematic variations were eliminated. All resulting microarray data were utilized as submitted to the National Center for Biotechnology Information’s Gene Expression Omnibus [22] (GSE2445). Genes with more than 30% missing values were removed, leaving a set of 4688 genes to be analysed. Because the correlation metrics used in network generation cannot tolerate missing data, the remaining instances of missing data were imputed using the k-nearest neighbours algorithm as implemented in significance analysis of microarrays (http://www-stat-class.stanford.edu/sam) [23]. The resulting matrix was then imported for reverse engineering into a bioinformatics platform (geWorkbench, http://gforge.nci.nih.gov/frs/?group_id=78) for the cellular network reconstruction procedure.
Cellular network reconstruction
For inference of the cellular network, we used an algorithm called ARACNe (algorithm for the reconstruction of accurate cellular networks). Applications of this algorithm have been published by others [7, 19, 24], and the detailed description of the cellular network reconstruction process has been published elsewhere [20]. Briefly, ARACNe is an information-theoretic, reverse-engineering algorithm introduced for the whole-genome inference of human cellular networks [19]. ARACNe computes regeneration of cellular networks in two distinct steps. In the first step, candidate interactions are identified by calculating the mutual information (I[gi;gj]) for all gene–gene pairs, retaining only those pairs exceeding a mutual information threshold, I0, corresponding to the desired P-value, P0, for rejecting the null hypothesis that the two genes are statistically independent, i.e. I[gi;gj]†I0. In the second step, ARACNe eliminates most indirect interactions by use of the data-processing inequality [25] (DPI), a property of information theory. The DPI states that if genes g1 and g3 interact only through a third gene, g2, the following relationship holds: I[g1;g3]≤ min(I[g1;g2], I[g2;g3]). ARACNe proceeds by identifying each three-edge loop from the first step, removing all (g1–g3) interactions that satisfy the previous DPI formula. Ultimately, ARACNe imputes a genetic ‘consensus’ network, which requires further exploration and validation.
Network visualization
Graphical representation of the reconstructed cellular network was created in Cytoscape [26]. Cytoscape is an open source bioinformatics software platform for visualizing molecular interaction networks and biological pathways and for integrating these networks with annotations, gene expression profiles and other data relating to the state of cells (http://www.cytoscape.org).
Identification of genes to focus the initial study
After reconstructing the cellular network, the first challenge is to determine how to approach its study. One approach to find a starting point is to focus on genes with a central role in general biology [27, 28]. Among these, transcription factor (TF) genes, which are highly connected and regulate numerous processes within the cell, are relevant to multiple processes. Therefore, we chose to focus on TFs. To identify the TFs within the microarray, we used the gene ontology term ‘TF activity’ (GO:0003700) according to the Gene Ontology Consortium classification [29]. The next question was how these TFs are differentially regulated during rejection or quiescence. We used ARACNE to perform cellular network reconstruction during rejection and quiescence (because of small sample size only in an exploratory intention) and used this information for our algorithm, as described below.
Exploration of the cellular network
To identify a candidate group of genes to focus on in our initial study, we created an empirical score which we called the ‘priority score’. This score is a quantitative measure that summarizes properties of the reconstructed network and integrates published biological information underlying a specific gene as it relates to the process under study. This priority score is estimated as follows:
For a given gene, the overlap term[1] measures the differential connectivity across both phenotypes; the connectivity term[2] measures the connectivity degree (number of first neighbours or connected edges) and the plausibility term[3] measures biological plausibility in immune response based on published literature. All three factors are scaled between 0 and 1 and are multiplied by a weighting factor to produce a total priority score that is also scaled between 0 and 1. The weighting factors allow flexibility in determining the relevance of each of these factors. In the overlap term [1], i is the number of overlapping connections between the two phenotypes of quiescence and rejection; a is the number of total connections for the quiescence phenotype and b is the number of total connections for the rejection phenotype. We use the minimum of a and b to weight the overlap, i, in order to scale the whole factor between 0 and 1. We use the rejection and quiescence networks to estimate which TF has the greatest differential connectivity. This allows us to identify TFs whose network might serve as important genetic modules mediating rejection versus quiescence. Thus, this score is higher for TF genes that undergo a significant change in connectivity between the two states.
In the connectivity term [2], k is the connectivity degree of the gene of interest, found by summing the number of targets across phenotypes and subtracting the overlap, such that only unique targets are counted towards the score. kmax is the maximum connectivity degree to establish that a TF is highly connected.
In the plausibility term [3], l is measured by the amount of relevant literature for the gene as it relates to rejection and immune system activation compared to the maximum amount found among TFs being studied. As it is difficult to measure the success of search recall (retrieval), this factor can not easily be determined. In this case, we used the GeneCards interface to determine the number, l, of relevant publications associated with the TF and normalized this by lmax, a constant representing the maximum amount of literature required to establish that a TF has been well studied. GeneCards is an integrated database of human genes that automatically mines genomic, proteomic and transcriptomic information and is publicly available at http://www.genecards.org/index.shtml. Through this tool, we searched PubMed using available aliases and the additional search terms, ‘immune’, ‘immunity’ and ‘inflammation’.
Validation
We performed two steps of validation: in silico using previously published data and in vitro using the chromatin immunoprecipitation (ChIP) assay.
Validation in silico
For validation in silico, the necessary information was available as published data from identified genes searched through PubMed using each gene name and ‘chromatin immunoprecipitation’ as key words. Among the genes identified as candidates (see the ‘Results’ section), information was available for the c-AMP response element binding protein (CREB). Therefore, for validation, we selected this gene, which is included in a target gene database available at http://natural.salk.edu/CREB/[30]. Genes that have been identified as first neighbours of CREB were compared with our findings by use of the Fisher exact test. A P-value <0.01 was considered significant.
Validation in vitro
ChIP assays were accomplished for PBMCs obtained from buffy coats (New York City Blood Bank, New York, NY, USA) and subjected to cross-linking with formaldehyde [31]. Briefly, cells were lysed and sonicated to generate small fragments of genomic DNA. After preparation of the cellular extract and chromatin fragmentation, proteins together with cross-linked DNA were immunoprecipitated. Protein-DNA cross-links in the immunoprecipitated material were then reversed, and the DNA fragments were purified. For association of a specific genomic region in vivo, DNA fragments of this region should be enriched in the immunoprecipitate compared to other portions of the genome. The presence of the relevant genomic regions in the immunoprecipitate was determined by PCR amplification with specific primers from the region in question and reference region [24]. Primers were designed using the Transcriptional Regulatory Element Database (http://rulai.cshl.edu/cgi-bin/TRED/tred.cgi?process=home) by searching 4000 bp upstream and 250 bp downstream from the transcription initiation site for the CREB-binding site (TGACGTCA) using geWorkbench v1.0.4 (http://gforge.nci.nih.gov/frs/). For all targets, the binding site with the highest probability of matching the putative binding site sequence was chosen. The primers flanking most closely to the binding site were chosen by Primer3 (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi), with the product size between 150 and 250 bp.
Results
Reconstruction of the cellular network
Reconstruction of the cellular network was accomplished in two steps. In the first step, a consensus network (phenotype independent) was established. In the second step, a phenotype-specific network was reconstructed in an exploratory intention. This network was constructed to predict independently the possible interactions of the genes as they relate to each phenotype. Following this procedure, ARACNe reconstructed the network using all microarray 4688 genes retained for the analysis, independent of the biology of the genes. For each gene, we obtained a list of ‘first-neighbour’ genes that, based on this algorithm, were predicted to interact directly with each gene. This network generation process is resource intense. It incorporates limited information about the biology of the gene and the behaviour of the gene in the presence or absence of rejection. Nevertheless, it reflects the so-called putative consensus or average network found for the combination of all the gene expression profiles, which we validated to assess its accuracy (see below). After assessing the reproducibility of the generated network, we explored it further.
Reconstruction of the TF network
We first focused on the group of genes with a high biological relevance and studied their surrounding networks. To do so, we first focused on the TF genes which we identified by the gene ontology classification. We recovered the cellular network for each TF across the quiescent and rejection samples in an exploratory intention. Focusing on the TFs is an intuitive first step toward two goals, identifying relevant genes that govern major cellular processes and reducing the complexity of the initial problem. ARACNe reconstructed hubs for 152 TFs, following the procedure summarized in Table 1. This allowed application of the DPI only to the subset of interactions in which all three interactions are transcriptional in nature, thus avoiding the elimination of transcriptional interactions by pairs of same-complex proteins, which typically have a highly correlated expression profile. In parent studies of the present analysis, this approach increased the statistical significance of the validated-target enrichment [20].
Differential TF networks
TF candidates which most likely represent key hubs in alloimmunity were identified by deriving separate networks for rejection and quiescence in an exploratory intention, ranking TFs for differential connectivity, and applying the priority score using the ‘quiescent’ (ISHLT grade 0 rejection) or ‘rejection’ (ISHLT grade ≥ 1A/1R) samples. This allowed the exploratory generation of separate ARACNe networks for each phenotype. The priority score was calculated based on these results. The priority score was applied to rank the TFs listed in Table S1 according to their biological role in rejection and quiescence. The results of the highest 10% scoring genes are given in Table 2 and the complete list of genes and scores in Table S2.
Table 2.
Highest ranking TFs
| TF gene | N nodes in Q | N nodes in R | N nodes overlapping | N articles found | Hub degree | PS |
|---|---|---|---|---|---|---|
| GABPB2 | 354 | 203 | 105 | 219,114 | 452 | 0.829877 |
| RERE | 113 | 60 | 28 | 1036 | 145 | 0.798333 |
| NFKB1 | 369 | 214 | 126 | 6188 | 457 | 0.783399 |
| NR4A3 | 43 | 42 | 20 | 5333 | 65 | 0.776946 |
| STAT3 | 147 | 165 | 52 | 627 | 260 | 0.76502 |
| RELA | 80 | 75 | 17 | 540 | 138 | 0.758773 |
| MYC | 188 | 169 | 62 | 545 | 295 | 0.741747 |
| NR3C1 | 167 | 121 | 55 | 596 | 233 | 0.701433 |
| STAT5B | 48 | 36 | 4 | 245 | 80 | 0.682093 |
| CREB1 | 151 | 136 | 46 | 299 | 241 | 0.669258 |
| STAT5A | 102 | 124 | 28 | 244 | 198 | 0.667587 |
| SMAD3 | 270 | 192 | 71 | 224 | 391 | 0.663352 |
| PPARG | 90 | 64 | 34 | 690 | 120 | 0.661664 |
| IRF1 | 66 | 66 | 22 | 346 | 110 | 0.659133 |
| RARA | 121 | 94 | 26 | 170 | 189 | 0.642897 |
TF: transcription factor; Q: quiescence; R: rejection.
Prioritization of TFs using the scoring system
By its priority score, genes which ranked high included for example NFKB1, genes of the signal transducer and activator of transcription (STAT) family, human c-myc gene (MYC), peroxisome proliferator-activated receptor gamma (PPARG) and interferon regulatory factor (IRF). The TF CREB ranked 10th of 152 TFs and 5th by hub connectivity degree. We chose CREB to do further analysis and validation. The reason why we chose CREB, out of the candidate TFs ranking in the top 10%, for further study was the significant amount of information available in the literature to pursue our validation using ChIP-on-chip data. Also, its role in the process of cellular proliferation and the lack of information in the field of transplantation made CREB an interesting candidate. However, several other genes could have been chosen with similar justification. As further elaborated in the ‘Discussion’ section, there are, with regard to CREB’s role in rejection, unanswered questions that can be further investigated based on improved knowledge of the gene’s regulatory activity. Additional high-ranking targets from this list may be selected for future studies.
Validation in silico of predicted first neighbours
After CREB first-neighbour reconstruction by ARACNe, we conducted a two-tiered validation procedure to confirm that the predicted target genes of CREB were indeed target genes. In our strategy, we conducted a literature search for published validated targets using the CREB target gene database available [30] at http:((natural.salk.edu(CREB(containing ChIP-on-chip information. Eleven (38%) of the 29 predicted first neighbours were previously reported as CREB binding targets (P= 0.000001, Fisher’s exact test); 18 constituted novel predictions. Previously validated genes include cytokine inducible SH2-containing protein (CISH), topoisomerase (TOP)1, MAP/microtubule affinity-regulating kinase (MARK)3, SON DNA binding protein (SON), FADD, tumor susceptibility gene (TSG)101, zinc finger protein (ZNF)146, HSPC111 (Hypothetical Protein), MDS025 (Hypothetical Protein), GA binding protein transcription factor, β subunit (GABPB)2 and methyl-CpG binding domain protein (MDB)4 (Fig. 1, left).
Fig 1.
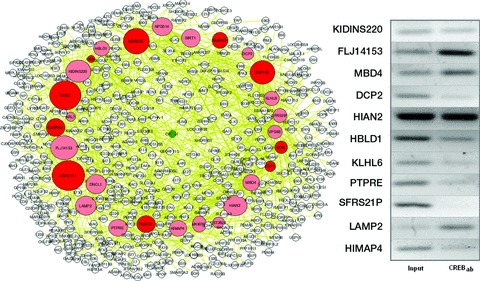
CREB First-/second-neighbour network. Left: A CREB-specific subnetwork was obtained by including all genes that have P < 1e-7 based on their pairwise mutual information with CREB. The CREB subnetwork includes 29 genes directly connected to CREB (first neighbours; represented by larger circles) and 491 genes connected through an intermediate (second neighbours). Red or pink nodes represent first-neighbour target genes for which ChIP data are available or not available, respectively; yellow and light yellow nodes represent second-neighbour target genes for which ChIP data are available or not available, respectively; CREB is shown in green; white nodes represent genes for which no CREB-related information is available. The complete list of genes, including gene symbol, ID, and LocusLink ID, is given in the Appendix. ChIP was accomplished for the CREB binding site (TGACGTCA) of 14 genes of the 18 non-validated putative CREB first neighbours. With 108 human leucocytes, 11 of 14 gene products were immunoprecipitated. Total chromatin before immunoprecipitation (input DNA) was used in a 1:1000 dilution as a positive control for PCR (negative controls not shown).
ChIP validation of predicted first neighbours that were not previously validated in silico
Additionally, we performed specific ChIP assays to validate the predicted targets that were not validated in silico. Of the 18 putative targets identified by ARACNe, we selected 14 genes whose promoter sequence could be retrieved from the TRE database and whose CREB-binding site (TGACGTCA) was identified within –4 kbp to +250 bp relative to the transcription initiation site for further biochemical validation by ChIP. Eleven of these 14 genes [MBD4, FLJ14153 (Hypothetical Protein), homolog of rat kinase D-interacting substance of 220 kDa (KIDINS220), decapping enzyme (DCP)2, human immune associated nucleotide (HIAN)2, HESB-like domain containing (HBLD)1, kelch-like 6 (Drosophila) (KLHL6), protein tyrosine phosphatase, receptor type E (PTPRE), splicing factor, arginine/serine-rich (SFRS)2IP, lysosomal-associated membrane protein (LAMP)2, HIMAP4 (immunity associated protein 2)] could be immunoprecipitated using a CREB antibody (sc-2027, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), showing that CREB binds to their promoter in vivo (Fig. 1, right). When CREB targets validated in silico and biochemically are considered together, 75% of ARACNe-inferred putative CREB targets (22 of 29) were validated, a higher fraction than predicted by chance (P < 0.0001, Fisher’s exact test). This confirmed the high accuracy of the ARACNe algorithm for the inference of human cellular networks.
Discussion
Knowledge of the complete sequences of genomes, together with technology that permits monitoring of information flow leading to specific cellular functions, set the stage for development of systems biology[14–18]. Accordingly, we have suggested that the complex regulation of the cardiac allograft rejection process can be better understood through application of a systems biology approach. Previous work involved data derived from a PBMC expression profile to generate a diagnostic test that would rule out acute cellular cardiac allograft rejection [9].
In this retrospective study, we demonstrate the feasibility of a systems biological approach, involving novel technologies and concepts, for evaluation of the rejection process. This approach involves (i) reconstruction of the cellular networks, (ii) a focus on relevant genes (such as TFs) likely to function in a regulatory network, (iii) application of an exploratory tool termed ‘priority score’, developed to inform the selection of candidate genes using information related to the characteristics of the network and published biology of the immune system and (iv) validation of the inferred consensus cellular network by use of previously published information [19, 30] and by ChIP assay [24].
We demonstrate that a putative PBMC regulatory network can be computationally inferred by use of a reverse engineering approach via a set of focused PBMC microarray expression profiles of heart transplant recipients obtained during both acute cellular cardiac allograft rejection and quiescence. Specifically, we aimed to develop a systematic strategy for reconstructing a cellular network from microarray analysis and to evaluate salient TF genes. From network and biological rationales, the priority score was developed as tool to analyse and integrate computational and biological information to narrow a list of candidate genes to a set of highly relevant genes. We combined information gathered from the computationally reconstructed network and published biological evidence, while allowing flexibility on the weight assigned to each variable to be applicable to different analytical approaches. To compute the priority score, we (i) analysed the connectivity (number of first neighbours) for each TF in the average network (connectivity term of the priority score), (ii) assessed the variation in the genes of the two subnetworks of rejection and quiescence (overlap term of the priority score) and (iii) incorporated a biological relevance criterion (plausibility term of the priority score) as it relates to rejection for each TF by assessing relevant literature for each TF using the GeneCards and PubMed interfaces. The priority score adequately identified TFs, such as GABP, NFKB and those of the STAT family, which have been extensively studied and related to the cellular immune response [32–34].
To validate the reconstructed network we used two procedures, in silico and in vitro. Although only 11 of the 29 genes (38%) inferred by ARACNe as target of CREB in the non-TF focused consensus network were previously validated by ChIP–chip data [30], additional 11 genes were validated in this study, bringing the total percent to 75%. This result reproduces the success rate of previous studies using this approach [19, 20] and confirms its value in inferring physical interaction among genes. In support of our results that ARACNe facilitates the prediction likelihood of hub genes of interest (75% validated putative CREB first neighbours) are the results of other groups [28]. Zhang et al., using a genome-wide approach to characterize target genes regulated by CREB, found 64 of 82 published genes (77%, P= 2 × 10−11) validated on the genome-level, suggesting that their used in silico methods were yielding biologically relevant sites. For a similar reason, we feel that the confirmation/validation of 75% of our ARACNe-generated putative CREB first-neighbour genes by the combination of in silico and in vitro methods is in the range of published standards. The benefit of using the ARACNe algorithm in conjunction with conventional strategies consists in a rapid and reliable identification of targets.
Our experiments answer, at least in part, our leading questions. First, we identified modules, in this case, each TF with their first neighbours (‘TF neighbourhood’). These modules are typically composed of genes which share similar functional characteristics or are biologically related to one another in such a way that they all participate in a specific coordinated response. Second, we identified properties of these modules which represent modulations of the activity of these genes in response to changes in the environmental context (i.e. rejection), inferred as directly interacting under one condition but not another.
Genes known to be involved in the pathology of rejection (such as GABP, NFKB and STAT) were identified as high scoring hub genes. The TF CREB which we elected to use for validation the computational inferred network, is a gene involved in cellular proliferation, a mechanism that is important in the immune response [35, 36]. Upon antigen presentation, T cells become activated, which implies release of calcium to the cytosol and activation, via c-AMP, of a pathway involving P56, protein kinase C (PKC), rat sarcoma (RAS), murine leukemia viral oncogene homolog (RAF)-1, Mitogen-activated protein kinase (MEK) and ribosomal S6 protein kinase 2 (RSK2) among others. The product of the activation of these pathways phosphorylates CREB which binds to CREB and travels into the nucleus as a complex. In the nucleus, it binds to CRE, inducing a transcriptional response leading to inhibition of c-jun, c-fos, Fra-2 and Fos B with consequent inhibition of apoptosis and promotion of T-cell proliferation [36, 37]. This mechanism has been proposed to support the role of CREB in regulating cellular immunity [37] but this has not been explored in the field of acute cellular cardiac allograft rejection.
Because of limitations with respect to of the sample material and sample size used in our study, these putative computational results could not be validated. More data generated by collaborative endeavours to collect the necessary events will be necessary to validate our findings [38]. This pilot study has additional limitations including its retrospective nature; the use of EMBs to classify acute cellular rejection, a procedure that has significant variability in diagnosis [39, 40]; the use of a focused, non-whole genome array; enriched for genes involved in leucocyte physiology and immune response; the approach is limited to analysing expression data with TF information (ignoring known pathway information from literature and other sources) and the heuristic nature of our TF priority score. Nevertheless, our findings are encouraging; they highlight our increasing ability to use the genome-wide reverse engineering of cellular networks in human cells to identify molecular targets of key genomic programs and to guide biomarker and molecular target discovery.
In summary, genomics and computational biology provide powerful tools for the reverse engineering of key regulatory hubs and modules that are candidates for phenotype-specific regulators. These can be further pursued as potential targets for designing novel molecular-pharmaceutical interventions for allograft rejection. For the identification of molecules that are important in the field of transplantation, our study is the first involving use of techniques that are being increasingly applied in the field of molecular biology.
Conflict of interest
The authors confirm that there are no conflicts of interest.
Supporting Information
List of transcription factors in the microarray (sorted alphabetically)
Table S2 Priority score for transcription factors.lmax = 1000, kmax = 500,w1 = 0.6, w2 = 0.3 andw3 = 0.1.
References
- 1.Stewart S, Winters GL, Fishbein MC, et al. Revision of the 1990 working formulation for the standardization of nomenclature in the diagnosis of heart rejection. J Heart Lung Transplant. 2005;24:1710–20. doi: 10.1016/j.healun.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 2.Hale DA. Basic transplantation immunology. Surg Clin North Am. 2006;86:1103–25. doi: 10.1016/j.suc.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 3.Kirklin JK, Young JB, McGiffin D. Heart transplantation. New York: Churchill Livingstone; 2002. [Google Scholar]
- 4.Rodriguez ER. The pathology of heart transplant biopsy specimens: revisiting the 1990 ISHLT working formulation. J Heart Lung Transplant. 2003;22:3–15. doi: 10.1016/s1053-2498(02)00575-2. [DOI] [PubMed] [Google Scholar]
- 5.Patel JK, Kobashigawa JA. Immunosuppression, diagnosis, and treatment of cardiac allograft rejection. Semin Thorac Cardiovasc Surg. 2004;16:378–85. doi: 10.1053/j.semtcvs.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 6.Downward J. Cancer biology: signatures guide drug choice. Nature. 2006;439:274–5. doi: 10.1038/439274a. [DOI] [PubMed] [Google Scholar]
- 7.Mani KM, Lefebvre C, Wang K, et al. A systems biology approach to prediction of oncogenes and molecular perturbation targets in B-cell lymphomas. Mol Syst Biol. 2008 doi: 10.1038/msb.2008.2. 10.1038/msb.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schena M, Shalon D, Davis RW, et al. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–70. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 9.Deng MC, Eisen HJ, Mehra MR, et al. Noninvasive discrimination of rejection in cardiac allograft recipients using gene expression profiling. Am J Transplant. 2006;6:150–60. doi: 10.1111/j.1600-6143.2005.01175.x. [DOI] [PubMed] [Google Scholar]
- 10.Simon R. Challenges of microarray data and the evaluation of gene expression profile signatures. Cancer Invest. 2008;26:327–32. doi: 10.1080/07357900801971032. [DOI] [PubMed] [Google Scholar]
- 11.Allison DB, Cui X, Page GP, et al. Microarray data analysis: from disarray to consolidation and consensus. Nat Rev Genet. 2006;7:55–65. doi: 10.1038/nrg1749. [DOI] [PubMed] [Google Scholar]
- 12.Ioannidis JP, Allison DB, Ball CA, et al. Repeatability of published microarray gene expression analyses. Nat Genet. 2009;41:149–55. doi: 10.1038/ng.295. [DOI] [PubMed] [Google Scholar]
- 13.Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–73. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 14.Bertalanffy Lv, LaViolette PA. A systems view of man. Boulder, Colo: Westview Press; 1981. [Google Scholar]
- 15.Oltvai ZN, Barabasi AL. Systems biology. Life’s complexity pyramid. Science. 2002;298:763–4. doi: 10.1126/science.1078563. [DOI] [PubMed] [Google Scholar]
- 16.Ideker T, Galitski T, Hood L. A new approach to decoding life: systems biology. Annu Rev Genomics Hum Genet. 2001;2:343–72. doi: 10.1146/annurev.genom.2.1.343. [DOI] [PubMed] [Google Scholar]
- 17.Hood L. Systems biology: integrating technology, biology, and computation. Mech Ageing Dev. 2003;124:9–16. doi: 10.1016/s0047-6374(02)00164-1. [DOI] [PubMed] [Google Scholar]
- 18.Hood L, Perlmutter RM. The impact of systems approaches on biological problems in drug discovery. Nat Biotechnol. 2004;22:1215–7. doi: 10.1038/nbt1004-1215. [DOI] [PubMed] [Google Scholar]
- 19.Basso K, Margolin AA, Stolovitzky G, et al. Reverse engineering of regulatory networks in human B cells. Nat Genet. 2005;37:382–90. doi: 10.1038/ng1532. [DOI] [PubMed] [Google Scholar]
- 20.Margolin AA, Nemenman I, Basso K, et al. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics. 2006;7:S7. doi: 10.1186/1471-2105-7-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Billingham ME, Cary NR, Hammond ME, et al. A working formulation for the standardization of nomenclature in the diagnosis of heart and lung rejection: Heart Rejection Study Group. The International Society for Heart Transplantation. J Heart Transplant. 1990;9:587–93. [PubMed] [Google Scholar]
- 22.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–10. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palomero T, Lim WK, Odom DT, et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc Natl Acad Sci USA. 2006;103:18261–6. doi: 10.1073/pnas.0606108103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han TS, Kobayashi K. Mathematics of information and coding. Providence, RI: American Mathematical Society; 2002. [Google Scholar]
- 26.Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jeong H, Mason SP, Barabasi AL, et al. Lethality and centrality in protein networks. Nature. 2001;411:41–2. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 28.Albert R, Jeong H, Barabasi AL. Error and attack tolerance of complex networks. Nature. 2000;406:378–82. doi: 10.1038/35019019. [DOI] [PubMed] [Google Scholar]
- 29.Creating the gene ontology resource: design and implementation. Genome Res. 2001;11:1425–33. doi: 10.1101/gr.180801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X, Odom DT, Koo SH, et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci USA. 2005;102:4459–64. doi: 10.1073/pnas.0501076102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weinmann AS. Novel ChIP-based strategies to uncover transcription factor target genes in the immune system. Nat Rev Immunol. 2004;4:381–6. doi: 10.1038/nri1353. [DOI] [PubMed] [Google Scholar]
- 32.Xue HH, Bollenbacher-Reilley J, Wu Z, et al. The transcription factor GABP is a critical regulator of B lymphocyte development. Immunity. 2007;26:421–31. doi: 10.1016/j.immuni.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 33.Goodnow CC. Pathways for self-tolerance and the treatment of autoimmune diseases. Lancet. 2001;357:2115–21. doi: 10.1016/s0140-6736(00)05185-0. [DOI] [PubMed] [Google Scholar]
- 34.Benczik M, Gaffen SL. The interleukin (IL)-2 family cytokines: survival and proliferation signaling pathways in T lymphocytes. Immunol Invest. 2004;33:109–42. doi: 10.1081/imm-120030732. [DOI] [PubMed] [Google Scholar]
- 35.Refojo D, Liberman AC, Giacomini D, et al. Integrating systemic information at the molecular level: cross-talk between steroid receptors and cytokine signaling on different target cells. Ann NY Acad Sci. 2003;992:196–204. doi: 10.1111/j.1749-6632.2003.tb03150.x. [DOI] [PubMed] [Google Scholar]
- 36.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 37.Kuo CT, Leiden JM. Transcriptional regulation of T lymphocyte development and function. Annu Rev Immunol. 1999;17:149–87. doi: 10.1146/annurev.immunol.17.1.149. [DOI] [PubMed] [Google Scholar]
- 38.Bansal M, Belcastro V, Ambesi-Impiombato A, di Bernardo D. How to infer gene networks from expression profiles. Mol Syst Biol. 2007;3:78. doi: 10.1038/msb4100120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marboe CC, Billingham M, Eisen H, et al. Nodular endocardial infiltrates (Quilty lesions) cause significant variability in diagnosis of ISHLT Grade 2 and 3A rejection in cardiac allograft recipients. J Heart Lung Transplant. 2005;24:S219–26. doi: 10.1016/j.healun.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 40.Crespo-Leiro MG, Schulz U, Vanhaecke J, et al. Inter-observer variability hi the interpretation of cardiac biopsies remains a challenge: results of the Cardiac Allograft Rejection Gene Expression Observational (CARGO) II Study. J Heart Lung Transplant. 2009;28:S230. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of transcription factors in the microarray (sorted alphabetically)
Table S2 Priority score for transcription factors.lmax = 1000, kmax = 500,w1 = 0.6, w2 = 0.3 andw3 = 0.1.