

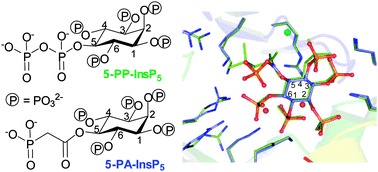
 Replacing the diphosphate (PP) motif of 5-PP-InsP5 with an α-phosphonoacetic acid (PA) ester gives 5-PA-InsP5, which is recognised by the catalytic domain of diphosphoinositol pentakisphosphate kinase 2.
Replacing the diphosphate (PP) motif of 5-PP-InsP5 with an α-phosphonoacetic acid (PA) ester gives 5-PA-InsP5, which is recognised by the catalytic domain of diphosphoinositol pentakisphosphate kinase 2.
Abstract
We synthesised analogues of diphosphoinositol polyphosphates (PP-InsPs) in which the diphosphate is replaced by an α-phosphonoacetic acid (PA) ester. Structural analysis revealed that 5-PA-InsP5 mimics 5-PP-InsP5 binding to the kinase domain of PPIP5K2; both molecules were phosphorylated by the enzyme. PA-InsPs are promising candidates for further studies into the biology of PP-InsPs.
myo-Inositol polyphosphates (InsPs) regulate many cellular processes.1 The chemical synthesis of InsPs, plus various analogues and probes, has been vital to the functional characterisation of enzymes and receptors involved in InsP metabolism and signalling.2 In the early 1990s, InsPs containing diphosphate groups were discovered.3 These diphosphoinositol polyphosphates (PP-InsPs, Fig. 1a) have been proposed to regulate a range of cellular functions including vesicular trafficking, apoptosis, DNA repair, telomere maintenance, insulin signalling and stress responses.4 However, little is known about the molecular mechanisms that underlie these effects. There is evidence that PP-InsPs can act through non-covalent interactions with receptor proteins.5 A more provocative mechanistic proposal involves non-enzymatic transfer of the β-phosphoryl of the diphosphate (PP) group in PP-InsPs to a phosphorylated serine residue in target proteins, to give a diphosphorylated protein.6
Fig. 1. (a) Origin of diphosphoinositol polyphosphates. IP5K, inositol pentakisphosphate 2-kinase; IP6K, inositol hexakisphosphate 5-kinase; PPIP5K, diphosphoinositol pentakisphosphate kinase. (b) Structures of phosphonoacetic acid ester (PA) analogues 1, 2 and 3. Bn = benzyl.

Several chemical syntheses of PP-InsPs have been published,7 although no analogues or probes based on PP-InsPs have been described. We set out to synthesise analogues that cannot transfer phosphoryl groups, yet can mimic non-covalent interactions of PP-InsPs with target proteins. Such stabilised analogues could potentially be used for the mechanistic dissection of PP-InsP signalling. They would also be open to synthetic modifications such as tagging with fluorophores or photoreactive groups and the synthesis of biotinylated and resin-immobilised versions.
Diphosphate mimics such as methylenebisphosphonates and imidodiphosphates, often used in non-hydrolysable analogues of nucleotide diphosphates,8 are not easily introduced onto the secondary hydroxyl groups of myo-inositol. We therefore chose to explore α-phosphonoacetic acid (phosphonoacetate, PA) esters9 as mimics of the PP motif in PP-InsPs. The PA ester can be introduced by straightforward O-acylation, without disturbing the myo-inositol stereochemistry. Due to the higher activation energy for cleavage of a C–P bond versus an O–P bond,10 it is not plausible that PA-InsPs could act as non-enzymatic phosphoryl donors. We identified 5-PA-InsP4 (1) and 5-PA-InsP5 (2) (Fig. 1b) as initial targets. Noting previous evidence that a 2-O-benzyl group enhances the anti-tumour and pro-apoptotic properties of InsP5,11 we also synthesised 2-O-benzyl-5-PA-InsP4 (3).
The syntheses of 1, 2 and 3 (Scheme 1) began with diol 4, directly available from myo-inositol in multi-gram quantities.12 For the synthesis of 1, alkylation of 4 was used to introduce a benzyl ether selectively at the axial O-2 atom, giving 5. The origin of the regioselectivity observed in this reaction is not clear. The free 5-OH group of 5 was then acylated using dibenzylphosphonoacetic acid and EDAC giving 6. Careful removal of the butane diacetal (BDA) groups using aqueous trifluoroacetic acid gave tetraol 7. Phosphitylation of 7 using bis(benzyloxy)diisopropylaminophosphine, followed by oxidation gave 8, and global deprotection by hydrogenolysis gave 5-PA-InsP4 (1). For the synthesis of 5-PA-InsP5 (2), regioselective carbodiimide-mediated acylation of the equatorial 5-hydroxyl group of diol 4 with dibenzylphosphonoacetic acid gave alcohol 9. Removal of the BDA groups to give pentaol 10, followed by phosphitylation/oxidation to give 11 and hydrogenolytic deprotection as before gave 5-PA-InsP5 (2).
Scheme 1. Synthesis of PA analogues 1, 2 and 3. Reagents and conditions: a. BnBr, NaH, DMF, 55%; b. (BnO)2P(O)CH2COOH, DCC, CH2Cl2, 73%; c. (BnO)2P(O)CH2COOH, EDAC, DMAP, CH2Cl2, 95%; d. TFA, H2O, 5 min, 50–62%; e. (i) (BnO)2PNPr2 i, 5-phenyl-1H-tetrazole, CH2Cl2; (ii) mCPBA, CH2Cl2, 84–95%; f. H2, Pd(OH)2/C, MeOH, H2O, 92%; g. H2, Pd(OH)2/C, MeOH, H2O, 50 psi, 79%; h. H2, Pd(OH)2/C, MeOH, aqueous triethylammonium bicarbonate, 79%. Bn = benzyl.
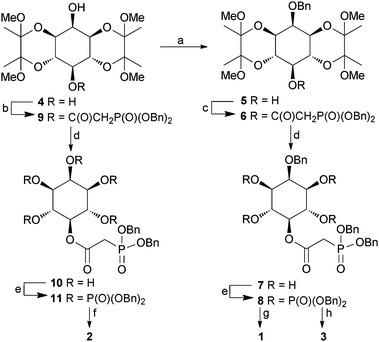
2-O-Bn-5-PA-InsP4 (3) was prepared by selective deprotection of 8. We have previously noted that triethylammonium ions suppress hydrogenolytic cleavage of 2-O-benzyl ethers in myo-inositol derivatives. For 8, we found that hydrogenolysis in the presence of aqueous triethylammonium bicarbonate cleaved the benzylphosphate esters within 30 minutes, while the 2-O-benzyl ether was unaffected, giving 3 as the triethylammonium salt.
To determine whether 1, 2 and 3 could act as mimics of 5-PP-InsP5, we examined whether they were recognized by the highly specific catalytic domain of human PPIP5K2 (PPIP5K2KD).13 To increase the throughput of these experiments, we took advantage of the “reversibility” of the kinases that synthesise PP-InsPs.14 We used luciferin/luciferase to assay the ATP that was formed when PPIP5K2KD was incubated with ADP and 1,5-[PP]2-InsP4. Analogues 1, 2 and 3 inhibited ATP formation (Fig. 2) indicating that they were accommodated within the enzyme's active site; 5-PA-InsP5 (2) showed the strongest inhibition, while 5-PA-InsP4 (1) was a ten-fold weaker inhibitor. This is consistent with the finding that 5-PP-InsP4 is a poor substrate for PPIP5K2.13 Unexpectedly, the 2-O-benzyl analogue 3 was only 3-fold less potent than 5-PA-InsP5 (2), suggesting that an axial 2-O-benzyl group can partly compensate for the loss of the axial 2-phosphate. This may have implications for inhibitor design.
Fig. 2. Inhibition of human PPIP5K2 by analogues 1, 2 and 3 in the presence of 100 nM 1,5-[PP]2-InsP4.

We obtained an X-ray structure of the most potent inhibitor, 5-PA-InsP5 (2) in complex with PPIP5K2KD (Fig. 3). Superposition of this structure with that of PPIP5K2KD in complex with 5-PP-InsP5 (PDB accession code ; 3T9D), yielded RMSDs (root mean square deviations) of 0.13 Å for the Cα atoms of the residues in the protein (Fig. 3a). A simulated-annealing omit map shows that 5-PA-InsP5 binds at the same site as 5-PP-InsP5 with a similar orientation (Fig. 3a) although there were some conformational changes of the phosphate groups on the side of inositol carbon positions C-4 to C-6. This reflects the substitution of a PA group for the diphosphate group at C-5; as a result of accommodating 5-PA-InsP5 in the active site rather than 5-PP-InsP5 a magnesium atom is lost and the side chains of residues Lys214, Arg262, Arg273 and Arg281 slightly adjust their orientations (Fig. 3a and b). On the other hand, the position of the 1-phosphate of 5-PA-InsP5 relative to the γ-phosphorus atom of AMPPNP and the key catalytic residue Lys248 is very similar to the 1-phosphate of 5PP-InsP5 in complex with PPIP5K2 [ref. 13 and Fig. 3]. Indeed, by using a gel electrophoresis method,15 phosphorylation of 5-PA-InsP5 could be detected (Fig. 3d) although, due to limitations of sensitivity, an accurate comparison of reaction rates with natural substrate, InsP6 (Fig. 1a), was not possible.
Fig. 3. Binding of 5-PA-InsP5 (2) to human PPIP5K2KD (a–c) and phosphorylation of 2 by PPIP5K2KD (d). (a) 5-PA-InsP5 (2, ball-and-stick model) and its interacting residues (stick model) for hPPIP5K2 are shown in light blue for carbon, blue for nitrogen, orange and red for phosphate groups. Residues that make contacts with 5-PP-InsP5 (green stick) are also shown in green carbon. Magnesium atoms are shown as spheres. C-4 and C-5 in the inositol ring are labelled. (b) Residues that interact with PA or PP are shown. Bond distances (dashed lines) are denoted in Å. (c) Key catalytic residues are shown in stick model. Inositol phosphates and nucleotide cofactors are shown in ball-and-stick model. Colours are as (a). Also shown are catalytic magnesium atoms (magenta spheres). C-1 of the inositol ring is labelled. Bond distances (dashed lines) are denoted in Å. (d) Phosphorylation of 5-PA-InsP5 by PPIP5K2KD.

In conclusion, PA esters 1, 2 and 3 may be regarded as first-generation synthetic mimics of PP-InsPs. Proof-of-principle that PA substitution need not prevent target recognition was obtained from studies using PPIP5K2. By using other protecting group arrangements, the synthetic methods can be extended to give other PA-InsPs including [PA]n-InsPs with multiple PA esters, e.g. as mimics of [PP]2-InsP4 isomers. By scaling up the enzymatic phosphorylation of 5-PA-InsP5 (Fig. 3d), it should be possible to prepare sufficient quantities of 1-PP-5-PA-InsP4 to dissect the separate roles of the two diphosphate groups in 1,5-[PP]2-InsP4. Stabilised PP-InsP mimics such as PA-InsPs could help to identify effects of PP-InsPs that do not involve protein dephosphorylation. They also have the potential for further synthetic elaboration, to provide new probes for the study of InsP and PP-InsP signalling.
We thank the Wellcome Trust (Programme Grant 082837 to AMR and BVLP) and NIEHS/NIH for support. X-ray Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-beamline at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
Footnotes
References
- Hatch A. J., York J. D. Cell. 2010;143:1030–1030.e1. doi: 10.1016/j.cell.2010.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best M. D., Zhang H., Prestwich G. D. Nat. Prod. Rep. 2010;27:1403–1430. doi: 10.1039/b923844c. [DOI] [PubMed] [Google Scholar]
- Europe-Finner G. N., Gammon B., Newell P. C., Menniti F. S., Miller R. N., Putney Jr. J. W., Shears S. B., Stephens L., Radenberg T., Thiel U., Vogel G., Khoo K. H., Dell A., Jackson T. R., Hawkins P. T., Mayr G. W. Biochem. Biophys. Res. Commun. J. Biol. Chem. J. Biol. Chem. 1991;1993;1993;81268268:191–196. 3850–3856, 4009–4015. [Google Scholar]
- For access to the literature, see; Saiardi A., in: Phosphoinositides II: The Diverse Biological Functions: Subcellular Biochemistry 59, ed. T. Balla, M. Wymann and J. D. York, Springer, Dordrecht, Heidelberg, London, New York, 2012, ch. 14, pp. 413–443 [Google Scholar]
- Lee Y. S., Huang K., Quiocho F. A., O'Shea E. K. Nat. Chem. Biol. 2008;4:25–32. doi: 10.1038/nchembio.2007.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandari R., Saiardi A., Ahmadibeni Y., Snowman A. M., Resnick A. C., Kristiansen T. Z., Molina H., Pandey A., Werner Jr J. K., Juluri K. R., Xu Y., Prestwich G. D., Parang K., Snyder S. H. Proc. Natl. Acad. Sci. U. S. A. 2007;104:15305–15310. doi: 10.1073/pnas.0707338104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck J. R., Reddy K. K., Ye J., Saady M., Mioskowski C., Shears S. B., Tan Z., Safrany S., Reddy K. M., Reddy K. K., Falck J. R., Albert C., Safrany S. T., Bembenek M. E., Reddy K. M., Reddy K. K., Falck J. R., Bröcker M., Shears S. B., Mayr G. W., Zhang H., Thompson J., Prestwich G. D. J. Am. Chem. Soc. Tetrahedron Lett. Biochem. J. Org. Lett. 1995;1997;1997;2009;1173832711:12172–12175. 4951–4952, 553–560, 1551–1554. [Google Scholar]
- Elliott T. S., Slowey A., Ye Y., Conway S. J. Med. Chem. Commun. 2012;3:735–751. [Google Scholar]
- Lambert R. W., Martin J. A., Thomas G. J., Duncan I. B., Hall M. J., Heimer E. P., Vincent S., Grenier S., Valleix A., Salesse C., Lebeau L., Mioskowski C. J. Med. Chem. J. Org. Chem. 1989;1998;3263:367–374. 7244–7257. doi: 10.1021/jm00122a014. [DOI] [PubMed] [Google Scholar]
- Quinn J. P., Kulakova A. N., Cooley N. A., McGrath J. W. Environ. Microbiol. 2007;9:2392–2400. doi: 10.1111/j.1462-2920.2007.01397.x. [DOI] [PubMed] [Google Scholar]
- Falasca M., Chiozzotto D., Godage H. Y., Mazzoletti M., Riley A. M., Previdi S., Potter B. V. L., Broggini M., Maffucci T. Br. J. Cancer. 2010;102:104–114. doi: 10.1038/sj.bjc.6605408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montchamp J. L., Tian F., Hart M. E., Frost J. W., J. Org. Chem., 1996, 61 , 3897 –3899Riley A. M., Jenkins D. J., Potter B. V. L., Carbohydr. Res., 1998, 314 , 277, –281Baeschlin D. K., Chaperon A. R., Green L. G., Hahn M. G., Ince S. J., Ley S. V., Chem.–Eur. J., 2000, 6 , 172 –186 , ; For an optimised method, see ESI . [DOI] [PubMed] [Google Scholar]
- Wang H., Falck J. R., Hall T. M., Shears S. B. Nat. Chem. Biol. 2012;8:111–116. doi: 10.1038/nchembio.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voglmaier S. M., Bembenek M. E., Kaplin A. I., Dorman G., Olszewski J. D., Prestwich G. D., Snyder S. H. Proc. Natl. Acad. Sci. U. S. A. 1996;93:4305–4310. doi: 10.1073/pnas.93.9.4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losito O., Szijgyarto Z., Resnick A. C., Saiardi A. PLoS One. 2009;4:e5580. doi: 10.1371/journal.pone.0005580. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.