

Tagging RNAs with biotin has numerous useful applications including affinity selections. This paper describes three simple and straightforward techniques for biotinylating the 3′ ends of RNA molecules.
Keywords: 3′-end modification, biotin, RNA tagging
Abstract
Biotinylation of RNA allows its tight coupling to streptavidin and is thus useful for many types of experiments, e.g., pull-downs. Here we describe three simple techniques for biotinylating the 3′ ends of RNA molecules generated by chemical or enzymatic synthesis. First, extension with either the Schizosaccharomyces pombe noncanonical poly(A) polymerase Cid1 or Escherichia coli poly(A) polymerase and N6-biotin-ATP is simple, efficient, and generally applicable independently of the 3′-end sequences of the RNA molecule to be labeled. However, depending on the enzyme and the reaction conditions, several or many biotinylated nucleotides are incorporated. Second, conditions are reported under which splint-dependent ligation by T4 DNA ligase can be used to join biotinylated and, presumably, other chemically modified DNA oligonucleotides to RNA 3′ ends even if these are heterogeneous as is typical for products of enzymatic synthesis. Third, we describe the use of ϕ29 DNA polymerase for a template-directed fill-in reaction that uses biotin-dUTP and, thanks to the enzyme's proofreading activity, can cope with more extended 3′ heterogeneities.
INTRODUCTION
Biotinylation of macromolecules is a powerful tool in molecular biology due to the almost irreversible interaction of biotin with streptavidin (Kd = 10−14 M) (Green 1990). For example, biotinylation of RNA is used for one-step enrichment of RNA–protein complexes from complex mixtures by binding to immobilized streptavidin (Ruby and Abelson 1988). Although biotinylated RNA can be generated by chemical synthesis, derivatization of enzymatically synthesized RNA is often necessary, for example, if long RNAs are to be used. Cotranscriptional biotinylation through incorporation of a biotinylated nucleotide is not site-specific, and the extent of biotinylation is not easy to control (Theissen et al. 1989). Biotin can also be incorporated at the 5′ end by enzymatic synthesis, but this precludes capping of the RNA (Huang et al. 2008). Specific biotinylation at the 3′ end can be achieved by NaIO4 oxidation followed by reaction of the resulting dialdehyde with a suitable biotin derivative (Qin and Pyle 1999; Willkomm and Hartmann 2005), but β-elimination is a competing reaction, resulting in the loss of the 3′ nucleotide (nt) and lowering the yield (Proudnikov and Mirzabekov 1996). Also, if the RNA is capped, the cap ribose will be biotinylated as well. T4-RNA ligase can join pCp derivatives, including biotinylated pCp, to the 3′ end of an acceptor RNA (Kore et al. 2009). However, the reaction is not very efficient, and the enzyme requires high concentrations of substrates for quantitative turnover.
In this study, we describe three simple enzymatic methods for efficient 3′ biotinylation of RNA.
RESULTS AND DISCUSSION
3′ Biotinylation by poly(A) polymerases
Poly(A) polymerases (PAPs) catalyze the extension of RNA 3′ ends with little or no specificity regarding the RNA sequence and have been used for end-labeling of RNA (Martin and Keller 1998). “Classical” PAPs have a pronounced specificity for the incorporation of AMP, using ATP as a precursor, but “noncanonical” PAPs extend the substrate spectrum (Schmidt and Norbury 2010). Two classical PAPs, the bovine (Wahle 1991) and the Saccharomyces cerevisiae enzyme (Lingner et al. 1991), the noncanonical PAP Cid1 from Schizosaccharomyces pombe (Rissland and Norbury 2007), and Escherichia coli PAP (Sippel 1973) were compared for their ability to extend an in vitro transcript with two different biotinylated ATP derivatives. On the basis of the mode of ATP binding seen in crystal structures of bovine PAP (PDB 1Q78) (Martin et al. 2000), N6- and 8-biotinylated ATP derivatives were chosen. Whereas the two classical PAPs failed to incorporate substantial amounts of either biotin-AMP (data not shown), both Cid1 and E. coli PAP, under optimized conditions (see Materials and Methods), incorporated N6-biotin-AMP but not 8-biotin-AMP (Fig. 1; data not shown). Whereas E. coli PAP incorporated a small number of biotin-AMP residues, each leading to a retardation in the gel comparable to the incorporation of several regular AMP residues, Cid1 unexpectedly polymerized the biotin-AMP to long tails (Fig. 1). PAP-dependent biotinylation was confirmed by binding to streptavidin (Fig. 2).
FIGURE 1.
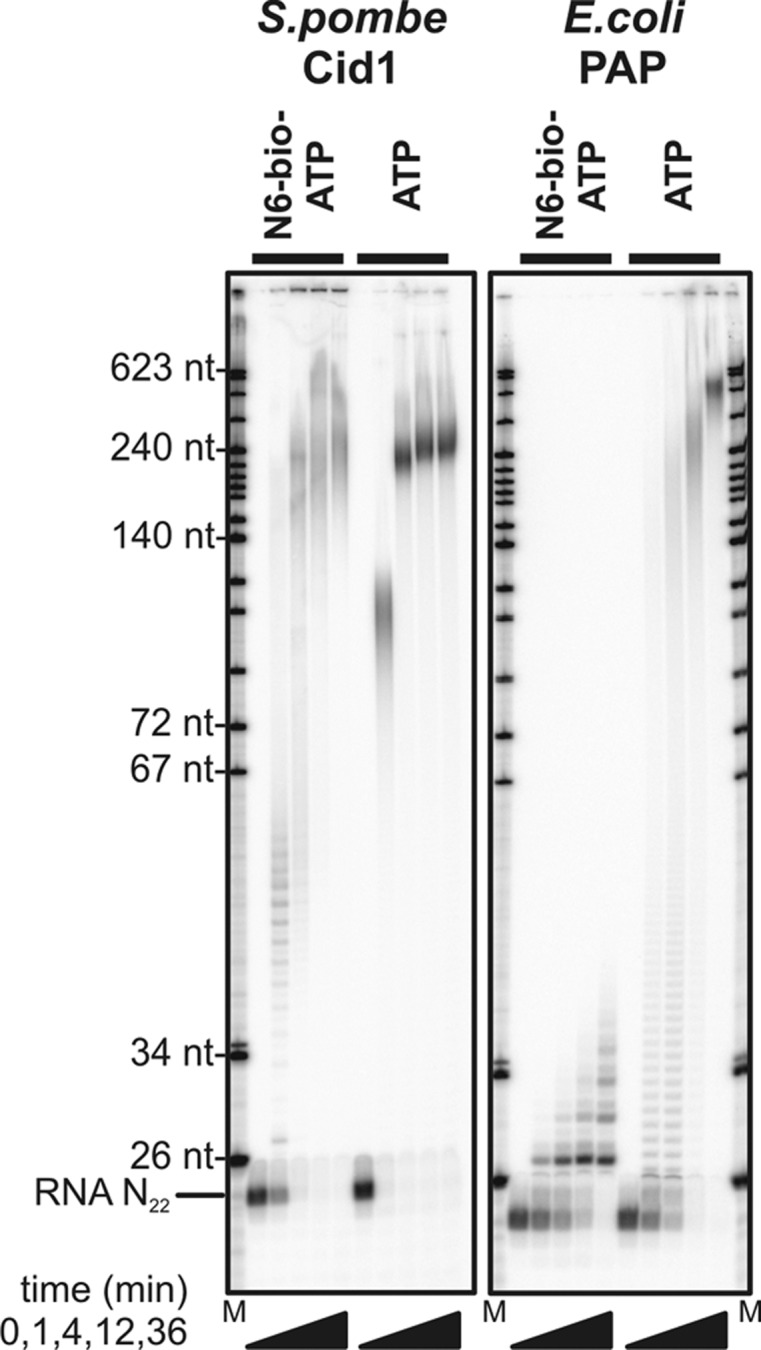
RNA tailing with biotin-ATP by S. pombe Cid1 and E. coli PAP. Two micromolar 5′ labeled RNA N22 was incubated with 0.5 mM ATP or N6-biotin-ATP as indicated and the following enzyme concentrations: 1 µM tCid1 (ATP reaction); 5 µM tCid1 (N6-biotin-ATP); 0.25 units/µL E. coli PAP (N6-biotin-ATP); and 0.025 units/µL E. coli PAP (ATP). After the incubation times indicated, aliquots were mixed with 8 M urea in 1× TBE and analyzed by denaturing polyacrylamide gel electrophoresis and phosphorimaging. DNA marker sizes are indicated.
FIGURE 2.

Streptavidin pull-down of enzymatically biotinylated RNAs. 5′-Labeled RNA N22 was biotinylated with E. coli PAP, and body-labeled RNA1600 was biotinylated with φ29 DNA polymerase. Both were purified and applied to streptavidin Sepharose beads or, as a control, to the same beads blocked with biotin (for details, see Materials and Methods). After two rounds of washing, radioactivity on the beads and all previously collected fractions was measured by liquid scintillation and plotted as the percentage of input. (fth) Flowthrough, supernatant of pelleted beads.
PAP-catalyzed biotinylation has several advantages: The same simple commercial source of biotin can be used for all kinds of RNA, and the enzymes can cope with 3′-sequence heterogeneity typical of in vitro transcripts and with high RNA concentrations: Up to 2 µM have been used, and higher concentrations are probably possible. In Figure 1, extension of the primer RNA with either polymerase was virtually complete (∼2.5% of substrate RNA left after 36 min), but, with longer RNAs, a relatively large fraction (∼20%–30%) was normally left unextended in Cid1 reactions (data not shown). We have also noticed that Cid1 is not very stable during storage, but a truncated version lacking the first 31 amino acids (tCid) (Rissland and Norbury 2007) was significantly more stable than the full-length enzyme. Stability could be further improved by choice of the storage buffer (see Materials and Methods). With Cid1, care has to be taken to avoid the synthesis of long biotinylated tails, which appear to cause aggregation problems. With E. coli PAP, the number of biotin residues incorporated per RNA primer can be kept very low (Fig. 1). However, depending on the purpose for which the RNA is to be used, the incorporation of more than one biotin residue might be undesirable, and achieving a specific monobiotinylation with a high yield would presumably be difficult.
Biotinylation with DNA ligase and degenerate splints
To incorporate single biotin residues into RNA, we explored the use of T4 DNA ligase. The enzyme can catalyze the ligation of single-stranded polynucleotides if these are joined by base-pairing to a “splint.” In this way, chemically synthesized DNA oligonucleotides carrying a variety of modifications, including biotinylation, can be ligated to RNA (Moore and Sharp 1992). T4 DNA ligase is very efficient even at low substrate concentrations. However, the enzyme's requirement for perfect junctions is a problem, as in vitro transcripts almost invariably have heterogeneous 3′ ends due to the tendency of RNA polymerases to incorporate one and sometimes more non-encoded nucleotides at the 3′ ends of their products. The sequence heterogeneity can be abolished by the incorporation of a self-cleaving ribozyme, but this entails additional complications, like removal of the 2′–3′ cyclic phosphate resulting from cleavage and the problem of controlling the efficiency of cleavage with long RNAs. It has also been reported that the use of PCR-derived templates carrying two 2′-O-methyl ribonucleotides instead of deoxynucleotides at the very 5′ end of the template reduces 3′-end addition of non-encoded nucleotides (Kao et al. 1999).
It should be possible to accommodate 3′-sequence variety of the RNA by using a combination of splints, one matching the 3′ end predicted by the template sequence plus a second, degenerate splint carrying a mixture of all four nucleotides at the position opposite the potential non-encoded 3′ nucleotide. The reaction would then have to be carried out under conditions allowing the rapid exchange of splints, as stable base-pairing independent of the degenerate position would be expected to block many RNA molecules in a nonligatable duplex. Therefore, conditions were sought that would minimize the stability of the splint–RNA hybrid but still allow ligation. The minimal length of the hybrid required for a two-way ligation at two different temperatures was found to be 6 bp; a 4-bp hybrid was unproductive, at least at the temperatures and with the nucleotide sequence tested (Fig. 3A).
FIGURE 3.

Tagging RNA by splint-dependent ligation. (A) 0.5 µM chemically synthesized, radiolabeled RNA N22 was ligated with 0.75 µM of different preannealed splint–donor duplex DNA combinations as indicated in the scheme at the top. Two different splints were used having a 6-nucleotide (nt) or 4-nt complementarity to the RNA 3′ end as indicated (splint 4 nt, splint 6 nt) (Table 1). Ligations were performed at two different temperatures for the times indicated. Reactions where stopped by mixing with 20 mM EDTA in formamide, and samples were analyzed by denaturing polyacrylamide gel electrophoresis. (B) The ligation reaction was carried out as in A with an enzymatically synthesized, radiolabeled RNA consisting of two species, 50 and 51 nt long. One splint (6 nt) (Table 1) had a 6-nt complementarity to the expected end of the 50-nt RNA, immediately followed by the sequence matching the tagging oligonucleotide. The second splint (6 ntN) (Table 1) had the same complementarity, followed by a position containing a mixture of all four nucleotides and then the sequence matching the tagging oligonucleotide. Ligations were carried out with either splint or the mixture of both as indicated. DNA marker sizes are indicated on the right.
A tagging oligonucleotide was then ligated to an in vitro transcript that consisted of the 50-nt species predicted by the template and a 51-nt species, presumably containing a mixture of nucleotides at the last position (Fig. 3B). Use of a perfect splint forming a 6-bp hybrid with the 50-nt species resulted in nearly quantitative but exclusive ligation of this shorter RNA, whereas use of a degenerate splint carrying an additional nucleotide solely guided ligation of the 51-nt RNA. The mixture of the perfect and the degenerate splint (i.e., five oligonucleotides, each at a concentration below the total transcript concentration) led to the almost quantitative ligation of both RNAs (Fig. 3B). Only 5% of the RNA species targeted by the splint oligonucleotides remained unmodified. Due to the presence of a small amount of even longer transcripts, 10% of the total amount of RNA escaped ligation.
3′-End modification by the ligation method is fast, simple, and very efficient. Transcript concentrations of up to 2 µM have been successfully ligated. Presumably, the method can be used to fuse the RNA of choice with tagging oligonucleotides carrying any desired chemical modification. A limitation is that in vitro transcripts sometimes carry up to three non-encoded nucleotides at the 3′ end. We doubt that ligation of such RNAs would be possible given the degeneracy of the splint that would be required to match the heterogeneity of the RNA. However, the added nucleotides are mostly A or C (Milligan et al. 1987), so the complexity of the splint mix might be reduced to drive ligation even of RNAs with more than one additional 3′-end nucleotide.
Biotinylation by ϕ29 DNA polymerase
DNA polymerases can use RNA as a primer. Thus, a DNA oligonucleotide can be annealed to the RNA 3′ end, and the DNA overhang will direct the extension of the RNA in a fill-in reaction (Huang and Szostak 1996). Moreover, the proofreading 3′-exonuclease activity associated with most DNA polymerases should also be able to remove any nucleotides at the RNA 3′ end not base-paired to the DNA oligonucleotide, for example, non-encoded nucleotides added during the transcription reaction. However, two DNA polymerases commonly used as reagents did not perform well in RNA extension: E. coli DNA polymerase I exhibited a low polymerization activity on the perfect RNA–DNA hybrids tested but was unable to proofread unpaired RNA 3′ ends. T4 DNA polymerase was better with respect to proofreading, as expected (Kornberg and Baker 1992), but worse in its polymerase activity (data not shown). Poor activity of DNA polymerase I on longer RNA primers has been noted before (Astatke et al. 1998).
Therefore, we turned to the DNA polymerase of bacteriophage ϕ29, which exhibits a robust exonucleolytic activity on RNA (Lagunavicius et al. 2008) and has previously been used for extension of long RNA primers (Jonstrup et al. 2006). A chemically synthesized RNA of 22 nt was annealed to either of two DNA oligonucleotides. Apart from a 3- or 4-nt 5′ overhang, the DNA had either a perfect match to the 3′ end of the RNA or a 2-nt mismatch (Fig. 4A). Extension of the RNA was fast and complete even with the mismatched DNA. Biotin-11-dUTP as a substitute for dTTP was easily accepted by ϕ29 DNA polymerase, as judged from a comparison of the product patterns with and without biotin-11-dUTP; the product containing the biotinylated nucleotide migrated more slowly during gel electrophoresis. The RNA was labeled nearly quantitatively (Fig. 4A). To determine whether ϕ29 DNA polymerase extends unpaired 3′ ends or resects them before elongation, we designed an RNA/DNA hybrid that permits the incorporation of biotin-11-dUTP only after previous exonucleolytic trimming of the RNA 3′ end (Fig. 4B). The product pattern obtained in the presence of the four regular dNTPs showed the anticipated net elongation of 2 nt. This was the result of trimming of the two mismatched nucleotides followed by the addition of 4 nt, because biotin was incorporated into the product when biotin-11-dUTP was present; incorporation was possible only at the first mismatch position. The product patterns were comparable to those obtained with a control hybrid that did not require proofreading for biotin incorporation (Fig. 4B). Thus, as expected, ϕ29 DNA polymerase proofreads a mismatched 3′ end before elongation.
FIGURE 4.

Tagging RNA by a fill-in reaction with φ29 DNA polymerase. (A) 0.5 µM chemically synthesized, radiolabeled RNA N22 was annealed to 0.75 µM of two different DNA oligonucleotides to form the hybrids indicated. One oligonucleotide (Table 1, “fill in”) was perfectly complementary to the RNA, the other (Table 1, “unpaired fill in”) had a 2-nt mismatch, as indicated in the scheme on top. (Arrow) The position where the incorporation of the biotinylated nucleotide is expected. The fill-in reaction was started by the addition of 7 units/µL φ29 DNA polymerase in the presence of 0.2 mM all four regular dNTPs or with dTTP substituted by 0.5 mM biotin-11-dUTP, as indicated. Samples where taken at the indicated time points, stopped by mixing with 20 mM EDTA in formamide and analyzed by denaturing polyacrylamide gel electrophoresis. DNA marker sizes are indicated on the right. The band labeled with a single dot corresponds to the addition of a nontemplated regular nucleotide; bands labeled with two dots correspond to the addition of a nontemplated biotinylated nucleotide. (B) A DNA polymerase reaction was carried out as in A but with different DNA oligonucleotides. The oligonucleotide shown in the scheme on the left (Table 1, “recessed fill in”) had a 2-nt mismatch to the RNA followed by an additional 2-nt 5′ overhang. The template position directing the incorporation of biotin-dUMP (labeled with an arrow) was “covered” by the mismatch, such that biotin incorporation required 3′ resection of the RNA by the proofreading activity of the DNA polymerase. The oligonucleotide shown on the right (Table 1, “fill in”) had a perfect match to the RNA 3′ end followed by a 4-nt 5′ overhang containing the position directing the incorporation of biotin-11-dUMP (marked with an arrow). DNA marker sizes are indicated on the right. Bands were labeled with one or two dots as in A. (C) A fill-in reaction was carried out as in A with four different DNA oligonucleotides differing in 2 nt at their 5′ ends (from left to right: “fill in,” “fill in” TG, “fill in” GG, “fill in” TT). Reactions were done in the presence of 0.2 mM all four dNTPs, i.e., biotin-11-dUTP was omitted. The DNA marker size is indicated on the left. (D) A fill-in reaction was carried out as in A including substitution of dTTP by biotin-11-dUTP. The concentration of the biotinylated nucleotide was varied as indicated. The regular dNTPs were present at 0.2 mM. The DNA marker sizes are indicated on the right. (E) Fill-in reactions were carried out as in A. An enzymatically synthesized, radiolabeled RNA (0.5 µM) of 200 nt was preincubated with 0.75 μM N200 “fill in” at 30°C for the times indicated. Amounts of φ29 DNA polymerase were varied as indicated. Biotin-11-dUTP was used at a concentration of 0.5 mM where indicated. The DNA marker sizes are indicated on the left.
In the DNA polymerase reactions shown so far, a large fraction of the products was elongated by an additional nontemplated nucleotide, either a “regular” nucleotide (product labeled with one dot in Fig. 4A,B) or a second biotinylated nucleotide (labeled with two dots in Fig. 4A,B). Evidence for the presence of a second biotinylated nucleotide was (i) the gel retardation it caused, (ii) absence of the band in reactions lacking biotin-dUTP, and (iii) its disappearance with lower concentrations of biotin-dUTP (see below). In attempts to reduce this undesired modification, the sequence of the template and the concentration of biotin-11-dUTP were altered. In Figure 4C, the results of fill-in reactions with different template ends can be compared. A 5′–TG–3′ sequence at the 5′ end of the template prevented the incorporation of an additional nucleotide at the 3′ end, and 5′–TT–3′ worked almost as well. The concentration of biotin-11-dUTP could be lowered substantially to reduce the amount of additionally incorporated biotin with no loss of fully elongated product (Fig. 4D). Thus, monobiotinylated RNA can be prepared by ϕ29 DNA polymerase with close to 100% yield if the nucleotide at the 5′ end of the template DNA is chosen well and the amount of the modified nucleotide in the reaction mixture is low. Figure 4E shows that a high efficiency of biotinylation is also possible with a longer in vitro transcript, although relatively high amounts of enzyme have to be used: 5% of synthetic RNA N22 and 15% of the in vitro transcript remained unmodified over the time course of the experiments (Fig. 4D,E). Biotinylation of the RNA N22 and another transcript of 1600 nt was also verified by binding to streptavidin (Fig. 2; data not shown).
Compared with the two other methods, templated 3′ elongation by ϕ29 DNA polymerase is as simple and efficient as the ligase method, and it is more versatile in its ability to cope with 3′-sequence heterogeneity. However, its extension to other chemical modifications may be less straightforward, as the modifications have to be incorporated by the polymerase reaction via a modified nucleotide. Given the quite high polymerase concentrations needed for appropriate elongation, the RNA concentration that can be labeled may be limited, but this has not been tested.
MATERIALS AND METHODS
Proteins
E. coli Bl21 (DE3) was transformed with an expression plasmid for tCid1, an N-terminally truncated variant of the S. pombe Cid1 enzyme (Rissland and Norbury 2007) (a kind gift of Chris Norbury, University of Oxford). Cells were grown in M9 minimal medium supplemented with 0.4% glucose and 0.8% peptone at 37°C to OD600 = 1. The culture was diluted with one volume of ice-cold medium supplemented with 1 mM IPTG and incubated overnight at 16°C. Cells were harvested, resuspended in lysis buffer (500 mM KCl, 20 mM HEPES at pH 7.4, 3 mM MgCl2, 10% glycerol) supplemented with DNase I (10 μg/mL), lysozyme (0.2 mg/mL) and PMSF (1 mM) and disrupted by a French press. The lysate was centrifuged at 30,000g for 1 h, and the supernatant was applied to a GSH-Sepharose (GE Healthcare) (0.5 mL beads per l culture medium). Elution was performed with 20 mM glutathione in lysis buffer. Eluates were supplemented with 1 mM EDTA and 100 units of PreScission protease (GE Healthcare), dialyzed against 150 mM potassium acetate, 20 mM MES-KOH (pH 6.5), and 10% glycerol and applied to a second GSH-Sepharose column. The flowthrough was immediately applied to a 1-mL MonoS column (GE Healthcare) and eluted with a linear gradient to 1 M potassium acetate in the same buffer. Pure protein was stored in 150 mM potassium acetate, 20 mM HEPES-KOH (pH 7.5), 1 mM magnesium acetate, 25% glycerol, and 0.05% (v/v) NP-40 at −80°C. NP-40 and increased glycerol concentration improved the stability of the enzyme during storage. Full-length Cid1 was obtained from NEB. Bovine poly(A) polymerase (C-terminal deletion variant PAP513) (Martin and Keller 1996) and yeast poly(A) polymerase were gifts of U. Kühn. E. coli PAP was obtained from NEB. ϕ29 DNA polymerase (1300 units/µL) was a kind gift from Arunas Lagunavicius (Fermentas).
RNA preparation
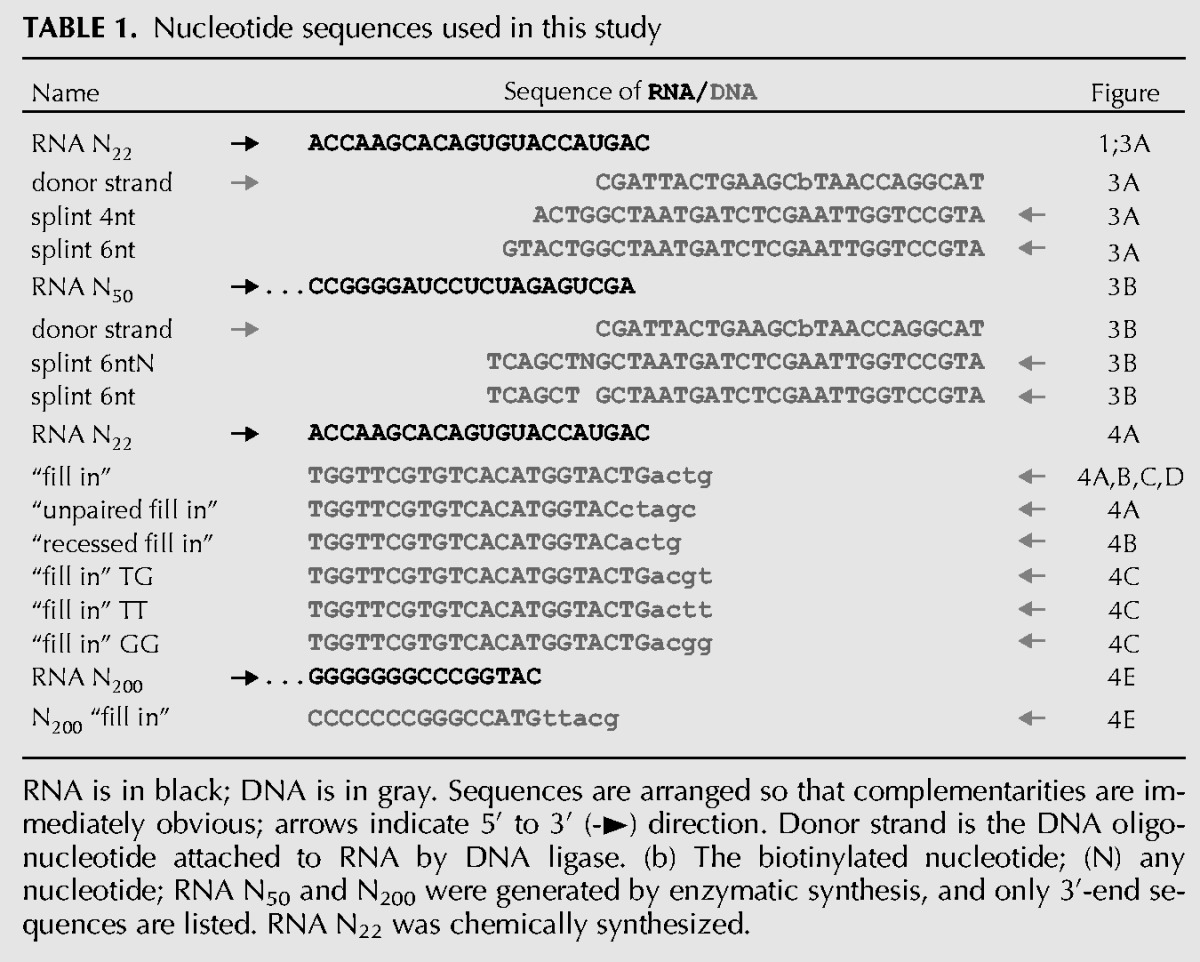
RNAs were synthesized with T3 RNA polymerase (Stratagene) according to the manufacturer's recommendation with 2 mM NTPs and trace-labeled with 20 μCi of [α-32P]UTP (Amersham Biosciences) per 25-μL reaction. After 2 h at 37°C, the template was digested with 1.5 units of RNase-free DNase I (Roche Applied Science). RNA was purified through a Sephadex G50 spin column, phenol-extracted, ethanol-precipitated, and dissolved in water. RNA 3′ sequences are listed in Table 1. Chemically synthesized RNA N22 (Table 1, biomers.net) was 5′-labeled with [γ-32P]ATP and T4 polynucleotide kinase (NEB).
TABLE 1.
Nucleotide sequences used in this study
Poly(A) polymerase reactions
Up to 2 µM RNA was incubated with 1–5 µM tCid1 in 100 mM potassium acetate, 20 mM Tris-HCl (pH 8.0), 2 mM magnesium acetate, 0.05% NP-40 at 38°C for the indicated time in the presence of 0.5 mM ATP or N6-ATP analog (N6-[(6-amino)hexyl]-amino-ATP-biotin) or 8-ATP analog (8-[(6-amino)hexyl]-amino-ATP-Biotin; both from Jena Bioscience, Germany). E. coli poly(A) polymerase (NEB) was used at 0.025–0.25 units/µL under the same conditions but at 37°C. No incorporation of biotin was observed in the reaction buffer supplied with the enzyme. Bovine and yeast poly(A) polymerase were used under the conditions described previously (Lingner et al. 1991; Wahle 1991).
φ29 DNA polymerase reactions
RNA substrate and DNA template were annealed in 1× ϕ29 buffer (Fermentas) at 37°C and cooled to 30°C. The reaction mix contained 0.2 mM dCTP, dATP, dGTP; and 0.5 mM γ-[N-(biotin-6-amino-hexanoyl)]-5-aminoallyl-dUTP (biotin-11-dUTP; Jena Bioscience). The reaction was started by the addition of 7 units/µL ϕ29 DNA polymerase unless otherwise indicated. Incubation was at 30°C.
Ligation reactions
Four hundred units of T4-DNA ligase (NEB) was used per 15-µL reaction under the conditions recommended by the manufacturer. Annealing of RNA, biotinylated DNA oligonucleotide, and splint was performed in 1× T4 ligase buffer by heating for 5 min at 65°C and cooling down to 30°C within 10 min. The reaction times and nucleic acid concentrations used are indicated in the figure legends.
Streptavidine pull-down
Biotinylated RNA was purified through a Sephadex G25 spin column, diluted with one volume of 2× PK-buffer (100 mM Tris-HCl at pH 7.6, 200 mM NaCl, 1% SDS, 2 mM EDTA) and treated with 0.1 mg/mL proteinase K (Merck), followed by phenol extraction, ethanol precipitation from 2 M ammonium acetate, solubilization, and ethanol precipitation from 0.3 M potassium acetate (pH 5.3). The RNA was dissolved in DEPC-treated water. For the pull-down, it was diluted to 10–50 nM in 150 µL of 500 mM potassium acetate, 20 mM Tris-HCl (pH 8.0), 5 mM DTT, 20 units of Ribolock (Fermentas), 0.1 mg/mL yeast total RNA (Roche), mixed with 20 µL of streptavidine beads (GE healthcare) and incubated for 15 min at 20°C. Beads were washed twice in 150 μL of binding buffer, and radioactivity in the various fractions was measured by liquid scintillation counting. To ascertain the specificity of binding, beads were pretreated with 150 µL of 1 mM biotin in a control reaction.
ACKNOWLEDGMENTS
We are most grateful to Uwe Kühn for the gift of purified S. cerevisiae and bovine poly(A) polymerases; to Arunas Lagunavicius for highly concentrated ϕ29 DNA polymerase and critical reading of an earlier version of the manuscript; and to Chris Norbury for the tCid1 expression clone. This work was supported by a grant from the Deutsche Forschungsgemeinschaft (DFG).
REFERENCES
- Astatke M, Ng K, Grindley NDF, Joyce CM 1998. A single side chain prevents Escherichia coli DNA polymerase I (Klenow fragment) from incorporating ribonucleotides. Proc Natl Acad Sci 95: 3402–3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green NM 1990. Avidin and streptavidin. Method Enzymol 184: 51–67 [DOI] [PubMed] [Google Scholar]
- Huang Z, Szostak JW 1996. A simple method for 3′-labeling of RNA. Nucleic Acids Res 24: 4360–4361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, He J, Zhang Y, Guo Y 2008. Synthesis of biotin–AMP conjugate for 5′ biotin labeling of RNA through one-step in vitro transcription. Nat Protoc 3: 1848–1861 [DOI] [PubMed] [Google Scholar]
- Jonstrup SP, Koch J, Kjems J 2006. A microRNA detection system based on padlock probes and rolling circle amplification. RNA 12: 1747–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao C, Zheng M, Rüdisser S 1999. A simple and efficient method to reduce nontemplated nucleotide addition at the 3′ terminus of RNAs transcribed by T7 RNA polymerase. RNA 5: 1268–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kore AR, Charles I, Yang L, Kuersten S 2009. Synthesis and activity of modified cytidine 5′-monophosphate probes for T4 RNA ligase 1. Nucleosides Nucleotides Nucleic Acids 28: 292–302 [DOI] [PubMed] [Google Scholar]
- Kornberg A, Baker TA 1992. DNA replication. Freeman, New York [Google Scholar]
- Lagunavicius A, Kiveryte Z, Zimbaite-Ruskuliene V, Radzvilavicius T, Janulatitis A 2008. Duality of polynucleotide substrates for φ29 DNA polymerase: 3′ → 5′ RNase activity of the enzyme. RNA 14: 503–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingner J, Radtke I, Wahle E, Keller W 1991. Purification and characterization of poly(A) polymerase from Saccharomyces cerevisiae. J Biol Chem 266: 8741–8746 [PubMed] [Google Scholar]
- Martin G, Keller W 1996. Mutational analysis of mammalian poly(A) polymerase identifies a region for primer binding and a catalytic domain, homologous to the family X polymerases, and to other nucleotidyl transferases. EMBO J 15: 2593–2603 [PMC free article] [PubMed] [Google Scholar]
- Martin G, Keller W 1998. Tailing and 3′-end labeling of RNA with yeast poly(A) polymerase and various nucleotides. RNA 4: 226–230 [PMC free article] [PubMed] [Google Scholar]
- Martin G, Keller W, Doublie S 2000. Crystal structure of mammalian poly(A) polymerase in complex with an analog of ATP. EMBO J 19: 4193–4203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan JF, Groebe DR, Witherell GW, Uhlenbeck OC 1987. Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res 15: 8783–8798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Sharp PA 1992. Site-specific modification of pre-mRNA: The 2′-hydroxyl groups at the splice sites. Science 256: 992–997 [DOI] [PubMed] [Google Scholar]
- Proudnikov D, Mirzabekov A 1996. Chemical methods of DNA and RNA fluorescent labeling. Nucleic Acids Res 24: 4535–4542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin PZ, Pyle AM 1999. Site-specific labeling of RNA with fluorophores and other structural probes. Methods 18: 60–70 [DOI] [PubMed] [Google Scholar]
- Rissland O, Norbury CJ 2007. Efficient RNA polyuridylation by noncanonical poly(A) polymerases. Mol Cell Biol 27: 3612–3624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby S, Abelson J 1988. An early hierarchic role of U1 small nuclear ribonucleoprotein in spliceosome assembly. Science 242: 1028–1035 [DOI] [PubMed] [Google Scholar]
- Schmidt M-J, Norbury CJ 2010. Polyadenylation and beyond: Emerging roles for noncanonical poly(A) polymerases. Wiley Interdiscip Rev RNA 1: 142–151 [DOI] [PubMed] [Google Scholar]
- Sippel AE 1973. Purification and characterization of adenosine triphosphate:ribonucleic acid adenyltransferase from Escherichia coli. Eur J Biochem 37: 31–40 [DOI] [PubMed] [Google Scholar]
- Theissen G, Richter A, Lukacs N 1989. Degree of biotinylation in nucleic acids estimated by a gel retardaton assay. Anal Biochem 179: 98–105 [DOI] [PubMed] [Google Scholar]
- Wahle E 1991. Purification and characterization of a mammalian polyadenylate polymerase involved in the 3′ end processing of messenger RNA precursors. J Biol Chem 226: 3131–3139 [PubMed] [Google Scholar]
- Willkomm DK, Hartmann RK 2005. 3′-Terminal attachment of fluorescent dyes and biotin. In Handbook of RNA biochemistry (ed. Hartmann RK, et al. ), pp. 86–94 Wiley-VCH, Weinheim, Germany [Google Scholar]