

Abstract
Methicillin-resistant Staphylococcus aureus (MRSA) is a major cause of a myriad of insidious and intractable infections in humans, especially in patients with compromised immune systems and children. Here, we report the apo- and CoA-bound crystal structures of a member of the galactoside acetyltransferase superfamily from methicillin-resistant S. aureus SACOL2570 which was recently shown to be down regulated in S. aureus grown in the presence of fusidic acid, an antibiotic used to treat MRSA infections. SACOL2570 forms a homotrimerin solution, as confirmed by small-angle X-ray scattering and dynamic light scattering. The protein subunit consists of an N-terminal alpha-helical domain connected to a C-terminal LβH domain. CoA binds in the active site formed by the residues from adjacent LβH domains. After determination of CoA-bound structure, molecular dynamics simulations were performed to model the binding of AcCoA. Binding of both AcCoA and CoA to SACOL2570 was verified by isothermal titration calorimetry. SACOL2570 most likely acts as an acetyltransferase, using AcCoA as an acetyl group donor and an as-yet-undetermined chemical moiety as an acceptor. SACOL2570 was recently used as a scaffold for mutations that lead the generation of cage-like assemblies, and has the potential to be used for the generation of more complex nanostructures.
Keywords: Galactoside acetyltransferases, GAT, Methicillin-resistant Staphylococcus aureus (MRSA) subsp. COL, Molecular dynamics, Small-angle X-ray scattering, Crystal structure
Introduction
Staphylococcus aureus strain COL [1] is a methicillin-resistant (MRSA) opportunistic human pathogen causing both community- and hospital-acquired infections. It is linked to skin infections (abscesses), bacteremia, central nervous system infections, necrotizing pneumonia, infec-tive endocarditis, osteomyelitis, urinary tract infections and chronic lung infections associated with cystic fibrosis. Exotoxins and enterotoxins produced by S. aureus cause food poisoning and toxic shock syndromes [2, 3]. This causes life-threatening illnesses and deaths and generates high hospital costs [4, 5]. MRSA is most commonly treated with vancomycin [5], however the recent emergence of vancomycin-resistant MRSA strains [6] calls for novel, innovative treatment strategies [7–9] or development of new antibiotics.
One of the central objectives of the Center for Structural Genomics of Infectious Diseases (CSGID) [10] is to elu-cidate high-resolution, three-dimensional structures of proteins from human pathogens in the NIAID Category A–C priority lists. SACOL2570, a putative galactoside O-acetyltransferase (GAT) protein from the MRSA strain S. aureus subsp. COL was chosen as a CSGID target for its potential involvement in the cellular processes of toxin production and antibiotic resistance.
Galactoside acetyltransferases (GAT, EC 2.3.1.18) are enzymes that transfer an acetyl group from acetyl coen-zyme A (AcCoA) to β-galactosides (Eq. 1) [11]. The enzymes have a broad substrate specificity and can acety-late many galactoside derivatives, including thiogalacto-sides and lactosides [12]. The precise physiological role of GAT is not well understood, but it was suggested to act as a detoxifying enzyme, acetylating non-metabolizable carbo-hydrates to prevent their re-entry into the cell [12, 13].
| (1) |
One of the best studied GATs is the enzyme from Escherichia coli (GATEC), for which several ligand bound structures have been determined [12]. GATEC contains an LβH (left-handed parallel β-helix) structural domain and forms a trimer that contains three substrate-binding sites located at the interface between adjacent LβH subunits. Kinetic studies demonstrated that GATEC adopts an ordered bi-bi ternary complex mechanism with AcCoA and CoA as the leading substrate and corresponding product, respectively [11, 14, 15].
GATs belong to the hexapeptide acyltransferase super-family of enzymes [16, 17] so named for the presence of tandem repeated, imperfect copies of a six-residue amino acid sequence motif called the hexapeptide repeat [18, 19]. The hexapeptide acyltransferases transfer acetate, succi-nate, or long chain fatty acyl groups from thioester donors to a variety of structurally dissimilar acceptors [16, 17]. Analysis of the available crystal structures reveals that the hexapeptide repeat motif directs folding of the character-istic coiled LβH structural domain [11]. Several crystal structures of such enzymes have been determined, includ-ing maltose acetyltransferase (MATGK; PDB code 2IC7) from Geobacillus kaustophilus [20], xenobiotic acetyl-transferase (XAT; PDB code 1XAT) from Pseudomonas aeruginosa[15] and serine acetyltransferase (SAT; PDB code: 1T3D) from E. coli [21]. The crystal structures, in conjunction with experimental data on enzymatic activity, imply that GATs and MATs are closely related and might share similar cellular functions [17].
In the present study, the crystal structure of an apo-form of a putative GAT SACOL2570 from S. aureus was determined at 1.6 Å resolution. The structural similarity of SACOL2570 and GATEC (in complex with AcCoA) prompted us to assess the substrate binding properties of SACOL2570. X-ray crystallography was used to examine the binding of CoA, AcCoA and a selection of carbohy-drates, potential substrates of SACOL2570. The structural studies were followed by isothermal titration calorimetry (ITC) testing of the binding of AcCoA, CoA and the sug-ars. Molecular dynamics (MD) simulations were used to predict the binding mode of AcCoA to SACOL2570, and to determine the structural basis of the AcCoA binding. In addition, the molecular mechanical and generalized Born/Surface Accessible (MM-GBSA) model [22, 23] was used to estimate the binding free energies between AcCoA and SACOL2570/GATEC. The MD-simulated model was in agreement with our experimental data.
Results and discussion
Overall structure of the apo-form of SACOL2570
A ribbon diagram of the SACOL2570 structure is shown in Fig. 1. The asymmetric unit of SACOL2570 crystals con-tains one protein monomer that includes nineteen β-strands and four α-helixes. The protein forms a trimer and the three-fold axis of the oligomer coincides with the crystal symmetry axis. The trimeric assembly in solution was confirmed by SAXS and DLS (see below). The monomer composed of 188 amino acids can be divided into an N-terminal alpha-helical region and a C-terminal LβH domain. The N-terminal domain, comprising residues 1–55, includes three α-helices (residues 2–9, 18–37, and 42–55) and one short β-strand (residues 13–15). This β-strand is absent in the N-terminal domain of GATEC[2, 12]. Residues 56–188 form the C-terminal LβH domain (the hexapeptide repeat motif) which includes eighteen β-strands and one α-helix (residues 117–123), whereas GATEC has one long loop (residues 112–131) protruding from the LβH domain [24].
Fig. 1.
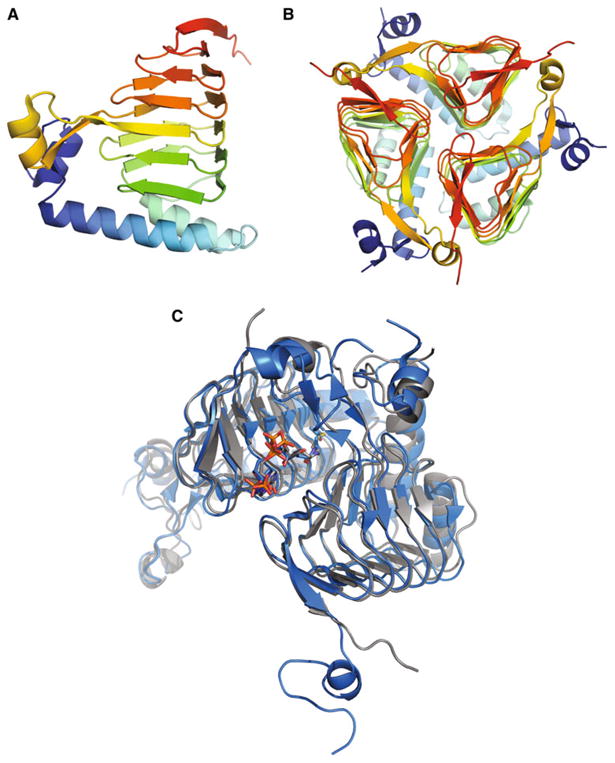
Ribbon diagram showing the apo-form of galactoside acetyltransferase SACOL2570 from S. aureus (a a monomer and b a trimer, a biological unit). c The molecular superposition of GATSA (the SACOL2570/AcCoA complex) in grey and GATEC (PDB code: 1KRR) in blue after molecular dynamics simulations. AcCoA and CoA are shown as sticks
The maltose acetyltransferase from G. kaustophilus (MATGK) is the protein of known structure most similar by sequence to SACOL2570 (43 % identity), followed by GATEC (42 % identity). A multiple structure-based sequence alignment of SACOL2570 to MATGK and GATEC is shown in Fig. 2. The RMSD of 182 aligned pairs of Cα atoms between SACOL2570 and GATEC using the SSM (secondary structure matching) algorithm [25] is 1.26 Å (Fig. 1c), while that of 182 aligned pairs of Cα atoms between SACOL2570 and MATGK is 1.04 Å.
Fig. 2.
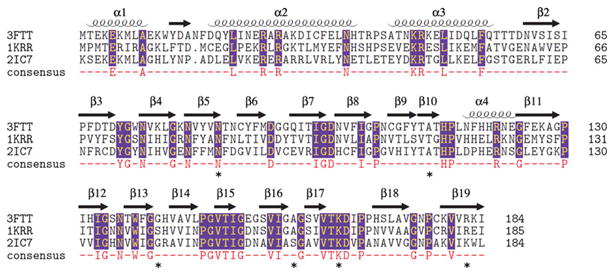
Multiple structure-based sequence alignment of the putative galactoside acetyltransferase SACOL2570 from S. aureus (PDB: 3FTT), galactoside acetyltransferase GATEC from E. coli (PDB: 1KRR), and maltose acetyltransferase from G. kaustophilus (PDB: 2IC7). The alignment was made with Multiprot (http://bioinfo3d.cs.tau.ac.il/MultiProt/). Residues participating in CoA binding are marked by an asterisk
SAXS and DLS solution studies
The goal of small-angle X-ray scattering (SAXS) and dynamic light scattering (DLS) solution studies was to determine the oligomeric state of the protein in solution. Three solutions of SACOL2570 at different concentrations were exposed twice to collect SAXS data (Supplementary Table 2). The radius of gyration (Rg) values calculated for different exposures as well as by different programs (Au-toRg [26] (PRIMUS and command line versions), Auto-GNOM [27] and via the Guinier region) varied somewhat both in value and precision, and the overall mean Rg over all exposures and analyses was 28 ± 2 Å. When the pro-tein was measured by DLS, the regularization algorithm of the DYNAMICS software (Wyatt Technology, Santa Barbara, CA) routinely returned a species distribution of one peak at an average hydrodynamic radius of 34.6 ± 0.2 Å with a polydispersity of 12.3 %. The molecular weights obtained from AutoPorod [27], SAXS MoW [28] and Wyatt Dynamics (Wyatt Technology) are approximately 66.5, 77.3, and 61.3 kDa respectively. These measurements were consistent with the trimeric state of the protein (66.9 kDa). The PDBePISA server [29] also predicted that the trimer was the most probable oligomeric form of the protein in solution out of all assemblies observed in the crystal lattice. Theoretical scattering curves for both the monomeric and trimeric structures of the protein calculated by the program CRYSOL [30] returned χ2 values of 6.13 and 1.53 respectively when compared with the experi-mental data. Figure 3 depicts the averaged experimental scattering data, the theoretical scattering curves (from monomer and trimer) and the particle distance distribution function.
Fig. 3.
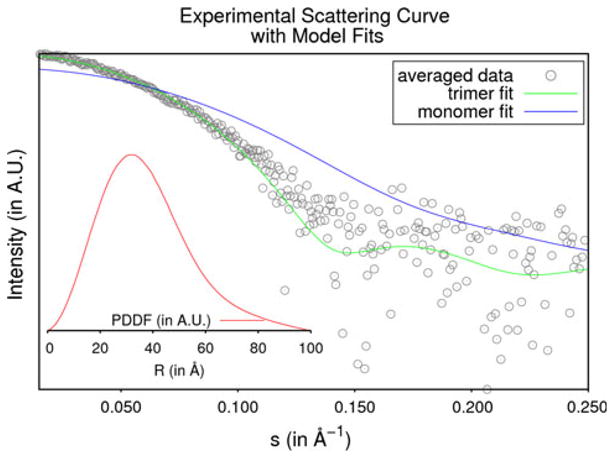
Small-angle X-ray scattering data of SACOL2570. The averaged experimental data for SACOL2570 are shown as grey circles. The theoretical scattering curves for the monomeric and trimeric models are shown in blue and green respectively. The inlaid figure shows the particle distance distribution function in red
SACOL2570 binds AcCoA and CoA in vitro
The binding of the putative acetyltransferase SACOL2570 to CoA and AcCoA was validated by X-ray crystallography after crystal soaking. In addition, the binding in solution was characterized using ITC.
The crystal structure was obtained for SACOL2570 with a bound CoA (PDB code: 3V4E). When SACOL2570 was soaked with AcCoA, only a CoA molecule was observed in the active site, likely due to hydrolysis (see below). Binding of CoA in the 3V4E crystal structure involves residues from two adjacent monomers in the homotrimeric assembly (Fig. 4a). Residues responsible for CoA binding by SACOL2570 are Asn84, Ala112*, Gly141, Lys165* and Arg182 (a star denotes residues contributed by a neighboring subunit in the trimer) (Fig. 4b). Residues responsible for the coordination of the phosphates groups of CoA are Lys165* and Arg182. The Asn84 side-chain forms a hydrogen bond interaction with a water molecule in SACOL2570, but in the structure of lactose operon acetyltransferase from E. coli the corresponding residue Asn85 coordinates the acetyl group (PDB code: 1KRR). The importance of this residue in substrate binding was also confirmed by our further MD analysis (see below).
Fig. 4.
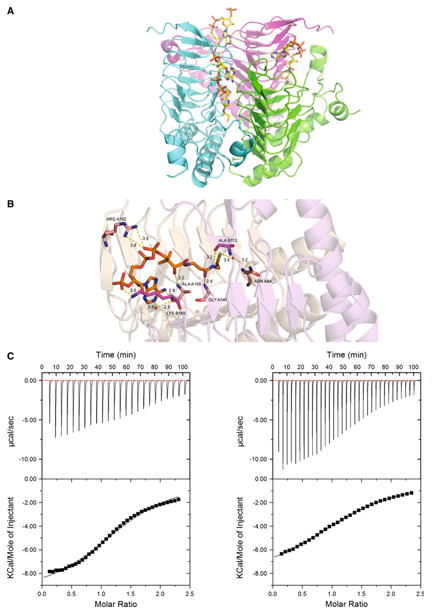
Acetyl coenzyme A (AcCoA) as a cofactor of SACOL2570. a The structure of SACOL2570 with bound CoA. CoA binding sites (in the trimeric assembly) are formed at the interface of two monomers. b Critical residues predicted by MD simulations to participate in AcCoA binding are present in the CoA bound crystal structure of SACOL2570. All distances are shown in Å. c Both AcCoA (left panel KD = 440 μM) and CoA (right panel KD = 660 μM) bind SACOL2570
ITC demonstrated that both AcCoA and CoA bind to SACOL2570 with 1:1 stoichiometry and KD values of 460 ± 35 and 660 ± 13 μM, ΔH of –9,823 ± 200 and –8,451 ± 60 cal/mol, and ΔS of –17.6 and –13.8 cal/mol/deg, respectively (Fig. 4c). As expected the binding of AcCoA is stronger than CoA. Thus, the data confirm that SACOL2570 binds AcCoA and suggest that the protein has acetyltransferase activity. The relatively low binding affinity of SACOL2570 to AcCoA could be related to the absence of a second substrate that might impact an active site organization.
SACOL2570 hydrolyzes AcCoA in the absence of a second substrate
AcCoA exposed to SACOL2570 in solution for 20 h was completely converted to CoA, as shown by mass spec-troscopy analysis (Supplementary Figure 2). Almost no conversion was observed in a control experiment in the absence of a protein. ITC titration (∼1 h) showed only the binding of AcCoA to SACOL2570 without a sign of a reaction. Thus, SACOL2570 appears to catalyze the hydrolysis of AcCoA to CoA, albeit slowly, and crystals soaked with AcCoA for ∼48 h are likely to contain bound CoA.
Search for the second substrate of SACOL2570
We attempted to identify a putative carbohydrate substrate of SACOL2570 by testing the interaction of the protein with a variety of carbohydrates and their derivatives by ITC and crystallography. Glucose, galactose, maltose, saccharose, lactose, mannose, arabinose, fucose, trehalose, xylitol, isopropyl, β-d-1-thiogalactopyranoside, p-nitro-phenyl-β-d-galactoside were tested. However, no binding of any tested compound to SACOL2570 was detected. In addition, the search for a substrate was also performed using glycan array screening by the Functional Glycomics Gateway (www.functionalglycomics.org). The affinity of SACOL2570 towards a diversified set of glycans was explored using an array containing 465 different glycans (Supplementary Table 1). Again, no significant binding was detected (Supplementary Figure 1). Bacterial lectin protein was used as a positive control and showed specific binding to certain glycans (bottom panel, Supplementary Figure 1).
MD simulations and MM-GBSA analysis
Since the SACOL2570 crystals soaked with AcCoA contained only CoA, we employed molecular dynamics simulations to describe the interaction of the protein and AcCoA and to explore the dynamic stability of the two systems (GATSA and GATEC). The RMSD values of backbone atoms relative to the initial X-ray structures were generated over 8 ns NPT trajectories and plotted in Fig. 5. The RMSD curves of the two systems indicated that the solvated systems achieved their equilibrium after 4 ns.
Fig. 5.
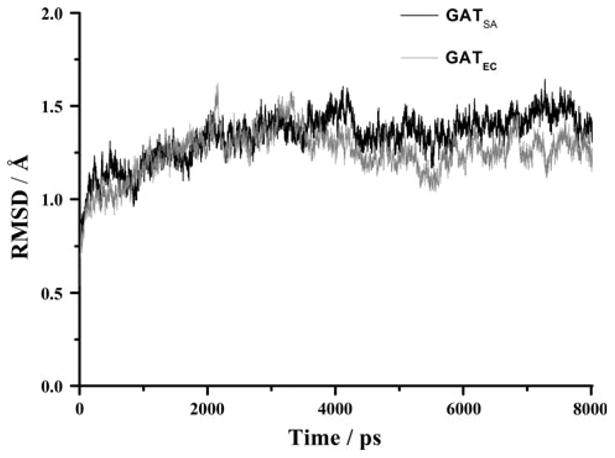
Plots of RMSD versus simulation time in the MD-simulated SACOL2570 GATSA(black) from S. aureus (PDB code: 3FTT) and galactoside acetyltransferase GATEC (grey) from E. coli (PDB code: 1KRR). RMSD represents the root mean-square deviation (Å) of the simulated positions of the backbone atoms (C, N, and Cα) of SACOL2570 and GATEC from those in the initial X-ray crystal structure
Based on the MD simulated-trajectories, the MM/GBSA method predicted binding free energies of –40.89 and –36.44 kcal/mol for AcCoA binding to GATSA and GATEC, respectively. This suggests that AcCoA binds to SACOL2570 with a higher affinity.
Figure 6 depicts the simulated models for the two sys-tems after 8 ns MD simulations. As shown in Fig. 6a, the binding site of AcCoA to SACOL2570 consists of nineteen residues, and several significant binding interactions can be observed. The phosphate group of AcCoA formed two hydrogen bonds of 2.97 and 3.07 Å to the NH1 and NH2 atoms of Arg A182. The N atom of Ala A159, the N atom of Gly A141, and the ND2 atom of Asn A84 make three hydrogen bonds (H1 = 2.95 Å, H2 = 3.17 Å, and H3 = 3.18 Å) with AcCoA. The O atoms of Ala 159 and Ala B112 accept hydrogen bonds (H4 = 3.31 Å and H5 = 3.05 Å) from the N3 and N7 atoms of AcCoA, respectively. A water molecule participates in stabilization of AcCoA binding in the active site of SACOL2570. A hydrogen bond is observed between the O atom of the water and the N1 atom of AcCoA. In addition, the water molecule forms a hydrogen bond to the closest residue, Ala A159. It is clear that eight hydrogen bond interactions between SACOL2570/water and AcCoA are the major forces in stabilizing the AcCoA binding pattern.
Fig. 6.
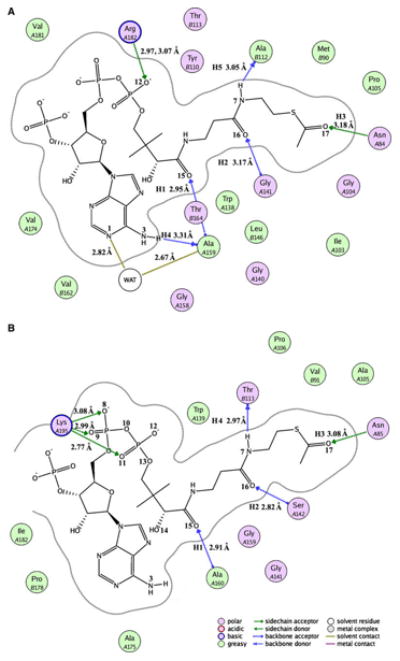
The binding model prepared with molecular operating environment (MOE) software version 2008.10 (http://www.chemcomp.com/software.htm) between acetyl coenzyme A and SACOL2570 (a) or GATEC (b). SACOL2570 and GATEC mean the (putative) galactoside acetyltransferase enzymes from S. aureus (PDB code: 3FTT) and E. coli (PDB code: 1KRR) with bound acetyl coenzyme A, respectively
Similar to the SACOL2570/AcCoA system, AcCoA binds in a long crevice between two adjacent LβH domains in the model of GATEC. As shown in Fig. 6B, the phos-phate groups accept three hydrogen bonds from Lys A195, which play an important role in the substrate binding pat-tern. In addition, four residues (Asn A85, Ser A142, Ala A160, and Thr B113) form four hydrogen bonds (H1 = 2.91 Å, H2 = 2.82 Å, H3 = 3.08 Å, H4 = 2.97 Å) with AcCoA. Excepting H5 (3.69 Å, it can be viewed as an electrostatic interaction), the other three hydrogen bonds are maintained in the crystal structure of GATEC. These five hydrogen bonds were identified in both GATEC and GATSA (as can be seen by comparing Figs. 6a, b). The conserved residues Asn A84, Gly A141, Ala A159, and Ala B112 were observed in the SACOL2570/AcCoA complex. The only interaction which appears to be important for recognition of CoA in SACOL2570 crystal structure which was not predicted by MD simulations is hydrogen bonding between Lys 165* and the phosphate group of CoA. These subtle differences in interactions may account for the slight differences in the predicted binding affinities of AcCoA to GATEC versus SACOL2570.
Implication of the biological role
The structural and biochemical characterization of SACOL2570 presented here strongly suggests that the protein possesses an acetyltransferase function. The ability of SACOL2570 to hydrolyze AcCoA was further confirmed by mass spectrometry measurements (Supplementary Figure 2). Soaking experiments yielded a structure with a bound CoA, and MD analysis completed this study with analysis of AcCoA binding and comparison with GATEC. Conserved residues of SACOL2570 are responsible for binding of AcCoA and CoA in the similar way as in GATEC: Asn84, Ala112*, Gly141, Lys165* and Arg182. The MD model based on the apo-structure implies that AcCoA can bind to the active site pockets of both struc-tures in a similar manner. In the AcCoA bound model of SACOL2570 four of the conserved residues (Asn84, Ala112*, Gly141, Arg182) form five hydrogen bonds with AcCoA stabilizing it in the active site and contributing to the predicted free energy of binding of –40.89 kcal/mol. The ITC experiments confirmed that SACOL2570 binds both CoA and AcCoA in vitro. Taken together, these data suggest that SACOL2570 acts as an acetyltransferase, using AcCoA as an acetyl group donor and an as-yet-undetermined chemical moiety as an acceptor.
The gene encoding SACOL2570 was recently identified by S. aureus pangenomic microarrays as one of the genes for which expression is altered in S. aureus cells grown in the presence of fusidic acid, an antibiotic used to treat MRSA and other Staphylococcus infections [31]. Upon fusidic acid treatment the level of expression of SA-COL2570 was down-regulated fivefold [31], which suggests that the putative galactoside O-acetyltransferase plays a role in cellular processes of resistance to antibiotics and other toxins [31, 32].
SACOL2570 was also recently used as a model protein for the development of a method to obtain self-assembling protein nanomaterials [33]. The natural trimeric form of SACOL2570 allowed for careful engineering of protein– protein interactions between monomers and subsequent formation of cage-like, higher-order assemblies [33]. Further research on utilization of protein trimeric assemblies in SACOL2570 and other structurally similar proteins could lead the design of functional molecular machines with a wide range of applications. Thus determination of the SA-COL2570 crystal structure is not only important for under-standing the molecular mechanisms of antibiotic resistance, but also for innovative approaches in nanotechnology.
Materials and methods
Protein cloning, expression, purification, and crystallization
The SACOL2570 gene was cloned into pMCSG7 vector as described previously [34]. The protein was expressed in E. coli BL21-CodonPlus(DE3)RIPL cells (Stratagene) in M9 minimal media (Shanghai Medicilion) at 37 °C. When OD600 reached 0.8 an amino acid cocktail (VILKTF + SeMet) was added, and after 20 min the culture was induced with isopropyl-1-thio-β-d-galactopyranoside (IPTG) added to a final concentration of 1 mM and grown over-night at 16 °C. Cells were harvested by centrifugation and the cell pellet was re-suspended in binding buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 5 mM imidazole, and 5 % glycerol) supplemented with Complete Protease inhibitors without EDTA (Roche). Resuspended cells were lysed by sonication. The soluble fraction was cleared by centrifu-gation and loaded onto Ni–NTA resin (Qiagen) pre-equil-ibrated with binding buffer. The resin was washed with 300 mL of wash buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 30 mM imidazole, and 5 % glycerol). Elution was performed with 10 mL of elution buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 250 mM imidazole, and 5 % glycerol). EDTA and TCEP were added immediately to the eluted fractions to final concentrations of 1 and 0.5 mM respectively. The His6 tag was removed by cleavage with His-tagged TEV protease at 4 °C overnight in dialysis buffer (50 mM HEPES pH 7.5, 500 mM NaCl, and 5 % glycerol). The cleaved tag and TEV protease were removed from the purified protein with a second Ni-NTA column. Additional purification by size-exclusion chromatography was performed using a Superdex-200 column and crystallization buffer (10 mM HEPES, pH 7.5, 500 mM NaCl). The identity and purity of the final protein samples were evaluated by mass spectrometry and SDS-PAGE. Protein samples were concentrated to 2.6 mg/mL and used for crystallization screening. Crystals of SACOL2570 were grown by hanging-drop vapor diffusion at 295 K. A drop consisting of 1 μL of protein solution (2.6 mg/mL Se-Met-SACOL2570 in 10 mM HEPES buffer, pH 7.5 and 500 mM NaCl) and 1 μL of the well solution (200 mM di-ammonium hydrogen citrate, 20 % w/v PEG3350 pH 5.0) was equilibrated against 1 mL well solution. Crystals selected for data collection were transferred to a paratone-N and flash-cooled in the liquid nitrogen at 100 K. For ligand–protein complex experiments, protein was overex-pressed in Luria-Broth media, purified as described above and crystallized in 200 mM NaH2PO4, 20 % w/v PEG 3350, pH 7.5 by hanging-drop vapor diffusion at 295 K. Obtained crystals were soaked with AcCoA. Crystals were taken directly from crystallization drops and flash-cooled in liquid nitrogen.
Data collection, structure determination, and refinement
Diffraction data from SACOL2570 crystals were collected at 100 K at beamline 19-ID [35] of the Structural Biology Center at the Advanced Photon Source. The processing and scaling as well as electron density map and preliminary model were obtained with HKL-3000 [36], which inte-grates with the family of SHELX programs [37], MLP-HARE [38], DM [39], SOLVE/RESOLVE [40], CCP4 [41], ARP/wARP [42], and COOT [43], The space group of the crystal was R32 with one molecule in the asymmetric unit. The resulting model was improved by cycles of manual model building in COOT followed by maximum-likelihood refinement with REFMAC [44], The TLSMD Web server [45] was used for generation of multigroup TLS models. MOLPROBITY [46] and the ADIT Web server [47] were used to validate the structure. The atomic coordinates together with the structure factors, were deposited in PDB with accession code 3FTT.
The diffraction data of CoA-bound crystal were col-lected at 100 K at beamline 21ID-D of the Life Sciences Collaborative Access Team (LS-CAT) at APS. The struc-ture was solved by molecular replacement using MOLREP [48] and the structure of apo-SACOL2570 as a search model. The HKL-3000 package was used, as well as COOT and REFMAC for further model building and refinement. The structure was deposited in PDB with accession code 3V4E. Data collection, structure determination, and refinement statistics for both apo- and CoA-bound structures are summarized in Table 1.
Table 1. Summary of data collection, phasing, and refinement sta-tistics for the putative acetyltransferase SACOL2570 from S. aureus.
| Organism |
S. aureus PDB code: 3FTT SACOL2570 |
S. aureus PDB code: 3V4E SACOL2570 |
|---|---|---|
| Crystal | ||
| Space group | R32 | P1 |
| Unit cell (Å) | a = 75.8, b = 75.8, c = 93.9 | a = 53.4, b = 53.5, c = 53.5 |
| (°) | α = 90.00, β = 90.00, γ = 120.00 | α = 89.7, β = 89.8, γ = 89.9 |
| No. of molecules in asymmetric unit | 1 | 3 |
| Data collection | ||
| Diffraction protocol | SAD | MR |
| Wavelength (Å) | 0.9794 | 0.9787 |
| Resolution (Å) | 50.0–1.60 | 50.0–1.95 |
| Highest resolution shell (Å) | 1.60–1.59 | 1.98–1.95 |
| Observed reflections | 27,020 (504) | 87,088 (1,293) |
| Unique reflections | 25,203 | 39,809 |
| Redundancy | 5.4 (3.6) | 2.2 (2.1) |
| Completeness (%) | 99.4 (75.4) | 92.5 (57.2) |
| I/σ(I) | 30.5 (4.1) | 31.8 (10.6) |
| Rmerge (%) | 9.3 (19.5) | 3.4 (7.5) |
| Refinement | ||
| Rwork (%) | 15.9 (16.1) | 18.0 (16.4) |
| Rfree (%) | 18.7 (21.5) | 22.2 (22.8) |
| RMSD for bond length (Å) | 0.020 | 0.018 |
| RMSD for bond angles (°) | 1.63 | 1.80 |
| Number of total atoms | 1,703 | 4,762 |
| Number of protein atoms | 1,485 | 4,396 |
| Number of water molecules | 218 | 200 |
| Average B factor (Å2) | 18.2 | 34.9 |
| Ramachandran statistics | ||
| Most favored regions (%) | 98.9 | 98.2 |
| Additional allowed regions (%) | 0.2 | 0.2 |
Data for the highest resolution shell are given in parentheses. Ramachandran statistics were calculated with MOLPROBITY
Small-angle X-ray scattering and dynamic light scattering
Purified protein was loaded onto a Sephadex G-200 column equilibrated with a buffer consisting of 150 mM NaCl and 10 mM HEPES pH 7.5 and eluted at the concentration of 5.3 mg/mL. Samples were diluted to one half or one quarter of that concentration. Three concentrations were used (5.3, 2.7 and 1.3 mg/mL). Each sample was passed through a 0.1 μm filter before SAXS and DLS trials. For each sample analyzed with SAXS, the polydispersity, hydrodynamic radius and molecular weight were measured using a Wyatt DynaPro Titan (Wyatt Technology) dynamic light scattering setup at 10 °C with the DYNAMICS soft-ware package (Wyatt Technology). Each sample was analyzed for at least two trials of twenty acquisitions, each lasting 20 s. For SAXS studies, each protein sample was analyzed at 4 °C for a total of 4 h including a check for radiation damage on a Rigaku S-MAX3000 (λ = 1.54 Å). Each sample was exposed twice, for 2 h followed by a second 2 h (to check for radiation damage). The buffer used for column equilibration (150 mM NaCl, 10 mM HEPES, pH 7.5) was analyzed twice, for 2 h each time. The SAXS GUI (JJ X-Ray Systems ApS, Lyngby, Den-mark; Rigaku IT Inc., Auburn Hills, MI) was used to combine detector readouts for each concentration and to remove dead pixels and those regions of the detector immediately behind the beam stop. Slight angular varia-tions in intensity immediately surrounding the beam stop were noted for the lowest and middle concentrations and one of the buffer detector readouts, and a range of 0.015 to 0.25 Å–1 was selected for further data processing of all sets. Scaling for beam intensity and buffer subtraction was handled by PRIMUS [26], The radius of gyration (Rg) was measured for each concentration and linearity for the Guinier region was verified using the AutoRg extension of PRIMUS. The Rg values for the individual exposures are listed in Supplementary Table 2. The automated indirect Fourier transform application AutoGNOM [27] was used to calculate the particle distance distribution function. The online tool SAXS MoW [28] and the ATSAS tool Au-toPorod [27] were each used to estimate the molecular weight of the protein. The monomer of the protein (PDB code: 3FTT) was submitted to the online service PDBeP-ISA [29] and the theoretical scattering curve of the sub-mitted and returned models was calculated and compared to the experimental data using CRYSOL [30].
MD simulations
The starting model of the SACOL2570/AcCoA complex was constructed based on the crystal structures of SACOL2570 and GATEC. A single active site is formed by residues from two neighboring subunits in GATEC. GATEC and SACOL2570 were structurally aligned, and then the substrate AcCoA in GATEC was extracted and merged into SACOL2570. This new complex which was composed of SACOL2570 and AcCoA from the original complex GATEC, was used as the starting model for the following simulations. A new system was prepared, named GATSA, which is SACOL2570 in complex with AcCoA. All hydrogen atoms were added using the Xleap tools from the AMBER10 package [49].
Based on the electrostatic potential calculations at the ab initio HF/6-31G* level [50], the partial atomic charges of AcCoA were determined by using the restricted elec-trostatic potential [51] fitting protocol implemented in the Antechamber module of the AMBER10 package. GAFF (general AMBER force field) parameters were used as the parameters for AcCoA, and the AMBER FF03 force field parameters were used for the receptor. Counter ions were added to maintain the electroneutrality of the two systems. Each system was subsequently solvated in a rectangular box of water molecules with solvent layers of 10 Å.
Conventional MD simulations on the aforementioned systems GATEC and GATSA were carried out with AMBER10. The two systems were submitted to energy minimization to remove unfavorable contacts. After the relaxation, each system was gradually heated from 0 to 300 K in 50 ps. A weak constraint of 10 kcal/mol/Å2 was employed to keep the protein constrained during the heating procedure. Finally, periodic boundary dynamics simulations of 8 ns were performed in an NPT (constant composition, pressure, and temperature) ensemble at 1 atm and 300 K. The temperature was kept at 300 K by means of the weak-coupling algorithm [52]. The SHAKE algorithm [53] was turned on for all bonds involving hydrogens with a tolerance of 1 × 10-5 Å Electrostatic interactions were calculated using the Particle-Mesh-Ewald method with a 10 Å non-bonded cutoff. The output trajectory files were saved every 1 ps for subsequent analysis.
MM-GBSA analysis
The MM-GBSA model, which is implemented in AMBER10, was applied to compute the binding free energy (ΔGbind) between a protein and a ligand [22, 23, 43]. A grid spacing of 0.5 Å was chosen, and the dielectric constant for the solute and solvent were set to 1 and 80, respectively. The optimized atomic radii set in AMBER 10 were used. A total of 100 snapshots were taken from 7 to 8 ns trajectory with an interval of 10 ps. All counterions and water molecules were stripped. In the present study, the binding free energies of the two systems were calculated using the MM-GBSA model. In most cases, different ligands binding to the same protein may give similar entropy values [54, 55]. In the present work, the entropy contribution was not included due to the same ligand AcCoA. Thus, the predicted binding free energies are based on a relative scale and can only be compared with each other to evaluate the relative binding affinity of AcCoA with SACOL2570 versus GATEC.
Isothermal titration calorimetry
The iTC200 calorimeter (MicroCal) was used for all iso-thermal titration calorimetry (ITC) experiments. The puri-fied protein sample was dialyzed overnight at 4 °C against 100 mM HEPES pH = 7.5 and 150 mM NaCl. All ligands were dissolved in the same buffer. Experiments were per-formed at 25 °C by titrating 1.8 mM SACOL2570 with a 20 mM ligand solution. ITC titration curves were collected and analyzed using the Origin software package.
Glycoarray binding assay
SACOL 2570 was diluted to 0.5 mg/mL in 200 mM NaCl, 10 mM HEPES pH 7.5 and sent to the Consortium for Functional Glycomics (CFG) for oligosaccharides binding assay. The glycoarray binding assay was employed in CFG facility with bacterial lectin used as a positive control. Two different methods of binding were applied: a sequential addition of the enzyme followed by anti-His antibody, and pre-complexing the enzyme with anti-His and anti-mouse IgG to increase the affinity and valency of binding.
Mass spectrometry
To test the ability of SACOL2570 to catalyze the hydro-lysis of AcCoA to CoA in the absence of substrate, two reactions samples were prepared: a test sample with 70 μM SACOL2570 and 2.5 mg/mL AcCoA, and a control sample with only 2.5 mg/mL AcCoA. Both samples also contained a buffer with 50 mM HEPES and 50 mM NaCl at a pH of 7.0. The samples were incubated for 20 h at 16 °C and analyzed by MALDI mass spectroscopy (MS). Due to requirements of MS analysis it was not possible to replicate all aspects of the soak solution, but the reactions were the same pH and temperature as the soaking conditions. The MALDI experiment was conducted in negative reflector mode, using the Bruker Microflex spectrometer in the W. M. Keck Biomedical Mass Spectrometry Laboratory at the University of Virginia.
Supplementary Material
Acknowledgments
We thank Matthew D. Zimmerman, David Cooper and Dominika Borek for their help in this study and for critical comments on the manuscript. This research was funded by the National Institute of Allergy and Infectious Diseases, National Insti-tutes of Health, Department of Health and Human Services under Contract No. HHSN272200700058C), Natural Science Foundation of China (21103234), and Fundamental Research Funds for the Central Universities of China (11ykzd05). The diffraction data were collected at the beam lines 19-ID, 19-BM, and 21-ID at Argonne National Laboratory. Argonne is operated by University of Chicago Argonne, LLC, for the US Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor for the support of this research program (Grant 085P1000817). We would like to acknowledge Dr. Nicholas E. Sherman, the director of the W. M. Keck Biomedical Mass Spectrometry Laboratory is funded by a grant from the University of Virginia Pratt Fund through the School of Medicine.
Abbreviations
- MRSA
Methicillin-resistant Staphylococcus aureus
- GAT
Galactoside acetyltransferase
- SAXS
Small-angle X-ray scattering
- DLS
Dynamic light scattering
- MD
Molecular dynamics
- PDB
Protein Data Bank
- CSGID
Center for Structural Genomics of Infectious Diseases
- ITC
Isothermal titration calorimetry
- RMSD
Root mean square deviation
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10969-013-9158-6) contains supplementary material, which is available to authorized users.
Contributor Information
Hai-Bin Luo, Department of Molecular Physiology and Biological Physics, University of Virginia, 1340 Jefferson Park Avenue, Charlottesville, VA 22908, USA; School of Pharmaceutical Sciences, Sun Yat-Sen University, East Campus, 510006 Guangzhou, China; Center for Structural Genomics of Infectious Diseases, Charlottesville, VA, USA.
Aleksandra A. Knapik, Department of Molecular Physiology and Biological Physics, University of Virginia, 1340 Jefferson Park Avenue, Charlottesville, VA 22908, USA; Center for Structural Genomics of Infectious Diseases, Charlottesville, VA, USA
Janusz J. Petkowski, Department of Molecular Physiology and Biological Physics, University of Virginia, 1340 Jefferson Park Avenue, Charlottesville, VA 22908, USA
Matthew Demas, Department of Molecular Physiology and Biological Physics, University of Virginia, 1340 Jefferson Park Avenue, Charlottesville, VA 22908, USA.
Igor A. Shumilin, Department of Molecular Physiology and Biological Physics, University of Virginia, 1340 Jefferson Park Avenue, Charlottesville, VA 22908, USA; Center for Structural Genomics of Infectious Diseases, Charlottesville, VA, USA
Heping Zheng, Department of Molecular Physiology and Biological Physics, University of Virginia, 1340 Jefferson Park Avenue, Charlottesville, VA 22908, USA; Center for Structural Genomics of Infectious Diseases, Charlottesville, VA, USA.
Maksymilian Chruszcz, Department of Molecular Physiology and Biological Physics, University of Virginia, 1340 Jefferson Park Avenue, Charlottesville, VA 22908, USA; Center for Structural Genomics of Infectious Diseases, Charlottesville, VA, USA; Department of Chemistry and Biochemistry, University of South Carolina, 631 Sumter Street, Columbia, SC 29208, USA.
Wladek Minor, Email: wladek@iwonka.med.virginia.edu, Department of Molecular Physiology1 and Biological Physics, University of Virginia, 1340 Jefferson Park Avenue, Charlottesville, VA 22908, USA; Center for Structural Genomics of Infectious Diseases, Charlottesville, VA, USA.
References
- 1.Kuroda M, et al. Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet. 2001;357(9264):1225–1240. doi: 10.1016/s0140-6736(00)04403-2. [DOI] [PubMed] [Google Scholar]
- 2.Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339(8):520–532. doi: 10.1056/NEJM199808203390806. [DOI] [PubMed] [Google Scholar]
- 3.Gill SR, et al. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-pro-ducing methicillin-resistant Staphylococcus epidermidis strain. J Bacteriol. 2005;187(7):2426–2438. doi: 10.1128/JB.187.7.2426-2438.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klein E, Smith DL, Laxminarayan R. Hospitalizations and deaths caused by methicillin-resistant Staphylococcus aureus, United States, 1999–2005. Emerg Infect Dis. 2007;13(12):1840–1846. doi: 10.3201/eid1312.070629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siegman-Igra Y, et al. The role of vancomycin in the persistence or recurrence of Staphylococcus aureus bacteraemia. Scand J Infect Dis. 2005;37(8):572–578. doi: 10.1080/00365540510038488. [DOI] [PubMed] [Google Scholar]
- 6.Howden BP, et al. Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin Microbiol Rev. 2010;23(1):99–139. doi: 10.1128/CMR.00042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bowling FL, Salgami EV, Boulton AJ. Larval therapy: a novel treatment in eliminating methicillin-resistant Staphylococcus aureus from diabetic foot ulcers. Diabetes Care. 2007;30(2):370–371. doi: 10.2337/dc06-2348. [DOI] [PubMed] [Google Scholar]
- 8.Soothill JS. Treatment of experimental infections of mice with bacteriophages. J Med Microbiol. 1992;37(4):258–261. doi: 10.1099/00222615-37-4-258. [DOI] [PubMed] [Google Scholar]
- 9.Matsuzaki S, et al. Experimental protection of mice against lethal Staphylococcus aureus infection by novel bacteriophage phi MR11. J Infect Dis. 2003;187(4):613–624. doi: 10.1086/374001. [DOI] [PubMed] [Google Scholar]
- 10.Anderson WF. Structural genomics and drug discovery for infectious diseases. Infect Disord Drug Targets. 2009;9(5):507–517. doi: 10.2174/187152609789105713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roderick SL. The lac operon galactoside acetyltransferase. C R Biol. 2005;328(6):568–575. doi: 10.1016/j.crvi.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 12.Wang XG, Olsen LR, Roderick SL. Structure of the lac operon galactoside acetyltransferase. Structure. 2002;10(4):581–588. doi: 10.1016/s0969-2126(02)00741-4. [DOI] [PubMed] [Google Scholar]
- 13.Wang XG, Roderick SL. Expression, purification, crys-tallization and preliminary x-ray data of Escherichia coli galac-toside acetyltransferase. Acta Crystallogr D Biol Crystallogr. 1999;55(Pt 11):1955–1957. doi: 10.1107/s0907444999011701. [DOI] [PubMed] [Google Scholar]
- 14.Musso RE, Zabin I. Substrate specificity and kinetic studies on thiogalactoside transacetylase. Biochemistry. 1973;12(3):553–557. doi: 10.1021/bi00727a031. [DOI] [PubMed] [Google Scholar]
- 15.Beaman TW, Sugantino M, Roderick SL. Structure of the hexapeptide xenobiotic acetyltransferase from Pseudomonas aeruginosa. Biochemistry. 1998;37(19):6689–6696. doi: 10.1021/bi980106v. [DOI] [PubMed] [Google Scholar]
- 16.Olsen LR, Vetting MW, Roderick SL. Structure of the E. coli bifunctional GlmU acetyltransferase active site with substrates and products. Protein Sci. 2007;16(6):1230–1235. doi: 10.1110/ps.072779707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lo Leggio L, et al. The structure and specificity of Esch-erichia coli maltose acetyltransferase give new insight into the LacA family of acyltransferases. Biochemistry. 2003;42(18):5225–5235. doi: 10.1021/bi0271446. [DOI] [PubMed] [Google Scholar]
- 18.Sugantino M, Roderick SL. Crystal structure of Vat(D): an acetyltransferase that inactivates streptogramin group A antibiotics. Biochemistry. 2002;41(7):2209–2216. doi: 10.1021/bi011991b. [DOI] [PubMed] [Google Scholar]
- 19.Olsen LR, et al. Structure of serine acetyltransferase in complexes with CoA and its cysteine feedback inhibitor. Bio-chemistry. 2004;43(20):6013–6019. doi: 10.1021/bi0358521. [DOI] [PubMed] [Google Scholar]
- 20.Liu ZJ, Li Y, Chen L, Zhu J, Rose JP, Ebihara A, Yokoyama S, Wang BC. Crystal structure of maltose transacetylase from Geobacillus kaustophilus at 1.78 angstrom resolution. 2006 PDB http://dx.doi.org/10.2210/pdb2ic7/pdb.
- 21.Pye VE, et al. The structure and mechanism of serine acetyltransferase from Escherichia coli. J Biol Chem. 2004;279(39):40729–40736. doi: 10.1074/jbc.M403751200. [DOI] [PubMed] [Google Scholar]
- 22.Bea I, et al. Chelate effect in cyclodextrin dimers: a com-putational (MD, MM/PBSA, and MM/GBSA) study. J Org Chem. 2006;71(5):2056–2063. doi: 10.1021/jo052469o. [DOI] [PubMed] [Google Scholar]
- 23.Massova I, Kollman PA. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect Drug Discov Des. 2000;18:113–135. [Google Scholar]
- 24.Lewendon A, Ellis J, Shaw WV. Structural and mecha-nistic studies of galactoside acetyltransferase, the Escherichia coli LacA gene product. J Biol Chem. 1995;270(44):26326–26331. doi: 10.1074/jbc.270.44.26326. [DOI] [PubMed] [Google Scholar]
- 25.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 26.Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI. PRIMUS: a Windows-PC based system for small-angle scattering data analysis. J Appl Cryst. 2003;36:1277–1282. [Google Scholar]
- 27.Petoukhov MV, et al. ATSAS 2.1—towards automated and web-supported small-angle scattering data analysis. J Appl Crystallogr. 2007;40:S223–S228. [Google Scholar]
- 28.Fischer H, de Oliveira Neto M, Napolitano HB, Polikarpov I, Craievich AF. Determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale. J Appl Cryst. 2010;43:101–109. [Google Scholar]
- 29.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372(3):774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 30.Svergun D, Barberato C, Koch MHJ. CRYSOL-a program to evaluate X-ray solution scattering of biological macromole-cules from atomic coordinates. J Appl Cryst. 1995;28:768–773. [Google Scholar]
- 31.Delgado A, et al. The fusidic acid stimulon of Staphylo-coccus aureus. J Antimicrob Chemother. 2008;62(6):1207–1214. doi: 10.1093/jac/dkn363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Howden BP, Grayson ML. Dumb and dumber–the poten-tial waste of a useful antistaphylococcal agent: emerging fusidic acid resistance in Staphylococcus aureus. Clin Infect Dis. 2006;42(3):394–400. doi: 10.1086/499365. [DOI] [PubMed] [Google Scholar]
- 33.King NP, et al. Computational design of self-assembling protein nanomaterials with atomic level accuracy. Science. 2012;336(6085):1171–1174. doi: 10.1126/science.1219364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stols L, et al. A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr Purif. 2002;25(1):8–15. doi: 10.1006/prep.2001.1603. [DOI] [PubMed] [Google Scholar]
- 35.Rosenbaum G, et al. The Structural Biology Center 19ID undulator beamline: facility specifications and protein crystallo-graphic results. J Synchrotron Radiat. 2006;13(Pt 1):30–45. doi: 10.1107/S0909049505036721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Minor W, et al. HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 8):859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 37.Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64(Pt 1):112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 38.Isomorphous replacement and anom-alous scattering. In: Otwinowski Z, editor; Wolf W, Evans PR, Leslie AGW, editors. Proceedings of the CCP4 study weekend. Daresbury Laboratory; Warrington, UK: 1991. pp. 80–86. [Google Scholar]
- 39.Cowtan KD, Zhang KY. Density modification for macro-molecular phase improvement. Prog Biophys Mol Biol. 1999;72(3):245–270. doi: 10.1016/s0079-6107(99)00008-5. [DOI] [PubMed] [Google Scholar]
- 40.Terwilliger T. SOLVE and RESOLVE: automated structure solution, density modification and model building. J Syn-chrotron Radiat. 2004;11(Pt 1):49–52. doi: 10.1107/s0909049503023938. [DOI] [PubMed] [Google Scholar]
- 41.Winn MD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6(5):458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 43.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 44.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(Pt 3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 45.Painter J, Merritt EA. TLSMD web server for the generation of multi-group TLS models. J Appl Crystallogr. 2006;39:109–111. [Google Scholar]
- 46.Lovell SC, et al. Structure validation by Calpha geometry: phi, psi and Cbeta deviation. Proteins. 2003;50(3):437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 47.Yang H, et al. Automated and accurate deposition of structures solved by X-ray diffraction to the Protein Data Bank. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 10):1833–1839. doi: 10.1107/S0907444904019419. [DOI] [PubMed] [Google Scholar]
- 48.Vagin AA, Isupov MN. Spherically averaged phased translation function and its application to the search for molecules and fragments in electron-density maps. Acta Crystallogr D Biol Crystallogr. 2001;57(Pt 10):1451–1456. doi: 10.1107/s0907444901012409. [DOI] [PubMed] [Google Scholar]
- 49.Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr. 2003;36:860–864. doi: 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Case DA, Darden T, Cheatham TE, III, Simmerling CL, Wang J. AMBER 10. University of California; San Francisco: 2008. [Google Scholar]
- 51.Frisch MJ, Trucks G, Schlegel HB, Scuseria GE, Robb MA. Gaussian 03, Revision D.01. Gaussian, Inc; Pittsburgh, PA: 2004. [Google Scholar]
- 52.Wang JM, Wang W, Kollman PA. Antechamber: an accessory software package for molecular mechanical calculations. Abstr Pap Am Chem Soc. 2001;222:U403–U403. [Google Scholar]
- 53.Berendsen HJC, et al. Molecular-dynamics with coupling to an external bath. J Chem Phys. 1984;81(8):3684–3690. [Google Scholar]
- 54.Darden T, York D, Pedersen L. Particle mesh Ewald—an N. Log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98(12):10089–10092. [Google Scholar]
- 55.Liu M, et al. Binding of curcumin with glyoxalase I: molecular docking, molecular dynamics simulations, and kinetics analysis. Biophys Chem. 2010;147(1–2):28–34. doi: 10.1016/j.bpc.2009.12.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.