

Background: Obesity-related diseases result from accumulation of lipids in nonadipose tissues.
Results: Mutations in 167 yeast genes confer fatty acid sensitivity. Loss of yeast and mammalian ARV1 results in pronounced lipid hypersensitivity, lipoapoptosis, and reduced triglyceride synthesis.
Conclusion: 75 evolutionarily conserved components of obesity-related disorders were identified.
Significance: Understanding lipid sensitivity may lead to treatment of numerous human metabolic diseases.
Keywords: Fatty Acid, Lipid Droplets, Lipid Metabolism, Lipotoxicity, Triglyceride, Yeast Genetics
Abstract
The toxic subcellular accumulation of lipids predisposes several human metabolic syndromes, including obesity, type 2 diabetes, and some forms of neurodegeneration. To identify pathways that prevent lipid-induced cell death, we performed a genome-wide fatty acid sensitivity screen in Saccharomyces cerevisiae. We identified 167 yeast mutants as sensitive to 0.5 mm palmitoleate, 45% of which define pathways that were conserved in humans. 63 lesions also impacted the status of the lipid droplet; however, this was not correlated to the degree of fatty acid sensitivity. The most liposensitive yeast strain arose due to deletion of the “ARE2 required for viability” (ARV1) gene, encoding an evolutionarily conserved, potential lipid transporter that localizes to the endoplasmic reticulum membrane. Down-regulation of mammalian ARV1 in MIN6 pancreatic β-cells or HEK293 cells resulted in decreased neutral lipid synthesis, increased fatty acid sensitivity, and lipoapoptosis. Conversely, elevated expression of human ARV1 in HEK293 cells or mouse liver significantly increased triglyceride mass and lipid droplet number. The ARV1-induced hepatic triglyceride accumulation was accompanied by up-regulation of DGAT1, a triglyceride synthesis gene, and the fatty acid transporter, CD36. Furthermore, ARV1 was identified as a transcriptional of the protein peroxisome proliferator-activated receptor α (PPARα), a key regulator of lipid homeostasis whose transcriptional targets include DGAT1 and CD36. These results implicate ARV1 as a protective factor in lipotoxic diseases due to modulation of fatty acid metabolism. In conclusion, a lipotoxicity-based genetic screen in a model microorganism has identified 75 human genes that may play key roles in neutral lipid metabolism and disease.
Introduction
Obesity-related diseases such as type 2 diabetes (T2D)6 and nonalcoholic fatty liver disease frequently arise from the ectopic deposition of fatty acids in tissues such as the pancreas, muscle, and liver. The limited lipid storage capacity of these tissues frequently results in cellular dysfunction and apoptotic initiation. This process of lipotoxicity is a common phenomenon, arising in many mammalian cell types (1–4) as well as in model eukaryotes such as Drosophila melanogaster (5) and Saccharomyces cerevisiae (6, 7). Despite this preponderance, the pathways that either sensitize or protect cells against lipid-induced cell death are relatively unexplored.
Saturated fatty acids (8–11) and unsaturated fatty acids (UFA) (12–17) induce cytotoxicity by different mechanisms but are normally accommodated in eukaryotic cells by esterification with alcohols to produce neutral lipids. Accordingly, DGAT1−/− murine fibroblasts, deficient in a diacylglycerol acyltransferase encoding the committed terminal step in triglyceride biosynthesis, are highly sensitive to UFA and saturated fatty acid (18). Similarly, although most strains of S. cerevisiae are resistant to lipid-induced cell death, deletion of all four neutral lipid synthesis enzymes (encoded by the ARE1, ARE2, DGA1, and LRO1 genes) results in severe sensitivity to monounsaturated fatty acids such as oleate (C18:1) and palmitoleate (C16:1) (6, 7).
In human cells, at least 11 distinct acyltransferase reactions promote the storage of fatty acids in the esterified form (19). This redundancy implies that variation in these genes will be a rare contributor to lipotoxicity in humans. Consequently, the accumulation of NL into lipid droplets has been studied in cultured mammalian cells, fruit flies, nematodes, and yeast (19, 20) as a pathological reporter of lipid toxicity and the associated pathologies. Accordingly, lipid droplet morphology screens and genome-wide association studies have identified several pathways involved in cellular lipid metabolism (21–25) or genetic variants associated with disease phenotypes (26–32). Surprisingly, these studies display little concordance, and very few of the outcomes pertain directly to lipotoxicity. To identify pathways that specifically protect eukaryotic cells against fatty acid overload, we performed a genome-wide mutagenesis screen in yeast for sensitivity to unsaturated fatty acids. We describe here 167 loci that are required for yeast cells to tolerate UFA such as palmitoleate. The most severely UFA-sensitive strain resulted from deletion of the “ARE2 required for viability” (ARV1) gene. The ARV1-encoded pathway was first identified in yeast as essential in cells lacking sterol esterification due to deletions in the two acyl-coenzyme A cholesterol acyltransferase-related enzymes, ARE1 and ARE2. Orthologs of yeast ARV1 have been identified in all eukaryotic organisms queried thus far (33–35). We describe here studies that implicate the ARV1 gene product as a modulator of cellular fatty acid levels, a process that plays a role in progression of many lipotoxic diseases.
EXPERIMENTAL PROCEDURES
Yeast General
Molecular biology and yeast procedures were performed conventionally (36). All yeast strains used in this study were derived from s288C (Open Biosystems), with the exception of SCY2021 (are1::HIS3 are2::LEU2 dga1::URA3 lro1::URA3), which is derived from W303 (37). Fatty acids were added to media in 0.6% ethanol/tyloxapol (5:1, v/v) at the designated molar concentration (10% w/v stock in ethanol). Liquid growth assays were initiated at 0.1 A600 nm on a Microbiology Workstation Bioscreen C (Thermo Electron Corp.) and analyzed with Research Express Bioscreen C software (Transgalactic Ltd.). Three isolates per genotype were normalized to an absorbance (A600) of 0.1, and 10 μl of each strain was added to 290 μl of media per well and grown at 30 °C for 4 days. Sensitivity assays on solid media contained 0.5 mm fatty acids (palmitoleate, oleate, and linoleate). Cells were plated as serial dilutions.
Genome-wide UFA Sensitivity Screen
The palmitoleate screen was carried out using the MATa haploid deletion strains (Open Biosystems, catalog no. YSC1053) and essential gene knockdown DAmP (decreased expression through mRNA perturbation) strains (provided by M. Schuldiner). Frozen stocks were thawed and inoculated into YPD + 200 mg/liter G418 in 96-well dishes using a singer RoToR HDA replicator robot. Cultures were grown for 2 days at 30 °C and diluted 1:100 in water. 1 μl of the culture was pinned onto SC + 2% dextrose agar plates, with or without 0.5 mm palmitoleate, and grown for 2 days at 30 °C. Growth on plates was quantified using ImageJ software. Growth normalization was done by dividing each colony size on YPD + PO by the average growth on YPD. The normalized growth of the quadruplicates was then averaged. Differential growth was determined by using the log2 ratio between control and experimental (+ PO) plate values. p values comparing these normalized growth variables were completed by Student's t test.
Screen Validation
Strains showing decreased growth in plate assays on palmitoleate containing media were confirmed in liquid SCD growth media for three isolates per genotype (Fig. 1A). An automated growth analysis program (Matlab R2007b version 7.5) was used to identify growth curve parameters for each mutant in SCD and SCD + palmitoleate. The maximum growth rate and the maximum A600 in palmitoleate-containing media were normalized to growth in FA-free SCD within each strain and then expressed as a ratio relative to control strains. By this manipulation, a higher rate ratio indicates slower growth of the mutant strain in fatty acid-containing media. Similarly, a higher absorbance ratio indicates a less saturated mutant culture due to FA supplementation. The last variable, a time lag, was calculated as the time at which maximum growth rate occurs. A normalized time value was then determined by subtracting the maximum rate time in SCD from the time in SCD + palmitoleate. This represents the time lag caused by fatty acid supplementation within each strain. Finally, this value was expressed as the difference between the normalized mutant time lag and the control time lag. A larger value indicates the deletion strain reaches log phase growth after controls. The three growth curve variables were then used to determine statistically significant changes in growth of the mutants, compared with a normal strain, using analysis of variance and t tests.
FIGURE 1.

Identification and analysis of gene deletions conferring sensitivity to palmitoleate in yeast. A, growth curve analysis. Values for each curve were calculated and normalized compared with wild type control. Strains were assigned a toxicity score calculated as a normalized sum relative to control strains of three growth curve parameters as follows: maximum growth rate, maximum cell density, and time to reach maximum density. The higher the score, the worse the growth compared with no treatment control strains. B, lipid droplet morphometry. Single deletion mutants were grown overnight in YPD and stained with Nile Red. Five or more images were captured to determine whether the strains had abnormal lipid droplet number or size. Four mutants representative of the spectrum of the observed morphologies are shown with their associated toxicity score in parentheses. Mutants were classified into three CLD groups as follows: class I (fewer CLD, magenta), class II (normal CLD, blue), and class III (more CLD, orange). C, gene deletion strains were categorized by cellular process and plotted according to their degree of sensitivity based on toxicity score (log2 scale, supplemental Table S1) and lipid droplet morphology (supplemental Table S2). In addition to this classification, 65 genes with human orthologs (filled circles, Table 1) are differentiated from those without known human orthologs (89 genes, open circles). The liporesistant control strain with a toxicity score of 2 and the liposensitive neutral lipid-deficient quadruple knock-out (are1Δ are2Δ dga1Δ lro1Δ, toxicity score of 103) are plotted in the last row. In each category, the most liposensitive deletion is identified. D, PO-sensitive strains induce lipid droplet stores in response to lipid exposure Seven FA-sensitive gene deletions (toxicity score >15) with normal basal CLD numbers were grown to log phase in YPD and subsequently incubated for 16 h in sublethal levels of fatty acids (0.05 mm PO). The area of the cell composed of CLDs was quantified using ImageJ analysis and expressed as a ratio of CLD area following PO incubation compared with CLD area in YPD (mean ± S.D, five fields per strain). Lipid synthesis in the same media conditions was measured by [3H]oleate incorporation into triacylglycerol, steryl ester, and phospholipids (TG, sterol ester, and PL, respectively) and expressed as a ratio of percent of total lipid species in YPD + PO compared with YPD. Asterisks denote statistical significance compared with the wild type control in the same condition (*, p < 0.05; **, p < 0.005).
Cell Culture
The MIN6 mouse insulinoma cell line (Alan Attie, Madison, WI) (38) was maintained in Dulbecco's modified Eagles medium (DMEM) containing 25 mm glucose, 15% heat-inactivated fetal bovine serum (FBS), 100 units/ml penicillin, 100 μg/ml streptomycin, 100 μg/ml l-glutamine, and 5 μl/liter β-mercaptoethanol. Human embryonic kidney 293 (HEK293) cells (ATCC, Manassas, VA) were cultured in DMEM + 10% FBS, l-glutamine, and antibiotics. Fatty acids were bound to 1% fatty acid-free bovine serum albumin (Invitrogen) in DMEM + 10% FBS containing oleate, palmitate, or palmitoleate (Sigma) or BSA alone, and cells were incubated for 0–20 h.
Fluorescence Microscopy
For lipid droplet analysis, yeast cells were grown to saturation, stained with Nile Red (1 μg/ml) (Sigma), and visualized with a long pass GFP filter (440 nm). We and others (39, 40) have demonstrated the utility of this fluor for detecting neutral lipids and thus organelles, such as the CLD, that are enriched for TG and sterol ester. For quantification, 10 confocal frames 0.25 μm apart were captured and displayed as maximum intensity projections. Lipid droplets were counted for six frames per strain, with 15–30 cells per frame. All microscopy was performed using a Zeiss Axiovert 200 M using a 63× oil immersion objective.
Lipid Analysis and Metabolic Labeling
Mammalian lipid labeling was performed on cells seeded overnight in a 6-well plate in serum-free DMEM. Cells were pulse-labeled with 0.5 μCi/ml [9,10-3H]oleic acid for 6 h with 1% essentially fatty acid-free BSA + 0.25 mm cold okadaic acid or with 3 μCi/ml [3H]glycerol for 8 h at 37 °C (41). Yeast metabolic labeling was accomplished as described previously (42). For lipid extraction, 2 ml of hexane/isopropyl alcohol (3:2) was added to each well and incubated at room temperature for 1 h, after which the supernatant was removed, and the extraction was repeated. Samples were dried with nitrogen gas and resuspended in chloroform/methanol (2:1), and lipids were resolved by TLC in petroleum ether/diethyl ether/acetic acid (84:15:1). Total cholesterol levels were measured by gas-liquid chromatography as described (43). TG concentrations were determined enzymatically (Trig/GB, Roche Applied Science). Lipid levels were quantified as described previously and normalized to protein using a Bradford protein quantification assay (Bio-Rad).
Apoptosis Assays
Yeast apoptosis was assessed using annexin V binding and propidium iodide staining using the FITC annexin V/dead cell apoptosis kit (Molecular Probes V13242), as described previously (44). Apoptosis levels in MIN6 cells were quantified using Invitrogen Vibrant Apoptosis Assay Kit number 5 (V13244). Positive cells from 10 frames per condition were counted.
shRNA and ASO Transfection
Murine ARV1-specific (GCACAGTCACTGCTCACATC and CCTTCTCTTTTGAGGTAGCT corresponding to nucleotides 821 and 872, respectively (35, 45)) and negative control ASOs were provided by Isis Pharmaceuticals, Inc. (Carlsbad, CA). Transient transfection experiments were conducted using Lipofectamine 2000 with 250 nm ASO in OptiMEM for 6 h, after which media were changed to DMEM with 10% FBS for 24–48 h. For gene knockdown stable cell line creation, cells were individually transfected with eight pSilencer-ARVi constructs (nucleotides 138–156, 282–300, 404–422, and 830–848 of ARV1 fused to two promoters) or a pSilencer-Scrambled control (Ambion, Austin, TX) using Lipofectamine 2000 and selection in 600 μg/ml geneticin. Fatty acids were added to cultures, 24 h after knockdown.
Immunoblots
Cells were washed twice with PBS, lysed using SDS buffer (2% SDS, 62.5 mm Tris-HCl (pH 6.8), 10% glycerol, 10% β-mercaptoethanol, and 0.01% bromphenol blue), and boiled for 5 min. Protein extracts were prepared in a lysis buffer containing 25 mm Tris-HCl (pH 7.4), 2 mm Na3VO4, 10 mm NaF, 10 mm Na4P2O7, 1 mm EGTA, 1 mm EDTA, 1% Nonidet P-40, 5 μg/ml leupeptin, 5 μg/ml aprotinin, 10 nm okadaic acid, and 1 mm phenylmethylsulfonyl fluoride. Equal amounts of protein extract were separated on a 12% SDS-polyacrylamide gel and electrotransferred to a 0.45-μm nitrocellulose membrane. Membranes were blocked for 1 h at room temperature with 5% nonfat milk in Tris-buffered saline with 0.1% Tween 20 (TBST) and incubated with primary antibodies overnight at 4 °C. Protein bands were detected with HRP-conjugated secondary antibodies and SuperSignal West Pico-enhanced chemiluminescent solution (Pierce).
Recombinant Adenovirus Construction
ARV1 adenovirus was made using Adeno-X Expression System (Clontech). In brief, a 1.5-kb EcoRI-NotI fragment containing full-length hARV1 was subcloned into pShuttle vector at EcoRI and NotI sites, followed by I-CeuI and PI-SceI double digestion. The released insert was ligated into Adeno-X viral DNA (provided by University of Pennsylvania Vector Core). The DNA products were amplified in Escherichia coli, purified, and linearized with PacI and used to transfect HEK293 cells to collect high titer recombinant adenoviruses (AdhARV1).
Animal Studies
Female C57BL/6 mice (6–8 weeks old) were obtained from The Jackson Laboratory (Bar Harbor, ME) and fed a chow diet. Male Pparα−/− mice, on an A129S4/SvJae strain background (provided by Frank Gonzalez, NCI, National Institutes of Health), were fed powdered cereal-based rodent diet (Teklad 7001), supplemented with nuclear hormone receptor agonists as described (46). All experiments were performed with the approval of the Institutional Animal Care and Use Committees of the University of Texas Southwestern Medical Center and the University of Pennsylvania Medical Center. In adenovirus-mediated overexpression experiments, C57BL/6 mice were injected intravenously via the tail vein with PBS, AdhARV1, or a control virus containing no transgene (AdNull) at 1 × 1011 particles per animal. For plasma lipid measurements, mice were fasted for 4 h and bled from the retro-orbital plexus using heparinized capillary tubes at day 0 and at day 4 following adenovirus injections. Serum was assayed for total cholesterol, triglycerides, and HDL as described previously (47, 48). After 4 days of injection, mice were sacrificed, and livers were perfused and collected for biochemical and histological analysis. Liver cryosections were stained with Oil Red O (0.5% in propylene glycol, 16 h) followed by counterstaining with Mayer's hematoxylin (13). Perfused livers from adenovirus-infected mice were homogenized with Polytron, and lipids were extracted and measured enzymatically (49). Cholesterol and triglyceride reagents were from ThermoTrace (Melbourne, Australia), or Wako Chemicals, Inc. (Wako, TX). Perfused livers from adenovirus-infected mice were Polytron-homogenized and lipids extracted and measured enzymatically (49). Approximately 100 mg of flash-frozen perfused livers were used for lipid analysis under contract to Lipomics (West Sacramento, CA).
Transcriptional Analysis
RNA was isolated using the Qiagen RNeasy kit and used to generate first strand cDNA (SuperScript First-strand Synthesis System, Invitrogen). Real time PCRs were performed with the MyiQ single color real time PCR detection system (Bio-Rad), SYBR Green 2× Supermix, and the indicated primers. Expression levels were calculated relative to mouse GAPDH using the MyiQ real time detection software (50). For Northern blot analysis, 10 μg of RNA was run on a 1.2% formaldehyde gel and transferred to a nylon membrane (43). Membranes were hybridized with PCR-generated hARV1 probe (Stratagene) and analyzed by phosphorimaging (GE Healthcare). The same blot was stripped and used for actin normalization. Total RNA was extracted from liver tissue using RNA STAT-60 (Tel-Test, Inc.), treated with DNase I (RNase-free), and reverse-transcribed with random hexamers using SuperScript II. Quantitative real time PCR was performed using an Applied BioSystems Prism 7900HT sequence detection system as described (51) using SYBR Green, and the results were evaluated by the comparative cycle number at the threshold method (52).
RNA prepared as above from indicated mice was used for double strand cDNA synthesis and oligonucleotide array hybridization using SuperScript Choice System for cDNA synthesis (Invitrogen) with GeneChip T7-Oligo(dT) promoter primers (Affymetrix, Santa Clara, CA). cRNA were synthesized and labeled with biotin using a BioArray high yield RNA transcript labeling kit (T7) (Enzo, Farmingdale, NY). Hybridization, washing, scanning, and analysis of the Affymetrix GeneChip MouseGenome 430 2.0 version (Affymetrix) were carried out as described. Data obtained from the microarray hybridizations were processed with MICROARRAY SUITE 5.0 software (Affymetrix) and Significance Analysis of Microarrays (Stanford University (53)).
RESULTS
Genome-wide Screen Identifies Fatty Acid-sensitive Mutants in Yeast
Cellular lipid accumulation induces metabolic dysfunction through a variety of insults, including elevated reactive oxygen species (9, 54), ceramide biosynthesis (10, 55), ER stress (56–58), and altered membrane fluidity (59). Previously, we demonstrated that complete loss of neutral lipid synthesis in yeast results in marked sensitivity to exogenous palmitoleate (C16:1), to a greater degree than any other fatty acid tested (linoleate (C18:2), oleate (C18:1), stearate (C18:0), myristate (C14:0), or palmitate (C16:0) (6)). Moreover, we discovered that the characteristics of death of mutant yeast cells in response to UFA are strikingly similar to the lipotoxic responses of mammalian cells. Thus, we hypothesized that sensitivity of mutant yeast strains to PO would identify pathways required for resistance to FA in yeast and metazoans. Our strategy included plating the haploid (MATa) yeast deletion collection (deletions in ∼4,800 nonessential genes) and ∼850 knockdown DAmP alleles of essential genes, in quadruplicate, to synthetic complete media containing 0.5 mm PO. The concentration of fatty acids used in this study is similar to that occurring in normal human serum (60). The screen was conducted under carbon-replete conditions (2% dextrose) such that utilization of the FA as an energy source was de-emphasized (β-oxidation of fatty acids in yeast is suppressed by glucose).
Growth sensitivity of the mutant collection to PO was initially determined by comparison of colony size on fatty acid-containing media with the no treatment media for each deletion strain. Consequently, 256 single deletion and 50 DAmP alleles were identified as conferring sensitivity to PO. For screen validation, the 306 strains were individually grown in rich liquid media (SCD) with or without 0.5 mm PO and monitored over a 3–4-day growth period. Three growth curves per strain were analyzed for three parameters as follows: maximum cell density (“Max Absorbance”), maximum rate of growth (“Max Rate” or A/h), and time at which the maximum rate of growth was reached (“Time lag” in hours) (Fig. 1A). Statistically significant growth defects (Student's t test) of each mutant in palmitoleate-containing media for any single growth parameter (Max Absorbance, Max Rate, or Time) identified 152 deletion mutants as liposensitive. Each strain was then ranked on a “toxicity score” calculated as the sum of the three growth parameters (“Time lag” + “Max Absorbance” + “Max Rate”), with high values indicating increased sensitivity to PO (Fig. 1C and supplemental Table S1A). By this analysis, growth similar to the control strain would have a time lag value close to 0, a Max Rate value of ∼1, a Max Absorbance value of ∼1, and thus a toxicity score around 2.0. Conversely, the acyltransferase-deficient, quadruple deletion strain (are1Δ are2Δ dga1Δ lro1Δ) had a toxicity score of 103.4 (Max Rate = 81.4 ± 33; Time lag = 20.7 ± 0.6; Max Absorbance = 1.3 ± 0.1). Toxicity score values for all 152 deletion strains are indicated in supplemental Table S1A and in Fig. 1 (categorized by cellular process). Out of 50 hypomorphic DAmP alleles analyzed, we similarly validated 15 essential genes as required for resistance to exogenous UFA (supplemental Table S1B).
Biological Pathways That Confer Palmitoleate Resistance in Yeast
Given the resistance of the majority of yeast strains to exogenous FA, we reasoned that PO-sensitive mutations identify pathways that normally confer liporesistance to control cells. Out of the 152 PO-sensitive strains, 48 bear deletions in genes encoding constituents of 12 protein complexes. Interestingly, half of these are involved in RNA and DNA metabolism suggesting an altered transcriptional or expression profile that underlies the observed lipotoxicity. This includes the chromatin remodeling complexes SWR-1/INO80 and RSC; the histone acetyl/deacetylation complexes, Rpd3L and SAGA-like (SILK), and the spliceosomal protein U6 small nuclear ribonucleoprotein complex. Yeast strains grown in UFAs, such as oleate, exhibit global changes in gene expression, particularly in genes involved in stress responses, cell organization, and subcellular transport (6, 61). This suggests that a transcriptional response is altered upon exposure to FA and that defects in this response could lead to increased FA sensitivity. Additionally, genes encoding components of the cellular respiration complexes III and IV, mitochondrial and cytoplasmic ribosomes, and two vesicular trafficking complexes (Golgi-associated ER trafficking and Golgi to ER trafficking) were also implicated in protection against PO-induced cell death. The independent identification of multiple members of the same complexes as required for resistance to FA increases our confidence in the screen and further validates the importance of each complex in manifestation of this phenotype.
Liposensitivity in Yeast Identifies Analogous Pathways in Human Cells
A primary goal of this study was to use a model system-based approach to identify mammalian genes that play a role in lipotoxic diseases such as type 2 diabetes. Using conventional sequence alignments (NCBI, Basic Local Alignment Search Tool), we determined that 45% of the 167 yeast genes identified by this screen are conserved throughout evolution; 75 genes have human and murine orthologs with a sequence conservation that ranges from 18 to 82.6% amino acid identity (Fig. 1 and supplemental Table S1). We hypothesize that mutations in many of these orthologs may confer analogous fatty acid sensitivity in mammalian cells and accordingly represent genetic risk factors for a variety of metabolic disorders. Human orthologs to 11 yeast genes identified here have been previously associated with metabolic diseases, diabetic phenotypes, or stress-induced apoptosis (Table 1). The diabetic phenotypes linked to these orthologs include defects in glucose homeostasis or altered insulin secretion in metazoan models. For example, ASNA1, an ortholog of GET3, a key component of the yeast Golgi to ER trafficking complex associated here with UFA resistance in yeast, was shown to positively regulate insulin secretion in both Caenorhabditis elegans and mammalian pancreatic β-cells (62). Similarly, the GLUT2 gene, a sequence ortholog of yeast ITR1, encodes an insulin-independent glucose transporter, altered expression of which results in pancreatic β-cell dysfunction (63).
TABLE 1.
Mammalian orthologs of yeast genes required for liporesistance, previously associated with glucose homeostasis, insulin secretion, dyslipidemia or oxidative stress
Associations determined through the National Center for Biotechnology (NCBI) database National Human Genome Research Institute catalog of published genome-wide association studies (genome.gov) and HuGE Navigator human genome database.
| Yeast gene | Human ortholog | Associated metabolic syndrome |
|---|---|---|
| GET3 | ASNA1 (46.5%) | ASNA-1 positively regulates insulin secretion in C. elegans and mammalian cells. (PMID 17289575) |
| PCP1 | PARL (32.3%) | L262V polymorphism of PARL is associated with earlier onset of T2D. (PMID 19185381) |
| MCK1 | GSK3β (43.5%) | Mice with β-cell overexpression of GSK3β have reduced β-cell mass and proliferation. (PMID 18219478) |
| ARV1 | ARV1 | Decreased expression of ARV1 results in cholesterol retention in the endoplasmic reticulum and abnormal bile acid metabolism. (PMID 20663892) |
| GCN5 | GCN5 (46%) | GCN5-mediated transcriptional control of the metabolic coactivator PGC1β through lysine acetylation. (PMID 19491097) |
| FPS1 | AQP9 (28%) | Gene expression of paired abdominal adipose AQP7 and liver AQP9 occurring in patients with morbid obesity and relationship with glucose abnormalities. (PMID 19615702) |
| IFM1 | MTIF2 (36%) | Genetic association analysis of 13 nucleus-encoded mitochondrial candidate genes with type II diabetes mellitus, the DAMAGE study. (PMID 19209188) |
| ITR1 | GLUT2 (27%) | GLUT2 was thus demonstrated to be required for maintaining normal glucose homeostasis and normal function and development of the endocrine pancreas. (PMID 9354799) |
| MDJ1 | DNAJB9 (46%) | MDG1/ERdj4 an ER-resident DnaJ family member suppresses cell death induced by ER stress. (PMID 12581160) |
| FEN1 | ELOVL6 (30.6%) | ELOVL6 genetic variation is related to insulin sensitivity. (PMID 21701577) |
| MGR2 | ROMO1 (43%) | Serum deprivation-induced reactive oxygen species production is mediated by Romo1. (PMID 19904609) |
| ETR1 | MECR (36.4%) | Structural enzymological studies are of 2-enoyl thioester reductase of the human mitochondrial FAS II pathway, and new insights into its substrate recognition properties are given. (PMID 18479707) |
Similarly, several human orthologs of the yeast genes identified in our screen have been genetically associated with T2D in human population studies (Table 1). Polymorphisms in PARL (presenilin-associated, rhomboid-like), MTIF2 (mitochondrial initiation factor 2), and ELOVL6 (elongation of very long chain fatty acids protein 6), the mammalian orthologs of yeast PCP1, IFM1, and FEN1, respectively, have been associated with increased risk for type 2 diabetes (27, 30). The identification of the yeast orthologs of these genes in a screen based on lipid sensitivity suggests an unsuspected connection between diabetes and cellular dysfunction due to increased lipid sensitivity.
Status of the Lipid Droplet Does Not Predict FA Sensitivity
Given that neutral lipid (triacylglycerol and steryl ester) storage in the form of lipid droplets is a cells primary response to increased fatty acid exposure (6, 18, 64), we performed a lipid droplet morphometric screen of the 152 palmitoleate-sensitive deletion mutants identified here. Each deletion mutant was grown overnight in rich media (YPD), stained with the lipophilic dye, Nile Red, and phenotypically designated by three blinded observers (Fig. 1B). 48 mutants were classified as showing decreased droplet number, whereas 15 mutants presented with increased lipid droplets (supplemental Table S2 and, Fig. 1C). Out of 152 FA-sensitive mutants, 11 were previously reported to exhibit aberrant lipid droplets (23–25). Six of these (get1Δ, ssn3Δ, vma1Δ, vps51Δ, vps64Δ, and vps69Δ) were confirmed in our Nile Red subscreen. The genes cluster within the droplet phenotype categories based on their biological process, validating the analysis of both data sets. For example, mutants that impact fatty acid synthesis, respiration, or vesicular transport generally had decreased droplet number. The majority of mutants (89 of the 152) had a normal number of droplets suggesting a lack of overlap between morphometric and viability screens. Indeed, loss of two well characterized genes, TGL3 (64, 65) and FLD1 (24), encoding yeast orthologs of adipose triglyceride lipase and Seipin, respectively, results in striking changes in lipid droplet morphology but no detectable impact on FA sensitivity. A one-way analysis of variance of the toxicity score across the 152 gene mutants showed no significant (p = 0.2804) differences in lipid toxicity between the droplet classes. Changes in CLD number or morphology do not apparently predict vulnerability of a cell to excess lipids.
Neutral lipids are clearly an important reservoir for limiting intracellular free FA and sterol accumulation. However as described here, the vast majority of pathways that altered sensitivity to FA, such as palmitoleate, did not affect basal levels of NL storage. To examine whether exogenous FA induces droplet number or lipid biosynthesis in these strains, we grew mutants with or without a sublethal concentration of palmitoleic acid (0.05 mm) for 16 h in the presence of radiolabeled FA ([3H]oleic acid). The percentage of the cell containing Nile Red-stained CLDs was assessed by fluorescence microscopy, and lipids were extracted to assess FA incorporation (Fig. 1D). We focused on seven mutants with elevated FA sensitivity but normal basal lipid droplets (arv1Δ, rpb9Δ, aro1Δ, vma7Δ, yml095cΔ, ies6Δ, and fps1Δ). The impact of FA incubation on these strains was expressed as the ratio of CLD levels in YPD + PO compared with YPD media alone. PO at this concentration had no impact on CLD formation or lipid synthesis in the control strain. By contrast, five of the seven liposensitive strains tested showed a statistically significant increase in CLD area following PO incubation compared with the control (Fig. 1D). This expansion of the lipid droplets was invariably associated with elevated neutral lipid (steryl ester or triacylglycerol) production, often at the expense of phospholipid synthesis. We surmise that these mutants compensate for fatty acid sensitivity by increasing neutral lipid stores when challenged with exogenous free fatty acid. Ultimately, however, increasing the NL pool size is a futile response to FA surplus in these strains.
Deletion of ARV1 Results in Fatty Acid Sensitivity
Of the 152 deletion strains identified in the palmitoleate sensitivity screen, the most sensitive strain based on growth curve analysis and colony density arose from deletion of the ARV1 gene. The arv1Δ mutant achieved a toxicity score of 47 (Fig. 1 and supplemental Table S1). This mutant was the only strain tested with comparable FA sensitivity to the are1Δ are2Δ dga1Δ lro1Δ neutral lipid-deficient yeast strain (Fig. 2A); arv1Δ and are1Δ are2Δ dga1Δ lro1Δ strains were markedly inhibited by 0.05 mm palmitoleate (a concentration 10-fold lower than used in the screen). The arv1Δ strain also has decreased growth in the presence of 0.5 mm oleate but is not sensitive to saturated fatty acids such as palmitate (0.5 mm; Fig. 2B).
FIGURE 2.

Deletion of ARV1 results in severe fatty acid sensitivity and increased lipid droplet number. A, control and arv1Δ strain growth in YPD liquid media with indicated concentrations of palmitoleate. n = 3 for each condition. B, growth of control, neutral lipid-deficient (4Δ; are1Δ are2Δ dga1Δ lro1Δ) and arv1Δ strains on YPD solid media with 0.5 mm of PO, oleate (OLT), or palmitate (PA) for 3 days at 30 °C. C, strains were pre-grown to logarithmic phase and exposed to 0.1 mm PO in YPD for 16 h, followed by staining for lipid droplets with Nile Red (1 μg/ml). Lipid droplets per cell were quantified for each strain and treatment (10 frames each). Asterisk denotes statistical significance compared with the wild type control in the same condition (*, p < 0.05). DIC, differential interference contrast.
Because NL synthesis is a yeast cell's primary defense against lipotoxicity (6), we considered whether increased fatty acid sensitivity was due to insufficient lipid storage. Our lipid droplet subscreen (Fig. 1 and supplemental Table S2) indicates the ARV1 mutant has a normal lipid droplet number under rich media growth conditions. Under lipotoxic conditions (YPD + 0.1 mm palmitoleate, 3 h), the average number of droplets per cell was significantly elevated in arv1Δ (8.06 droplets/cell), compared with control (5.23 droplets/cell) (Fig. 2C). This is consistent with the 16 h, 0.05 mm PO incubation study (Fig. 1D) that demonstrated a significant increase in free fatty acid-induced CLD area in arv1Δ control and shows that this change occurs quickly upon FA exposure. A similar increase in lipid droplet number was seen in cholesterol-treated ARV1 mutants (45, 66).
To determine whether the UFA-induced sensitivity was due to apoptosis or necrosis, we grew control and ARV1 deletion strains to exponential stage in FA-free media followed by incubation with 0.1 mm palmitoleate for 16 h, after which cells were stained with annexin V and propidium iodide (PI) (44). In YPD media alone, arv1Δ had significantly higher levels of annexin V and PI staining compared with the control, indicating that ablation of ARV1 results in increased apoptosis under basal conditions. Consistent with its growth sensitivity phenotype, we saw multiple annexin-positive and PI-positive apoptotic cells in the ARV1 deletion after palmitoleate treatment, which did not occur in the control strain (Fig. 3).
FIGURE 3.
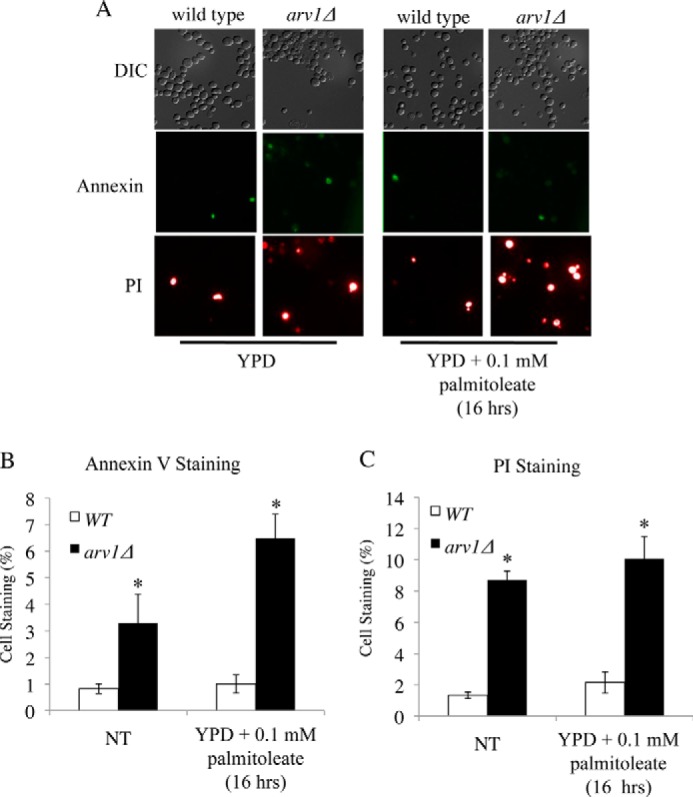
Deletion of ARV1 increases fatty acid-induced apoptosis in yeast. Annexin V/PI staining of control and arv1Δ cells after incubation in 0.1 mm palmitoleate for 16 h (A) and quantified (B and C) based on proportion of total cells stained for externalization of phosphatidylserine (stained with annexin V and quantified in B) and propidium iodide internalization (PI fluorescence and, quantified in C). Asterisks denote statistical significance of mean number of stained mutant cells (± S.E, 10 fields of cells, >60 cells per field) compared with the control strain (p < 0.05). NT denotes the no treatment control condition of FA-free media. DIC, differential interference contrast
Decreased Expression of Mammalian ARV1 Induces Lipotoxicity in Pancreatic β-Cell Lines
Human ARV1 is a functional and structural ortholog of yeast ARV1 and as such is able to rescue all growth and lipid phenotypes observed in yeast arv1Δ strains (33, 66). The identification of ARV1 in a yeast lipotoxicity screen suggests that mammalian ARV1 may also play an integral role in FA-induced cell death and lipotoxic disease progression. We hypothesized that altering expression levels of ARV1 in mammalian systems would affect FA sensitivity, similarly to that seen in yeast. To elucidate the role of ARV1 in β-cell health, we used the MIN6 mouse pancreatic β-cell line to test for levels of lipoapoptosis upon specific gene knockdown. Cells were treated with murine ARV1-specific antisense oligonucleotides (ASOs (35)) for 48 h to induce an ∼40% decrease in ARV1 expression compared with control ASO treatment (Fig. 4A). Levels of apoptosis were measured after 16 h of fatty acid treatment using the fluorescent apoptotic marker stain, Hoechst 33342, and the cell death marker, PI (Fig. 4D). Apoptotic cells were identified as cells staining positive for Hoechst 33342 and negative for PI (67). ARV1 knockdown caused a significant increase in the apoptotic cell percentage compared with control ASO-treated cells (Fig. 4D). Treatment of MIN6 cells with the saturated fatty acid palmitate (at 0.1 and 0.5 mm) and the UFA palmitoleate (0.5 mm) induced a significant increase in apoptosis in both control and ARV1 siRNA groups compared with FA-free conditions (Fig. 4). Similar levels of apoptosis have been demonstrated in pancreatic β-cell models following chronic fatty acid exposure (68, 69). When comparing control and ARV1 siRNA treatments, we saw a significant increase in apoptosis in ARV1-deficient cells following the addition of palmitate (at 0.1 and 0.5 mm) and 0.5 mm palmitoleate (Fig. 4D). Knockdown of ARV1 clearly sensitizes MIN6 cells to FA-induced apoptosis, which is consistent with the FA sensitivity phenotype in yeast and congruent with our original prediction.
FIGURE 4.

ARV1 knockdown in MIN6 pancreatic β-cells results in decreased triglyceride and increased fatty acid-induced apoptosis. A, mouse ARV1 mRNA expression levels with 0.25 μm ASO treatment for 48 h measured by quantitative RT-PCR. Lipid synthesis measured by [3H]oleate incorporation into TG (B) and PL (C) as a percentage of total lipid species. No significant changes were seen in any other lipid species. Asterisks denote statistical significance (p < 0.05) compared with control ASO, as determined by unpaired t test (mean ± S.D., n = 3). D, quantification of the average number of apoptotic cells based on Hoechst/PI staining after 16 h treatment with indicated concentrations of oleate (18:1), palmitoleate (16:1), or palmitate (16:0) conjugated to fatty acid-free BSA. Asterisks denote statistical significance (p < 0.05) of ARV1-ASO-treated cells (mean ± S.E., 7 fields of 70–200 cells per field) compared with control ASO treatment.
Neutral Lipid Synthesis Is Directly Correlated with ARV1 Expression Levels
A reduction in NL synthesis and storage in both yeast and mammalian systems is associated with increased levels of fatty acid-induced apoptosis, ER stress, and cellular dysfunction (6, 18). Obesity-related diseases such as T2D are due in part to these lipid-induced cellular defects (70, 71). As an alternative strategy to the aforementioned murine ARV1-specific ASOs, we chose to suppress ARV1 expression in human HEK293 cell lines using eight ARV1-specific shRNA constructs (four regions of ARV1 fused to two different promoters). Stable cell lines from two independent shARV1 constructs with a >85% reduction in ARV1 expression were chosen for subsequent experiments (Fig. 5A). Knockdown of human ARV1 resulted in an approximate 40% decrease in [3H]glycerol incorporation into triglyceride (Fig. 5B) implicating a generalized role of ARV1 in the regulation of NL synthesis. Similarly, we measured lipid synthesis in MIN6 cells following knockdown of ARV1 expression after a 4-h label. Triglyceride synthesis was decreased with ARV1 knockdown compared with control (Fig. 4B). All other lipid species measured (free fatty acid, diacylglycerol, and cholesterol ester) had normal incorporation with the exception of phospholipids, which showed a significant increase (Fig. 4C). Consistent with these results, Tong et al. (35) previously demonstrated that liver TG was significantly reduced in mice with decreased hepatic ARV1 expression.
FIGURE 5.

Triglyceride synthesis is correlated with ARV1 expression in HEK293 cells. Human hARV1 expression was suppressed using small hairpin RNA-mediated transcript degradation. HEK293 stable cell lines with altered expression were created through transfection and antibiotic selection and assessed for hARV1 mRNA level by Northern blot (A). Relative abundance of ARV1 mRNA was expressed as a ratio to glyceraldehyde-3-phosphate dehydrogenase (G3PDH). ARV1 knockdown resulted in reduced levels of triglyceride (B) with no detectable change in phospholipid synthesis (C), as assessed by [3H]glycerol incorporation. Stable HEK293 cell lines with increased expression of human ARV1 were created through transfection with hARV1-EGFP, maintained in DMEM + 400 μg/ml geneticin, and assessed by immunoblotting of total extracts with antisera against GFP (D). Control cells were transfected with an enhanced GFP empty vector. hARV1-EGFP cells have increased triglyceride and phospholipid levels (E and F) compared with control. Asterisks denote statistical significance (*, p < 0.05) as determined by unpaired t test (mean ± S.D., n = 3).
Overexpression of Human ARV1 Elevates Triglyceride Synthesis
To further elucidate the role of ARV1 in mammalian lipid metabolism, we used a constitutively active human ARV1 expression vector to determine the impact of elevated ARV1 expression upon lipid synthesis. Stable HEK293 cell lines transfected with human ARV1 (hARV1-EGFP) were selected using fluorescent microscopy and Western blot analysis to identify clones with elevated hARV1-EGFP expression (Fig. 5D). Metabolic incorporation of [3H]glycerol into TG and phospholipid was increased in hARV1-EGFP-+transfected cells compared with control (163 and 109%, respectively, Fig. 5, E and F). Radiolabeled acetate and oleate corroborated the ARV1-induced TG accumulation. Together with the aforementioned impact of ARV1 knockdown, these results further indicate that ARV1 expression correlates with levels of triglyceride synthesis.
Overexpression of Human ARV1 in Mice Increases Hepatic Triglyceride Levels
We propose that ARV1 is an important regulator of lipotoxic pathways through its role in lipid trafficking. To assess the role of ARV1 in mammalian physiology, we induced ARV1 overexpression in mouse liver and monitored hepatic and plasma lipid levels. Hepatic ARV1 levels were increased through injection of a human ARV1-specific adenovirus (Ad-hARV1) into C57BL/6 female mice for 4 days, after which serum was collected and tested for total cholesterol, triglyceride, and HDL, and mice were sacrificed for hepatic lipid analysis (47, 72). As expected, mRNA expression levels of hepatic hARV1 were consistently increased in ARV1 adenovirus-injected mice compared with the control adenovirus injection (Fig. 6A). This resulted in a significant increase in hepatic TG and PL compared with control (Fig. 6B and Table 2) but did not alter the serum lipid profile between the two groups (Table 3). This is consistent with our in vitro studies showing increased TG synthesis with higher ARV1 expression (Fig. 5E). Both free cholesterol and phospholipids also showed a significant increase in the livers of the Ad-hARV1-injected mice compared with control, with a 33 and 21% increase, respectively (Fig. 6B). Lipid profiling of the Ad-hARV1-infected livers showed an increase in triglyceride and phospholipid and a reduction in free fatty acids compared with control (Table 4). To confirm the hepatic lipid accumulation, we stained the Ad-hARV1-infected livers with Oil Red O and observed elevated numbers of larger neutral lipid droplets relative to the control AdNull-infected animals (Fig. 6C).
FIGURE 6.
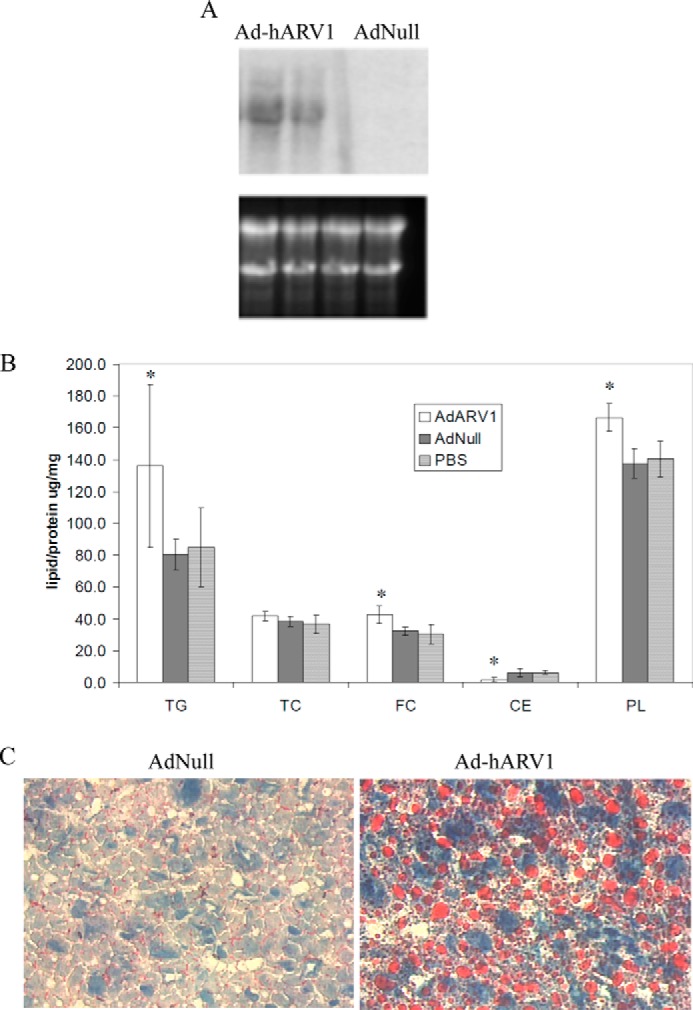
Overexpression of human ARV1 in mice causes fatty liver. C57BL/6 female mice were injected with Ad-hARV1 or AdNull for 4 days, sacrificed, and livers perfused and homogenized. A, mRNA levels of hARV1 gene expression in mouse livers were assessed by Northern blot and ethidium bromide staining of rRNA to show loading of RNA samples. B, liver lipids as indicated (TG, TC, free cholesterol, cholesterol ester, and PL) were measured enzymatically as per “Experimental Procedures.” Asterisks denote statistical significance compared with the AdNull control in the same condition (*, p < 0.05). C, neutral lipid staining in livers with Oil-Red-O stain.
TABLE 2.
Lipid profiling of livers from adenovirus-infected mice
Female control mice (C57BL/6, 6–8 weeks old) were injected with Ad-hARV1 (n = 4) or AdNull (n = 4). On day 4, mice were bled and livers harvested and analyzed for lipid levels.
a Liver lipids in μg/mg tissue are at p < 0.05.
b Values are p < 0.01.
TABLE 3.
Lipid profiling of plasma from adenovirus-infected mice
Female control mice (C57BL/6, 6–8 weeks old) were injected with Ad-hARV1 (n = 4) or AdNull (n = 4). On day 4, mice were bled, and plasma was analyzed for lipid levels. Plasma lipids are given in mg/dl.
| Total cholesterol | TG | HDL | Non-HDL | PL | |
|---|---|---|---|---|---|
| AdNull | 64.3 | 52.8 | 53.5 | 10.8 | 136.5 |
| Ad-hARV1 | 66.0 | 45.8 | 53.5 | 12.5 | 140.5 |
TABLE 4.
Lipomic profiling of livers from adenovirus-infected mice
Lipomic profiling is shown of livers from adenovirus-infected female control mice (C57BL/6, 6–8 weeks old) (AdhARV1 (n = 3), AdNull (n = 2)). Lipids are represented as pmol/mg tissue.
| Sample | CE | DAG | TG | FFA | PL | FC |
|---|---|---|---|---|---|---|
| Ad-hARV1–1 | 5636.7 | 909.9 | 47012.7 | 933.8 | 57560.3 | 6103 |
| Ad-hARV1–2 | 4778.8 | 880.8 | 43781.2 | 809.1 | 53443.8 | 5296 |
| Ad-hARV1–3 | 5130.1 | 822.0 | 39736.4 | 1003.5 | 49694.4 | 5328 |
| AdNull-1 | 4395.0 | 691.9 | 24001.0 | 2884.4 | 33095.9 | 5026 |
| AdNull-2 | 4364.5 | 852.4 | 27226.9 | 1615.3 | 36452.9 | 5276 |
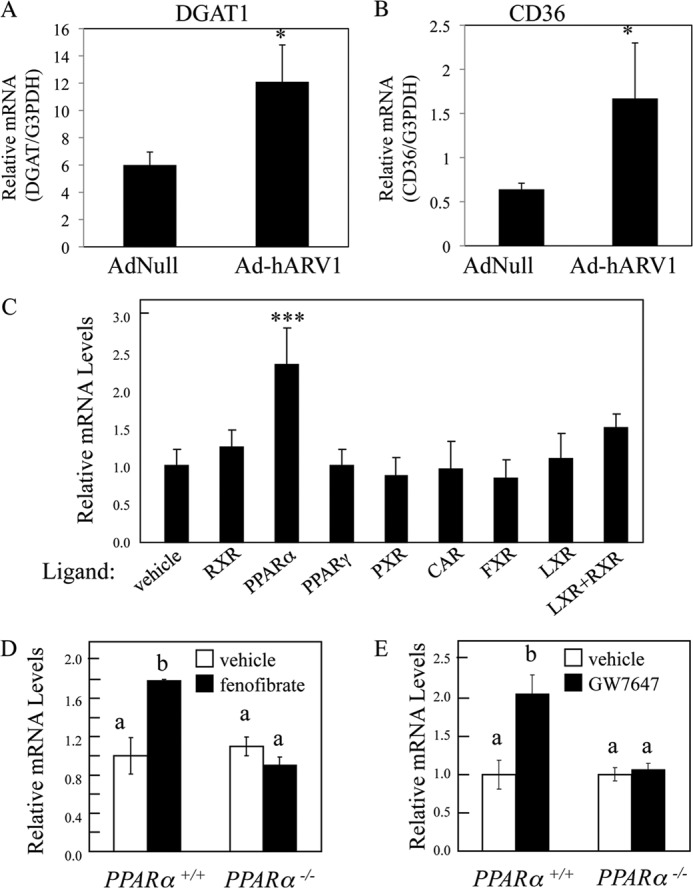
Increased liver TG is often associated with hepatic insulin resistance and ER stress and occurs due to an imbalance between TG liver influx (de novo biosynthesis, diet, or adipose tissue) and removal (secretion or oxidation) (73). To determine potential mechanisms for the lipid changes observed in Ad-hARV1-infected mice, we used gene expression profiling. Using highly stringent criteria (Student's t test p < 0.001 between Ad-hARV1 and AdNull), we identified 27 up-regulated genes and 9 down-regulated genes in the Ad-hARV1 livers (Table 5). The triglyceride synthesis gene, DGAT1, was found to be up-regulated 1.6-fold in AdhARV1 livers, and this increase was confirmed by real time PCR (Fig. 7A). Gene expression of the fatty acid transporter, CD36, was also significantly increased in the Ad-hARV1-injected livers compared with control (Fig. 7B).
TABLE 5.
Genes with altered expression in AdhARV1-treated mice
Up-regulated (fold change 1.5 or more p < 0.001) and down-regulated (fold change 0.5 or less, p < 0.001) genes in livers with hARV1 overexpression. Results were calculated based on data of six sets of Affymetrix oligonucleotide arrays. Genes were categorized by GO process and ranked within these groups by fold change.
| Process | Gene | -Fold change |
|---|---|---|
| Lipid metabolism | DGAT1 | 1.6 |
| DNA replication and repair | POLE2 | 2.2 |
| MCM4 | 2.2 | |
| UNG | 2.2 | |
| NASP | 1.7 | |
| Protein phosphorylation | BUB1 | 2.6 |
| PBK | 1.8 | |
| PLK4 | 1.6 | |
| BRD4 | 1.8 | |
| STK25 | 0.391 | |
| RNA transport | NUP35 | 1.8 |
| FMR1 | 1.7 | |
| Chromatin modification | ASF1B | 2.7 |
| KDM2B | 1.7 | |
| Miscellaneous | DET1 | 9.9 |
| COX6B2 | 4.6 | |
| GPR98 | 3.3 | |
| GSTM3 | 2.3 | |
| MMP14 | 2.2 | |
| SULT1C1 | 2.2 | |
| ITIH5 | 2.1 | |
| DCAKD | 1.8 | |
| NT5DC2 | 1.6 | |
| ABHD10 | 1.6 | |
| PPM1D | 1.5 | |
| EIFAD | 1.5 | |
| ZFP871 | 0.486 | |
| CAR5 | 0.412 | |
| TTC39C | 0.409 | |
| LCN13 | 0.369 | |
| Uncharacterized | 4933439C10Rik | 6.2 |
| EST X83313 | 5.7 | |
| RIKEN cDNA 1810010H24 | 0.479 | |
| RIKEN cDNA 5730414N17 | 0.441 | |
| AW549877 | 0.422 |
FIGURE 7.
Regulation of hepatic ARV1 RNA levels in the mouse. A, DGAT1; B, CD36 expression in AdhARV1-injected mouse livers compared with AdNull control. C, male A129/SvEv strain mice were provided overnight (12 h) with low fat rodent diet supplemented with various nuclear hormone receptor agonists as follows: RXR, 30 mg/kg LG268; PPARα, 0.5% fenofibrate; PPARγ, 150 mg/kg troglitazone; PXR, 0.05% pregnenolone α-carbonitrile; CAR, 3 mg/kg TCPOBOP; FXR, 0.5% chenodeoxycholic acid; LXR, 50 mg/kg T0901317; LXR + RXR, 30 mg/kg LG268 + 50 mg/kg T0901317, or vehicle (0.9% carboxymethylcellulose, 9% PEG, 0.05% Tween), n = 4 per group. Male A129 wild type (Pparα+/+) and Pparα−/− mice were provided diets with and without 0.5% fenofibrate (D) for 7 days, n = 4/group, or 5 mg/kg GW7647 or vehicle twice by oral gavage over 14 h, n = 5 per group. RNA levels were measured by quantitative RT-PCR using cyclophilin as the invariant housekeeping gene. Values depict mean ± S.E. Results were analyzed by one-way unfolded protein response using (A–C) Dunnett's post hoc comparison., *, p < 0.05; ***, p < 0.001 versus control group. D and E, Neuman-Keul's post hoc comparison, groups with different letters are significantly different at p < 0.05.
The known relationship between peroxisome proliferator-activated receptor α (PPARα) and transcriptional regulation of key players in TG homeostasis, such as DGAT1 and CD36 (74), led us to question whether ARV1 expression was also regulated through orphan receptor activation. We included specific activators of the RXR, PPARα, PPARγ, PXR, CAR, FXR, LXR, and orphan receptors (LG268, fenofibrate, troglitazone, pregnenolone α-carbonitrile, TCPOBOP; chenodeoxycholic acid, and T0901317, respectively) in low fat rodent diet to male A129/SvEv mice for 12 h and assessed ARV1 expression (Fig. 7C). We determined that murine ARV1 expression is specifically driven by PPARα activation, unlike the other nuclear receptors tested (RXR, PPARγ, PXR, CAR, FXR, and LXR, Fig. 7C). In confirmation of this, treatment of wild type mice with the PPARα agonists, GW7647 and fenofibrate, induced an increase in ARV1 gene expression, which did not occur in PPARα knock-out mice (Fig. 7, D and E). In these samples, DGAT1 expression was regulated in the same fashion (data not shown). These results indicate that murine ARV1 is a PPARα target and likely plays an integral role in the maintenance of cellular TAG homeostasis alongside other PPARα targets.
DISCUSSION
We undertook a study on lipotoxicity in yeast that we reasoned would provide a novel outlook on eukaryotic lipid metabolism and diseases such as obesity and diabetes. Multiple genome-wide screens have been completed in several model organisms focusing on altered lipid droplet morphology (21–25). To date, however, no screen for mutants with increased susceptibility to fatty acid-induced cell death has been completed in any organism. Our screen was further unique in that we focused on sensitivity to lipids in rich media containing glucose and fatty acids. This strategy mitigates pathways that relate to utilization of FA for energy. In this same context of FA surplus, we previously observed severe cytotoxicity in the NL-deficient mutant (are1Δ are2Δ dga1Δ lro1Δ). This finding persists in mammalian cells whereby ablation of triglyceride synthesis, through deletion of DGAT1 in mouse fibroblasts, results in sensitivity to unsaturated fatty acids (6, 7, 18). This analogy led us to confidently expect outcomes from our screen that would be relevant to a host of human diseases that arise from lipid overload. Our study not only identified a novel pathway involved in lipotoxic disease (mediated by the ARV1 gene) but also exemplifies the utility of yeast genetics in the identification of human disease candidate genes.
As the most FA-sensitive yeast deletion identified by our screen, we went on to clarify how ARV1 is involved in eukaryotic triglyceride synthesis and lipotoxicity in yeast and mammalian cells. We demonstrate that the impact of loss of ARV1 is conserved between these organisms spanning ∼2 billion years of evolution. We observed a significant increase in apoptosis in response to fatty acid treatment in the yeast ARV1 deletion compared with control (Fig. 3) and that knockdown of ARV1 in MIN6 pancreatic β-cells also increased lipoapoptosis (Fig. 4). Moreover, ARV1 expression was positively correlated with TG synthesis in three mammalian models as follows: MIN6 pancreatic β-cells, HEK-293 embryonic kidney cells, and mouse liver. At the transcriptional level, DGAT1, CD36, and ARV1 are coordinately regulated in murine cells by the same orphan receptor (PPARα). These results implicate the ARV1 gene product as a modulator of cellular NL synthesis, a process that plays a role in progression of many lipotoxic diseases.
The role of neutral lipid synthesis as a protective mechanism against the formation of toxic lipid metabolites is well established (6, 7, 18, 75). It is therefore not surprising that decreased TG synthesis in MIN6 cells upon ARV1 knockdown increases cell sensitivity to FA-induced apoptosis (Fig. 3). Yeast ARV1 mutants have a multitude of lipid-related phenotypes, many of which involve defects in sterol and ceramide metabolism (33, 66). Strains lacking ARV1 have increased sensitivity to the polyene antibiotic nystatin, a decrease in sterol uptake, and an increase in the pro-apoptotic sphingolipid ceramide (33, 66). In addition, deletion of ARV1 results in a significant increase in ER stress and in the unfolded protein response under basal conditions (45). Ceramide production (55, 76–78) and induction of an ER stress response (56, 79) play an integral role in pathways of lipotoxicity and insulin resistance. The accumulation of ceramide and the constitutively active unfolded protein response, characteristic of ARV1 mutants, are two potential mechanisms explaining FA sensitivity in yeast arv1Δ strains. Interestingly, activation of ER stress in yeast through tunicamycin or brefeldin A treatment results in lipid droplet accumulation (81). Because ARV1 deletion strains have a constitutively active unfolded protein response (45), the lipid challenge occurring with palmitoleate treatment may result in increased CLD number through a similar ER stress induction and/or altered ER architecture. In addition to its role in protection from FA-induced cell death, neutral lipid synthesis in nonadipose tissue, such as the liver, is associated with fatty acid-induced cellular dysfunction, through increased toxic fatty acid metabolites (i.e. ceramides, free fatty acid, and diacylglycerol) (4, 71, 82–84). Hepatic ARV1 overexpression in Ad-hARV1-injected mice resulted in lipid droplet accumulation (Fig. 6) and up-regulation of the TG synthesis gene DGAT1 (Table 5 and Fig. 7A). Elevated DGAT1 expression has been associated with fatty liver phenotypes in rodent models and human subjects. Accordingly, DGAT1 deficiency in hepatocytes or pharmacological inhibition of the Dgat1 enzyme protected mice against hepatic steatosis and high fat diet-induced fatty liver (85).
A unifying concept regarding the pleiotropy of ARV1 modulation in numerous cell types would be a primary role in fatty acid homeostasis. Previous studies have focused on sterol/steryl ester-related phenotypes resulting from a reduction in hepatic ARV1 expression (35) or deletion of yeast ARV1 (66). Indeed, in contrast to some liposensitive strains, the major response of arv1Δ cells to nonlethal levels of FA is to induce steryl ester not TG synthesis (Fig. 1D). The defects in TG homeostasis resulting from altered ARV1 expression may thus be secondary to its role as a possible sterol transporter (although this role has recently been questioned (80)). Alternatively, the global changes in lipid composition and architecture of multiple cellular membranes that arise in ARV1-deficient states may disturb their function to the extent that the additional stress of FA overload or impaired esterification is lethal. In both cases, the ARV1 gene product functions to detoxify the ER and perhaps the PM, although the precise mechanism still remains elusive.
In conclusion, we identified 167 yeast lipotoxicity alleles (supplemental Table S1), of which 75 genes represent potential targets for intervention in human lipotoxic diseases by virtue of their evolutionary conservation. As a proof of principle to this premise, altered expression of ARV1 causes major changes in liposensitivity, neutral lipid synthesis, and overall lipid homeostasis. This suggests that changes in ARV1 expression in humans will predispose a variety of obesity-related disorders. Consequently, in future studies, we propose to pursue a role for mammalian ARV1 in the progression of two major lipotoxic disorders, nonalcoholic fatty liver disease and lipid-induced pancreatic β-cell failure.
This work was supported, in whole or in part, by National Institutes of Health Grants DK54320 (to S. L. S.) and DK078592 (to J. R.). This work was also supported by the American Diabetes Association, the American Heart Association, and the Ara Parseghian Medical Research Foundation.

This article contains supplemental Tables S1 and S2.
- T2D
- type 2 diabetes
- Ad
- adenovirus
- ASO
- antisense oligonucleotide
- DAmP
- decreased expression through mRNA perturbation
- DGAT
- diacylglycerol acyltransferase
- ER
- endoplasmic reticulum
- FA
- fatty acid
- CLD
- cytoplasmic lipid droplet
- NL
- neutral lipid
- PL
- phospholipid
- PPAR
- peroxisome proliferator-activated receptor
- PO
- palmitoleate
- TG
- triglyceride
- UFA
- unsaturated fatty acid
- hARV1
- human ARV1
- PI
- propidium iodide
- EGFP
- enhanced GFP.
REFERENCES
- 1. Lee Y., Hirose H., Ohneda M., Johnson J. H., McGarry J. D., Unger R. H. (1994) Beta-cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: impairment in adipocyte-beta-cell relationships. Proc. Natl. Acad. Sci. U.S.A. 91, 10878–10882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Briaud I., Harmon J. S., Kelpe C. L., Segu V. B., Poitout V. (2001) Lipotoxicity of the pancreatic beta-cell is associated with glucose-dependent esterification of fatty acids into neutral lipids. Diabetes 50, 315–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wei Y., Wang D., Topczewski F., Pagliassotti M. J. (2006) Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am. J. Physiol. Endocrinol. Metab. 291, 275–281 [DOI] [PubMed] [Google Scholar]
- 4. Trauner M., Arrese M., Wagner M. (2010) Fatty liver and lipotoxicity. Biochim. Biophys. Acta 1801, 299–310 [DOI] [PubMed] [Google Scholar]
- 5. Lim H. Y., Bodmer R. (2011) Phospholipid homeostasis and lipotoxic cardiomyopathy: a matter of balance. Fly 5, 234–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Garbarino J., Padamsee M., Wilcox L., Oelkers P. M., D'Ambrosio D., Ruggles K. V., Ramsey N., Jabado O., Turkish A., Sturley S. L. (2009) Sterol and diacylglycerol acyltransferase deficiency triggers fatty acid-mediated cell death. J. Biol. Chem. 284, 30994–31005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Petschnigg J., Wolinski H., Kolb D., Zellnig G., Kurat C. F., Natter K., Kohlwein S. D. (2009) Good fat, essential cellular requirements for triacylglycerol synthesis to maintain membrane homeostasis in yeast. J. Biol. Chem. 284, 30981–30993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gwiazda K. S., Yang T. L., Lin Y., Johnson J. D. (2009) Effects of palmitate on ER and cytosolic Ca2+ homeostasis in beta-cells. Am. J. Physiol. Endocrinol. Metab. 296, E690–E701 [DOI] [PubMed] [Google Scholar]
- 9. Listenberger L. L., Ory D. S., Schaffer J. E. (2001) Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J. Biol. Chem. 276, 14890–14895 [DOI] [PubMed] [Google Scholar]
- 10. Summers S. A. (2006) Ceramides in insulin resistance and lipotoxicity. Prog. Lipid Res. 45, 42–72 [DOI] [PubMed] [Google Scholar]
- 11. Unger R. H., Clark G. O., Scherer P. E., Orci L. (2010) Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim. Biophys. Acta 1801, 209–214 [DOI] [PubMed] [Google Scholar]
- 12. Schwarz S., Hufnagel B., Dworak M., Klumpp S., Krieglstein J. (2006) Protein phosphatase type 2Cα and 2Cβ are involved in fatty acid-induced apoptosis of neuronal and endothelial cells. Apoptosis 11, 1111–1119 [DOI] [PubMed] [Google Scholar]
- 13. Zhu Y., Schwarz S., Ahlemeyer B., Grzeschik S., Klumpp S., Krieglstein J. (2005) Oleic acid causes apoptosis and dephosphorylates Bad. Neurochem. Int. 46, 127–135 [DOI] [PubMed] [Google Scholar]
- 14. Malhi H., Barreyro F. J., Isomoto H., Bronk S. F., Gores G. J. (2007) Free fatty acids sensitise hepatocytes to TRAIL mediated cytotoxicity. Gut 56, 1124–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. De Gottardi A., Vinciguerra M., Sgroi A., Moukil M., Ravier-Dall'Antonia F., Pazienza V., Pugnale P., Foti M., Hadengue A. (2007) Microarray analyses and molecular profiling of steatosis induction in immortalized human hepatocytes. Lab. Invest. 87, 792–806 [DOI] [PubMed] [Google Scholar]
- 16. Cury-Boaventura M. F., Gorjão R., de Lima T. M., Newsholme P., Curi R. (2006) Comparative toxicity of oleic and linoleic acid on human lymphocytes. Life Sci. 78, 1448–1456 [DOI] [PubMed] [Google Scholar]
- 17. Cury-Boaventura M. F., Pompéia C., Curi R. (2004) Comparative toxicity of oleic acid and linoleic acid on Jurkat cells. Clin. Nutr. 23, 721–732 [DOI] [PubMed] [Google Scholar]
- 18. Listenberger L. L., Han X., Lewis S. E., Cases S., Farese R. V., Jr., Ory D. S., Schaffer J. E. (2003) Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. U.S.A. 100, 3077–3082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ruggles K. V., Turkish A., Sturley S. L. (2013) Making, baking, and breaking: the synthesis, storage, and hydrolysis of neutral lipids. Annu. Rev. Nutr. 33, 413–451 [DOI] [PubMed] [Google Scholar]
- 20. Goodman J. M. (2008) The gregarious lipid droplet. J. Biol. Chem. 283, 28005–28009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guo Y., Walther T. C., Rao M., Stuurman N., Goshima G., Terayama K., Wong J. S., Vale R. D., Walter P., Farese R. V. (2008) Functional genomic screen reveals genes involved in lipid-droplet formation and utilization. Nature 453, 657–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Beller M., Sztalryd C., Southall N., Bell M., Jäckle H., Auld D. S., Oliver B. (2008) COPI complex is a regulator of lipid homeostasis. PLoS Biol 6, e292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fei W., Alfaro G., Muthusamy B. P., Klaassen Z., Graham T. R., Yang H., Beh C. T. (2008) Genome-wide analysis of sterol-lipid storage and trafficking in Saccharomyces cerevisiae. Eukaryot. Cell 7, 401–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fei W., Shui G., Gaeta B., Du X., Kuerschner L., Li P., Brown A. J., Wenk M. R., Parton R. G., Yang H. (2008) Fld1p, a functional homologue of human seipin, regulates the size of lipid droplets in yeast. J. Cell Biol. 180, 473–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Szymanski K. M., Binns D., Bartz R., Grishin N. V., Li W. P., Agarwal A. K., Garg A., Anderson R. G., Goodman J. M. (2007) The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc. Natl. Acad. Sci. U.S.A. 104, 20890–20895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cauchi S., Choquet H., Gutiérrez-Aguilar R., Capel F., Grau K., Proença C., Dina C., Duval A., Balkau B., Marre M., Potoczna N., Langin D., Horber F., Sørensen T. I., Charpentier G., Meyre D., Froguel P. (2008) Effects of TCF7L2 polymorphisms on obesity in European populations. Obesity 16, 476–482 [DOI] [PubMed] [Google Scholar]
- 27. Hatunic M., Stapleton M., Hand E., DeLong C., Crowley V. E., Nolan J. J. (2009) The Leu262Val polymorphism of presenilin associated rhomboid like protein (PARL) is associated with earlier onset of type 2 diabetes and increased urinary microalbumin creatinine ratio in an Irish case-control population. Diabetes Res. Clin. Pract. 83, 316–319 [DOI] [PubMed] [Google Scholar]
- 28. Magré J., Laurell H., Fizames C., Antoine P. J., Dib C., Vigouroux C., Bourut C., Capeau J., Weissenbach J., Langin D. (1998) Human hormone-sensitive lipase: genetic mapping, identification of a new dinucleotide repeat, and association with obesity and NIDDM. Diabetes 47, 284–286 [DOI] [PubMed] [Google Scholar]
- 29. Marchetti P., Lupi R., Federici M., Marselli L., Masini M., Boggi U., Del Guerra S., Patanè G., Piro S., Anello M., Bergamini E., Purrello F., Lauro R., Mosca F., Sesti G., Del Prato S. (2002) Insulin secretory function is impaired in isolated human islets carrying the Gly(972) → Arg IRS-1 polymorphism. Diabetes 51, 1419–1424 [DOI] [PubMed] [Google Scholar]
- 30. Morcillo S., Martín-Núñez G. M., Rojo-Martínez G., Almaraz M. C., García-Escobar E., Mansego M. L., de Marco G., Chaves F. J., Soriguer F. (2011) ELOVL6 genetic variation is related to insulin sensitivity: a new candidate gene in energy metabolism. PLoS One 6, e21198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tsunoda K., Sanke T., Nakagawa T., Furuta H., Nanjo K. (2001) Single nucleotide polymorphism (D68D, T to C) in the syntaxin 1A gene correlates to age at onset and insulin requirement in Type II diabetic patients. Diabetologia 44, 2092–2097 [DOI] [PubMed] [Google Scholar]
- 32. Blakemore A. I., Froguel P. (2008) Is obesity our genetic legacy? J. Clin. Endocrinol. Metab. 93, S51–56 [DOI] [PubMed] [Google Scholar]
- 33. Swain E., Stukey J., McDonough V., Germann M., Liu Y., Sturley S. L., Nickels J. T., Jr. (2002) Yeast cells lacking the ARV1 gene harbor defects in sphingolipid metabolism. Complementation by human ARV1. J. Biol. Chem. 277, 36152–36160 [DOI] [PubMed] [Google Scholar]
- 34. Forés O., Arró M., Pahissa A., Ferrero S., Germann M., Stukey J., McDonough V., Nickels J. T., Jr., Campos N., Ferrer A. (2006) Arabidopsis thaliana expresses two functional isoforms of Arvp, a protein involved in the regulation of cellular lipid homeostasis. Biochim. Biophys. Acta 1761, 725–735 [DOI] [PubMed] [Google Scholar]
- 35. Tong F., Billheimer J., Shechtman C. F., Liu Y., Crooke R., Graham M., Cohen D. E., Sturley S. L., Rader D. J. (2010) Decreased expression of ARV1 results in cholesterol retention in the endoplasmic reticulum and abnormal bile acid metabolism. J. Biol. Chem. 285, 33632–33641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ausubel F. M., Brent R., Kingston R. E., Moore D. D., Seidman J. G., Smith J. A., Struhl K. (eds) (1998) Current Protocols in Molecular Biology, John Wiley & Sons, New York [Google Scholar]
- 37. Orr-Weaver T. L., Szostak J. W., Rothstein R. J. (1981) Yeast transformation: a model system for the study of recombination. Proc. Natl. Acad. Sci. U.S.A. 78, 6354–6358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ishihara H., Asano T., Tsukuda K., Katagiri H., Inukai K., Anai M., Kikuchi M., Yazaki Y., Miyazaki J. I., Oka Y. (1993) Pancreatic beta cell line MIN6 exhibits characteristics of glucose metabolism and glucose-stimulated insulin secretion similar to those of normal islets. Diabetologia 36, 1139–1145 [DOI] [PubMed] [Google Scholar]
- 39. Greenspan P., Mayer E. P., Fowler S. D. (1985) Nile red: a selective fluorescent stain for intracellular lipid droplets. J. Cell Biol. 100, 965–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Adeyo O., Horn P. J., Lee S., Binns D. D., Chandrahas A., Chapman K. D., Goodman J. M. (2011) The yeast lipin orthologue Pah1p is important for biogenesis of lipid droplets. J. Cell Biol. 192, 1043–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu P., Ying Y., Zhao Y., Mundy D. I., Zhu M., Anderson R. G. (2004) Chinese hamster ovary K2 cell lipid droplets appear to be metabolic organelles involved in membrane traffic. J. Biol. Chem. 279, 3787–3792 [DOI] [PubMed] [Google Scholar]
- 42. Yang H., Bard M., Bruner D. A., Gleeson A., Deckelbaum R. J., Aljinovic G., Pohl T. M., Rothstein R., Sturley S. L. (1996) Sterol esterification in yeast: a two-gene process. Science 272, 1353–1356 [DOI] [PubMed] [Google Scholar]
- 43. Seo T., Oelkers P. M., Giattina M. R., Worgall T. S., Sturley S. L., Deckelbaum R. J. (2001) Differential modulation of ACAT1 and ACAT2 transcription and activity by long chain free fatty acids in cultured cells. Biochemistry 40, 4756–4762 [DOI] [PubMed] [Google Scholar]
- 44. Madeo F., Fröhlich E., Fröhlich K. U. (1997) A yeast mutant showing diagnostic markers of early and late apoptosis. J. Cell Biol. 139, 729–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shechtman C. F., Henneberry A. L., Seimon T. A., Tinkelenberg A. H., Wilcox L. J., Lee E., Fazlollahi M., Munkacsi A. B., Bussemaker H. J., Tabas I., Sturley S. L. (2011) Loss of subcellular lipid transport due to ARV1 deficiency disrupts organelle homeostasis and activates the unfolded protein response. J. Biol. Chem. 286, 11951–11959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Cha J. Y., Repa J. J. (2007) The liver X receptor (LXR) and hepatic lipogenesis. The carbohydrate-response element-binding protein is a target gene of LXR. J. Biol. Chem. 282, 743–751 [DOI] [PubMed] [Google Scholar]
- 47. Jaye M., Lynch K. J., Krawiec J., Marchadier D., Maugeais C., Doan K., South V., Amin D., Perrone M., Rader D. J. (1999) A novel endothelial-derived lipase that modulates HDL metabolism. Nat. Genet. 21, 424–428 [DOI] [PubMed] [Google Scholar]
- 48. Tsukamoto K., Tangirala R. K., Chun S., Usher D., Puré E., Rader D. J. (2000) Hepatic expression of apolipoprotein E inhibits progression of atherosclerosis without reducing cholesterol levels in LDL receptor-deficient mice. Mol. Ther. 1, 189–194 [DOI] [PubMed] [Google Scholar]
- 49. Wetterau J. R., Gregg R. E., Harrity T. W., Arbeeny C., Cap M., Connolly F., Chu C. H., George R. J., Gordon D. A., Jamil H., Jolibois K. G., Kunselman L. K., Lan S. J., Maccagnan T. J., Ricci B., Yan M., Young D., Chen Y., Fryszman O. M., Logan J. V., Musial C. L., Poss M. A., Robl J. A., Simpkins L. M., Slusarchyk W. A., Sulsky R., Taunk P., Magnin D. R., Tino J. A., Lawrence R. M., Dickson J. K., Jr., Biller S. A. (1998) An MTP inhibitor that normalizes atherogenic lipoprotein levels in WHHL rabbits. Science 282, 751–754 [DOI] [PubMed] [Google Scholar]
- 50. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔC(T)) Method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 51. Kurrasch D. M., Huang J., Wilkie T. M., Repa J. J. (2004) Quantitative real-time polymerase chain reaction measurement of regulators of G-protein signaling mRNA levels in mouse tissues. Methods Enzymol. 389, 3–15 [DOI] [PubMed] [Google Scholar]
- 52. Schmittgen T. D., Livak K. J. (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108 [DOI] [PubMed] [Google Scholar]
- 53. Tusher V. G., Tibshirani R., Chu G. (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U.S.A. 98, 5116–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schrauwen P., Schrauwen-Hinderling V., Hoeks J., Hesselink M. K. (2010) Mitochondrial dysfunction and lipotoxicity. Biochim. Biophys. Acta 1801, 266–271 [DOI] [PubMed] [Google Scholar]
- 55. Chavez J. A., Summers S. A. (2010) Lipid oversupply, selective insulin resistance, and lipotoxicity: molecular mechanisms. Biochim. Biophys. Acta 1801, 252–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Borradaile N. M., Han X., Harp J. D., Gale S. E., Ory D. S., Schaffer J. E. (2006) Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J. Lipid Res. 47, 2726–2737 [DOI] [PubMed] [Google Scholar]
- 57. Diakogiannaki E., Welters H. J., Morgan N. G. (2008) Differential regulation of the endoplasmic reticulum stress response in pancreatic beta-cells exposed to long-chain saturated and monounsaturated fatty acids. J. Endocrinol. 197, 553–563 [DOI] [PubMed] [Google Scholar]
- 58. Pineau L., Colas J., Dupont S., Beney L., Fleurat-Lessard P., Berjeaud J. M., Bergès T., Ferreira T. (2009) Lipid-induced ER stress: synergistic effects of sterols and saturated fatty acids. Traffic 10, 673–690 [DOI] [PubMed] [Google Scholar]
- 59. Moffitt J. H., Fielding B. A., Evershed R., Berstan R., Currie J. M., Clark A. (2005) Adverse physicochemical properties of tripalmitin in beta cells lead to morphological changes and lipotoxicity in vitro. Diabetologia 48, 1819–1829 [DOI] [PubMed] [Google Scholar]
- 60. Roden M., Price T. B., Perseghin G., Petersen K. F., Rothman D. L., Cline G. W., Shulman G. I. (1996) Mechanism of free fatty acid-induced insulin resistance in humans. J. Clin. Invest. 97, 2859–2865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Smith J. J., Ramsey S. A., Marelli M., Marzolf B., Hwang D., Saleem R. A., Rachubinski R. A., Aitchison J. D. (2007) Transcriptional responses to fatty acid are coordinated by combinatorial control. Mol. Syst. Biol. 3, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kao G., Nordenson C., Still M., Rönnlund A., Tuck S., Naredi P. (2007) ASNA-1 positively regulates insulin secretion in C. elegans and mammalian cells. Cell 128, 577–587 [DOI] [PubMed] [Google Scholar]
- 63. Guillam M. T., Hümmler E., Schaerer E., Yeh J. I., Birnbaum M. J., Beermann F., Schmidt A., Dériaz N., Thorens B., Wu J. Y. (1997) Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat. Genet. 17, 327–330 [DOI] [PubMed] [Google Scholar]
- 64. Kurat C. F., Natter K., Petschnigg J., Wolinski H., Scheuringer K., Scholz H., Zimmermann R., Leber R., Zechner R., Kohlwein S. D. (2006) Obese yeast: triglyceride lipolysis is functionally conserved from mammals to yeast. J. Biol. Chem. 281, 491–500 [DOI] [PubMed] [Google Scholar]
- 65. Athenstaedt K., Daum G. (2003) YMR313c/TGL3 encodes a novel triacylglycerol lipase located in lipid particles of Saccharomyces cerevisiae. J. Biol. Chem. 278, 23317–23323 [DOI] [PubMed] [Google Scholar]
- 66. Tinkelenberg A. H., Liu Y., Alcantara F., Khan S., Guo Z., Bard M., Sturley S. L. (2000) Mutations in yeast ARV1 alter intracellular sterol distribution and are complemented by human ARV1. J. Biol. Chem. 275, 40667–40670 [DOI] [PubMed] [Google Scholar]
- 67. Wrede C. E., Dickson L. M., Lingohr M. K., Briaud I., Rhodes C. J. (2002) Protein kinase B/Akt prevents fatty acid-induced apoptosis in pancreatic beta-cells (INS-1). J. Biol. Chem. 277, 49676–49684 [DOI] [PubMed] [Google Scholar]
- 68. Green C. D., Olson L. K. (2011) Modulation of palmitate-induced endoplasmic reticulum stress and apoptosis in pancreatic β-cells by stearoyl-CoA desaturase and Elovl6. Am. J. Physiol. Endocrinol. Metab. 300, E640–E649 [DOI] [PubMed] [Google Scholar]
- 69. Sargsyan E., Bergsten P. (2011) Lipotoxicity is glucose-dependent in INS-1E cells but not in human islets and MIN6 cells. Lipids Health Dis. 10, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kusminski C. M., Shetty S., Orci L., Unger R. H., Scherer P. E. (2009) Diabetes and apoptosis: lipotoxicity. Apoptosis 14, 1484–1495 [DOI] [PubMed] [Google Scholar]
- 71. Cusi K. (2009) Lessons learned from studying families genetically predisposed to type 2 diabetes mellitus. Curr. Diab. Rep. 9, 200–207 [DOI] [PubMed] [Google Scholar]
- 72. Tsukamoto H., Yamamoto T., Nishigaki T., Sakai N., Nanba E., Ninomiya H., Ohno K., Inui K., Okada S. (2001) SSCP analysis by RT-PCR for the prenatal diagnosis of Niemann-Pick disease type C. Prenat. Diagn. 21, 55–57 [DOI] [PubMed] [Google Scholar]
- 73. Cohen M. H., French A. L., Benning L., Kovacs A., Anastos K., Young M., Minkoff H., Hessol N. A. (2002) Causes of death among women with human immunodeficiency virus infection in the era of combination antiretroviral therapy. Am. J. Med. 113, 91–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Duval C., Müller M., Kersten S. (2007) PPARα and dyslipidemia. Biochim. Biophys. Acta 1771, 961–971 [DOI] [PubMed] [Google Scholar]
- 75. Koliwad S. K., Streeper R. S., Monetti M., Cornelissen I., Chan L., Terayama K., Naylor S., Rao M., Hubbard B., Farese R. V., Jr. (2010) DGAT1-dependent triacylglycerol storage by macrophages protects mice from diet-induced insulin resistance and inflammation. J. Clin. Invest. 120, 756–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Holland W. L., Brozinick J. T., Wang L. P., Hawkins E. D., Sargent K. M., Liu Y., Narra K., Hoehn K. L., Knotts T. A., Siesky A., Nelson D. H., Karathanasis S. K., Fontenot G. K., Birnbaum M. J., Summers S. A. (2007) Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab. 5, 167–179 [DOI] [PubMed] [Google Scholar]
- 77. Park T. S., Hu Y., Noh H. L., Drosatos K., Okajima K., Buchanan J., Tuinei J., Homma S., Jiang X. C., Abel E. D., Goldberg I. J. (2008) Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J. Lipid Res. 49, 2101–2112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Haus J. M., Kashyap S. R., Kasumov T., Zhang R., Kelly K. R., Defronzo R. A., Kirwan J. P. (2009) Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 58, 337–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wang H., Kouri G., Wollheim C. B. (2005) ER stress and SREBP-1 activation are implicated in beta-cell glucolipotoxicity. J. Cell Sci. 118, 3905–3915 [DOI] [PubMed] [Google Scholar]
- 80. Georgiev A. G., Johansen J., Ramanathan V. D., Sere Y. Y., Beh C. T., Menon A. K. (2013) Arv1 regulates PM and ER membrane structure and homeostasis but is dispensable for intracellular sterol transport. Traffic 14, 912–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Fei W., Wang H., Fu X., Bielby C., Yang H. (2009) Conditions of endoplasmic reticulum stress stimulate lipid droplet formation in Saccharomyces cerevisiae. Biochem. J. 424, 61–67 [DOI] [PubMed] [Google Scholar]
- 82. Anderson N., Borlak J. (2008) Molecular mechanisms and therapeutic targets in steatosis and steatohepatitis. Pharmacol. Rev. 60, 311–357 [DOI] [PubMed] [Google Scholar]
- 83. Mei S., Ni H. M., Manley S., Bockus A., Kassel K. M., Luyendyk J. P., Copple B. L., Ding W. X. (2011) Differential roles of unsaturated and saturated fatty acids on autophagy and apoptosis in hepatocytes. J. Pharmacol. Exp. Ther. 339, 487–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. van Herpen N. A., Schrauwen-Hinderling V. B. (2008) Lipid accumulation in non-adipose tissue and lipotoxicity. Physiol. Behav. 94, 231–241 [DOI] [PubMed] [Google Scholar]
- 85. Villanueva C. J., Monetti M., Shih M., Zhou P., Watkins S. M., Bhanot S., Farese R. V., Jr. (2009) Specific role for acyl CoA:Diacylglycerol acyltransferase 1 (Dgat1) in hepatic steatosis due to exogenous fatty acids. Hepatology 50, 434–442 [DOI] [PMC free article] [PubMed] [Google Scholar]