

The C2A and C2B domains of Doc2 have markedly distinct Ca2+-sensing and lipid-binding properties. When expressed in wild-type neurons, a Ca2+-ligand mutant form of Doc2, that lacks apparent Ca2+-binding activity and is constitutively bound to the plasma membrane, results in an anomalous enhancement of evoked and spontaneous synaptic transmission.
Abstract
Double C2-domain protein (Doc2) is a Ca2+-binding protein implicated in asynchronous and spontaneous neurotransmitter release. Here we demonstrate that each of its C2 domains senses Ca2+; moreover, the tethered tandem C2 domains display properties distinct from the isolated domains. We confirm that overexpression of a mutant form of Doc2β, in which two acidic Ca2+ ligands in the C2A domain and two in the C2B domain have been neutralized, results in markedly enhanced asynchronous release in synaptotagmin 1–knockout neurons. Unlike wild-type (wt) Doc2β, which translocates to the plasma membrane in response to increases in [Ca2+]i, the quadruple Ca2+-ligand mutant does not bind Ca2+ but is constitutively associated with the plasma membrane; this effect is due to substitution of Ca2+ ligands in the C2A domain. When overexpressed in wt neurons, Doc2β affects only asynchronous release; in contrast, Doc2β Ca2+-ligand mutants that constitutively localize to the plasma membrane enhance both the fast and slow components of synaptic transmission by increasing the readily releasable vesicle pool size; these mutants also increase the frequency of spontaneous release events. Thus, mutations in the C2A domain of Doc2β that were intended to disrupt Ca2+ binding result in an anomalous enhancement of constitutive membrane-binding activity and endow Doc2β with novel functional properties.
INTRODUCTION
Ca2+ signaling plays a crucial role in neuronal communication at chemical synapses. Proteins with Ca2+-binding motifs (i.e., C2 domains, E-F hands) are integral components of this system, serving as Ca2+ sensors that regulate numerous steps in the synaptic vesicle cycle, including priming, fusion, recycling, and replenishment (Burgoyne and Morgan, 2007; Haucke et al., 2011). Synaptotagmin 1 (syt1) is a tandem C2-domain protein that has been proposed to serve as a Ca2+ sensor for fast, synchronous release of neurotransmitter (Brose et al., 1992; Littleton et al., 1993; Broadie et al., 1994). In the mouse, the large synchronous component of synaptic vesicle (SV) release is absent in syt1-knockout (KO) neurons, but the slow asynchronous component persists (Figure 1A; Geppert et al., 1994; Maximov and Sudhof, 2005). Moreover, it is now apparent that the slow phase of transmission is actually up-regulated in the KOs (Nishiki and Augustine, 2004b; Liu et al., 2009). The kinetic differences observed for synchronous and asynchronous release can involve the proximity between Ca2+ sensors for release and Ca2+ channels (Rosenmund et al., 2003; Hu et al., 2013). However, recent studies using caged Ca2+ also support a dual Ca2+ sensor model in which slow transmission is regulated by a second sensor (or sensors) with slower kinetics and a higher sensitivity for Ca2+ (Burgalossi et al., 2010).
FIGURE 1:
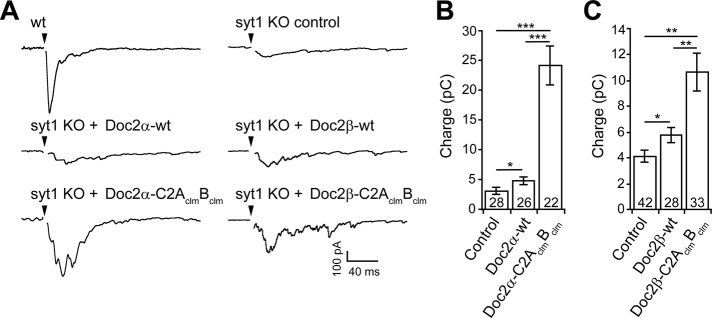
Expression of the C2AclmBclm mutant form of Doc2α or Doc2β enhances asynchronous release in syt1-KO hippocampal neurons. (A) Representative traces of evoked EPSCs recorded from wt, syt1-KO, and syt1-KO neurons expressing wt or the C2AclmBclm mutant form of Doc2α or Doc2β. (B) In syt1-KO neurons, which exhibit only asynchronous release, the total EPSC charge transfer (3.1 ± 0.6 pC, n = 28) was slightly increased by expression of Doc2α-wt (4.9 ± 0.6 pC, n = 26) but was markedly increased by expression of Doc2α-C2AclmBclm (24.1 ± 3.2 pC, n = 22). (C) Similarly, expression of Doc2β-wt (5.8 ± 0.6 pC, n = 28) slightly increased the total charge transfer, whereas Doc2β-C2AclmBclm (10.7 ± 1.5 pC, n = 33) had a larger effect as compared with the noninfected control (4.1 ± 0.4 pC, n = 42). Data are represented as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, Student's t test. The number of cells, n, is indicated in the bar graphs. In all cases recordings were obtained using neurons from three or more independent litters of mice.
We recently reported that two closely related isoforms (α and β) of the double C2-domain protein (Doc2) regulate asynchronous release (Yao et al., 2011). Both isoforms bind the soluble N-ethylmaleimide–sensitive factor attachment protein receptor (SNARE) proteins syntaxin 1A and SNAP-25B and promote vesicle fusion in vitro in response to Ca2+ (Groffen et al., 2010; Yao et al., 2011). Moreover, Ca2+•Doc2 interacts with membranes on time scales consistent with asynchronous release (Yao et al., 2011). Indeed, modulation of the levels of Doc2α in neurons specifically affected the slow component of neurotransmitter release without affecting fast release. Overexpression of a mutant form of Doc2α in which two Ca2+ ligands had been neutralized in each of its C2-domains, C2A and C2B, resulted in enhanced asynchronous release (Yao et al., 2011). In principle, this result could be explained by recent studies indicating that the same Ca2+-ligand mutations in the C2A domain of Doc2β increase, rather than impair, the apparent affinity for Ca2+, resulting in activation of the protein and translocation to the plasma membrane at resting levels of Ca2+ (Groffen et al., 2006; Friedrich et al., 2008).
The α and β isoforms of Doc2 harbor a set of 10 conserved acidic residues shown to coordinate Ca2+ in other C2-domain–containing proteins (Nalefski and Falke, 1996). A third isoform, Doc2γ, lacking three of the five acidic residues in the C2A domain, does not appear to bind Ca2+ (Fukuda and Mikoshiba, 2000). The Ca2+-dependent lipid-binding properties of the C2A domain of Doc2β are well established (Kojima et al., 1996; Fukuda and Mikoshiba, 2000; Friedrich et al., 2008; Groffen et al., 2010). However, it remains unclear whether the C2B domain also binds Ca2+: there are conflicting reports on whether C2B can bind to 1,2-dioleoyl-sn-glycero-3-[phospho-l-serine] (PS)–containing liposomes in response to Ca2+ (Kojima et al., 1996; Groffen et al., 2010).
Here we report that both C2 domains of Doc2β sense Ca2+ and bind to membranes. Remarkably, only the isolated C2B was able to bind Ca2+ in the absence of anionic phospholipids; Ca2+ binding to C2A was apparent only in the presence of these lipids. In addition, we observed striking biochemical differences between the tandem and isolated C2 domains of Doc2β, suggesting that intramolecular interactions modulate the Ca2+-sensing properties of the protein. We found that like Doc2α, a mutant form of Doc2β harboring Ca2+-ligand mutations in both C2A and C2B enhances asynchronous neurotransmitter release when expressed in syt1-KO neurons, but here we show this mutant fails to bind Ca2+. Unlike wild-type (wt) Doc2β, expression of this mutant in wt neurons affects both the fast and slow components of SV release, apparently via an increase in the size of the readily releasable pool (RRP) of vesicles. Overexpression of this mutant also resulted an increase in the frequency of spontaneous fusion events. By analyzing additional Ca2+-ligand mutants, we found that the observed gain of function for evoked and spontaneous release mapped to the C2A domain and was correlated with the ability of the mutants to constitutively localize to the plasma membrane. Of importance, the C2A mutations described in this study, which have been widely used to study Doc2, do not disrupt activity of the protein but instead result in an anomalous gain of function.
RESULTS
Ca2+-ligand mutations in Doc2α and β enhance asynchronous neurotransmitter release
Syt1-KO neurons were used to investigate the role of Doc2 in asynchronous neurotransmission without contamination by the fast phase of release. In the first series of experiments, hippocampal neurons from syt1-KO mice were infected with lentivirus expressing wt Doc2α or β or mutant forms of each in which two aspartic acid residues in each C2 domain (D181, D183, D342, and D344 in Doc2α or D218, D220, D357, and D359 in Doc2β), predicted to coordinate Ca2+, were mutated to asparagines. Because these are presumptive Ca2+-ligand mutations (clms), these constructs were designated Doc2-AclmBclm.
Consistent with previous results (Yao et al., 2011), expression of Doc2α-wt in syt1-KO neurons enhanced the magnitude of the excitatory postsynaptic current (EPSC) charge transfer (Figure 1, A and B), and expression of Doc2α-C2AclmBclm further increased the extent of asynchronous release (Figure 1, A and B). Similarly, Doc2β expression resulted in an increase in asynchronous release (Figure 1, A and C), and expression of the Doc2β-C2AclmBclm mutant further enhanced release. In addition, the delay between stimulation and the peak of the EPSC (i.e., time to peak) was not affected by the expression of any of the four Doc2 constructs (Supplemental Table S1), indicating that the synchronous component was not rescued by expression of wt Doc2α or β or the respective C2AclmBclm mutants. These observations confirm the gain of function of these Ca2+-ligand mutations during asynchronous transmission and extend this observation to both α and β isoforms (Yao et al., 2011).
Ca2+-, lipid-, and target-SNARE–binding properties of Doc2β
The ability of Doc2β to bind Ca2+ was directly measured by isothermal titration calorimetry (ITC). For these experiments, truncated forms of Doc2β consisting of the tandem or isolated C2 domains were used. We began by analyzing the tandem C2 domains (Figure 2A) and observed that C2AB and C2AclmB (D218N, D220N) displayed robust exothermic Ca2+-binding activity (Figure 2B). These data were fitted and analyzed using a “one set of sites” binding model (Supplemental Figure S1), and the results were unexpected; Doc2β C2AB and C2AclmB were nearly identical in their thermodynamic properties, indicating that the mutations within C2A did not significantly alter the Ca2+-binding characteristics of Doc2β under these assay conditions (i.e., in the absence of anionic phospholipids). Conversely, analogous mutations in the C2B domain (Doc2β C2ABclm) virtually abolished the apparent Ca2+-binding activity.
FIGURE 2:

Unusual Ca2+- and membrane-binding properties of the C2 domains of Doc2β. (A) Doc2β and rabphilin 3A share significant sequence identity (>65%); therefore the C2A (Protein Data Bank 2K3H) and C2B (Protein Data Bank 2CM6) structures for rabphilin 3A were used for illustrative purposes to represent Doc2β. The linker between the C2 domains is unstructured and was added to the illustration. The blue spheres represent Ca2+ ions found in the rabphilin structures. The mutated amino acids are shown below each structure; the absence of blue spheres represents the predicted loss of Ca2+-binding activity. (B, E) Representative ITC titrations for each construct; the data from three separate experiments are summarized in Supplemental Table S1. (C, F) The ability of each Doc2 mutant to bind to protein-free liposomes as a function of [Ca2+] was measured via cosedimentation assays. The supernatant and pellet fractions were collected and separated by SDS–PAGE. Proteins stained with Coomassie blue. (D, G) The supernatant signal is normalized to the 0 Ca2+ lane, and mean ± SEM is plotted vs. [Ca2+]; n ≥ 3.
The Ca2+-dependent interaction of Doc2β with membranes requires the presence of anionic phospholipids (e.g., phosphatidylserine [PS]; Kojima et al., 1996; Yao et al., 2011; see Figure 5A later in this article), and negatively charged lipids are likely to alter the Ca2+-sensing activity of the protein, as documented for syt1 (Brose et al., 1992). To address this issue, we measured the ability of Doc2β to cosediment with liposomes composed of 25% PS, 30% 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (PE), and 45% 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (PC) as a function of [Ca2+] (Figure 2C). Consistent with the ITC data, wt C2AB and C2AclmB had the greatest Ca2+ sensitivity for binding to lipids. Conversely, C2ABclm, which showed little to no apparent Ca2+-binding activity via ITC, bound efficiently to liposomes in response to Ca2+; the apparent affinity for Ca2+ was only slightly diminished by the mutations (Figure 2D and Supplemental Figure S3A). C2AclmBclm did not bind to liposomes in a Ca2+-dependent manner, but analysis of the pellet fraction revealed a significant amount of Ca2+-independent membrane-binding activity, suggestive of some degree of constitutive activation of the protein. Indeed, all of the Doc2β mutants tested—C2AclmB, C2ABclm, and C2AclmBclm—displayed this property to different degrees; we return to this point later. The appearance of protein in the pellet fraction was largely dependent on the presence of liposomes and was not the product of protein aggregation (Supplemental Figure S2).
FIGURE 5:

Ca2+-independent binding of wt and mutant forms of Doc2β to membranes composed of complex lipid mixtures. (A) The lipid requirements for Ca2+-dependent binding of Doc2β to liposomes were examined using different lipid mixtures. In each sample, 4 μM protein was incubated with liposomes (4 mM lipid) in the absence (–, 0.2 mM BAPTA) or presence (+) of 300 μM free Ca2+ and subjected to centrifugation. Supernatant (S) and pellet (P) fractions were collected and proteins separated by SDS–PAGE and stained with Coomassie blue. (B) Protein intensities normalized to total input signal (unpublished data) and plotted as mean ± SD, n = 2.
We then analyzed the isolated C2 domains of Doc2β. Surprisingly, C2A displayed no apparent Ca2+-binding activity (Figure 2E), but, consistent with a previous study (Kojima et al., 1996), this domain displayed robust Ca2+-dependent membrane-binding activity (Figure 2, F and G, and Supplemental Figure S3B). Note that ITC analysis predicted ∼1 Ca2+-binding site for C2AB and C2AclmB (Supplemental Figure S1C; N value); however, the Hill slope calculated from the Ca2+-dependent lipid binding curve was >1, indicative of cooperative binding (Supplemental Figure S3C). Together with the ITC findings, these data suggest a novel mechanism in which the C2A domain has an absolute requirement for PS (or some anionic lipid) in order to bind Ca2+. In marked contrast, the C2B domain readily bound Ca2+ in the absence of lipids (Figure 2E), but this activity did not result in efficient Ca2+-dependent interactions with membranes (Figure 2, F and G). Mutating Ca2+ ligands in the isolated C2B domain abolished its ability to sense Ca2+ as measured by ITC (Figure 2, E and F). Identical mutations in the C2A domain reduce, but do not abolish, Ca2+-dependent membrane-binding activity (Figure 2, F and G). Analysis of the Ca2+ titrations revealed that the responses of isolated C2Aclm and C2B became saturated at lower [Ca2+] than wt isolated C2A; however, this apparent increase in Ca2+ sensitivity does not result in efficient membrane interactions (Supplemental Figure S3). Together these data reveal that both C2 domains of Doc2 operate as Ca2+-sensing modules but have distinct Ca2+-binding requirements and properties.
The ability of Doc2 to bind target (t)-SNAREs in a Ca2+-promoted manner has been established (Friedrich et al., 2008; Fukuda et al., 2009; Groffen et al., 2010; Sato et al., 2010; Yao et al., 2011; Yu et al., 2013), but it is unclear whether both C2 domains contribute to this Ca2+ response. To address this question, we incubated Doc2β Ca2+-ligand mutants with PS-free proteoliposomes harboring the t-SNARE proteins syntaxin-1A and SNAP-25B and subjected them to centrifugation through a density gradient. All four Doc2β variants bound t-SNAREs weakly in the absence of Ca2+. Ca2+ promoted the binding of Doc2β C2AB, C2AclmB, and C2ABclm to t-SNAREs but had no effect on the C2AclmBclm mutant (Figure 3A). The extent of Doc2 binding to t-SNARES was determined by normalizing the intensity of the Doc2 band to the syntaxin signal in each lane (Figure 3B). C2AB and C2ABclm bound with a stoichiometry of >1:1, suggesting that multiple copies of Doc2β may interact with each t-SNARE heterodimer. Both isolated C2 domains interacted with t-SNAREs; binding of C2A, but not C2B, was enhanced by Ca2+ (Supplemental Figure S4A).
FIGURE 3:

Doc2β-C2AclmBclm does not bind t-SNAREs or accelerate membrane fusion in vitro in response to Ca2+. (A) Representative gel of a t-SNARE coflotation assay. Liposomes bearing syntaxin 1A and SNAP-25B were incubated with 0.2 mM BAPTA and 30 μM wt and mutant forms of C2AB (from Doc2β) in the absence (–; 0.2 mM BAPTA) or presence (+) of 0.3 mM free Ca2+ and floated through a density gradient. The vesicles were collected and the proteins separated via SDS–PAGE and stained with Coomassie blue. (B) The Doc2β signals were normalized to the syntaxin signals. (C) In vitro fusion assays: 3 μM of the indicated Doc2β proteins were added to reactions containing 0.2 mM BAPTA, preassembled t-SNARE vesicles, and vesicle-SNARE vesicles. Samples were incubated for 10 min at 37°C before injection of Ca2+ (arrow); the final free [Ca2+] was 300 μM. (D, F) Percentage Fmax determined at t = 60 min. (E, F) Experiments repeated using “split t-SNAREs,” with syntaxin reconstituted and 1 μM SNAP-25 added as soluble protein. Representative data from ≥3 independent trials are shown in A, C, and E; the mean ± SEM for each experiment is displayed in B, D, and F.
Next we compared wt and mutant forms of Doc2β in in vitro fusion assays. Initially, proteins were tested for their ability to stimulate fusion in response to Ca2+ in a standard assay in which t-SNARE vesicles were prepared using preassembled syntaxin1A:SNAP-25B heterodimers (Figure 3, C and D). Consistent with the t-SNARE and membrane-binding experiments, C2AB, C2AclmB, and C2ABclm were able to stimulate fusion upon addition of Ca2+; only C2AclmBclm was inactive. The cytosolic domain of VAMP2 (cdV) blocked fusion, confirming that lipid mixing was mediated by trans-SNARE pairing (Supplemental Figure S4B).
Given that molecules that aggregate SNARE-bearing proteoliposomes stimulate fusion in the standard assay (Hui et al., 2011), we also tested the Doc2 constructs in a “split” t-SNARE fusion assay in which syntaxin had been reconstituted into the t-SNARE vesicles but SNAP-25 was added in-trans in a soluble form. In this variant of the fusion assay, Doc2, or syt1, must first fold SNAP-25 onto syntaxin to drive fusion (Bhalla et al., 2006; Hui et al., 2011; Yao et al., 2011), and proteoliposome aggregation alone is without effect (Hui et al., 2011). Again, we observed the same pattern for Ca2+-stimulated fusion; C2AB, C2AclmB, and C2ABclm, but not C2AclmBclm, were able to promote lipid mixing (Figure 3, E and F).
The inability of C2AclmBclm to stimulate fusion in either assay system suggests that, in vivo, C2AclmBclm can regulate release via interactions with other effector molecules that are not present in the in vitro fusion assay.
Cellular localization of wt and Ca2+-ligand mutant forms of Doc2
Doc2β is located in the cytosol at resting [Ca2+]i and rapidly translocates to the plasma membrane via two independent mechanisms: indirectly, via protein–protein interactions with Munc13-1 after phorbol ester treatment (Duncan et al., 1999), or directly, via Ca2+-triggered binding of its tandem C2 domains to anionic phospholipids upon depolarization and influx of Ca2+ (Groffen et al., 2004, 2006; Malkinson and Spira, 2006). It has been observed that Doc2β-C2AclmB localizes to the plasma membrane in chromaffin cells under resting conditions ([Ca2+]i ∼ 50 nM), and Groffen et al. (2006) proposed that this was due to enhanced Ca2+ sensitivity imparted by the mutations. This interpretation was supported by Ca2+-dependent liposome aggregation assays using isolated C2Aclm (Friedrich et al., 2008).
Because our data are not consistent with the idea that resting [Ca2+]i can activate any of the Doc2β constructs examined, we postulated that the C2AclmB mutant might localize to the plasma membrane of cells via a novel constitutive interaction with plasma membrane–bound Munc13-1 (Orita et al., 1997). The in vitro assays described earlier (ITC, cosedimentation, and proteoliposome fusion assays) were performed using truncated Doc2 proteins lacking the Munc13-1–interacting domain (MID). To test whether Munc13 played a role in constitutive plasma membrane localization in cells, we generated a series of full-length Doc2β Ca2+-ligand mutants in which the MID was deleted or scrambled (Figure 4A; to distinguish the truncated C2-domain proteins used for in vitro experiments from the wt or MID mutants used in cell-based experiments, the latter are denoted with a Doc2β prefix). Doc2β-wt or -MID mutants were fused to green fluorescent protein (GFP), transfected into PC12 cells, and visualized via confocal microscopy (Figure 4B).
FIGURE 4:

Constitutive plasma membrane localization of mutant forms of Doc2β. (A) The wt and Ca2+-ligand mutant forms of Doc2β were modified by deleting or scrambling the MID. (B) These constructs were fused to GFP, transfected into PC12 cells, and imaged using confocal microscopy before and after depolarization with 60 mM KCl. (C) Increases in [Ca2+]i were monitored using the high-affinity Ca2+-sensor GCaMP-6F. GCaMP-6F was expressed in PC12 cells, and the fluorescence intensity was monitored before and after depolarization with KCl. (D) Live-cell images of HEK 293 cells expressing the indicated GFP-Doc2β fusion protein.
To stimulate translocation of Doc2β to the plasma membrane of PC12 cells, we depolarized the cells with 60 mM KCl (1.2 mM [Ca2+]o); images were obtained before and after stimulation (Figure 4B). Influx of Ca2+ after KCl depolarization was confirmed using the high-affinity Ca2+ sensor GCaMP-6F (Chen et al., 2013; Figure 4C). The two constructs that bore mutations in the C2A domain, Doc2β-C2AclmB and Doc2β-C2AclmBclm, were localized to the plasma membrane under resting conditions. To determine whether these constructs bound to the plasma membrane in a Ca2+-dependent manner, we simultaneously treated cells with 10 mM ethylene glycol tetraacetic acid (EGTA), 50 μM 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA)–acetoxymethyl ester (AM), and 30 μM cyclopiazonic acid (CPA) to remove extracellular Ca2+ and deplete internal stores. Based on the use of Fura2-AM, the resting [Ca2+]i was determined to be ∼90 nM; after treatment the [Ca2+]i was <20 nM (i.e., well below the 140 nM Kd of Fura2). Under these stringent conditions Doc2β-C2AclmB dissociated only slightly from the plasma membrane and Doc2β-C2AclmBclm remained tightly bound (Supplemental Figure S5). These data are qualitatively consistent with our in vitro observations that C2AclmBclm and C2AclmB constitutively bind to membranes in a Ca2+-independent manner.
In contrast, Doc2β-C2AB and Doc2β-C2ABclm were localized to the cytosol under resting conditions and translocated to the plasma membrane after depolarization with KCl. Deletion or scrambling of the MID domain did not alter the membrane localization of any of the Doc2β constructs examined. These results rule out interactions with Munc13-1 at the plasma membrane as the mechanism underlying the constitutive membrane localization observed for Doc2β-C2AclmB and Doc2β-C2AclmBclm. Of importance, these experiments indicate that binding of these mutants to the plasma membrane is not mediated by resting levels of [Ca2+]i; instead, these interactions appear to be constitutive and independent of Ca2+. Moreover, Doc2β-C2AclmBclm and Doc2β-C2AclmB do not exhibit enhanced interactions with t-SNAREs under any conditions (Figures 2 and 3), so these interactions cannot underlie the constitutive localization of these mutants at the plasma membrane. In addition, we found that Doc2β-C2AclmB and Doc2β-C2AclmBclm were also constitutively associated with the plasma membrane in HEK 293 cells (Figure 4D), suggesting that the binding partner for these constructs is not cell-type specific.
In an effort to identify the mechanism underlying the constitutive membrane association of Doc2β-C2AclmBclm and Doc2β-C2AclmB, we screened the Ca2+-independent and -dependent binding properties of each Ca2+ ligand mutant in cosedimentation assays using alternative lipid mixtures: 0% PS, 30% PE, 70% PC; 25% PS, 30% PE, 45% PC; total brain extract; or polar brain extract (Figure 5). Assays were performed in the presence or absence of 300 μM free Ca2+ (Figure 5A), and the protein band intensities were normalized to the total protein (unpublished data) and plotted (Figure 5B). Of interest, when complex lipid mixtures were used, the amount of constitutive binding increased for each of the Doc2β proteins tested. This effect was most pronounced for the three mutant forms of the protein, which all exhibited similar degrees of Ca2+-independent liposome-binding activity. However, the pattern observed in these cosedimentation assays does not perfectly correlate with that observed in PC12 cells; that is, Doc2β-C2AclmB and Doc2β-C2AclmBclm, but not Doc2β-C2ABclm, were constitutively associated with the plasma membrane in PC12 cells. We have not been able to reconstitute the exact same pattern of Ca2+-independent binding for the Doc2 mutants using artificial vesicles in vitro, either because cells contain lipids that are not properly reconstituted in vitro (e.g., because they are labile or are not correctly clustered or organized) or because our artificial vesicles lack necessary protein components.
Asynchronous neurotransmitter release in neurons expressing wt and mutant forms of Doc2
To characterize the role of Ca2+-binding to each C2-domain of Doc2β, we recorded evoked EPSCs from syt1-KO hippocampal neurons infected with lentivirus encoding the C2AclmB or C2ABclm mutant forms of Doc2β; wt and C2AclmBclm served as controls (Figure 6A). Expression of Doc2β-C2AclmB or Doc2β-C2AclmBclm significantly enhanced EPSC charge transfer (Figure 6B). Cells expressing Doc2β-C2AB and Doc2β-C2ABclm, which translocate to the plasma membrane of PC12 cells only after depolarization (Figure 4B), displayed limited increases in asynchronous release (Figure 6B). Remarkably, the two mutant forms of Doc2 that were constitutively bound to the plasma membrane, Doc2β-C2AclmB and Doc2β-C2AclmBclm (Figure 4B), were the only two constructs that gave rise to robust increases in asynchronous release (Figure 6). Thus the molecular interactions that mediate constitutive association of the two mutants with the membrane potentially underlie their ability to enhance asynchronous release.
FIGURE 6:

Ca2+-ligand mutations in C2A underlie Doc2β gain of function independent of interactions with Munc13-1. (A) Representative evoked EPSCs recorded from syt1-KO neurons expressing wt-C2AB, C2AclmB, C2ABclm, or C2AclmBclm mutant forms of Doc2β. For each of these constructs, the MID domain was intact (wt), scrambled, or deleted. (B) Bar graph showing the total EPSC charge transfer recorded from syt1-KO neurons expressing each construct in A. Data are represented as mean ± SEM. N.S., p > 0.05; *p < 0.05; Student's t test. The number of cells, n, is indicated in the bar graphs; in all cases recordings were obtained using neurons from three or more independent litters of mice.
For completeness, we also measured asynchronous release from cells expressing each Doc2β construct but with scrambled or deleted MIDs (Figure 6A). Neither scrambling nor deletion of the MID led to any significant difference in EPSC charge transfer (Figure 6B), indicating that the Munc13-1•Doc2β complex is unlikely to mediate enhanced asynchronous release. These findings further support the existence of an unidentified Doc2β binding partner. We note that endogenous wt Doc2 is present in the neurons used for these experiments, and our binding assays indicate that more than one Doc2 molecule can bind to t-SNAREs, so it is possible that Doc2-C2AclmBclm could recruit endogenous Doc2 to the plasma membrane.
In wt neurons, overexpression of Doc2 Ca2+-ligand mutants enhances evoked synchronous and asynchronous transmission and increases the frequency of spontaneous fusion events
To further characterize the properties of Doc2β-C2AclmBclm, we expressed it in wt hippocampal neurons and recorded evoked EPSCs (Figure 7A). Surprisingly, and in sharp contrast to the wt protein (Yao et al., 2011), overexpression of Doc2β-C2AclmBclm significantly increased the peak amplitude and total charge transfer as compared with control neurons (Figure 7, B and C).
FIGURE 7:

Expression of Doc2β-C2AclmBclm, but not Doc2β-wt, enhances both synchronous and asynchronous release in wt hippocampal neurons. (A) Average evoked EPSCs recorded from control neurons and wt neurons expressing Doc2β-wt or Doc2β-C2AclmBclm. (B) In wt neurons, the EPSC peak amplitude (control: 481 ± 34 pA, n = 38) was increased by expression of Doc2β- C2AclmBclm (744 ± 120 pA, n = 34); Doc2β-wt was without effect (430 ± 31 pA, n = 32). (C) The total EPSC charge transfer (control: 8.3 ± 0.8 pC, n = 38) was slightly increased by expression of Doc2β-wt (9.4 ± 0.9 pC, n = 32) and strongly increased by expression of Doc2β-C2AclmBclm (14.4 ± 2.6 pC, n = 34). (D) Average normalized cumulative EPSC charge transfer over 0.5 s from wt neurons with and without expression of Doc2β-wt or Doc2β-C2AclmBclm. (E–H) Double-exponential fitting of cumulative EPSC charge transfer traces. The fast charge (control: 6.3 ± 0.7 pC, n = 38) was increased by expression of Doc2β-C2AclmBclm (11.7 ± 2.2 pC, n = 34) but not affected by Doc2β-wt (6.6 ± 0.7 pC, n = 22) (E). The slow charge (1.3 ± 0.1 pC, n = 38) was slightly increased by expression of Doc2β-wt (2.0 ± 0.3 pC, n = 32) and markedly increased by expression of Doc2β- C2AclmBclm (3.9 ± 0.9 pC, n = 34) (G). Time constants (τ) for the fast (F) and slow (H) phases were the same in all three groups. In all bar graphs, data are represented as mean ± SEM. *p < 0.05, **p < 0.01, Student's t test. The number of cells, n, is indicated in the bar graphs; in all cases recordings were obtained using neurons from four or more independent litters of mice.
To determine whether Doc2β-C2AclmBclm preferentially affects either the fast or slow component of transmission in wt neurons, we plotted the cumulative charge transfer versus time (Figure 7D) and quantified the size and time constants of each component by fitting each curve with a double-exponential function (Figure 7, E–H). Consistent with previous results, neurons infected with virus expressing Doc2β-wt did not display an altered fast component of release (Figure 7, E and F) but did exhibit significant increases in the amplitude of the slow component (Figure 7, G and H). In contrast, expression of Doc2β-C2AclmBclm greatly increased both the fast and slow phases of transmission (Figure 7, E–H). Neither Doc2β-wt nor Doc2β-C2AclmBclm led to significant changes in the time constants for fast (Figure 7F) or slow (Figure 7H) release. The effect of Doc2β-C2AclmBclm on the asynchronous component (approximate twofold increase) was more pronounced than on the synchronous component (approximate onefold increase), resulting in charge transfer kinetics that were slower than in the control or Doc2β-wt conditions (Figure 7D). In summary, Doc2β-wt and Doc2β-C2AclmBclm differentially affect evoked neurotransmission; the effect of overexpression of Doc2-wt is restricted to the asynchronous phase of evoked SV release, whereas Doc2β-C2AclmBclm affects both synchronous and asynchronous release.
We next determined the effect of the Doc2β mutants on the frequency of miniature EPSCs (mEPSCs) upon overexpression in wt neurons. As shown in Figure 8, overexpression of Doc2β-wt did not significantly increase the frequency of mEPSCs, consistent with previous results (Groffen et al., 2010). In contrast, Doc2β-C2AclmBclm resulted in significantly higher mEPSC frequencies compared with noninfected control neurons. In a previous study, Doc2β-C2AclmB, the other Doc2 Ca2+-ligand mutant that was constitutively bound to the plasma membrane, was also found to increase mEPSC frequency (Groffen et al., 2010). These findings suggest that the function of Doc2β-C2AclmBclm and Doc2β-C2AclmB during spontaneous neurotransmitter release differs yet again from that of the wt protein. In contrast, others reported that another Doc2 mutant with Ca2+ ligand mutations in both C2 domains can fully rescue the loss of spontaneous release in Doc2/rabphilin-knockdown neurons (Pang et al., 2011) and concluded that Doc2-regulated spontaneous release is via a Ca2+-independent mechanism. Further studies are needed to determine whether this is actually a gain-of-function mutant, as described for the mutant forms of Doc2 studied here.
FIGURE 8:
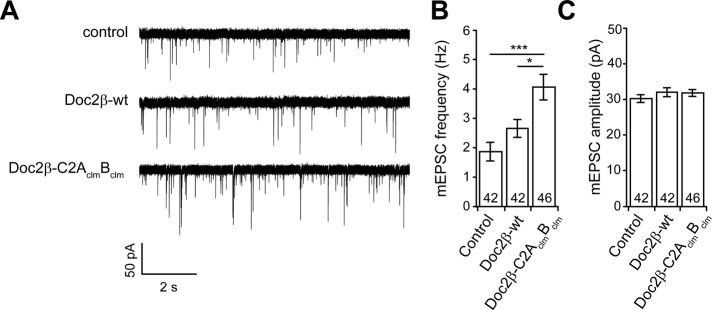
Expression of Doc2-C2AclmBclm enhances spontaneous release in wt hippocampal neurons. (A) Representative traces of mEPSCs recorded from wt neurons with and without expression of Doc2β-wt or Doc2β-C2AclmBclm. (B) The frequency of mEPSCs (control: 1.9 ± 0.3 Hz, n = 42) was not significantly increased by expression Doc2β-wt (2.7 ± 0.3 pC, n = 42) but was significantly increased by Doc2β-C2AclmBclm (4.1 ± 0.4 pC, n = 46). (C) mEPSC amplitude was not affected by expression of Doc2β-wt or Doc2β-C2AclmBclm. Data are represented as mean ± SEM. *p < 0.05, ***p < 0.001, Student's t test. The number of cells, n, is indicated in the bar graphs; in all cases recordings were obtained using neurons from four or more independent litters of mice.
Overexpression of Doc2β Ca2+-ligand mutants in wt neurons increases the size of the readily releasable pool of vesicles
The increase in evoked SV release could be due to an increase in the RRP of SVs, the release probability (Pvr), or both. There are two standard methods to determine RRP size and Pvr. The first is to calculate the RRP size from the phasic EPSC charge transfer evoked by high-frequency stimulation (Schneggenburger et al., 1999; Hosoi et al., 2007; Stevens and Williams, 2007), and the second is by directly releasing the entire RRP using hypertonic sucrose (Rosenmund and Stevens, 1996). Once the RRP size is determined, Pvr can be calculated by normalizing the total charge of a single EPSC to the size of the RRP.
We began by recording synaptic responses from neurons stimulated by a train of action potentials (20 Hz, 2 s; Figure 9A). As shown in Figure 8B, neurons that expressed Doc2β-C2AclmBclm exhibited larger EPSCs than control neurons or neurons expressing Doc2β-wt. When normalized to the amplitude of the first spike, the trends in the EPSC peak amplitudes were almost identical in all three groups (Figure 9C), suggesting that neither wt nor the C2AclmBclm mutant form of Doc2β affected the rate of synaptic depression. To estimate the size of the RRP, we plotted the cumulative phasic charge transfer versus time and fitted the last 10 data points (1.5–2.0 s) with a linear function (Figure 9D). Because the responses from the last 10 stimuli of the train reached steady state (Figure 9D, inset), we estimated the size of the RRP by extrapolating the linear fit and determining the y-intercept (Schneggenburger et al., 1999; Liu et al., 2009). Using this measurement, we found that expression of Doc2β-C2AclmBclm, but not Doc2β-wt, enhanced the size of the RRP (Figure 9E). Doc2β-C2AclmBclm did not affect the RRP replenishment rate (measured by normalizing the slope of the linear fit to the RRP size; Figure 9F) when compared with control neurons or neurons expressing Doc2β-wt. The unaffected release probability and replenishment rate are consistent with the unchanged rate of synaptic depression (Figure 9C).
FIGURE 9:

Expression of Doc2β-C2AclmBclm enhanced the size of the RRP and did not affect SV replenishment in wt hippocampal neurons. (A) Representative EPSCs evoked by a train of 40 action potentials (20 Hz) recorded from wt neurons with and without overexpression of Doc2β-wt or Doc2β-C2AclmBclm. (B) The amplitude of each peak vs. time. (C) The data in B normalized to the amplitude of the first peak. (D) Cumulative phasic charge transfer plotted vs. time. Data points from 1.5 to 2.0 s were fitted with a linear function, revealing a steady-state of SV release (inset). (E) The RRP size was determined by extrapolating the linear fit through the y-intercept shown in D. The RRP size was increased by expression of Doc2β-C2AclmBclm but not Doc2β-wt. (F) The replenishment rate was measured by normalizing the slope of the linear fitting trace (as shown in D) to the RRP size obtained in E. The replenishment rate was unaffected by expression of Doc2β-wt or Doc2β- C2AclmBclm. (G) Representative hypertonic sucrose responses recorded from wt neurons and neurons expressing Doc2β-wt or Doc2β-C2AclmBclm. (H) The RRP size was measured by integrating the sucrose responses and was, again, increased by expression of Doc2β-C2AclmBclm but not Doc2β-wt. (I) SV release probability (Pvr) calculated by normalizing the total EPSC charge transfer to the RRP size measured using 20-Hz train stimulation from E. Data are represented as mean ± SEM. *p < 0.05, Student's t test. The number of cells, n, is indicated in the bar graphs; in all cases recordings were obtained using neurons from three or more independent litters of mice.
Finally, we evaluated the size of the RRP via perfusion of neurons with hypertonic sucrose (Figure 9G). Although this method yields much larger values (Moulder and Mennerick, 2005), the results obtained were consistent with our findings using high-frequency stimulation; Doc2β-C2AclmBclm gave rise to a significant increase in the size of the RRP (Figure 9H). In addition, the Pvr (total EPSC charge in Figure 7C normalized to the size of the RRP in Figure 9E) was unaffected by overexpression of either Doc2β-wt or the C2AclmBclm mutant (Figure 9I). These observations further confirm that Doc2β-C2AclmBclm enhances neurotransmitter release by increasing the size of the RRP without affecting Pvr. Again, these effects are distinct from Doc2β-wt, which does not affect either of these aspects of neurotransmission.
DISCUSSION
Recently we identified Doc2α and β as potential Ca2+ sensors for the slow phase of synaptic transmission in central neurons. Of interest, neutralization of acidic Ca2+ ligands in both C2 domains of the protein resulted in markedly enhanced asynchronous release (Yao et al., 2011). These results were initially interpreted in light of earlier studies indicating that Ca2+-ligand mutations in the C2A domain did not diminish Ca2+-binding activity but actually increased the Ca2+ sensitivity of Doc2 for membrane binding (Groffen et al., 2006; Friedrich et al., 2008) and could thus underlie the observed gain of function. However, this is no longer a satisfying explanation, since we show in the present study that these mutations abolish the apparent Ca2+-sensing properties of Doc2β. These new observations led to a revised view of Doc2 structure–function relationships based on Ca2+-ligand mutations.
Both C2 domains of Doc2 sense Ca2+ but in different ways
The question of whether the C2B domain of Doc2β can bind Ca2+ and lipids has been the subject of debate. An early study reported that only the isolated C2A domain from Doc2β could bind PS-containing liposomes in response to Ca2+ (Kojima et al., 1996). In contrast, more recent studies provided evidence that the isolated C2B domain can sense Ca2+ and bind to liposomes harboring anionic lipids (Groffen et al., 2010). It was also reported that the fluorescence emission intensity of the endogenous aromatic residues in the isolated C2B domain markedly decrease in response to Ca2+, suggesting that a conformational change occurs upon binding (Pang et al., 2011). However, we could not reproduce the robust fluorescence change (∼30% decrease) observed in that study (Supplemental Figure S6). Therefore the question of whether isolated C2B binds Ca2+ has not been sufficiently resolved.
Using ITC, we were able to directly measure Ca2+ binding to the C2 domains of Doc2β (Figure 2). Surprisingly, we observed that the isolated C2B domain, but not the C2A domain, bound Ca2+ in the absence of lipids. These findings are supported by a recent report on Doc2β structure published during preparation of this article (Giladi et al., 2013). This appears to also be the case for the tandem C2 domains, as C2AclmB, but not C2ABclm, bound Ca2+. To reconcile these observations with the well-established finding that C2A senses Ca2+, we measured the Ca2+-dependent lipid-binding characteristics of each construct in parallel. Indeed, in the presence of PS-bearing liposomes, the Ca2+-binding properties of some of the Doc2β constructs were dramatically different. Namely, the isolated C2A domain clearly responded to Ca2+, as evidenced by its robust Ca2+-dependent PS binding activity. Conversely, the C2B domain, which exhibits robust Ca2+-binding activity in solution, inefficiently binds to liposomes in response to Ca2+. Our findings regarding C2A provide the first example of a C2 domain that is apparently unable to bind Ca2+ to any measurable degree in the absence of effector (i.e., anionic phospholipids). Consistent with this model, C2ABclm, which exhibited little to no Ca2+-binding activity in solution, was able to bind efficiently to liposomes in response to Ca2+.
The properties of the C2B domain appear to be affected by its proximity to the C2A domain. Namely, the isolated C2B domain of Doc2β binds membranes inefficiently in response to Ca2+; however, when positioned next to the C2A domain in a tandem structure, C2B avidly binds to membranes, even when the membrane-binding activity of C2A has been impaired via mutations (Figure 2, C and F). These findings are reminiscent of syt1, which also exhibits biochemical functions that would not be expected based on studies of its isolated C2 domains (Bai et al., 2002; Hui et al., 2006). The observed differences in Ca2+-sensing properties of the isolated versus tandem C2-domain fragments suggest that intramolecular interactions may occur between the C2 domains of Doc2β, thus influencing one another.
Gain-of-function mutant forms of Doc2 bind constitutively to the plasma membrane
Ca2+-ligand mutations in the C2A domain of Doc2β result in plasma membrane localization of the protein in chromaffin cells (Groffen et al., 2004; Friedrich et al., 2008). However, this was not observed when specific Ca2+-ligand mutants were expressed in adipocytes (Fukuda et al., 2009), suggesting that constitutive membrane localization required factors that were specific for neuroendocrine cells. However, in transfected HEK 293 cells, which are derived from kidney, Doc2β-C2AclmB and Doc2β-C2AclmBclm were constitutively associated with the plasma membrane (Figure 4D). To determine whether the constitutive membrane localization in neuroendocrine cells was due to interactions with Munc13-1, we expressed Ca2+-ligand mutants that harbored a scrambled or deleted MID in PC12 cells (Figure 4). We confirmed the plasma membrane localization of Doc2β-C2AclmB, and in our experiments Doc2β-C2AclmBclm was the only other construct constitutively associated with the plasma membrane. Targeting to the plasma membrane was unaffected by disruption of the Munc13-1–binding domain and was thus mediated via alternative interactions. Similarly, in vitro binding assays ruled out t-SNAREs as the constitutive binding partner for C2AclmB and C2AclmBclm at the plasma membrane (Figure 3).
We then explored the possibility that lipids mediate constitutive binding of Doc2β-C2AclmB and Doc2β-C2AclmBclm to the plasma membrane and observed that each of the three Doc2β Ca2+-ligand mutants bound PS-harboring liposomes to various extents in the absence of Ca2+. Of interest, as the lipid composition increased in complexity, the Ca2+ ligand mutants displayed enhanced Ca2+-independent membrane-binding activity (Figure 5); wt C2AB exhibited a more modest increase in Ca2+-independent binding compared with the mutants. However, the pattern of membrane binding in vitro did not precisely correlate with the PC12 membrane localization experiments. Namely, the C2ABclm mutant displays significant Ca2+-independent binding to liposomes prepared from total brain extract but is not constitutively localized to the plasma membrane in PC12 cells.
Of interest, among the constructs expressed in syt1-KO neurons, only Doc2β-C2AclmB and Doc2β-C2AclmBclm displayed large increases in asynchronous release (Figure 6), and only these constructs were constitutively localized to the plasma membrane in PC12 cells (Figure 4). Together these data suggest that the molecular interactions that draw Doc2β-C2AclmB and Doc2β-C2AclmBclm to the membrane also underlie the dramatic effects of these two mutants on the asynchronous component of release. As discussed earlier, the enhanced activity of the two mutant forms of Doc2β appears to be independent of interactions with known effectors (i.e., anionic lipids, t-SNAREs, and Munc13-1) and is evidently mediated through interactions with as-yet-unidentified molecules at the plasma membrane. However, we cannot completely rule out lipids, as labile species might be lost in the total brain lipid extract or the organization of lipids in the bilayer might not be properly recapitulated.
Ca2+-ligand mutations endow Doc2 with novel functions
We extended the electrophysiology experiments to include the overexpression of the Doc2β constructs in wt neurons. Surprisingly, we found that Doc2β-C2AclmBclm increased both synchronous and asynchronous evoked transmission (Figure 7) and even spontaneous release (Figure 8). In contrast, overexpression of Doc2β-wt in wt neurons had no effect on either synchronous or spontaneous release but did, as reported previously, result in a modest increase in asynchronous release (Yao et al., 2011). Further work indicated that the overall enhancement of exocytosis was most likely due to an increase in the size of the RRP (Figure 9). Consistent with this interpretation, overexpression of Doc2β-wt had no effect on the size of the RRP. In addition, both Doc2β-C2AclmB (Groffen et al., 2010) and Doc2β-C2AclmBclm (Figure 8) were found to enhance the frequency of spontaneous release events when overexpressed in wt neurons, whereas Doc2β-wt showed no effect. These results underscore the idea that the two Doc2β mutants Doc2β-C2AclmBclm and Doc2β-C2AclmB, which constitutively bind to the plasma membrane, function in a manner distinct from Doc2β-wt. Note that Doc2β-C2AclmBclm did not exhibit equal effects on the fast and slow components of synaptic transmission; its effect on the slow phase of release was more robust than on the fast phase. Of interest, a recent study revealed the existence of a SV pool, marked by VAMP4, which specifically maintains asynchronous release (Raingo et al., 2012); it is possible that Doc2β preferentially regulates this pool of vesicles.
The data reported here highlight the complexity of Ca2+-ligand mutations in C2 domains. In the case of Doc2β, these mutations appear to result in enhanced activity of the protein during multiple forms of synaptic transmission in wt neurons. These properties are likely to involve more than just the constitutive activation of the protein, as they endow Doc2β with novel functions. This complexity is further highlighted by studies of other C2-domain proteins. For example, neutralization of acidic residues in the C2A domain of syt1 abolish the Ca2+-dependent lipid-binding activity of the isolated domain in vitro but either had no effect or enhanced the apparent Ca2+ affinity of the full-length protein during synaptic transmission in neurons (Fernandez-Chacon et al., 2002; Robinson et al., 2002; Stevens and Sullivan, 2003; Pang et al., 2006). Further Ca2+-ligand mutations in the C2A domain of syt1 have also been reported to inhibit the function of the protein (Shin et al., 2009), so a clear view of these mutations has yet to emerge. In sharp contrast, neutralization of acidic Ca2+ ligands in the C2B domain of syt1 clearly result in a nearly complete loss of function in vivo and endow syt1 with dominant-negative activity (Mackler et al., 2002; Nishiki and Augustine, 2004a).
Although numerous questions remain concerning the mechanism by which C2 domains are regulated by Ca2+, it is known that binding of Ca2+ causes significant changes in electrostatic potential within the Ca2+- and membrane-binding loops of these motifs. For instance, binding of Ca2+ to the C2 domain of phospholipase C-δ1 stabilizes the loops and drives reorientation of the peptide backbone, resulting in exposure of lysine side chains (Grobler et al., 1996). For syt1, the binding of Ca2+ is predicted to shift the electrostatic charge of the binding loops from highly negative to positive, allowing the molecule to interact with anionic phospholipids (Chapman and Davis, 1998; Murray and Honig, 2002).
We postulate that mutations in the C2A domain of Doc2β (i.e., Doc2βC2AclmB and Doc2β-C2AclmBclm) alter its electrostatic interactions with effectors, resulting in constitutive activation and Ca2+-independent interactions with one or more binding partners in the plasma membrane. Studies are underway to understand the role of each Ca2+ ligand in sensing Ca2+, binding to effectors, and regulating SV exocytosis. Although specific mutations in C2A give rise to an anomalous gain of function, we predict that substitution of other Ca2+ ligands will disrupt the function of the protein, as described for the C2B domain of syt1.
The increase in release that results from expression of Doc2β-C2AclmBclm, in either wt or syt1-KO neurons, is still Ca2+ dependent, so under these conditions other Ca2+-binding proteins must sense Ca2+ to drive transmission. In the neurons used for these experiments, endogenous wt Doc2 is present and can contribute to release, so it will be interesting to determine whether the Doc2 gain-of-function mutants can still enhance release in neurons lacking the wt protein. Given that loss of Doc2 does not appear to disrupt all asynchronous release, there are likely to be additional molecules that drive this phase of transmission.
MATERIALS AND METHODS
cDNA constructs
cDNA encoding the tandem C2 domains (C2AB) of Doc2α (amino acids [aa] 88–400) and Doc2β (aa125–412) were provided by M. Verhage (Amsterdam, Netherlands). Full-length human Doc2α and Doc2β cDNA were provided by Y. Takai (Kobe, Japan). The bacterial expression vector for full-length mouse synaptobrevin 2 (VAMP2; pTW2), the cytoplasmic domain of VAMP2 (pET-rsybCD), and the full-length rat syntaxin 1A/mouse SNAP-25B t-SNARE heterodimer (pTW34) were provided by J. E. Rothman (New Haven, CT). cDNA encoding human SNAP-25B, provided by M. C. Wilson (Albuquerque, NM), and full-length rat syntaxin 1A were subcloned into pTrcHis to generate individual N-terminal hexahistidine-tagged fusion proteins for expression in Escherichia coli. The expression vector for GCaMP-6F was provided by L. L. Looger (Janelia Farm, Howard Hughes Medical Institute). Doc2β MID mutants were generated by splice overlap extension PCR (SOE-PCR); recoded N-terminal segments of rat Doc2β were synthesized (IDT, Inc.) to encode wt, scrambled, or deleted MID sequences. Full-length MID: 5′-gga tcc atg acc ctc cga aga cgt gga gag aag gct aca atc agc atc caa gag cat atg gca atc gac gtg tgt cct gga cct atc cga cct atc aag cag atc tcc gat tac ttc cca cga ttc cct cgt gga ctc cca cca aca gct gca cca cgt gct tca gca cct cca gac gct cct gca cga tcg cca gca gct acc gca ggt cct cgt agc ccc tcc gac gga gca cgc gac gac gac gaa gat gtg gac cag ctc ttc gga gcc tac ggt gcc agc cca ggc ccc agc ccc gga ccc agc ccc gtg agg ccg ccg gcc aag ccc ccc gag gac gaa ccg gac gcc gac ggc tac gag gcg gcc gc-3′. Scrambled MID: 5′-gga tcc atg acc ctc cga aga cgt gga gag aag gct aca atc agc atc tgc ccg att cag cgc cat aac ggc gcg ttc ttt gcg aac gaa tat att ttt gaa att cgc att atg gcg cca cga ttc cct cgt gga ctc cca cca aca gct gca cca cgt gct tca gca cct cca gac gct cct gca cga tcg cca gca gct acc gca ggt cct cgt agc ccc tcc gac gga gca cgc gac gac gac gaa gat gtg gac cag ctc ttc gga gcc tac ggt gcc agc cca ggc ccc agc ccc gga ccc agc ccc gtg agg ccg ccg gcc aag ccc ccc gag gac gaa ccg gac gcc gac ggc tac gag tca gac gac tgc acc gcc ctg ggt aca ctg gcg gcc gc-3′. Deleted MID: 5′-gga tcc atg acc ctc cga aga cgt gga gag aag gct aca atc agc atc cca cga ttc cct cgt gga ctc cca cca aca gct gca cca cgt gct tca gca cct cca gac gct cct gca cga tcg cca gca gct acc gca ggt cct cgt agc ccc tcc gac gga gca cgc gac gac gac gaa gat gtg gac cag ctc ttc gga gcc tac ggt gcc agc cca ggc ccc agc ccc gga ccc agc ccc gtg agg ccg ccg gcc aag ccc ccc gag gac gaa ccg gac gcc gac ggc tac gag tca gac gac tgc acc gcc ctg ggt aca ctg gcg gcc gc-3′. The cDNA encoding the N-terminal portion of Doc2 was amplified using PCR and appended to a fragment encoding the C-terminal C2AB, C2AclmB, C2ABclm, or C2AclmBclm fragments of rat Doc2β via SOE-PCR. These constructs were subcloned into pAcGFP1-C1 to generate N-terminal GFP fusion proteins for transfection or into pLOX (Syn-DsRed(W)-Syn-GFP(W)) to generate lentiviral particles. Ca2+-ligand mutations were introduced using overlapping primers and the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA).
Protein expression/purification
SNAP-25B, VAMP2, cdV, and the preassembled t-SNARE heterodimer composed of full-length syntaxin 1A and SNAP-25B were purified as described previously (Tucker et al., 2004). The isolated and tandem C2 domains of wt Doc2β and each of the Ca2+ ligand mutants were expressed as glutathione S-transferase fusion proteins and purified using standard procedures. Briefly, 2–3 l of Luria broth was inoculated and grown overnight at 37°C until an OD600 of 0.8 was obtained. The incubation temperature was then reduced to 30°C, and protein expression was induced by addition of 400 μM isopropyl-β-d-thiogalactoside. After 4 h the cells were collected by centrifugation and the pellets stored at −30°C. Bacterial pellets were resuspended in 0.1 M phosphate-buffered saline (PBS) containing 10% glycerol and 1 mM Tris (2-carboxyethyl) phosphine hydrochloride (TCEP; thrombin cleavage buffer [TCB]), and bacteria were lysed by sonication. The sonicated material was solubilized with 1% Triton X-100 for 30 min, insoluble material was removed by centrifugation, and the resulting supernatant was incubated with glutathione–Sepharose beads and 0.35 μg of RNase/DNase overnight at 4°C on a rotator. Beads were washed with 50 volumes of TCB containing 1 M NaCl, followed by an additional three washes with 50 volumes of TCB. Protein was removed from the beads by cleavage with thrombin for 3 h at 20 °C; thrombin was inactivated by addition of 0.5 mM phenylmethylsulfonyl fluoride. After elution, the proteins were analyzed by SDS–PAGE and stained with Coomassie blue, and concentrations were determined using a bovine serum albumin standard curve.
Isothermal calorimetry
Purified proteins were extensively dialyzed against 100 mM NaCl, 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)–KOH, pH 7.4, and 10% glycerol (ITC buffer). All Ca2+ stocks were prepared fresh for each experiment with the buffer used for dialysis. Using a MicroCal ITC200 holding a constant temperature of 25°C with 1000-rpm sample mixing, we added 20 1.5-μl aliquots of 1 mM Ca2+ in succession to 100 μM protein in the reaction chamber. For background correction, Ca2+ was added to buffer alone and these signals were subtracted from the Doc2 data. The ΔH was integrated and plotted as a function of the molar ratio of Doc2:Ca2+. The data were fitted using a “one set of sites” binding model. The results are provided in Supplemental Figure S1.
Protein•liposome cosedimentation assays
Protein-free liposomes composed of 25% PS, 30% PE, 45% PC, and 0.2% rhodamine-PE were prepared by extrusion through 100-nm filters in 100 mM NaCl, 25 mM HEPES-KOH, 1 mM TCEP, and 10% glycerol. We prepared 100-μl reactions containing 4 μM of each protein, 4 mM lipid, and increasing amounts of Ca2+ in the same buffer but without glycerol to facilitate liposome sedimentation. Samples were incubated for 15 min at room temperature with shaking and centrifuged at 270,000 × g for 30 min; the efficiency of liposome sedimentation was determined by measuring the residual rhodamine fluorescence in the supernatant of each reaction. Fifty microliters of the supernatant from each sample was collected and mixed with 25 μl of SDS sample buffer (120 mM Tris-HCl, pH 6.8, 30% glycerol, 15 mM TCEP, and 125 mM SDS). The pellet was solubilized in 150 μl of SDS sample buffer. Samples were boiled for 1 min, and 15-μl aliquots were analyzed by SDS–PAGE and stained with Coomassie blue.
Protein reconstitution
Proteoliposomes harboring syntaxin 1A·SNAP-25B heterodimers or syntaxin 1A alone were prepared at 100 copies/vesicle as described previously (Tucker et al., 2004). Briefly, t-SNAREs were reconstituted into vesicles containing 25% PS, 30% PE, and 45% PC, purified by ultracentrifugation through a density gradient, and collected from the 0–30% Accudenz interface. In cases in which PS was omitted from the vesicles the percentage PC was increased to 70%. Syb2 was reconstituted into vesicles containing 25% PS, 27% PE, 45% PC, 1.5% NBD-PE, and 1.5% rhodamine-PE. As negative controls for membrane fusion, protein-free liposomes lacking VAMP2 or t-SNARES were prepared in parallel.
In vitro membrane fusion assays
Fusion assays were carried out in white-bottom 96-well plates, in total reaction volumes of 100 μl, in a BioTek Synergy HT plate reader equipped with 460/40-nm excitation and 530/25-nm emission filters. Each reaction contained 4.5 μl of purified t-SNARE vesicles, syntaxin 1A vesicles, or protein-free vesicles, 0.5 μl of purified NBD-rhodamine–labeled vesicle-SNARE vesicles, and 0.2 mM BAPTA. Doc2 was added to each reaction as indicated in the figures. For t-SNARE assembly fusion experiments, 2 μM soluble SNAP-25B was included in each reaction. To block SNARE-mediated fusion, the cytoplasmic domain of VAMP2 (cdV) was included in selected reactions at 2 μM. Samples were incubated at 37°C for 20 min, followed by injection of Ca2+ to give a final free concentration of 0.3 mM, unless otherwise indicated; reactions were monitored for an additional 60 min. The maximum fluorescence signal was obtained by addition of n-dodecyl β-d-maltoside to each reaction well. The data were normalized by setting the initial signal, before the addition of Ca2+, to 0% and the maximal fluorescence signal obtained using detergent to 100%. Data were plotted using Prism 4.0 software (GraphPad, La Jolla, CA).
Coflotation assays
For coflotation assays, 100-μl reactions were prepared in reconstitution buffer containing 0.2 mM BAPTA, 30 μM Doc2β, and 45 μl of PS-free t-SNARE-bearing or protein-free vesicles in the presence or absence of 0.3 mM free Ca2+. Samples were incubated at room temperature for 15 min with shaking. Vesicles were floated through a step gradient, collected, and resolved by SDS–PAGE, and the proteins were stained with Coomassie blue. Gels were scanned at 600 dpi using a Bio-5000 gel scanner (MicroTek, Santa Fe Springs, CA) and analyzed using GelEval 1.22 (FrogDance Software) and Prism 6 (GraphPad) software.
PC12 translocation experiments
PC12 cells were cultured in DMEM supplemented with 5% horse serum, 5% calf serum, and 100 μg/ml penicillin and streptomycin. Cells were maintained at 37°C with 10% CO2. Before transfection, cells were plated onto acid-washed glass coverslips coated with poly-d-lysine and collagen IV. When the cells were 70–80% confluent they were transfected with GFP-Doc2 expression vectors using Lipofectamine LTX reagent (Life Technologies, Carlsbad, CA). Cells were imaged 24–48 h after transfection using an Olympus FV1000 confocal microscope. The coverslips were transferred to 30-mm culture dishes containing 2 ml of imaging buffer (145 mM NaCl, 2.8 mM KCl, 1 mM MgCl2, 1.2 mM CaCl2, 10 mM glucose, and 10 mM HEPES-NaOH, pH 7.3). Individual cells were identified and imaged. To stimulate translocation, 2 ml of 2× depolarization buffer (27.8 mM NaCl, 120 mM KCl, 1 mM MgCl2, 1.2 mM CaCl2, 10 mM glucose, and 10 mM HEPES-NaOH, pH 7.3) was added manually. To lower the resting [Ca2+]i, cells were treated for 30 min with 10 mM EGTA, 50 μM BAPTA-AM, and 30 μM CPA to remove extracellular and intracellular Ca2+. To measure [Ca2+]i, cells were loaded with 5 μM of Fura2-AM (Life Technologies) for 45 min, and fluorescent signals were recorded using a photometry system (TILL Photonics, Munich, Germany). The [Ca2+]i was estimated using the equation [Ca2+]i = Keff(R − Rmin)/(Rmax − R), where R is the ratio of fluorescence intensity resulting from excitation at 340 and 380 nm (F340/F380), respectively; Keff, Rmin, and Rmax are constants that were obtained via in vitro calibration experiments.
Hippocampal neuronal culture and viral infection
Syt1-KO mice were obtained from Jackson Laboratory (Bar Harbor, ME). Hippocampal neurons were prepared from newborn pups (postnatal day 0) as described previously (Liu et al., 2009) in accordance with the guidelines of the National Institutes of Health, as approved by the Animal Care and Use Committee of the University of Wisconsin–Madison. Briefly, hippocampi were isolated from mouse brain, washed with Hank's buffered salt solution (Corning, Corning, NY), and digested in 0.25% trypsin-EDTA (Corning) at 37°C for 30 min. After digestion, the tissue was mechanically dissociated in DMEM with 20 mM d-glucose (Corning) and 10% fetal bovine serum (ThermoScientific, Waltham, MA). Cells were plated at 25,000–40,000 cells/cm2 on 12-mm poly-d-lysine–precoated glass coverslips (Warner Instruments, Hamden, CT). Cultures were maintained in Neurobasal-A culture medium (Life Technologies) supplemented with 2% B27 and 2 mM GlutaMAX at 37°C in a 5% CO2 humidified incubator.
cDNA constructs were subcloned into a pLOX vector that coexpresses GFP. The recombinant pLOX plasmid was cotransfected into HEK 293T cells containing two viral packaging vectors (vesicular stomatitis virus G glycoprotein and Delta 8.9) to obtain lentivirus particles. After 2 d, virus particles were harvested by ultracentrifugation at 25,000 rpm for 2 h using an SW-28 rotor (Beckman Coulter, Indianapolis, IN). Hippocampal neurons were infected with virus at the day 5 in vitro (5 DIV) and used for patch-clamp recordings at 12–17 DIV.
Electrophysiology
Whole-cell patch-clamp recordings of EPSCs were performed using a MultiClamp 700B amplifier (Molecular Devices, Sunnyvale, CA) or an EPC-10 double amplifier (HEKA Elektronik, Bellmore, NY). Hippocampal neurons were continuously perfused with bath solution consisting of 128 mM NaCl, 30 mM glucose, 5 mM KCl, 5 mM CaCl2, 1 mM MgCl2, 50 mM D-AP5, 20 mM bicuculline, and 25 mM HEPES, pH 7.3. Recording pipettes with resistances of 3–5 MΩ were filled with pipette solution consisting of 130 mM K-gluconate, 1 mM EGTA, 5 mM Na-phosphocreatine, 2 mM Mg-ATP, 0.3 mM Na-GTP, 5 mM QX-314, and 10 mM HEPES, pH 7.3.
Neurons were voltage clamped at −70 mV. Only cells with series resistances <15 MΩ were used for recording. Between 70 and 80% of the series resistance was compensated. For evoked EPSCs, randomly selected presynaptic neurons were always 10–100 μm away from the whole-cell patched postsynaptic neuron. Release was stimulated with a voltage step from 0 to 20–30 V for 1 ms, using a bipolar electrode pulled from theta glass capillary tubing (Warner Instruments) and filled with bath solution. The tip of the stimulation pipette gently touched the surface of the soma of the presynaptic neuron. To avoid stimulating multiple neurons, cells in clusters were never used. The infection efficiency was high (∼90%), and only neuron pairs in which both the presynaptic and postsynaptic cells were successfully infected were used for recording evoked EPSCs. For the hypertonic sucrose experiments, the whole-cell patched neuron was locally perfused with bath solution plus 500 mM sucrose for 10 s using a Picospritzer III (Parker, Cleveland, OH). For mEPSC recordings, 1 μM tetrodotoxin was added to the bath solution: the Ca2+ concentration was 2 mM. Data were acquired using PatchMaster (HEKA Elektronik) or pClamp software (Molecular Devices), sampled at 10 kHz, and filtered at 2.8 kHz. All electrophysiology experiments were performed at room temperature. Data were analyzed using Clampfit (Molecular Devices) and Igor (WaveMetrics, Portland, OR) software.
D-AP5, bicuculline, QX-314, and tetrodotoxin were purchased from TOCRIS Bioscience (R&D Systems, Minneapolis, MN).
Supplementary Material
Acknowledgments
We thank D. Kim, L. Looger, and the GENIE Project of the Howard Hughes Medical Institute Janelia Farm for providing the gCaMP-6F construct. We also thank R. Fettiplace and members of the Chapman lab for helpful discussions and comments. This study was supported by a grant from the National Institutes of Health (MH 61876). E.R.C. is an Investigator of the Howard Hughes Medical Institute.
Abbreviations used:
- AM
acetoxymethyl ester
- BAPTA
1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- clm
calcium-ligand mutation
- CPA
cyclopiazonic acid
- Doc2
double C2-domain protein
- EGTA
ethylene glycol tetraacetic acid
- EPSC
excitatory postsynaptic current
- ITC
isothermal titration calorimetry
- KO
knockout
- mEPSC
miniature excitatory postsynaptic current
- MID
Munc13-1–interacting domain
- PC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- PE
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine
- PS
1,2-dioleoyl-sn-glycero-3-[phospho-l-serine]
- RRP
readily releasable pool
- SNARE
soluble N-ethylmaleimide–sensitive factor attachment protein receptor
- SV
synaptic vesicle
- syt1
synaptotagmin 1
- TCEP
Tris (2-carboxyethyl) phosphine hydrochloride
- wt
wild type
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E13-10-0571) on December 19, 2013.
*These authors contributed equally to the work done in this study.
REFERENCES
- Bai J, Wang P, Chapman ER. C2A activates a cryptic Ca(2+)-triggered membrane penetration activity within the C2B domain of synaptotagmin I. Proc Natl Acad Sci USA. 2002;99:1665–1670. doi: 10.1073/pnas.032541099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhalla A, Chicka MC, Tucker WC, Chapman ER. Ca(2+)-synaptotagmin directly regulates t-SNARE function during reconstituted membrane fusion. Nat Struct Mol Biol. 2006;13:323–330. doi: 10.1038/nsmb1076. [DOI] [PubMed] [Google Scholar]
- Broadie K, Bellen HJ, DiAntonio A, Littleton JT, Schwarz TL. Absence of synaptotagmin disrupts excitation-secretion coupling during synaptic transmission. Proc Natl Acad Sci USA. 1994;91:10727–10731. doi: 10.1073/pnas.91.22.10727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose N, Petrenko AG, Sudhof TC, Jahn R. Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science. 1992;256:1021–1025. doi: 10.1126/science.1589771. [DOI] [PubMed] [Google Scholar]
- Burgalossi A, et al. SNARE protein recycling by alphaSNAP and betaSNAP supports synaptic vesicle priming. Neuron. 2010;68:473–487. doi: 10.1016/j.neuron.2010.09.019. [DOI] [PubMed] [Google Scholar]
- Burgoyne RD, Morgan A. Membrane trafficking: three steps to fusion. Curr Biol. 2007;17:R255–R258. doi: 10.1016/j.cub.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Chapman ER, Davis AF. Direct interaction of a Ca2+-binding loop of synaptotagmin with lipid bilayers. J Biol Chem. 1998;273:13995–14001. doi: 10.1074/jbc.273.22.13995. [DOI] [PubMed] [Google Scholar]
- Chen TW, et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan RR, Betz A, Shipston MJ, Brose N, Chow RH. Transient, phorbol ester-induced DOC2-Munc13 interactions in vivo. J Biol Chem. 1999;274:27347–27350. doi: 10.1074/jbc.274.39.27347. [DOI] [PubMed] [Google Scholar]
- Fernandez-Chacon R, Shin OH, Konigstorfer A, Matos MF, Meyer AC, Garcia J, Gerber SH, Rizo J, Sudhof TC, Rosenmund C. Structure/function analysis of Ca2 +binding to the C2A domain of synaptotagmin 1. J Neurosci. 2002;22:8438–8446. doi: 10.1523/JNEUROSCI.22-19-08438.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich R, Groffen AJ, Connell E, van Weering JR, Gutman O, Henis YI, Davletov B, Ashery U. DOC2B acts as a calcium switch and enhances vesicle fusion. J Neurosci. 2008;28:6794–6806. doi: 10.1523/JNEUROSCI.0538-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda N, Emoto M, Nakamori Y, Taguchi A, Miyamoto S, Uraki S, Oka Y, Tanizawa Y. DOC2B: a novel syntaxin-4 binding protein mediating insulin-regulated GLUT4 vesicle fusion in adipocytes. Diabetes. 2009;58:377–384. doi: 10.2337/db08-0303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M, Mikoshiba K. Doc2gamma, a third isoform of double C2 protein, lacking calcium-dependent phospholipid binding activity. Biochem Biophys Res Commun. 2000;276:626–632. doi: 10.1006/bbrc.2000.3520. [DOI] [PubMed] [Google Scholar]
- Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Sudhof TC. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell. 1994;79:717–727. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- Giladi M, Michaely L, Almagor L, Bar-On D, Buki T, Ashery U, Khananshvili D, Hirsch JA. The C2B domain is the primary Ca sensor in DOC2B: a structural and functional analysis. J Mol Biol. 2013;425:4629–4641. doi: 10.1016/j.jmb.2013.08.017. [DOI] [PubMed] [Google Scholar]
- Grobler JA, Essen LO, Williams RL, Hurley JH. C2 domain conformational changes in phospholipase C-delta 1. Nat Struct Biol. 1996;3:788–795. doi: 10.1038/nsb0996-788. [DOI] [PubMed] [Google Scholar]
- Groffen AJ, Brian EC, Dudok JJ, Kampmeijer J, Toonen RF, Verhage M. Ca(2+)-induced recruitment of the secretory vesicle protein DOC2B to the target membrane. J Biol Chem. 2004;279:23740–23747. doi: 10.1074/jbc.M400731200. [DOI] [PubMed] [Google Scholar]
- Groffen AJ, Friedrich R, Brian EC, Ashery U, Verhage M. DOC2A and DOC2B are sensors for neuronal activity with unique calcium-dependent and kinetic properties. J Neurochem. 2006;97:818–833. doi: 10.1111/j.1471-4159.2006.03755.x. [DOI] [PubMed] [Google Scholar]
- Groffen AJ, et al. Doc2b is a high-affinity Ca2+ sensor for spontaneous neurotransmitter release. Science. 2010;327:1614–1618. doi: 10.1126/science.1183765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haucke V, Neher E, Sigrist SJ. Protein scaffolds in the coupling of synaptic exocytosis and endocytosis. Nat Rev Neurosci. 2011;12:127–138. doi: 10.1038/nrn2948. [DOI] [PubMed] [Google Scholar]
- Hosoi N, Sakaba T, Neher E. Quantitative analysis of calcium-dependent vesicle recruitment and its functional role at the calyx of Held synapse. J Neurosci. 2007;27:14286–14298. doi: 10.1523/JNEUROSCI.4122-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E, Bai J, Chapman ER. Ca2+-triggered simultaneous membrane penetration of the tandem C2-domains of synaptotagmin I. Biophys J. 2006;91:1767–1777. doi: 10.1529/biophysj.105.080325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E, Gaffaney JD, Wang Z, Johnson CP, Evans CS, Chapman ER. Mechanism and function of synaptotagmin-mediated membrane apposition. Nat Struct Mol Biol. 2011;18:813–821. doi: 10.1038/nsmb.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Tong XJ, Kaplan JM. UNC-13L, UNC-13S, and Tomosyn form a protein code for fast and slow neurotransmitter release in Caenorhabditis elegans. eLife. 2013;2:e00967. doi: 10.7554/eLife.00967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima T, Fukuda M, Aruga J, Mikoshiba K. Calcium-dependent phospholipid binding to the C2A domain of a ubiquitous form of double C2 protein (Doc2 beta) J Biochem. 1996;120:671–676. doi: 10.1093/oxfordjournals.jbchem.a021464. [DOI] [PubMed] [Google Scholar]
- Littleton JT, Stern M, Schulze K, Perin M, Bellen HJ. Mutational analysis of Drosophila synaptotagmin demonstrates its essential role in Ca(2+)-activated neurotransmitter release. Cell. 1993;74:1125–1134. doi: 10.1016/0092-8674(93)90733-7. [DOI] [PubMed] [Google Scholar]
- Liu H, Dean C, Arthur CP, Dong M, Chapman ER. Autapses and networks of hippocampal neurons exhibit distinct synaptic transmission phenotypes in the absence of synaptotagmin I. J Neurosci. 2009;29:7395–7403. doi: 10.1523/JNEUROSCI.1341-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackler JM, Drummond JA, Loewen CA, Robinson IM, Reist NE. The C(2)B Ca(2+)-binding motif of synaptotagmin is required for synaptic transmission in vivo. Nature. 2002;418:340–344. doi: 10.1038/nature00846. [DOI] [PubMed] [Google Scholar]
- Malkinson G, Spira ME. Calcium concentration threshold and translocation kinetics of EGFP-DOC2B expressed in cultured Aplysia neurons. Cell Calcium. 2006;39:85–93. doi: 10.1016/j.ceca.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Maximov A, Sudhof TC. Autonomous function of synaptotagmin 1 in triggering synchronous release independent of asynchronous release. Neuron. 2005;48:547–554. doi: 10.1016/j.neuron.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Moulder KL, Mennerick S. Reluctant vesicles contribute to the total readily releasable pool in glutamatergic hippocampal neurons. J Neurosci. 2005;25:3842–3850. doi: 10.1523/JNEUROSCI.5231-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray D, Honig B. Electrostatic control of the membrane targeting of C2 domains. Mol Cell. 2002;9:145–154. doi: 10.1016/s1097-2765(01)00426-9. [DOI] [PubMed] [Google Scholar]
- Nalefski EA, Falke JJ. The C2 domain calcium-binding motif: structural and functional diversity. Protein Sci. 1996;5:2375–2390. doi: 10.1002/pro.5560051201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiki T, Augustine GJ. Dual roles of the C2B domain of synaptotagmin I in synchronizing Ca2+-dependent neurotransmitter release. J Neurosci. 2004a;24:8542–8550. doi: 10.1523/JNEUROSCI.2545-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiki T, Augustine GJ. Synaptotagmin I synchronizes transmitter release in mouse hippocampal neurons. J Neurosci. 2004b;24:6127–6132. doi: 10.1523/JNEUROSCI.1563-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orita S, Naito A, Sakaguchi G, Maeda M, Igarashi H, Sasaki T, Takai Y. Physical and functional interactions of Doc2 and Munc13 in Ca2+-dependent exocytotic machinery. J Biol Chem. 1997;272:16081–16084. doi: 10.1074/jbc.272.26.16081. [DOI] [PubMed] [Google Scholar]
- Pang ZP, Bacaj T, Yang X, Zhou P, Xu W, Sudhof TC. Doc2 supports spontaneous synaptic transmission by a Ca(2+)-independent mechanism. Neuron. 2011;70:244–251. doi: 10.1016/j.neuron.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang ZP, Shin OH, Meyer AC, Rosenmund C, Sudhof TC. A gain-of-function mutation in synaptotagmin-1 reveals a critical role of Ca2+-dependent soluble N-ethylmaleimide-sensitive factor attachment protein receptor complex binding in synaptic exocytosis. J Neurosci. 2006;26:12556–12565. doi: 10.1523/JNEUROSCI.3804-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raingo J, et al. VAMP4 directs synaptic vesicles to a pool that selectively maintains asynchronous neurotransmission. Nat Neurosci. 2012;15:738–745. doi: 10.1038/nn.3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson IM, Ranjan R, Schwarz TL. Synaptotagmins I and IV promote transmitter release independently of Ca(2+) binding in the C(2)A domain. Nature. 2002;418:336–340. doi: 10.1038/nature00915. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Rettig J, Brose N. Molecular mechanisms of active zone function. Curr Opin Neurobiol. 2003;13:509–519. doi: 10.1016/j.conb.2003.09.011. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- Sato M, Mori Y, Matsui T, Aoki R, Oya M, Yanagihara Y, Fukuda M, Tsuboi T. Role of the polybasic sequence in the Doc2alpha C2B domain in dense-core vesicle exocytosis in PC12 cells. J Neurochem. 2010;114:171–181. doi: 10.1111/j.1471-4159.2010.06739.x. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/s0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Shin OH, Xu J, Rizo J, Sudhof TC. Differential but convergent functions of Ca2+ binding to synaptotagmin-1 C2 domains mediate neurotransmitter release. Proc Natl Acad Sci USA. 2009;106:16469–16474. doi: 10.1073/pnas.0908798106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Sullivan JM. The synaptotagmin C2A domain is part of the calcium sensor controlling fast synaptic transmission. Neuron. 2003;39:299–308. doi: 10.1016/s0896-6273(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Williams JH. Discharge of the readily releasable pool with action potentials at hippocampal synapses. J Neurophysiol. 2007;98:3221–3229. doi: 10.1152/jn.00857.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker WC, Weber T, Chapman ER. Reconstitution of Ca2+-regulated membrane fusion by synaptotagmin and SNAREs. Science. 2004;304:435–438. doi: 10.1126/science.1097196. [DOI] [PubMed] [Google Scholar]
- Yao J, Gaffaney JD, Kwon SE, Chapman ER. Doc2 is a Ca2+ sensor required for asynchronous neurotransmitter release. Cell. 2011;147:666–677. doi: 10.1016/j.cell.2011.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Rathore SS, Davis EM, Ouyang Y, Shen J. Doc2b promotes GLUT4 exocytosis by activating the SNARE-mediated fusion reaction in a calcium- and membrane bending-dependent manner. Mol Biol Cell. 2013;24:1176–1184. doi: 10.1091/mbc.E12-11-0810. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.