

Identification of the 5′ splice site by group II introns involves a base-pairing interaction between the exon-binding site EBS1 and the intron-binding site IBS1. This study presents an NMR structure of this interaction. This structure reveals that the EBS1•IBS1 interaction presents the scissile phosphate at the 5′ splice site in a position that is structurally unique.
Keywords: group II intron, splicing, EBS1•IBS1, Mg2+ binding, NMR
Abstract
A crucial step of the self-splicing reaction of group II intron ribozymes is the recognition of the 5′ exon by the intron. This recognition is achieved by two regions in domain 1 of the intron, the exon-binding sites EBS1 and EBS2 forming base pairs with the intron-binding sites IBS1 and IBS2 located at the end of the 5′ exon. The complementarity of the EBS1•IBS1 contact is most important for ensuring site-specific cleavage of the phosphodiester bond between the 5′ exon and the intron. Here, we present the NMR solution structures of the d3′ hairpin including EBS1 free in solution and bound to the IBS1 7-mer. In the unbound state, EBS1 is part of a flexible 11-nucleotide (nt) loop. Binding of IBS1 restructures and freezes the entire loop region. Mg2+ ions are bound near the termini of the EBS1•IBS1 helix, stabilizing the interaction. Formation of the 7-bp EBS1•IBS1 helix within a loop of only 11 nt forces the loop backbone to form a sharp turn opposite of the splice site, thereby presenting the scissile phosphate in a position that is structurally unique.
INTRODUCTION
Group II introns are ribozymes occurring in bacterial and organellar genomes that catalyze their own splicing from primary RNA transcripts (Peebles et al. 1986; Schmelzer and Schweyen 1986; van der Veen et al. 1986). By reversing the splicing reaction, group II introns may also invade DNA and RNA sites (Augustin et al. 1990; Mörl et al. 1992; Yang et al. 1996; Lambowitz and Zimmerly 2011). This genomic mobility is supposed to have rendered group II introns key determinants in the evolution of eukaryotic genomes (Mattick and Gagen 2001; Martin and Koonin 2006). It has been proposed that group II introns are the ancestors of the eukaryotic spliceosome (Sharp 1985; Cavalier-Smith 1991), non-LTR retroelements (Curcio and Belfort 1996), and pre-mRNA introns in general, and thus might have tremendously promoted the development of genomic and transcript diversity in eukaryotes.
Group II introns are large RNAs that need to attain a complex fold in order to be catalytically active. A scheme of the secondary structure of a group IIB intron is depicted in Figure 1A. The intron comprises six domains (DI–DVI) arranged like the spokes of a wheel. Domain one is the largest and most complex one of the six domains and contains four major branches, a–d. Assembly to a complex tertiary structure ensures the correct arrangement of the catalytically relevant domains for catalysis. These relevant domains are DI and DV, which are essential (Koch et al. 1992; Michels and Pyle 1995), and DVI, containing the bulged adenosine that supplies the nucleophilic OH-group for the first step of splicing (Peebles et al. 1986; van der Veen et al. 1986).
FIGURE 1.
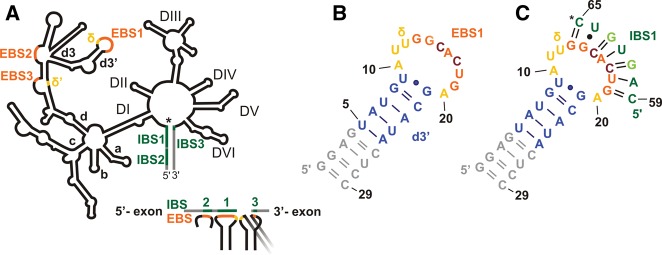
Secondary structures of a group IIB intron and the constructs under investigation. (A) Schematic representation of a group II intron with its six domains (labeled DI–VI) arranged around a central wheel. In DI, the four main branches a–d are labeled; the d3′ hairpin containing EBS1 is a substructure of branch d. The intron is shown in black, and the 3′ and 5′ exons are shown in gray. (*) The 5′ splice site. (Orange) EBS sequences; (green) IBS sequences; (yellow) the two nucleotides (nt) forming the δ–δ′ base pair. Below, the base-pairing scheme of the three EBS•IBS contacts and the δ–δ′ contact is shown. The latter is required to ensure that the 5′ and 3′ exons are bound close to each other in the active site of the intron (see also text). Secondary structures of d3′EBS1 (B) and d3′EBS1•IBS1 (C). (*) The location of the 5′ splice site. (Gray) Additional nucleotides added for better transcription yields; (blue) the helical stem of the native d3′ sequence; (yellow) the unpaired nucleotides; (orange) EBS1 (nucleotides G13–G19); (green) IBS1 (C59–C65). Light green letters in IBS1 and dark red letters in EBS1 mark mutations introduced for the sake of stability (see text and Kruschel and Sigel 2008).
Splicing involves two sequential phosphotransesterification reactions (Peebles et al. 1986; Schmelzer and Schweyen 1986; van der Veen et al. 1986). To initiate the first step, the 5′ exon has to be recognized by the intron to locate the cleavage site at the exon–intron junction precisely. This is achieved through base-pairing interactions of two different sequences within the intron, called exon-binding sites 1 and 2 (EBS1 and EBS2), with the intron-binding sites (IBS1 and IBS2) in the last 10–15 nt of the 5′ exon (Fig. 1A,C; Jacquier and Michel 1987; Michel et al. 1989). EBS1 and EBS2 are located in DI separated by several stem–loop structures. The 3′ splice site at the junction between the intron and the 3′ exon is bound to DI by a single base pair between EBS3 and IBS3 (Costa et al. 2000). To align the 5′ and 3′ splice sites in the active site, the δ–δ′ interaction is formed. δ–δ′ is a single base pair between different subdomains of DI (marked yellow in Fig. 1A), which helps to fix EBS1 and EBS3 and hence the two exons close to each other (Costa et al. 2000; Lencastre et al. 2005; Barrientos-Durán et al. 2011). The intron then cleaves the bond to the 5′ exon directly behind the last nucleotide of IBS1 using the OH-group of a bulged adenosine for nucleophilic attack. In a second step, the intron catalyzes the nucleophilic attack of the 3′-OH of the last nucleotide of the 5′ exon to cleave the bond between the intron and the 3′ exon. Thereby the intron is excised and the two exons are ligated.
Although EBS1•IBS1 lacks phylogenetic conservation of its sequence, both splicing and reverse splicing are highly site-specific reactions. It was shown that the nucleotides in IBS1 and EBS1 only need to be complementary (Jacquier and Michel 1987; Michel et al. 1989; Qin and Pyle 1999) for splicing to proceed correctly. Even more remarkably, group II introns are capable of cleaving any RNA or DNA sequence in trans and to reinsert into these sequences by reverse splicing (Augustin et al. 1990; Mörl et al. 1992; Guo et al. 2000; Mohr et al. 2000) as long as the target provides the correct IBS1 and IBS2 sequences (Eskes et al. 1997; Xiang et al. 1998). Hence, the cleavage site is not determined through recognition of specific nucleotides, as is the case for most other ribozymes like, e.g., the group I intron and the hammerhead and hairpin ribozymes (Ruffner et al. 1990; Berzal-Herranz et al. 1993; Pyle et al. 1994). Based on mutational studies of EBS1•IBS1, Su et al. (2001) have suggested that the Sc.ai5γ intron recognizes the helical part of the EBS1•IBS1 interaction and cleaves at the junction between single- and double-stranded nucleotides. They proposed that the specification of the cleavage site is defined by the most stable EBS1•IBS1 interaction (Su et al. 2001). However, there is no three-dimensional (3D) structure of the splice site of a group IIB intron available to explain how the identification of the most stable contact is structurally achieved and how the splice sites are recognized by DV and DVI of the intron, which are involved in catalysis but not in binding the exon–intron junctions. At present, structural information of a group II intron splice site is available only from crystal structures of an intact class IIC intron from Oceanobacillus iheyensis that was crystallized in the absence of the flanking exons (Toor et al. 2008a), and in the presence of an oligonucleotide that forms base pairs to EBS1 and EBS3 (Toor et al. 2008b). Further crystal structures captured the intron in its pre-catalytic state (Chan et al. 2012) and at different intermediate steps of splicing with different metal ions (Marcia and Pyle 2012). In group IIC introns, which represent the smallest class of group II introns, EBS1 comprises only 4 nt (instead of 5–7 nt in group IIB introns), and EBS2 is absent. However, the arrangement of EBS1, EBS3, and δ–δ′ equals the one of group IIB introns. The crystal structure of the post-splicing intron with a substrate reveals that EBS1 and EBS3 are stacking on top of each other, positioned close to the catalytically active nucleotides in DV (Toor et al. 2008b). Furthermore, it has been proposed that contacts between DV and EBS1•IBS1 in the Pl.LSU/2 intron greatly stabilize their interaction (Costa and Michel 1999; Costa et al. 2000).
The presence of Mg2+ ions is of crucial importance for the formation of the splice site. Mg2+ is required both to drive correct folding of the intron and to stabilize the tertiary structure (Swisher et al. 2002; Sigel 2005; Su et al. 2005), but it also participates directly in catalysis (Gordon et al. 2007; Marcia and Pyle 2012). In agreement with these findings, several metal ion-binding sites have been located in catalytically relevant positions of DV and DVI (Sigel et al. 2000, 2004; Erat et al. 2007; Toor et al. 2008b; Marcia and Pyle 2012). Su et al. (2001) observed that the identity of the divalent metal ion affects the cleavage site selection: adding Mn2+ to Mg2+ in the reaction buffer leads to a loss of fidelity in trans-cleavage assays. Similar findings have been reported for cleavage reactions catalyzed by RNase P, another large, metal-dependent ribozyme (Branvall and Kirsebom 1999). These data suggest that metal ion coordination plays an important role in forming the proper ribozyme cleavage sites. Tb3+ cleavage experiments on the Sc.ai5γ intron revealed a strong metal ion-binding site just at the 5′ end of EBS1 (Sigel et al. 2000). Also in the above-mentioned crystal structure of the O. iheyensis intron (Toor et al. 2008b), three Mg2+ ions are bound close to the splice site. All these data fuel the hypothesis that a Mg2+-binding site at the EBS1•IBS1 duplex might be relevant for group II intron fidelity.
In this study, we focus on the formation and structural features of the 5′ splice site of the group IIB intron Sc.ai5γ from mitochondrial transcripts of Saccharomyces cerevisiae and give a detailed analysis of the conformational changes undergone by EBS1 upon binding of IBS1. We solved the solution structure of IBS1-bound and -unbound EBS1 and studied its Mg2+-binding sites by NMR spectroscopy. The structure of the EBS1-containing loop changes substantially upon IBS1 binding, and an additional metal ion-binding site forms between EBS1 and IBS1. Based on our data, we discuss which aspects of the structure and metal ion binding of the unbound and IBS1-bound d3′EBS1 are relevant for a stable EBS1•IBS1 interaction and for specificity of the splicing reaction.
RESULTS
Design of the constructs d3′EBS1, d3′EBS1•IBS1, and d3′-TL
EBS1 of the Sc.ai5γ intron is 7 nt long and located within the 11-nt-long loop of the d3′ hairpin in DI (nucleotides 321–341) (Fig. 1A,B). EBS1 forms base pairs with IBS1 located at the 3′ end of the 5′ exon (Fig. 1A,C; Jacquier and Michel 1987; Michel et al. 1989). To ensure high RNA yields from in vitro transcriptions (Milligan et al. 1989) and to make the short d3′ stem more stable, all d3′ constructs were extended by a GGAG sequence at the 5′ end and the complementary nucleotides at the 3′ end (gray in Fig. 1B,C). Since the EBS1 and IBS1 sequences are not conserved (Jacquier and Michel 1987; Michel et al. 1989; Su et al. 2001), two A•U base pairs in EBS1•IBS1 were changed to G•Cs to stabilize the interaction for NMR studies (see light green and dark red letters in Fig. 1C). Single-turnover cleavage assays have proven that this mutation does not interfere with splicing activity (Kruschel and Sigel 2008). For more information on the design of the d3′EBS1•IBS1 construct, see Kruschel and Sigel (2008). The final construct of d3′-containing EBS1 used in our studies is shown in Figure 1B and is referred to as “d3′EBS1” throughout the text. The construct d3′EBS1•IBS1 denotes d3′EBS1 with IBS1 base-paired to it and is shown in Figure 1C.
EBS1 is largely unstructured in the absence of IBS1
The proposed secondary structure of d3′EBS1 (Fig. 1B) could be verified in a straightforward way using [1H,1H]-NOESY, [1H,15N]-HSQC, and JNN-HNN-COSY experiments. The formation of eight Watson–Crick base pairs and one G•U wobble pair was confirmed. The lack of observable NOE correlations between protons of the 11-nt loop in d3′EBS1 indicates that the loop does not contain any stable canonical base pairs.
In [1H,1H]-NOESY spectra of d3′EBS1 acquired in D2O, the sequential walk through the loop residues is difficult to follow due to nearly all signals being found in a rather small spectral region (Fig. 2) and some internucleotide cross peaks being very weak. Also, uracil and cytosine H5–H6 cross peaks of the loop residues appear very broad with relation to those of the stem. These spectral features indicate that the loop is flexible. Raising the KCl concentration from 10 to 110 mM did not improve the spectral dispersion, nor did it yield extra correlations. To clearly discriminate resonances of the helical d3′ stem from those of the loop, we used a construct, d3′-TL (pdb entry 2K66, BMRB entry 15859), that has the same stem sequence as d3′EBS1 but a much simpler GAAA tetraloop whose chemical shifts are available (Sigel et al. 2004). The assignment of this construct was straightforward and thus enabled us to assign the remaining d3′EBS1 resonances belonging to the loop (Fig. 2). The NMR structure of d3′-TL is given in Supplemental Figure S1 and Supplemental Table S1.
FIGURE 2.

[1H,1H]-NOESY spectrum of d3′EBS1 recorded at 293 K in D2O; a partially deuterated sample was used. Shown is the sequential walk region containing NOE cross peaks between the base protons H6 or H8 and H1′ sugar protons, which are traced by black lines. Peaks are labeled with the residue number close to their intraresidue cross peak. (Gray arrows) Internucleotide cross peaks of very low intensity between resonances of the loop protons.
The solution structure of d3′EBS1 (Fig. 3A) was calculated using the restraints listed in Table 1. d3′EBS1 adopts a hairpin structure, which is closed on one end by the 11-nt loop comprising EBS1. In the absence of IBS1, very few restraints, especially long-range NOE restraints, could be obtained for the loop region (Table 1), i.e., 12.5 NOEs per residue in contrast to the helical part, where 20.7 NOEs per residue were identified (Table 1). This scarcity of restraints, in turn, is probably the result of the loop being at least partly flexible, consistent with the observations from the NMR spectra (see above). Consequently, the fit to the experimental data does not yield one defined structure (Fig. 3A,C). For these reasons, we refrained from including any RDC data from loop nucleotides and only included seven 1H-13C RDC restraints from the helical stem into the calculation. This resulted in lower final energies but RMSD values that are slightly increased (with averages within the error limits) for the loop and the overall structure.
FIGURE 3.
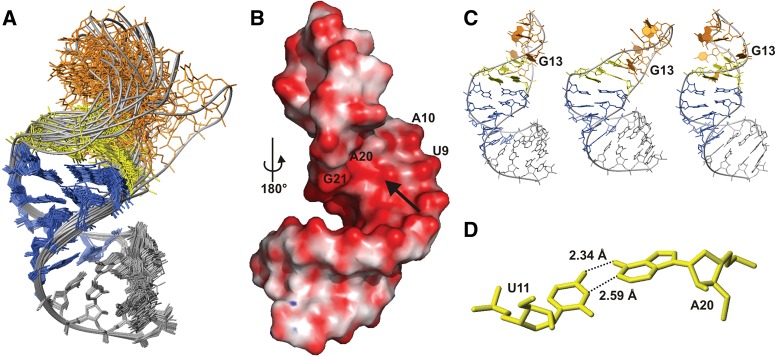
NMR structure of d3′EBS1. (A) Superposition of all heavy atoms in the stem (nucleotides 1–9 and 21–29) of d3′-EBS1 of the 20 lowest-energy structures. The coloring scheme corresponds to that of the secondary structure in Figure 1B. (B) Calculated surface potential of d3′EBS1. (Red) Negative (−682 mV) and (blue) positive (25 mV) potential. d3′EBS1 is turned by 180° with respect to A, thus showing the high density in negative potential on the major groove side at A20 and A10 marked by an arrow. (C) Three of the 20 lowest-energy structures showing possible orientations of the loop. (D) The relative orientation of U11 and A20 observed in some of the 20 lowest-energy conformers allows for hydrogen-bond formation between the two bases.
TABLE 1.
NMR restraints and structural statistics for the d3′EBS1 and d3′-EBS1•IBS1 structures
In spite of the low definition of the 11-nt loop, common structural features are observed in the 20 lowest-energy structures (Fig. 3C). The bases belonging to EBS1 protrude mostly into the solvent. This is in accordance with the absence of observable imino proton resonances (see above). However, the orientation of the EBS1 bases is not entirely random. G14–G19 (residues 2–7 of EBS1) are almost unanimously directed toward the major groove of the nearest stem base pair that closes the loop (U9•G21). The only base deviating from this orientation in the majority of the 20 lowest-energy structures is G13, the first residue of EBS1, which rather points into the loop or toward the minor groove of U9•G21 (Fig. 3C). The bases of A10 and A20, being the first and the last nucleotides of the loop, are pointing toward each other on the inside of the loop (Fig. 3C). A10 is partly stacked onto U9 of the helical stem as is A20 onto G21 and G19. The arrangement of A10 and A20 is additionally confirmed by the observation of several cross peaks between A10H2 and A20H2 as well as A10H2 and G21H1′ in the [1H,1H]-NOESY spectrum. A20 and U11 are in plane in some structures, and their Watson–Crick edges are facing each other (Fig. 3D). It is thus possible that the lower part of the loop is stabilized by hydrogen bonds between these two bases.
A stable d3′EBS1•IBS1 adduct is formed in solution
Binding of IBS1 to EBS1 strongly changes the NOE signature of the nucleotides of EBS1 and the entire loop region in the [1H,1H]-NOESY spectrum (cf. Figs. 2 and 4). The seven extra base pairs formed between EBS1 and its binding partner IBS1 (Fig. 1C) are confirmed by the respective intra- and interstrand cross peaks in [1H,1H]-NOESY spectra in 90% H2O/10% D2O and by JNN-HNN-COSY and F1-filtered NOESY-HSQC spectra of d3′EBS1•IBS1 (Supplemental Fig. S2). In contrast to free d3′EBS1, all 1H resonances of the non-exchangeable protons of d3′EBS1•IBS1 are strongly dispersed in the [1H,1H]-NOESY spectrum (Fig. 4) and could be assigned in a straightforward and unambiguous way. In the [1H,1H]-NOESY spectra, cross peaks between several resonances of C59 to resonances of A10, U11, and U12 are observed, indicating that C59 is embedded within the loop pointing into the direction of the three unpaired nucleotides A10, U11, and U12.
FIGURE 4.

[1H,1H]-NOESY spectrum of the sequential walk region of d3′EBS1•IBS1 recorded at 298 K in D2O. (Black lines) The sequential NOE correlations for the nucleotides of d3′EBS1; (dashed lines) the correlations for the nucleotides of IBS1 (see also Supplemental Fig. S3). Each residue number is given close to the intraresidue H1′-H6/H8 cross peak.
To obtain a more detailed view on the influence of IBS1 binding on the loop structure, the chemical shift changes (Δδ) upon IBS1 binding were mapped for the d3′EBS1 nucleotides (Fig. 5A). The majority of the EBS1 protons from G14 to G19 and protons of A20 experience an upfield shift, whereas those of the single-stranded nucleotides A10–U12 and G13 of EBS1 shift downfield. Although there are many caveats to interpreting chemical shift changes, an upfield shift often suggests that stacking interactions of the bases are increased. Accordingly, the strong upfield shifts (up to 0.567 ppm) of A16–U18 resonances of EBS1 are in good agreement with the formation of a helix between EBS1 and IBS1 leading to stacking interactions of the EBS1 nucleobases. The upfield shift of the A20 protons suggests that A20 stacks between G19 in the EBS1•IBS1 helix and G21 in the stem helix.
FIGURE 5.

Chemical shift changes Δδ of (A) d3′EBS1 proton resonances upon binding of IBS1 and (B) upon addition of 3 mM Mg2+ for protons in the loop region of d3′EBS1 (left) and of d3′EBS1•IBS1 (right). Chemical shift changes for aromatic protons are given in the top panels and the ones of H1′ sugar protons in the bottom panels. Coloring of the boxes corresponds to that of the secondary structures in Figure 1, B and C.
In contrast to the increased stacking on the 3′ end of EBS1, resonances of U11, U12, and G13 protons (at the 5′ end of EBS1) move downfield upon IBS1 addition, indicating that IBS1 binding reduces stacking interactions of these nucleotides. As a consequence, a marked difference in the loop structure on the 5′ end and on the 3′ end of EBS1 must be expected. The helical stem of d3′EBS1 is the part that is the least influenced by IBS1 binding, showing minute or no chemical shift changes. Only the U9•G21 base pair that closes the loop is affected by the presence of IBS1.
In summary, there is multiple spectral evidence that IBS1 binds to EBS1 in a stable and defined manner. As expected, the loop nucleotides are the part of the hairpin that is most affected by IBS1 addition. In the structure formed, the stacking interactions of G14–A20 are increased upon IBS1 binding, whereas U11–G13 seem to unstack.
IBS1 binding restructures the loop and abolishes its flexibility
The solution structure of d3′EBS1•IBS1 (Fig. 6A) comprises two well-defined A-form helices, the stem–helix that assumes essentially the same structure as in the absence of IBS1 (cf. Figs. 3A and 6A) and the short EBS1•IBS1 helix. The structure calculation of d3′EBS1•IBS1 is based on 747 conformationally restrictive NOE distance restraints and 20 1H-13C RDCs (Table 1). The unpaired nucleotides linking the stem and EBS1•IBS1 are rather well converged in the 20 lowest-energy structures and point inward into the joint between the two helices in a similar orientation as in the absence of IBS1 (cf. Figs. 3C and 6D). The structure of the unbound nucleotides probably facilitates binding of IBS1 to EBS1. As proposed already in the unbound d3′EBS1, the Watson–Crick edges of U11 and A20 are close enough to each other to allow for hydrogen bonding. They thus form a platform to accommodate the 5′ end of IBS1. Indeed, consistent with the NMR-spectral data, C59 of IBS1 is embedded within the unpaired nucleotides U11, U12, and A20 of d3′EBS1 (Fig. 6D) and is stacked with U11. This conformation ensures hydrogen bonding with G19, while the IBS1 backbone stays on the surface of the structure (Fig. 6A), leaving enough space for the exon nucleotides preceding C59 in a natural substrate (cf. Fig. 1A). A20 assumes a special geometry: It is in anti-conformation and has a C3′-endo-like sugar pucker that conforms to an A-form-helical geometry but displays unusual α and γ (−117° and 131°) backbone angles. As such, it maintains stacking interactions and a (slightly distorted) helical geometry throughout the terminal bases of the stem and EBS1 (Fig. 6E), another aspect that likely promotes EBS1•IBS1 duplex formation.
FIGURE 6.

NMR structure of d3′EBS1•IBS1. (A) Superposition of all heavy atoms in d3′EBS1•IBS1 of the 20 lowest-energy structures. The coloring scheme corresponds to that of the secondary structures in Figure 1C. (B,C) Calculated surface potential of d3′EBS1•IBS1. (Red) Negative (−642 mV), and (blue) positive (103 mV) potential. (Arrows) A high negative charge density (B) in the EBS1•IBS1 major groove and (C) near the 5′ end of IBS1 and the single-stranded nucleotides A20 and A10. (D) Section of d3′EBS1•IBS1 showing C59 (green) of IBS1 embedded between G19 (orange) of EBS1 and the unpaired nucleotides A10, U11, U12, and A20. The coloring scheme corresponds to that of the secondary structure in Figure 1C. (E) Superposition of nucleotides C17–C22 of the 10 lowest-energy structures of d3′-EBS1•IBS1 illustrating stacking interactions between G19, A20, and G21. (F) The orientation of U12, U18, and A60 is shown illustrating the putative major-groove triple base pair.
The three bases U12, U18, and A60 lie almost in one plane in an orientation of a UAU base triple in a cis-Hoogsteen–Watson–Crick fashion (Fig. 6F). A60 and U18 thereby form the Watson–Crick base pair, and U12 and A60 a cis-Hoogsteen base pair. Such noncanonical base pairs are now widely accepted to stabilize secondary as well as tertiary structural elements in RNAs (Saenger 1984; Hermann and Patel 1999; Nagaswamy et al. 2002). However, as U12 would be engaged in the δ–δ′ base pair in the folded full-length group II intron, this interaction is unlikely to occur at least while both exons are bound.
The stabilization of EBS1•IBS1 on the 5′ end of IBS1 provided by the surrounding loop nucleotides appears even more important in comparison to the 3′ end of IBS1 (C65, the splice site), which is completely exposed. C65 sticks out of the structure giving room for the following nucleotides of the intron strand and most importantly, for the remaining components of the splice site, EBS3, IBS3, DV, and DVI (Fig. 1A).
The isolated position of C65 is the result of a pronounced kink in the loop backbone (Fig. 6A) between δ/U12 and G13. This is the result of the position of EBS1 in the loop. The first three loop bases on the 5′ side of EBS1, A10–U12, take more space than A20 on the 3′ end of EBS1 but are not a long enough linker to allow coaxial stacking of EBS1•IBS1 on the stem. Consequently, the formation of the EBS1•IBS1 helix puts conformational strain on the sugar-phosphate backbone of the loop and causes the formation of the kink, which positions the negatively charged phosphate oxygens of U12 and G14 in close proximity, which is electrostatically unfavorable, and causes δ/U12 and G13 to orient perpendicular to each other. This suggests that the δ–δ′ base pair is formed at a certain distance from the splice site and in another direction than the EBS1•IBS1 contact.
When comparing the structures of d3′EBS1 and d3′EBS1•IBS1, two things become evident. Firstly, the flexibility of the loop is entirely abolished by the binding of IBS1, resulting in one, defined orientation of the EBS1•IBS1 helix and hence of the whole 5′ splice site throughout all 20 conformers. Secondly, the EBS1 nucleotides have to rearrange substantially to bind IBS1. As mentioned before in the free d3′EBS1, all EBS1 bases except for G13 point toward the major groove of the upper base pairs of the helical stem (U9•G21). IBS1, however, binds on the opposite side of the loop (above the minor groove of the hairpin stem), which forces G14 to G19 to flip to this side. These observations are well in line with the chemical shift changes (see previous paragraph and Fig. 5A). The EBS1 nucleotides display major upfield chemical shift changes that result from the increased stacking and from base-pairing while U12 and G13 experience partial unstacking through this rearrangement, thus displaying downfield shifts.
A second metal ion is coordinated upon IBS1 binding
Mg2+ ions are crucial for a stable formation of secondary and tertiary structure of RNA (Sigel 2005; Freisinger and Sigel 2007; Sigel and Pyle 2007) and are directly involved in the catalytic mechanism of most large ribozymes. It has been shown by Tb3+ cleavage experiments that a metal ion-binding site is located at the 5′ splice site recognition complex (Sigel et al. 2000; Sigel 2005) (corresponding to A10 and U11 in our construct) and that the identity of the divalent metal ion may influence fidelity of the intron (Su et al. 2001). To examine and compare the influence of Mg2+ binding on the structure of d3′EBS1 in the absence and presence of IBS1, we performed NMR titration studies on d3′EBS1 and d3′EBS1•IBS1 and monitored the chemical shift changes of the H1′ and aromatic protons as a function of Mg2+ concentration by recording [1H,1H]-NOESY spectra. Since Mg2+ addition did not cause any other changes in the NMR spectra than progressive chemical shift changes and line broadening, it can be assumed that Mg2+ binding does not lead to major structural changes, neither in d3′EBS1 nor d3′EBS1•IBS1, but occurs in regions that are already structured in the presence of monovalent ions.
Potential metal ion coordination sites in RNA are mostly the N7 of purines and the phosphate oxygens, but also other sites like the ribose 2′-OH are feasible (Schnabl and Sigel 2010; Al-Sogair et al. 2011). Mg2+ binding to RNA leads to changes in the chemical shifts (Δδ) of proton resonances near the coordinating atoms. These changes, however, can result from either metal ion coordination at the residue of the affected proton or from subtle structural changes induced by coordination of the metal (Sigel et al. 2004; Erat and Sigel 2011) at an adjacent residue. Consequently, only a binding region can be defined but usually not the exact atoms involved in coordination.
Chemical shift mapping indicates Mg2+ binding within the d3′ stem between the G4•C26 and A6•U24 base pairs irrespective of the presence or absence of IBS1 (Supplemental Fig. S4). G•C base pairs but also A•U tracks have been shown previously to bind divalent ions (Korth and Sigel 2012; Sigel and Sigel 2013). In the following, we now focus our analysis of the chemical shift changes on the 11-nt loop region and their comparison in the absence and in the presence of IBS1 (Fig. 5B). In both cases, A10, U11, and A20 being the first two and the last nucleotides of the loop are strongly affected by Mg2+. Also proton resonances of U9 and G21 of the stem base pair closing the loop display rather large Δδ values (Fig. 5B). In the presence of IBS1, the H1′ and H5 resonances of C59 shift strongly upon addition of Mg2+. These chemical shift changes are an indicator of a metal ion-binding site between the helical stem and the lower loop region both in d3′EBS1 and d3′EBS1•IBS1. This assumption is in accordance with the electrostatic surface potentials (Chin et al. 1999) of both d3′EBS1 and d3′EBS1•IBS1, which contain a cluster of high negative potential density on the major-groove side of the stem (see arrows in Figs. 3B and 6C). This cluster, however, lies in slightly different positions in the absence and in the presence of IBS1. In the free d3′EBS1 (Fig. 3B), the last stem base pair U9•G21 and A20 form a cluster of negative potential, while in the presence of IBS1 (Fig. 6C) the cluster of negative potential is shifted toward the loop at G19•C59, the last EBS1•IBS1 base pair, and A60. O4 of U11 and U12 and N7 of A10 and A20 (in both structures), as well as G19O6 and N7 (in d3′EBS1•IBS1) and G21O6 and N7 and U9O4 (in d3′EBS1) are atoms able to attract a Mg2+ ion to this site. The rather strong Δδ values of U18 and G19 resonances at the 3′ end of EBS1 might be due to a rigidification of the lower loop upon Mg2+ binding rather than a direct effect of coordination as U18 and G19 are in a flexible region and do not form a site of particularly negative potential.
In d3′EBS1•IBS1, there is evidence for a second binding site formed very close to the splice site between IBS1 and EBS1. G13 proton resonances at the 5′ end of EBS1 experience the most pronounced shifts. In the same region, resonances of U12 and G14 surrounding G13 and C65 and U64 on the opposite IBS1 strand are also influenced by Mg2+ binding rather strongly. The effects on the two first EBS1•IBS1 base pairs G13•C65 and G14•U64 clearly suggest the binding of a second Mg2+ ion. Also, the second binding site between EBS1 and IBS1 is in accordance with the electrostatic surface potential. In Figure 6B, the major groove of the EBS1•IBS1 helix is visible as a tunnel of dense negative potential, which makes it an appealing candidate for metal ion binding with N7 and O6 of G14 and G63 and O4 of U64 being possible coordinating atoms. Indeed, G•U wobble pairs such as G14•U64 often attract Mg2+ ions to bind in their major groove (Allain and Varani 1995; Sigel and Sigel 2013). The location of the Mg2+-binding sites was confirmed by performing chemical shift mapping of Cd2+ titrations and line broadening studies by Mn2+ titrations (data not shown). Both ions bind to nucleic acids in a similar way as Mg2+.
DISCUSSION
The position of EBS1 in the 11-nt loop of d3′ is important for positioning the cleavable bond
The free d3′EBS1 structure reveals three regions with different degrees of conformational freedom: (i) The backbone of EBS1 itself is very flexible. (ii) The loop nucleotides surrounding EBS1 (A10, U11, U12, and A20) are loosely restricted in their orientations by stacking interactions to each other or to the U9•G21 base pair in the stem, and in the case of U11 and A20, by putative formation of a hydrogen bond. They roughly maintain their positions upon binding of IBS1 and thereby probably facilitate binding of its 5′ end as described above. (iii) The d3′ stem possesses a rigid helical fold rooting EBS1 within the intron structure. This gradient in stability thus positions EBS1 in a region that is accessible to the strand containing IBS1 but leaves EBS1 flexible and adaptable for IBS1 binding. Previously, we have shown that ΔG37 for the melting of the isolated EBS1•IBS1 is ∼6.5 kJ/mol smaller than for melting of EBS1•IBS1 when EBS1 is part of d3′ (Kruschel and Sigel 2008). Although this difference is not large, it indicates that the structure of the d3′ loop indeed facilitates IBS1 binding to EBS1.
In the absence of IBS1, most bases of EBS1 point toward the major groove of the d3′ stem. Upon binding of IBS1 on the opposite side, the bases of EBS1 have to flip to the minor-groove side, and a sharp kink is introduced between U12 and G14, bringing their phosphate oxygens in rather close contact. The formation of EBS1•IBS1 is thus disfavored in both electrostatic terms and entropic terms as the large flexible loop containing EBS1 is frozen in a rigid conformation. To achieve this unfavorable rearrangement of the loop region, EBS1 and IBS1 have to form a continuous and stable helix to achieve the bending of the backbone and the exposure of the scissile bond between C65 and the last nucleotide of the intron. This observation is well in line with the very low tolerance of the EBS1•IBS1 pairing for mismatches and with the fact that any complementary EBS1 and IBS1 sequences can efficiently promote splicing (Jacquier and Michel 1987; Michel et al. 1989; Xiang et al. 1998; Qin and Pyle 1999). The particular geometry of the splice site and its orientation with respect to the stem is the result of the interplay between the steric demands of the EBS1•IBS1 helix itself and of the interactions of the four asymmetrically distributed loop nucleotides before and after EBS1. In the light of these findings, it is interesting to compare how many nucleotides precede and follow EBS1 in the different structural subdivisions of group IIB introns and introns with B-like hybrid structures (see the group II intron data base; http://www.fp.ucalgary.ca/group2introns/) (Dai et al. 2003; Candales et al. 2012). Indeed, it seems that in order to accommodate and orient the EBS1•IBS1 helix and the δ–δ′ base pair (Fig. 1A), most introns contain at least one and not more than two single-stranded nucleotide(s) before δ, while on the 3′ side of EBS1, one single-stranded nucleotide may or may not be present. This finding supports the notion that the extra single-stranded nucleotides between the helical stem and the δ-EBS1 sequence are critical for the correct positioning of the scissile bond but also illustrates that this can be achieved with slightly different distributions of nucleotides in the d3′ loop.
EBS1•IBS1 formation entails unstacking of G13 (bound to 65 at the splice site) from U12, which prevents the δ–δ′ base pair from obstructing the splice site (and vice versa). It has previously been reported that 3′ elongation of IBS1 with nucleotides complementary to the single-stranded nucleotides before EBS1 does not alter the cleavage site (Jacquier and Jacquesson-Breuleux 1991; Michels and Pyle 1995; Su et al. 2001). Costa et al. (2000) have shown that formation of the δ–δ′ base pair in the Pl.LSU/2 group II intron enhances stability of the interaction between the intron and the 5′ exon. These findings corroborate the idea that the kink is stable in the entire intron structure and together with the δ–δ′ base pair positions EBS1 for binding IBS1 but prevents the loop nucleotides upstream of EBS1 from pairing with the substrate.
The role of Mg2+ binding in splice site definition
The Mg2+-titration data of d3′EBS1•IBS1 reveal one binding site at the central base pairs of the d3′ stem as well as one on each side of the EBS1•IBS1 helix. In the free d3′EBS1, one major-groove Mg2+-binding site is located at the joint between the helical stem and the loop. In this region, a metal ion-binding site was already predicted by Tb3+ cleavage experiments (Sigel et al. 2000). Probably the Mg2+ ion bound here plays a role in stabilizing the loop nucleotides surrounding EBS1 (see above), thereby helping to pre-orient the loop for IBS1 binding. In contrast to this, circular dichroism studies of d3′EBS1 (Kruschel and Sigel 2008) revealed a slight increase in content of single-stranded nucleotides in d3′EBS1 upon addition of Mg2+. This prompted the hypothesis that base-pair formation within the loop (e.g., between U12 and G19), which would hamper IBS1 binding, is prevented by Mg2+ binding. However, we detected no evidence of such base pairs in the NMR data. Upon binding of IBS1, the Mg2+-binding site between the stem and loop is shifted from the end of the d3′ stem toward the loop region, now involving G19 and C59 of IBS1. This shift is probably caused by a different local geometry and the availability of extra ligand atoms supplied by the 5′ end of IBS1 and a generally more rigid structure compared with the unbound loop.
d3′EBS1•IBS1 clearly provides a second Mg2+-binding region close to the 5′ end of EBS1, probably situated at the end of the tunnel-shaped major grove of EBS1•IBS1 displaying a generally negative surface potential. Binding of Mg2+ to these two sides increases the affinity of IBS1 for d3′EBS1 as was indicated by UV melting experiments (Kruschel and Sigel 2008) and recently in more detail also by single-molecule Förster resonance energy transfer studies of d3′EBS1•IBS1 (data not shown). Both binding sites observed in the loop containing EBS1•IBS1 are too far away from the scissile bond to take part in the cleavage mechanism of the intron. However, we propose that the stabilizing effect of Mg2+ binding to EBS1•IBS1 might be relevant for the rejection of mismatched targets. If, due to a mismatch in the middle or on the 3′ end of IBS1, a regular helix is not formed or distorted by bulges, the major groove cannot form the negatively charged tunnel that attracts Mg2+, while mismatches at the 5′ end of IBS1 would probably prevent the binding site in the lower loop region from shifting. As a consequence, the stabilization of EBS1•IBS1 by Mg2+ would not occur, and thus the dissociation of the mismatched contact would be favored. To prove this hypothesis, however, more detailed investigation of the structure and metal interaction of mismatched d3′EBS1•IBS1 pairings would be required but are difficult to conduct due to the low stability of such contacts.
Parallels to the crystal structure of the O. iheyensis group IIC intron
To compare the d3′EBS1•IBS1 solution structure with d3′EBS1•IBS1 in a structural context, we compared it with the crystal structure of the group IIC intron of O. iheyensis, being the only one of a full-length group II intron ribozyme—and the only structure of a group II intron splice site—available. We used for comparison the structure of the spliced intron with a model substrate bound. EBS1 in this subgroup of group IIC introns comprises only 4 nt (AUAA) and is located in a loop of 8 nt in length and EBS2 is absent. Although this represents a simpler and differently organized splice site, the relative arrangement of EBS1, EBS3, and the δ–δ′ contact is essentially the same as in the IIB class (Fig. 1A). Figure 7 shows details of the crystal structure of the group IIC intron from O. iheyensis (Marcia and Pyle 2012), namely, the hairpin containing EBS1, the 6-nt substrate containing IBS1 and IBS3 joined on one strand and parts of domain I containing EBS3 and the δ nucleotide (for comparison, the orientation is the same as for d3′EBS1•IBS1 in Fig. 6A). Also, DV with the bulged nucleotides (purple in Fig. 7) and the catalytic triad of DV, which are known to play a major role in catalysis (Chanfreau and Jacquier 1994; Konforti et al. 1998; Sigel et al. 2004), is shown. The intron was crystallized in the presence of Ca2+ ions, which inhibit the splicing reaction (Erat and Sigel 2008) and represent the conformation before the first splicing step.
FIGURE 7.
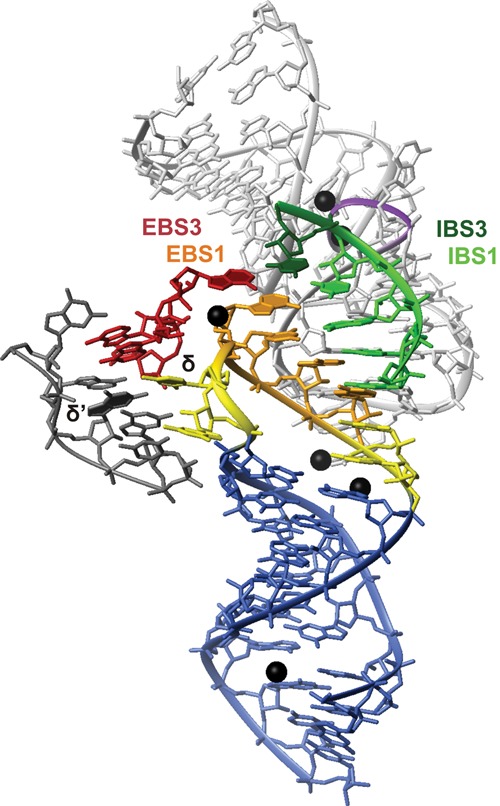
Detail of the splice site components including parts of DI and DV from the crystal structure of the group II intron from O. iheyensis (Marcia and Pyle 2012). (Blue) The hairpin containing EBS1 (stem nucleotides); (yellow) single-stranded and δ nucleotides; (orange) EBS1 nucleotides. The IBS1–IBS3 substrate is shown in green (IBS1) and dark green (IBS3). (Light gray) DV; (purple) the backbone at the bulge; (dark red) the sequence from DI that contains EBS3; (dark gray) the sequence containing the δ′ nucleotide; (black spheres) Ca2+ ions. Their positions are very similar to the ones of Mg2+ in a previous crystal structure (Toor et al. 2008b). The figure was prepared with MOLMOL (Koradi et al. 1996) from pdb entry 4E8K (Marcia and Pyle 2012).
Although this structure is naturally quite different from the one presented here, there are important similarities between the two. A turn in the backbone between δ and the first EBS1 nucleotide causes unstacking of their bases and a direction change of the backbone as observed in the d3′EBS1•IBS1 solution structure. The crystal structure beautifully shows that this enables EBS3 to stack onto EBS1 in order to bind IBS3–IBS1 close to each other. In contrast to our structure, the δ base is flipped to expose its Watson–Crick edge to δ′ and rotated out of the helix. It is reasonable to assume a similar orientation of δ in the full-length Sc.ai5γ intron, although δ/U12 is in our structure obstructed by C59 and A60 of IBS1 on one side and by the EBS1 backbone on the other side. This is a more favorable arrangement in the solution structure where no external binding partner is available but an UAU (UAδ) base triple can be formed (see Results; Fig. 6F). Although there is evidence for the formation of the δ–δ′ base pair in the absence of a splicing substrate (Costa et al. 2000; Marcia and Pyle 2012), it is possible that the UAδ base triple is formed during previous steps of the splicing reaction, where only the 5′ exon is bound. This could enhance flexibility within DI without destabilizing the EBS1•IBS1 pairing. Judging from the relative positions of the splice site and U12/δ in the crystal structure and in the solution structure, it seems probable that DV and the bulge containing δ′ and EBS3 are arranged in a similar way around d3′EBS1•IBS1 in Sc.ai5γ as they are in the O. iheyensis intron. In the crystal structure, the bulged nucleotides of DV contact the backbone of the IBS1–IBS3 substrate, and two Ca2+ ions (presumably in place of the two catalytic Mg2+ ions) (Toor et al. 2008b; Marcia and Pyle 2012) are coordinated between DV and the substrate.
On the 3′ end of EBS1, the two unpaired nucleotides in the loop stack between EBS1 and the d3′ stem like A20 in our structure. Additionally, this region is stabilized by metal ion binding on the major-groove side equaling the lower Mg2+-binding site of d3′EBS1•IBS1 presented here. This supports the idea that the nucleotides following EBS1 play a role in stabilizing the 5′ end of IBS1. In the crystal structure, two Ca2+ ions are located in the major groove coordinated to the EBS1 phosphate oxygens of the nucleotides that base-pair to the 5′-end nucleotides of IBS1. This binding site corresponds quite well to the region of negative surface potential in the major groove near U11 and G19, i.e., the last base pair of the EBS1•IBS1 helix, found in our d3′EBS1•IBS1 structure. The small deviation in binding of ∼1 nt is well explained by the difference in local geometry caused by the shorter EBS1•IBS1 helix in the O. iheyensis intron. Accordingly, divalent metal ion binding in the major groove at the transition from the EBS1•IBS1 helix to the d3′ stem seems to be common to group IIB and IIC introns, probably directly stabilizing the EBS1•IBS1 interaction.
It is obvious from previous studies and from the crystal structures of the O. iheyensis intron that the exact position of the 5′ splice site is only achieved in the ensemble of DV, DI, and the substrate and their various tertiary interactions (Pyle 2010; Lambowitz and Zimmerly 2011). Nevertheless, the comparison of the solution structure to the crystal structure essentially demonstrates that some structural elements that are important for recognition of the 5′ exon—such as the direction change between δ and EBS1 and stabilization of the 5′ end of IBS1 by the unpaired loop nucleotides on the 3′ end of EBS1—are common to the Sc.ai5γ group IIB and the O. iheyensis group IIC intron and that in Sc.ai5γ they do not require the presence of any other parts of the intron to form.
CONCLUSIONS
In this study, we characterize the structure of the d3′ hairpin containing EBS1 from the group IIB intron Sc.ai5γ without and with IBS1 bound. The correct positioning of IBS1 for cleavage is largely determined by the position of EBS1 in the 11-nt loop and by the interactions between the unpaired nucleotides on the 5′ and the 3′ sides of EBS1. The considerable structural changes undergone upon binding of IBS1 and the formation of two stabilizing metal ion-binding sites on each end of the EBS1•IBS1 helix might serve as a checkpoint for whether the correct IBS1 sequence is bound or not. They thus contribute to the understanding of the remarkably high fidelity of group II intron splicing despite the lack of sequence conservation of the splice sites.
MATERIALS AND METHODS
Materials
Nucleoside 5′-triphosphates (NTPs) were purchased from GE Healthcare and Acros-Organics. Partially deuterated NTPs were purchased from Cambridge Isotope Laboratories and 13C,15N-labeled nucleotides from Silantes GmbH. The 99.9% D2O used for NMR sample preparation was purchased from Armar Chemicals. T7 RNA-polymerase was made in-house. Synthetic double-stranded DNA templates for transcription were obtained from Microsynth. The 7-nt-long IBS1 was purchased HPLC-purified from Curevac. Pf1 filamentous bacteriophage was obtained from ASLA Ltd.
NMR sample preparation
d3′EBS1 and d3′-TL were synthesized by in vitro transcription with T7 RNA polymerase from synthetic double-stranded DNA templates as described in Gallo et al. (2005). Natural abundance, partially deuterated and uniformly 13C,15N-labeled d3′EBS1 was transcribed using the respective NTPs. The transcription mixtures contained 5 mM each NTP, 0.9 μM the double-stranded DNA template, and 35 mM MgCl2. The amount of T7 RNA-polymerase was adapted according to the activity of each enzyme batch. Transcription reactions were allowed to proceed for 12–14 h at 37°C. The RNA was then purified by denaturing 15% polyacrylamide gel electrophoresis, UV-shadowed, and excised from the gel and recovered by electroelution (Whatman). To avoid duplex formation, the RNA was annealed by dissolving it in 100 mL of 85°C double-distilled water, and, after 1 min, cooling on ice. The RNA was concentrated and desalted using Vivaspin 20 concentrators with a 3000-Da molecular weight cut-off (MWCO; VivaScience). The purchased short oligonucleotide IBS1 was dialyzed three times against 300 mL of 100 mM KCl and three times against 300 mL of double-distilled water using 500 MWCO dialysis membranes (Spectrum labs). The dialysis was performed as previously described (Sodhi and Rajput 2003).
The concentration of the RNA was determined by UV spectroscopy using the extinction coefficients ε260 325.8 mM−1 cm−1 (d3′EBS1), 257.7 mM−1 cm−1 (d3′-TL), and 73.6 mM−1 cm−1 (IBS1). Samples of d3′EBS1 and d3′-TL contained 0.4–1.2 mM RNA, 10 mM KCl, and 10 µM EDTA, while d3′EBS1•IBS1 samples were prepared by mixing equimolar amounts of d3′EBS1 and IBS1 and contained 0.5–0.9 mM complex, 110 mM KCl, and 10 μM EDTA. Prior to NMR data acquisition, all RNA samples were lyophilized and resuspended in either 99.9% D2O or 90% H2O/10% D2O. The pD or pH of the samples was adjusted to 6.8. The pD was determined by adding 0.4 to the pH meter reading (Lumry et al. 1951; Glasoe and Long 1960).
NMR spectroscopy
NMR spectra were recorded on a Bruker DRX 500 MHz spectrometer using a 5-mm BBI probe head, on a Bruker AV600 MHz spectrometer with a CP-TCI z-axis pulsed-field gradient CryoProbe or on a Bruker AV700 MHz spectrometer equipped with a CP-TXI z-axis pulsed-field gradient CryoProbe. The 1H chemical shift of DSS (4,4-dimethyl-4-silapentane-1-sulfonic acid) served as a direct reference for 1H chemical shifts and as an indirect reference for 13C and 15N chemical shifts (Markley et al. 1998). Resonances of exchangeable protons of d3′EBS1, d3′EBS1•IBS1, and d3′-TL were assigned using [1H,1H]-NOESY spectra acquired in 90% H2O/10% D2O at 278 K, 283 K, and 293 K (150 msec mixing time, WATERGATE pulse sequence for H2O suppression) and [1H,15N]-HSQC experiments at 278 K. The base-pairing scheme in all RNAs was confirmed by two-dimensional (2D) JNN-HNN-COSY spectra (Luy and Marino 2000). Resonances of non-exchangeable protons were assigned using [1H,1H]-NOESY spectra (mixing times 60, 120, and 250 msec) and [1H,1H]-TOCSY spectra (45 msec mixing time) of natural abundance and partially deuterated RNAs (99.9% D2O, 288 K, 293 K, 298 K, and 303 K). The residual HDO signal was suppressed with a low-power presaturation pulse. Aromatic carbon resonances were assigned from standard [1H,13C] HSQC spectra, and imino and amino proton resonances were assigned using standard [1H,15N] HSQC spectra. NOESY-HSQC spectra with a 15N filter in F1 (with WATERGATE water suppression) were recorded at 278 K of d3′EBS1•IBS1, where d3′EBS1 was uniformly 13C- and 15N-labeled and IBS1 at natural abundance to confirm duplex formation between IBS1 and EBS1. In addition, phase-sensitive ge-2D F1,F2-[13C,15N]-filtered NOESY and TOCSY experiments with a WATERGATE scheme for water suppression at 298 K (Ogura et al. 1996; Zwahlen et al. 1997; Breeze 2000; Iwahara et al. 2001) were recorded of the same sample to verify the assignment of IBS1 resonances (Supplemental Fig. S3). In such an experiment, only protons of the natural abundance molecule—in our case, IBS1—are detected.
Partial alignment of d3′EBS1 and d3′EBS1•IBS1 for the residual dipolar coupling (RDC) measurements was achieved by adding 25.6 mg/mL Pf1 filamentous bacteriophage (ASLA Ltd.) to the 13C,15N-labeled samples (Zhou et al. 1999; Hansen et al. 2000). Deuterium splitting was 24.2 Hz at 700 MHz. One-bond 1H-13C RDCs were measured from the splitting of the peak along the carbon dimension of [1H,13C]-HSQCs at 298 K in isotropic (unaligned) and Pf1-containing (aligned) solution. NMR data were processed using XWINNMR, Topspin 1.3, and 2.0, from Bruker BioSpin. Resonance assignments were made in Sparky (http://www.cgl.ucsf.edu/home/sparky/). NOE peak volumes were integrated with the Gaussian peak-fitting function in Sparky.
Structure calculations and analysis
NOE distances were estimated from the integrated peak volumes from [1H,1H]-NOESY spectra at 250 msec mixing time. Distances were calibrated relative to the NOE intensity of pyrimidine H5–H6 cross peaks (distance 2.4 Å) using the CALIBA macro in DYANA (Güntert et al. 1997). The NOEs were grouped into four categories, corresponding to strong (1.8–3.0 Å), medium (1.8–4.5 Å), weak (3.0–6.0 Å), and very weak (4.0–7.0 Å). [1H,1H]-TOCSY experiments with 45 msec mixing time were used to analyze sugar pucker conformations (28). Nucleotides with strong H1′–H2′ and H1′–H3′ cross peaks (A10, U11, and U12 for d3′-EBS1•IBS1; and A11 for d3′-TL) were restrained to be in S-type (C2′-endo) range (δ = 145° ± 30°, ν1 = 25°, ν2 = −35°, ±15°), those with absent H1′–H2′ cross peaks to be in N-type (C3′-endo) range (δ = 85°, ν1 = −25°, ν2 = 37°, ±30°), and nucleotides with intermediate cross-peak intensities (G1, A10–A20, and C29 for d3′-EBS1; G1, C29, and C65 for d3′-EBS1•IBS1; and G1, G10, A12, A13, and C22 for d3′-TL) were left unrestrained (Sigel et al. 2004). Analysis of the peak volumes of intranucleotide H1′-H6/8 NOEs from [1H,1H]-NOESY spectra with 60 msec mixing time revealed that all nucleotides in each of the constructs were in an anti conformation; thus, the torsion angle χ was restrained to −160° ± 20°. The other backbone dihedral angles were set to standard A-form values (α = −62°, β = −180°, γ = 48°, ε = −152°, ζ = −74°, ±10°) in the helical region of the structures (G2–U9, G21–C28 [G14–C21 in d3′-TL], G13–G19, and C59–U64 [in d3′EBS1•IBS1]), and otherwise left unrestrained. For the single-stranded loop residues A10, U11, U12, and A20, α and ζ were restrained loosely to exclude the trans range (0° ± 120°) since there were no 31P resonances outside of a region between −15 and 0 ppm (Varani et al. 1996). RDCs for d3′EBS1 and d3′EBS1•IBS1 were measured using Sparky by determining the difference between 1H–13C couplings for isotropic and partially aligned samples. PALES software (Zweckstetter and Bax 2000; Zweckstetter 2008) was used to estimate values for the axial (Da) and rhombic (R) components of the alignment tensor from low-energy structures calculated in the absence of RDCs. The PALES predicted values were optimized with a grid search (Clore et al. 1998) using XPLOR-NIH (Schwieters et al. 2003). In the grid search, lowest overall energies were used as a target criterion, and the identified optimal values were Da = −37 Hz/−35 Hz and R = 0.3/0.1 for d3′EBS1 and d3′EBS1•IBS1, respectively.
CNS 1.2 (Brünger et al. 1998) was used to calculate 200 initial structures from an extended structure with random initial velocities using NOE distance, dihedral, and H-bond restraints. A high-temperature stage of 40 psec at 20,000 K is followed by two cooling stages of 90 psec in torsional space and 30 psec in Cartesian space. The 20 lowest-energy structures obtained were subsequently refined in XPLOR-NIH (Schwieters et al. 2003) by slowly cooling from 3000 to 50 K. In case of d3′EBS1 and d3′EBS1•IBS1, RDCs were included in the refinement, and their force constant was gradually increased from 0.01 kcal mol−1 Hz−2 to 1 kcal mol−1 Hz−2. For all base pairs, planarity was enforced during calculations, and hydrogen bonds were maintained by short distance restraints. After refinement, the 20 lowest-energy structures out of 200 calculated, none of which contained NOE (>0.2 Å) or dihedral angle (>5°) violations, were visualized and analyzed using MOLMOL (Koradi et al. 1996). The electrostatic surface potentials of d3′EBS1 and d3′EBS1•IBS1 were calculated with the PDB2PQR version 1.8 webserver (http://nbcr-222.ucsd.edu/pdb2pqr_1.8/) (Dolinsky et al. 2004) and visualized with the APBS Tools2 plugin (Baker et al. 2001) for PYMOL (http://www.pymol.org).
Metal ion titration studies
Mg2+ line broadening and chemical shift changes of aromatic and sugar protons were monitored in a series of [1H,1H]-NOESY spectra recorded at 298 K as described earlier (Erat and Sigel 2011; Pechlaner and Sigel 2012). MgCl2 was added in steps of 0, 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 6.5, and 7 mM to d3′EBS1 and in steps of 0, 0.5, 1, 2 3, 4, 5, 6, 7, 8, and 10 mM to d3′EBS1•IBS1. An [1H,1H]-NOESY spectrum was recorded for each concentration of MgCl2. Chemical shift differences were analyzed by creating bar plots in Origin (OriginLab).
DATA DEPOSITION
Structure coordinates have been deposited to the Protein Data Bank (PDB) with the accession codes 2M24, 2M23, and 2K66. Chemical shift data and NMR restraint files have been deposited to the Biological Magnetic Resonance Bank (BMRB) with the accession codes 18894, 18893, and 15859.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article as pdf: Supplemental Results on d3′-TL, Supplemental Table S1 on the NMR structure of d3′-TL, Supplemental Figures S1–S4 (d3′-TL structure, F1 and F1/F2 –[13C,15N]-filtered NOESY-HSQC of d3′EBS1•IBS1, and chemical shift changes in the d3′-stem upon addition of 3 mM Mg2+).
ACKNOWLEDGMENTS
Financial support by the Boehringer Ingelheim Fonds (B.I.F. FUTURA 2/2005, 112 to D.K.), the Swiss National Science Foundation (200021-124834 to R.K.O.S.), the European Research Council (ERC Starting Grant MIRNA to R.K.O.S.), and the University of Zurich is gratefully acknowledged. We thank Dr. Maria Pechlaner and Dr. Silke Johannsen for carefully reading the manuscript and for their valuable comments.
REFERENCES
- Allain FHT, Varani G 1995. Divalent metal ion binding to a conserved wobble pair defining the upstream site of cleavage of group I self-splicing introns. Nucleic Acids Res 23: 341–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sogair FM, Operschall BP, Sigel A, Sigel H, Schnabl J, Sigel RKO 2011. Probing the metal-ion-binding strength of the hydroxyl group. Chem Rev 111: 4964–5003 [DOI] [PubMed] [Google Scholar]
- Augustin S, Müller MW, Schweyen RJ 1990. Reverse self-splicing of group II intron RNAs in vitro. Nature 343: 383–386 [DOI] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA 2001. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci 98: 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrientos-Durán A, Chillón I, Martínez-Abarca F, Toro N 2011. Exon sequence requirements for excision in vivo of the bacterial group II intron RmInt1. BMC Mol Biol 12: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berzal-Herranz A, Joseph S, Chowrira BM, Butcher SE, Burke JM 1993. Essential nucleotide sequences and secondary structure elements of the hairpin ribozyme. EMBO J 12: 2567–2573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branvall M, Kirsebom LA 1999. Manganese ions induce miscleavage in the Escherichia coli RNase P RNA–catalyzed reaction. J Mol Biol 292: 53–63 [DOI] [PubMed] [Google Scholar]
- Breeze AL 2000. Isotope-filtered NMR methods for the study of biomolecular structure and interactions. Prog Nucl Magn Reson Spectrosc 36: 323–372 [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang J-S, Kuszewski J, Nilges M, Pannu NS, et al. 1998. Crystallography & NMR System: A new software suite for macromolecular structure determination. Acta Crystallogr D54: 905–921 [DOI] [PubMed] [Google Scholar]
- Candales MA, Duong A, Hood KS, Li T, Neufeld RAE, Sun R, McNeil BA, Wu L, Jarding AM, Zimmerly S 2012. Database for bacterial group II introns. Nucleic Acids Res 40: D187–D190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalier-Smith T 1991. Intron phylogeny: A new hypothesis. Trends Genet 7: 145–148 [PubMed] [Google Scholar]
- Chan RT, Robart AR, Rajashankar KR, Pyle AM, Toor N 2012. Crystal structure of a group II intron in the pre-catalytic state. Nat Struct Mol Biol 19: 555–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanfreau G, Jacquier A 1994. Catalytic site components common to both splicing steps of a group II intron. Science 266: 1383–1387 [DOI] [PubMed] [Google Scholar]
- Chin K, Sharp KA, Honig B, Pyle AM 1999. Calculating the electrostatic properties of RNA provides new insights into molecular interactions and function. Nat Struct Mol Biol 6: 1055–1061 [DOI] [PubMed] [Google Scholar]
- Clore GM, Gronenborn AM, Tjandra N 1998. Direct structure refinement against residual dipolar couplings in the presence of rhombicity of unknown magnitude. J Magn Reson 131: 159–162 [DOI] [PubMed] [Google Scholar]
- Costa M, Michel F 1999. Tight binding of the 5′ exon to domain I of a group II self-splicing intron requires completion of the intron active site. EMBO J 18: 1025–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa M, Michel F, Westhof E 2000. A three-dimensional perspective on exon binding by a group II self-splicing intron. EMBO J 19: 5007–5018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curcio MJ, Belfort M 1996. Retrohoming: cDNA-mediated mobility of group II introns requires a catalytic RNA. Cell 84: 9–12 [DOI] [PubMed] [Google Scholar]
- Dai L, Toor N, Olson R, Keeping A, Zimmerly S 2003. Database for mobile group II introns. Nucleic Acids Res 31: 424–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA 2004. PDB2PQR: An automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res 32: W665–W667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erat MC, Sigel RKO 2008. Divalent metal ions tune the self-splicing reaction of the yeast mitochondrial group II intron Sc.ai5γ. J Biol Inorg Chem 13: 1025–1036 [DOI] [PubMed] [Google Scholar]
- Erat MC, Sigel RKO 2011. Methods to detect and characterize metal ion binding sites in RNA. Met Ions Life Sci 9: 37–100 [DOI] [PubMed] [Google Scholar]
- Erat MC, Zerbe O, Fox T, Sigel RKO 2007. Solution structure of domain 6 from a self-splicing group II intron ribozyme: A Mg2+ binding site is located close to the stacked branch adenosine. ChemBioChem 8: 306–314 [DOI] [PubMed] [Google Scholar]
- Eskes R, Yang J, Lambowitz AM, Perlman PS 1997. Mobility of yeast mitochondrial group II introns: Engineering a new site specificity and retrohoming via full reverse splicing. Cell 88: 865–874 [DOI] [PubMed] [Google Scholar]
- Freisinger E, Sigel RKO 2007. From nucleotides to ribozymes—a comparison of their metal ion binding properties. Coord Chem Rev 251: 1834–1851 [Google Scholar]
- Gallo S, Furler M, Sigel RKO 2005. In vitro transcription and purification of RNAs of different size. Chimia 59: 812–816 [Google Scholar]
- Glasoe PK, Long FA 1960. Use of glass electrodes to measure acidities in deuterium oxide. J Phys Chem 64: 188–190 [Google Scholar]
- Gordon PM, Fong R, Piccirilli JA 2007. A second divalent metal ion in the group II intron reaction center. Chem Biol 14: 607–612 [DOI] [PubMed] [Google Scholar]
- Güntert P, Mumenthaler C, Wüthrich K 1997. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol 273: 283–298 [DOI] [PubMed] [Google Scholar]
- Guo H, Karberg M, Long M, Jones JP III, Sullenger B, Lambowitz AM 2000. Group II introns designed to insert into therapeutically relevant DNA target sites in human cells. Science 289: 452–457 [DOI] [PubMed] [Google Scholar]
- Hansen MR, Hanson P, Pardi A 2000. Filamentous bacteriophage for aligning RNA, DNA, and proteins for measurement of nuclear magnetic resonance dipolar coupling interactions. Methods Enzymol 317: 220–240 [DOI] [PubMed] [Google Scholar]
- Hermann T, Patel DJ 1999. Stitching together RNA tertiary architectures. J Mol Biol 294: 829–849 [DOI] [PubMed] [Google Scholar]
- Iwahara J, Wojciak JM, Clubb RT 2001. Improved NMR spectra of a protein–DNA complex through rational mutagenesis and the application of a sensitivity optimized isotope-filtered NOESY experiment. J Biomol NMR 19: 231–241 [DOI] [PubMed] [Google Scholar]
- Jacquier A, Jacquesson-Breuleux N 1991. Splice site selection and role of the lariat in a group II intron. J Mol Biol 219: 415–428 [DOI] [PubMed] [Google Scholar]
- Jacquier A, Michel F 1987. Multiple exon-binding sites in class II self-splicing introns. Cell 50: 17–29 [DOI] [PubMed] [Google Scholar]
- Koch JL, Boulanger SC, Dib-Hajj SD, Hebbar SK, Perlman PS 1992. Group II introns deleted for multiple substructures retain self-splicing activity. Mol Cell Biol 12: 1950–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konforti BB, Abramovitz DL, Duarte CM, Karpeisky A, Beigelman L, Pyle AM 1998. Ribozyme catalysis from the major groove of group II intron domain 5. Mol Cell 1: 433–441 [DOI] [PubMed] [Google Scholar]
- Koradi R, Billeter M, Wüthrich K 1996. MOLMOL: A program for display and analysis of macromolecular structures. J Mol Graph 14: 51–55 [DOI] [PubMed] [Google Scholar]
- Korth MMT, Sigel RKO 2012. Unusually high-affinity Mg2+ binding at the AU-rich sequence within the antiterminator hairpin of a Mg2+ riboswitch. Chem Biodivers 9: 2035–2049 [DOI] [PubMed] [Google Scholar]
- Kruschel D, Sigel RKO 2008. Divalent metal ions promote the formation of the 5′-splice site recognition complex in a self-splicing group II intron. J Inorg Biochem 102: 2147–2154 [DOI] [PubMed] [Google Scholar]
- Lambowitz AM, Zimmerly S 2011. Group II introns: Mobile ribozymes that invade DNA. Cold Spring Harb Perspect Biol 3: 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lencastre Ad, Hamill S, Pyle AM 2005. A single active-site region for a group II intron. Nat Struct Mol Biol 12: 626–627 [DOI] [PubMed] [Google Scholar]
- Lumry R, Smith E, Glantz R 1951. Kinetics of carboxypeptidase action. I. Effect of various extrinsic factors on kinetic parameters. J Am Chem Soc 73: 4330–4340 [Google Scholar]
- Luy B, Marino JP 2000. Direct evidence for Watson–Crick base pairs in a dynamic region of RNA structure. J Am Chem Soc 122: 8095–8096 [Google Scholar]
- Marcia M, Pyle AM 2012. Visualizing group II intron catalysis through the stages of splicing. Cell 151: 497–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markley JL, Bax A, Arata Y, Hilbers CW, Kaptein R, Sykes BD, Wright PE, Wüthrich K 1998. Recommendations for the presentation of NMR structures of proteins and nucleic acids. IUPAC-IUBMB-IUPAB Inter-Union Task Group on the Standardization of Data Bases of Protein and Nucleic Acid Structures Determined by NMR Spectroscopy. J Biomol NMR 12: 1–23 [DOI] [PubMed] [Google Scholar]
- Martin W, Koonin EV 2006. Introns and the origin of nucleus–cytosol compartmentalization. Nature 440: 41–45 [DOI] [PubMed] [Google Scholar]
- Mattick JS, Gagen MJ 2001. The evolution of controlled multitasked gene networks: The role of introns and other noncoding RNAs in the development of complex organisms. Mol Biol Evol 18: 1611–1630 [DOI] [PubMed] [Google Scholar]
- Michel F, Umesono K, Ozeki H 1989. Comparative and functional anatomy of group II catalytic introns—a review. Gene 82: 5–30 [DOI] [PubMed] [Google Scholar]
- Michels WJ Jr, Pyle AM 1995. Conversion of a group II intron into a new multiple-turnover ribozyme that selectively cleaves oligonucleotides: Elucidation of reaction mechanism and structure/function relationships. Biochemistry 34: 2965–2977 [DOI] [PubMed] [Google Scholar]
- Milligan JF, Uhlenbeck OC, James E, Dahlberg JNA 1989. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol 180: 51–62 [DOI] [PubMed] [Google Scholar]
- Mohr G, Smith D, Belfort M, Lambowitz AM 2000. Rules for DNA target-site recognition by a lactococcal group II intron enable retargeting of the intron to specific DNA sequences. Genes Dev 14: 559–573 [PMC free article] [PubMed] [Google Scholar]
- Mörl M, Niemer I, Schmelzer C 1992. New reactions catalyzed by a group II intron ribozyme with RNA and DNA substrates. Cell 70: 803–810 [DOI] [PubMed] [Google Scholar]
- Nagaswamy U, Larios-Sanz M, Hury J, Collins S, Zhang Z, Zhao Q, Fox GE 2002. NCIR: A database of non-canonical interactions in known RNA structures. Nucleic Acids Res 30: 395–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura K, Terasawa H, Inagaki F 1996. An improved double-tuned and isotope-filtered pulse scheme based on a pulsed field gradient and a wide-band inversion shaped pulse. J Biomol NMR 8: 492–498 [DOI] [PubMed] [Google Scholar]
- Pechlaner M, Sigel RKO 2012. Characterization of metal ion–nucleic acid interactions in solution. Met Ions Life Sci 10: 1–42 [DOI] [PubMed] [Google Scholar]
- Peebles CL, Perlman PS, Mecklenburg KL, Petrillo ML, Tabor JH, Jarrell KA, Cheng HL 1986. A self-splicing RNA excises an intron lariat. Cell 44: 213–223 [DOI] [PubMed] [Google Scholar]
- Pyle AM 2010. The tertiary structure of group II introns: Implications for biological function and evolution. Crit Rev Biochem Mol Biol 45: 215–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle AM, Moran S, Strobel SA, Chapman T, Turner DH, Cech TR 1994. Replacement of the conserved G•U with a G–C pair at the cleavage site of the Tetrahymena ribozyme decreases binding, reactivity, and fidelity. Biochemistry 33: 13856–13863 [DOI] [PubMed] [Google Scholar]
- Qin PZ, Pyle AM 1999. Antagonistic substrate binding by a group II intron ribozyme. J Mol Biol 291: 15–27 [DOI] [PubMed] [Google Scholar]
- Ruffner DE, Stormo GD, Uhlenbeck OC 1990. Sequence requirements of the hammerhead RNA self-cleavage reaction. Biochemistry 29: 10695–10702 [DOI] [PubMed] [Google Scholar]
- Saenger W 1984. Principles of nucleic acid structure. Springer Verlag, New York [Google Scholar]
- Schmelzer C, Schweyen RJ 1986. Self-splicing of group II introns in vitro: Mapping of the branch point and mutational inhibition of lariat formation. Cell 46: 557–565 [DOI] [PubMed] [Google Scholar]
- Schnabl J, Sigel RKO 2010. Controlling ribozyme activity by metal ions. Curr Opin Chem Biol 14: 269–275 [DOI] [PubMed] [Google Scholar]
- Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM 2003. The Xplor-NIH NMR molecular structure determination package. J Magn Reson 160: 65–73 [DOI] [PubMed] [Google Scholar]
- Sharp PA 1985. On the origin of RNA splicing and introns. Cell 42: 397–400 [DOI] [PubMed] [Google Scholar]
- Sigel RKO 2005. Group II intron ribozymes and metal ions—a delicate relationship. Eur J Inorg Chem 2005: 2281–2292 [Google Scholar]
- Sigel RKO, Pyle AM 2007. Alternative roles for metal ions in enzyme catalysis and the implications for ribozyme chemistry. Chem Rev 107: 97–113 [DOI] [PubMed] [Google Scholar]
- Sigel H, Sigel RKO 2013. Metal ion interactions with nucleic acids and their constituent. In Comprehensive inorganic chemistry II (ed. Poepelmeier K, Reedjik J), pp. 623–660 Elsevier, Oxford, UK [Google Scholar]
- Sigel RKO, Vaidya A, Pyle AM 2000. Metal ion binding sites in a group II intron core. Nat Struct Mol Biol 7: 1111–1116 [DOI] [PubMed] [Google Scholar]
- Sigel RKO, Sashital DG, Abramovitz DL, Palmer AG III, Butcher SE, Pyle AM 2004. Solution structure of domain 5 of a group II intron ribozyme reveals a new RNA motif. Nat Struct Mol Biol 11: 187–192 [DOI] [PubMed] [Google Scholar]
- Sodhi R, Rajput YS 2003. Method for dialysis of samples in microliter volumes. Anal Biochem 315: 141–142 [DOI] [PubMed] [Google Scholar]
- Su LJ, Qin PZ, Michels WA, Pyle AM 2001. Guiding ribozyme cleavage through motif recognition: The mechanism of cleavage site selection by a group II intron ribozyme. J Mol Biol 306: 655–668 [DOI] [PubMed] [Google Scholar]
- Su LJ, Waldsich C, Pyle AM 2005. An obligate intermediate along the slow folding pathway of a group II intron ribozyme. Nucleic Acids Res 33: 6674–6687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swisher JF, Su LJ, Brenowitz M, Anderson VE, Pyle AM 2002. Productive folding to the native state by a group II intron ribozyme. J Mol Biol 315: 297–310 [DOI] [PubMed] [Google Scholar]
- Toor N, Keating KS, Taylor SD, Pyle AM 2008a. Crystal structure of a self-spliced group II intron. Science 320: 77–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toor N, Rajashankar K, Keating KS, Pyle AM 2008b. Structural basis for exon recognition by a group II intron. Nat Struct Mol Biol 15: 1221–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veen R, Arnberg AC, van der Horst G, Bonen L, Tabak HF, Grivell LA 1986. Excised group II introns in yeast mitochondria are lariats and can be formed by self-splicing in vitro. Cell 44: 225–234 [DOI] [PubMed] [Google Scholar]
- Varani G, Aboul-ela F, Allain FH-T 1996. NMR investigation of RNA structure. Prog Nucl Magn Reson Spectrosc 29: 51–127 [Google Scholar]
- Xiang Q, Qin PZ, Michels WJ, Freeland K, Pyle AM 1998. Sequence specificity of a group II intron ribozyme: Multiple mechanisms for promoting unusually high discrimination against mismatched targets. Biochemistry 37: 3839–3849 [DOI] [PubMed] [Google Scholar]
- Yang J, Zimmerly S, Perlman PS, Lambowitz AM 1996. Efficient integration of an intron RNA into double-stranded DNA by reverse splicing. Nature 381: 332–335 [DOI] [PubMed] [Google Scholar]
- Zhou H, Vermeulen A, Jucker FM, Pardi A 1999. Incorporating residual dipolar couplings into the NMR solution structure determination of nucleic acids. Biopolymers 52: 168–180 [DOI] [PubMed] [Google Scholar]
- Zwahlen C, Legault P, Vincent SJF, Greenblatt J, Konrat R, Kay LE 1997. Methods for measurement of intermolecular NOEs by multinuclear NMR spectroscopy: Application to a bacteriophage λ N-peptide/boxB RNA complex. J Am Chem Soc 119: 6711–6721 [Google Scholar]
- Zweckstetter M 2008. NMR: Prediction of molecular alignment from structure using the PALES software. Nat Protoc 3: 679–690 [DOI] [PubMed] [Google Scholar]
- Zweckstetter M, Bax A 2000. Prediction of sterically induced alignment in a dilute liquid crystalline phase: Aid to protein structure determination by NMR. J Am Chem Soc 122: 3791–3792 [Google Scholar]