

Abstract
Throughout life, animals face a variety of challenges such as developmental growth, the presence of toxins, or changes in temperature. Neuronal circuits and synapses respond to challenges by executing an array of neuroplasticity paradigms. Some paradigms allow neurons to up- or downregulate activity outputs, while countervailing ones ensure that outputs remain within appropriate physiological ranges. A growing body of evidence suggests that homeostatic synaptic plasticity (HSP) is critical in the latter case. Voltage-gated calcium channels gate forms of HSP. Presynaptically, the aggregate data show that when synapse activity is weakened, homeostatic signaling systems can act to correct impairments, in part by increasing calcium influx through presynaptic CaV2-type channels. Increased calcium influx is often accompanied by parallel increases in the size of active zones and the size of the readily releasable pool of presynaptic vesicles. These changes coincide with homeostatic enhancements of neurotransmitter release. Postsynaptically, there is a great deal of evidence that reduced network activity and loss of calcium influx through CaV1-type calcium channels also results in adaptive homeostatic signaling. Some adaptations drive presynaptic enhancements of vesicle pool size and turnover rate via retrograde signaling, as well as de novo insertion of postsynaptic neurotransmitter receptors. Enhanced calcium influx through CaV1 after network activation or single cell stimulation can elicit the opposite response—homeostatic depression via removal of excitatory receptors. There exist intriguing links between HSP and calcium channelopathies—such as forms of epilepsy, migraine, ataxia, and myasthenia. The episodic nature of some of these disorders suggests alternating periods of stable and unstable function. Uncovering information about how calcium channels are regulated in the context of HSP could be relevant toward understanding these and other disorders.
Keywords: homeostatic synaptic plasticity, VGCCs, CaV1 channels, CaV2 channels, neurotransmitter release, synaptic scaling, synaptic growth, calcium channelopathies
Introduction
Many forms of neuroplasticity drive changes in synaptic outputs, and they are thought to underlie fundamental neurological phenomena, like learning. At the same time, stabilizing forms of neuroplasticity—collectively termed homeostatic synaptic plasticity (HSP)—work to ensure that neuronal outputs are maintained within physiologically appropriate levels. The study of HSP has shed considerable light on how neuronal stability is maintained. Perturbations of synaptic function can trigger homeostatic modulations in activity parameters such as presynaptic neurotransmitter release, neurotransmitter receptor expression, ion channel density, or conductance properties (Pérez-Otaño and Ehlers, 2005; Davis, 2006, 2013; Marder and Goaillard, 2006; Turrigiano, 2008; Maffei and Fontanini, 2009; Pozo and Goda, 2010; Watt and Desai, 2010; Marder, 2012; Turrigiano, 2012). The underlying molecular mechanisms that enable such change are critical (Lazarevic et al., 2013). How exactly do synapses detect challenges to their activity and then engage biological homeostats to correct errors? Some progress has been made in answering this question using a variety of models such as Drosophila melanogaster (Davis, 2013; Frank, 2014), crustaceans (Marder and Goaillard, 2006; Marder and Bucher, 2007; Marder, 2012), and rodents (Chen et al., 2014; Lee et al., 2014; Pribiag and Stellwagen, 2014; Thalhammer and Cingolani, 2014; Wenner, 2014; Whitt et al., 2014). In recent years, voltage-gated calcium channels (VGCCs or CaV channels) have emerged as critical for homeostatic control of synapse function in several experimental contexts.
When considering how CaV channels control any process (such as HSP), it is important to consider auxiliary interacting proteins, cellular context, and the high degree to which CaV-driven processes are conserved across metazoans. It is known that neuronal CaV channels mediate cellular calcium entry and regulate activity-dependent processes such as neurotransmission, gene transcription, and intracellular signaling cascades (Catterall, 2000; Zamponi, 2005; Tedford and Zamponi, 2006; Catterall and Few, 2008; Dolphin, 2009; Currie, 2010; Lipscombe et al., 2013). Control of CaV function occurs at many levels, starting with the regulation of channel subunit mRNA splicing and channel subunit trafficking (Lipscombe et al., 2013). Much has also been learned about the targeting and cellular distribution of CaV channels (Herlitze et al., 2003), as well as G-Protein-mediated inhibition of presynaptic CaV channels (Tedford and Zamponi, 2006; Currie, 2010; Zamponi and Currie, 2013). Across metazoans as diverse as nematodes and humans, cytoplasmic, calcium-binding regulatory messengers such as calmodulin integrate cytoplasmic calcium entry with activation of downstream targets, such as calcium/calmodulin-dependent protein kinases (e.g., CaMK, CaMKII) (Liu et al., 2007)—or even inhibition or facilitation of CaV channels themselves (Halling et al., 2006; Dunlap, 2007; Catterall and Few, 2008; Minor and Findeisen, 2010; Christel and Lee, 2012). Mitochondria and the endoplasmic reticulum also play conserved modulatory roles at many model synapses, acting as calcium buffers and intracellular sources of calcium (Verkhratsky and Petersen, 1998; Collin et al., 2005; Liu et al., 2005; Williams et al., 2013). Additionally, genetic mutations and toxins that impair CaV channel function cause numerous cellular defects, including impairment of neurotransmission and alterations of forms of neuroplasticity (Catterall and Few, 2008; Norton and McDonough, 2008). These topics have been studied and reviewed thoroughly elsewhere (including a comprehensive book, Zamponi, 2005).
With the backdrop of this considerable knowledge, we consider the emerging roles that VGCCs play in HSP. We review electrophysiological, biochemical, and imaging data that have established the important roles CaV channels play in multiple forms of HSP across diverse experimental systems (Figures 1, 2). We also briefly consider the idea that CaV channels might link HSP to disorders in which underlying neuronal stability is lost.
Figure 1.
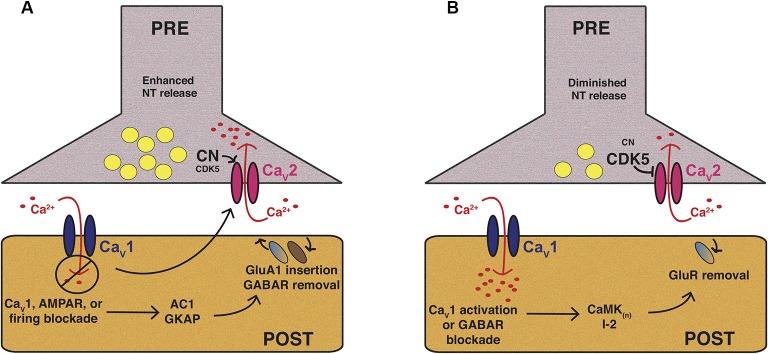
Mammalian CaV1 and CaV2 channels play central roles in forms of homeostatic plasticity. These two cartoons attempt to synthesize knowledge of mammalian pre- and postsynaptic homeostatic plasticity mechanisms involving CaV1 and CaV2 calcium channels in preparations like cultured hippocampal neurons. The cartoons are not intended to depict a single synaptic preparation or universally conserved mechanism, though some molecular responses may be widely conserved. (A) Homeostatic potentiation of synapse function. Inhibition of synaptic activity or postsynaptic CaV1 calcium influx results in multiple changes, including postsynaptic signaling through molecules like adenylate cyclase 1 (AC1) or guanylate kinase-associated protein (GKAP) to drive activating mechanisms, such as glutamate receptor insertion. Trans-synaptic signaling controlled by factors like Target of Rapamycin (TOR) and Brain-Derived Neurotrophic Factor (BDNF) can trigger enhanced presynaptic release probability. From a variety of systems there is evidence for enhanced presynaptic calcium influx through CaV2—which may require diminishment of cyclin-dependent kinase 5 (CDK5) function—as well as an enhanced readily releasable pool of presynaptic vesicles. (B) Homeostatic downscaling of synapse function. Synaptic activation (e.g., through GABA receptor blockade) and/or enhanced postsynaptic calcium influx through CaV1 results in the activation of diverse pathways, such as those mediated by calcium/calmodulin-dependent kinases (CaMK), as well as the Protein Phosphatase 1 (PP1) inhibitor, I-2. This can result in removal of excitatory glutamate receptors from the synapse. Presynaptically, there is evidence of diminished calcium influx through CaV2, and thus, diminished evoked presynaptic release.
Figure 2.
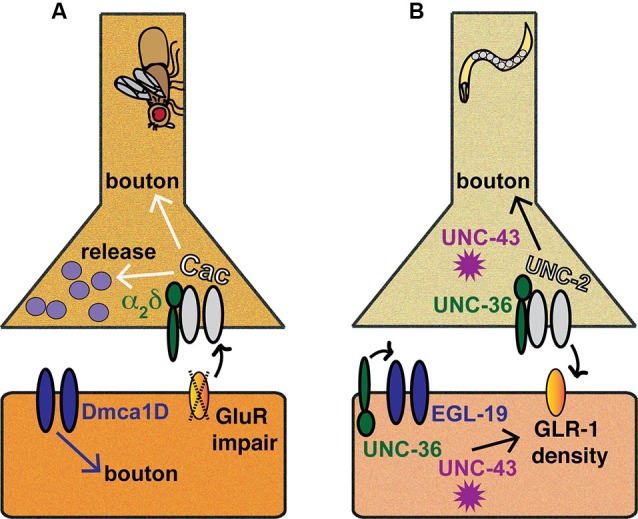
Invertebrate models of CaV-directed homeostatic plasticity. Inspection of the Drosophila melanogaster NMJ has provided a wealth of information regarding the roles VGCCs and associated molecules play in homeostatic plasticity, as has examination of C. elegans preparations, such as the ventral nerve cord (VNC) or the NMJ. (A) Drosophila NMJ. Pharmacological or genetic impairment of postsynaptic glutamate receptors triggers a retrograde signaling process that results in enhanced presynaptic Cac/CaV2 function and increased neurotransmitter release. Additionally, Cac/CaV2, α2δ, and Dmca1D/CaV1 all affect synaptic bouton development or maturation at the NMJ. (B) C. elegans synapses. At the C. elegans VNC, the coordinated functions of UNC-2/CaV2, EGL-19/CaV1, UNC-36/α2δ and UNC-43/CaMKII ensure proper coupling of GLR-1 glutamate receptor density to developmental growth.
CAV2 channels and presynaptic homeostatic synaptic plasticity
CaV2-type calcium channels function in the presynaptic nervous systems of nearly all animals. There are several CaV2 subtypes, classified primarily by sensitivity to different toxins; they include the P/Q- (CaV2.1), N- (CaV2.2), and R-type (CaV2.3) calcium channels (Catterall et al., 2005; Zamponi, 2005). CaV2 channels generally mediate fast neurotransmission, gating presynaptic calcium influx upon cellular depolarization (Catterall et al., 2005; Zamponi, 2005). In turn, calcium-sensing molecules such as Synaptotagmin I trigger rapid evoked neurotransmitter release (Giraudo et al., 2006; Schaub et al., 2006; Tang et al., 2006). Here we review data that demonstrate roles for presynaptic CaV2 channels in HSP at fruit fly and mammalian synapses. In particular, several forms of homeostatic plasticity appear to govern alterations in presynaptic neurotransmitter release by directly targeting the amount of terminal calcium influx through CaV2-type channels.
Homeostatic plasticity and CAV2 at the drosophila neuromuscular synapse
The Drosophila melanogaster larval neuromuscular junction (NMJ) is a glutamatergic synapse that exhibits a strong capacity for homeostatic regulation (Frank, 2014; Figure 2A). Several studies have established that the NMJ retains normal levels of postsynaptic evoked excitation, even when it is challenged by chronic or acute perturbations to excitability (Petersen et al., 1997; Davis et al., 1998; DiAntonio et al., 1999; Paradis et al., 2001; Frank et al., 2006). For instance, genetic or pharmacological impairment of muscle glutamate receptors results in decreased sensitivity to single vesicles of glutamate (Petersen et al., 1997; Frank et al., 2006). A homeostatic, retrograde, muscle-to-nerve signaling process helps to offset this decreased sensitivity by inducing a presynaptic increase in vesicle release. This increase has been measured by multiple electrophysiological means (Petersen et al., 1997; Frank et al., 2006). We recently reviewed our understanding of mechanisms underlying homeostatic plasticity at the Drosophila NMJ, including investigations to uncover the unknown retrograde signal(s) and the central roles played by CaV2 channels and presynaptic calcium influx (Frank, 2014). For completeness, some of that same information is reiterated here, but it is updated with newly published data.
Cacophony/CaV2
The pore-forming α1 subunit of Drosophila CaV2 channels is called Cacophony (Cac; Smith et al., 1996). Loss-of-function mutations in the cac gene were originally found in the 1970s from genetic approaches to identify fruit flies with visual defects and defects in mating behavior; indeed, partial loss-of-function cac mutant males buzz their wings with a defective, “cacophonous” mating song (Kulkarni and Hall, 1987; Smith et al., 1998). Cac is required throughout much of development. Null cac mutant embryos fail to hatch (Kulkarni and Hall, 1987; Kurshan et al., 2009), and cac mRNA expression is prominent in the nervous system (Smith et al., 1996). Partial loss-of-function cac hypomorphs are viable and fertile, permitting synaptic analyses at larval and adult stages (Smith et al., 1998; Kawasaki et al., 2000; Brooks et al., 2003). Hypomorphic cac mutations cause pronounced neurotransmission defects (Kawasaki et al., 2000; Brooks et al., 2003; Frank et al., 2006), and at the third instar larval stage of development, some hypomorphs display mild NMJ growth defects (Rieckhof et al., 2003; Xing et al., 2005). Neuronally expressed Cac-Green Fluorescent Protein (GFP) rescues the lethality of nulls and co-localizes with presynaptic active zone markers (Kawasaki et al., 2004). Transgenic Cac-GFP has proven to be a useful tool in examining active zones and associated proteins (as in Fouquet et al., 2009).
CaV2-type calcium channels also play a critical role in Drosophila NMJ homeostasis. When the NMJs of cac hypomorphs are challenged with a pharmacological or genetic impairment of postsynaptic glutamate receptor function, there is no increase in presynaptic neurotransmitter release (Frank et al., 2006). Therefore, not only do cac mutants have neurotransmission defects, but the NMJ also fails to maintain muscle excitation at baseline levels (i.e., the already impaired cac mutant levels). This phenotype constitutes a block of synaptic homeostasis. One concern about this finding is that any mutant with baseline neurotransmission defects might also have defects in the ability to respond to homeostatic challenges. However, it has been demonstrated that several Drosophila mutations cause baseline NMJ neurotransmission defects without impairing homeostatic plasticity (Goold and Davis, 2007; Dickman and Davis, 2009; Younger et al., 2013). Conversely, other mutations or gene knockdowns impair homeostatic plasticity without causing baseline neurotransmission defects (Dickman and Davis, 2009; Marie et al., 2010; Müller et al., 2011; Dickman et al., 2012).
The requirements for Cac/CaV2 during NMJ homeostasis correspond to measureable changes in presynaptic calcium influx. Direct measurement of evoked presynaptic calcium transients reveals that chronic and acute glutamate receptor impairments induce significant (23–30%) increases in presynaptic calcium influx, presumably through Cac/CaV2 channels (Müller and Davis, 2012). A hypomorphic cac point mutation, cacS, blocks this increase (Müller and Davis, 2012). In addition, pharmacological inhibition of the glutamate receptors results in a swift increase in expression of the presynaptic active zone protein Bruchpilot (an ELKS/CAST homolog), as well as an increase in the active zone cytomatrix structure (Weyhersmüller et al., 2011). Together, these results suggest that CaV2 and other active zone proteins are important targets for homeostatic signaling processes. This is logical because small changes in presynaptic calcium influx are well known to correspond to large changes in vesicle release and neurotransmission (Katz and Miledi, 1970).
What might enhance CaV2 function during HSP at the NMJ? Genetic, electrophysiological, and calcium imaging data suggest that presynaptic CaV2 may be targeted by several signaling paradigms that reside on both sides of the synapse. This is not surprising, given that retrograde, muscle-to-nerve signaling governs synaptic homeostasis at the NMJ. What is surprising is how many pieces of this signaling puzzle have emerged in a few short years. Linking those pieces together into a logical framework will be critical as research moves forward.
Postsynaptic signaling and CaV2
Following genetic glutamate receptor impairment, a protein-translation-dependent signaling process in the muscle is driven by Drosophila Target of Rapamycin (TOR) and S6 kinase (S6K) to potentiate presynaptic release (Penney et al., 2012). By contrast, two negative postsynaptic signaling factors are the nuclear receptor Importin 13 (Imp13) and the muscle cytoskeletal/matrix support molecule Dystrophin. Impairments of Imp13 or Dystrophin function cause an increase in presynaptic vesicle release (Giagtzoglou et al., 2009; Pilgram et al., 2010, 2011). Further, postsynaptic imp13 loss of function causes an increase in presynaptic intracellular calcium levels (Giagtzoglou et al., 2009). It is not known if S6K/TOR-, Dystrophin-, and Imp13-mediated signaling events directly interact with one another to regulate homeostatic changes in neurotransmitter release. Given the extensive Drosophila genetic toolkit, this issue is imminently addressable.
Presynaptic signaling and CaV2
On the presynaptic side of the synapse, several molecules are hypothesized to directly or indirectly enhance CaV2 function after glutamate receptor impairment. Among these is a signaling system driven by the Drosophila Eph receptor tyrosine kinase, the cytoplasmic guanine exchange factor Ephexin, and Rho-type GTPases (Frank et al., 2009). Loss-of-function mutations in components of this signaling system impair synaptic homeostasis and interact genetically with cac mutations (Frank et al., 2009).
The kinesin super family member Khc-73 may work in a similar fashion as Eph/Ephexin. khc-73 loss-of-function mutations completely impair synaptic homeostasis (Tsurudome et al., 2010), and electron microscopy data demonstrate that Khc-73 contributes to enhancement of active zone components at the NMJ (Tsurudome et al., 2010). A negative regulator of khc-73 gene expression is the miR-310 micro RNA cluster. Loss of the miR-310 genetic locus phenocopies transgenic overexpression of khc-73 and shows enhanced presynaptic evoked calcium transients, possibly due to enhanced CaV2 activity (Tsurudome et al., 2010).
Finally, a factor that interacts with calcium channels and assists their localization to the active zone is Drosophila Rab3 Interacting Molecule (RIM; Graf et al., 2012). rim loss-of-function mutations block synaptic homeostasis at the NMJ, but not because of a failure to upregulate presynaptic calcium influx. Instead, rim mutations occlude important increases in the size of the readily releasable pool (RRP) of presynaptic vesicles (Müller et al., 2012).
New data: ENaC and CaV2
A very recent study offers compelling insights into how CaV2 function may be potentiated during synaptic homeostasis. This study demonstrates that presynaptic Epithelial Sodium Channel (ENaC) components encoded by the pickpocket11 (ppk11) and pickpocket16 (ppk16) genes are both required for synaptic homeostasis (Younger et al., 2013). They are also required for the accompanying enhanced presynaptic calcium influx (Younger et al., 2013). These results are drawn not only from genetic mutant data, but also from application of the drug benzamil to impair ENaC (Younger et al., 2013). Based on these new data and previously published data about ENaC function as a voltage-insensitive cation channel (Schild, 2010), one compelling model is that PPK11- and PPK16-ENaC channels act to depolarize presynaptic membrane voltage (Younger et al., 2013). As a result, depolarized resting presynaptic voltage may enhance presynaptic CaV2 activity during homeostatic plasticity.
Arclight is a new genetically encoded voltage sensor that shows robust utility in Drosophila to measure membrane voltage in response to action potentials and sub threshold events (Cao et al., 2013). An attractive possibility for future research is to design new tools—similar to Arclight—that could reliably measure alterations in resting membrane voltage. Such tools could be utilized to directly test the voltage/CaV2 hypothesis for ENaC and possibly to hunt for other factors that control neurotransmission and HSP upstream of CaV2 function.
CAV2 and homeostatic synaptic plasticity at mammalian central synapses
There are similarities in CaV2-gated HSP between the Drosophila NMJ (Figure 2A) and mammalian central synapses (Figure 1). Cultured rodent neurons possess a strong ability to maintain a set point of activity through a homeostatic process termed synaptic scaling (Turrigiano, 2012; Chen et al., 2014; Lee et al., 2014; Pribiag and Stellwagen, 2014; Siddoway et al., 2014; Thalhammer and Cingolani, 2014). It is known that the drugs tetrodotoxin (TTX), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), and 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX) abolish neuronal firing and transmission. TTX blocks sodium channels (Narahashi, 2008), while CNQX and NBQX block postsynaptic AMPA glutamate receptors (Honore et al., 1988; Sheardown, 1993). Conversely, drugs like bicuculline or gabazine inhibit γ-aminobutyric acid (GABA) receptor-mediated inhibitory neurotransmission, and thus, increase activity (Curtis et al., 1970; Uchida et al., 1996). A landmark finding was that chronic application (>48 h) of TTX, CNQX, or bicuculline to cultured rodent visual cortical neurons or to spinal cord neurons elicits a scaling up (TTX and CNQX) or scaling down (bicuculline) of spontaneous miniature amplitudes (O’Brien et al., 1998; Turrigiano et al., 1998). Additionally, drug applications change firing rates in the short run, but upon TTX/CNQX washout or prolonged exposure to bicuculline, activity parameters in exposed neurons homeostatically drive firing rates back in the opposite direction (Turrigiano et al., 1998).
Much work has illuminated the postsynaptic events that accompany scaling—namely, altered postsynaptic neurotransmitter receptor composition and sensitivity to glutamate (O’Brien et al., 1998; Turrigiano et al., 1998; Wierenga et al., 2005; Sutton et al., 2006; Turrigiano, 2012). However, there has also been a related body of work demonstrating that some central synapses can respond to chronic drug applications in multiple ways. Depending upon the particular preparation or experimental condition, the synaptic response may also be governed through presynaptic changes in RRP size and/or transmitter release (Murthy et al., 2001; Burrone et al., 2002; Thiagarajan et al., 2005). These presynaptic changes do not have to be induced via network-wide perturbations. For example, silencing individual neurons with the inwardly rectifying potassium channel Kir2.1 after synapse formation causes a homeostatic increase of synaptic inputs (Burrone et al., 2002). But what exactly are the presynaptic events that control these responses? Recent results are shedding light on these phenomena.
Presynaptic calcium indicators
Presynaptic examinations have been illuminating in defining a role for CaV2 channels in HSP. For example, researchers utilizing a calcium reporter directly localized to synaptic vesicles (SyGCaMP2) demonstrated homeostatic changes in presynaptic calcium influx at rodent hippocampal neurons (Zhao et al., 2011). Synaptic boutons pretreated with TTX for 2 days show a marked increase in presynaptic calcium influx after action potential delivery (Zhao et al., 2011). By contrast, gabazine treatment to inhibit inhibitory GABAA receptors results in decreased presynaptic calcium influx (Zhao et al., 2011). These findings are strikingly reminiscent of those at the Drosophila NMJ (Müller and Davis, 2012). They are also consistent with microscopy data from cultured rodent cortical neurons and the aforementioned Drosophila NMJ, demonstrating that challenges to synaptic activity can result in profound homeostatic changes to components of the presynaptic release machinery (Lazarevic et al., 2011; Weyhersmüller et al., 2011).
Retrograde signaling: mTORC1, BDNF, and CaV2
Other results examining hippocampal cultures corroborate this notion. NBQX- or CNQX-mediated blockade of AMPA receptors in the absence of TTX induces two main homeostatic responses: (1) incorporation of new, GluA2-lacking AMPA receptors postsynaptically; and (2) a retrograde signaling process that results in increased presynaptic release properties, such as spontaneous miniature frequency (Thiagarajan et al., 2005; Gong et al., 2007). Similar to the Drosophila system, postsynaptic mammalian Target of Rapamycin Complex1 (mTORC1) drives this retrograde signaling process—albeit through release of Brain-Derived Neurotrophic Factor (BDNF), which is not found in Drosophila (Henry et al., 2012). Also resonant with the Drosophila NMJ, coincident application of a cocktail of N- and P/Q-type calcium channel blockers ω-conotoxin GVIA and ω-agatoxin IVA (CTx/ATx cocktail) completely abolishes the enhanced presynaptic activity induced by 3 h of CNQX exposure (Jakawich et al., 2010). This result supports the idea that presynaptic CaV2 function is required for this form of homeostatic plasticity.
Cyclin-dependent kinase 5 (CDK5) and calcineurin
Key components of CaV2.2 (N-type) regulation during HSP appear to be the cyclin-dependent kinase CDK5 and the α isoform of the phosphatase Calcineurin A (CNAα). Inhibition of CDK5 function in CA1-CA3 hippocampal cultures with the drug roscovitine results in enhanced action potential-evoked release and access to the resting synaptic vesicle pool for release (Kim and Ryan, 2010). Consistently, biochemical assays show that chronic silencing of synapses with TTX leads to a measurable decrease of CDK5 (Kim and Ryan, 2010). CDK5 exerts its control of synaptic activity by balancing an opposing function of CNAα (Kim and Ryan, 2010, 2013). The ability of either enzyme to exert control over action potential-driven exocytosis is reliant upon N-type calcium channels, as the N-type blocker ω-conotoxin GVIA occludes their effects (Kim and Ryan, 2013).
CAV1 channels and postsynaptic homeostatic synaptic plasticity
In contrast to CaV2 channels, L-type CaV1 channels are localized to both presynaptic and postsynaptic structures in neurons, as well as non-neuronal excitable tissues—such as skeletal and cardiac muscle—where they are required for excitation-contraction coupling (Hell et al., 1993; Lipscombe et al., 2004; Obermair et al., 2004). Calcium entry though L-type channels activates a variety of downstream calcium-sensitive signaling cascades and gene regulation programs (Lipscombe et al., 2004). There are several sub-types of L-type CaV1 channels, classified by the α1 subunit incorporated into the channel: CaV1.1, 1.2, 1.3, and 1.4 (mammalian α1 subunit genes CACNA1S, CACNA1C, CACNA1D, and CACNA1F respectively). The kinetics of CaV1 activation can be fast (Lipscombe et al., 2004; Helton et al., 2005), however, it is generally thought that slow L-type deactivation allows sustained current, and thus, calcium signaling (McCobb and Beam, 1991; Perrier et al., 2002; Helton et al., 2005). CaV1-type calcium channels are sensitive to dihydropyridine antagonists such as nifedipine or nimodipine, or to phenylalkylamines like verapamil. Neuroscientists typically apply these drugs to electrophysiological preparations in order to assess the results of impaired CaV1 function. These drugs have been used to great effect to show that L-type channels are critical for the expression of a variety of forms of neuroplasticity (Murphy et al., 1991; Magee and Johnston, 1997; Hardingham et al., 1998; Weisskopf et al., 1999; Mermelstein et al., 2000; Brosenitsch and Katz, 2001; Dolmetsch et al., 2001; Lei et al., 2003; Pak and Sheng, 2003)—as well as forms of HSP, detailed below.
Pioneering findings: chronic blockade of hippocampal neuron activity
In the same study that examined NBQX-induced blockade and the resulting incorporation of GluA2-lacking AMPA receptors in hippocampal cultures, it was reported that nifedipine application mimics NBQX blockade (Thiagarajan et al., 2005). This result suggests that loss of calcium influx through CaV1 channels could gate the mechanism of adding GluA1 homomeric (GluA2-lacking) AMPA receptors to the synapse. To test if CaV1 blockade and AMPA receptor blockade activate a shared mechanism, nifedipine and NBQX were applied concurrently. Dual drug application was indistinguishable from single drug application by biochemical and electrophysiological assays, consistent with a model in which AMPA blockade results in a loss of CaV1 activity (Thiagarajan et al., 2005). In turn, loss of CaV1 activity results in a postsynaptic increase in GluA1 homomers as well as an increase in presynaptic release properties (Thiagarajan et al., 2005). But does CaV1-mediated plasticity occur exclusively after AMPA receptor blockade? No. For example, potentiation of postsynaptic miniature excitatory postsynaptic current (mEPSC) amplitudes also occurs when hippocampal neurons are silenced for 24 h with TTX and allowed to recover in TTX-free saline for a short time before recording (Sokolova and Mody, 2008). In this case, however, TTX/TTX washout-induced potentiation is occluded by nifedipine (Sokolova and Mody, 2008).
How CaV1 silencing induces homeostatic synaptic plasticity
What molecular mechanisms underlie silencing-induced metaplasticity downstream of CaV1? Recent research has implicated many classical calcium-sensitive cytoplasmic signaling molecules and processes. Through biochemistry and electrophysiology, one investigation demonstrated that downstream of CNQX-mediated AMPAR blockade (or blockade of CaV1), adenylate cyclase 1 activates nuclear transcription of new GluA1 subunits, which are then trafficked and incorporated as homomers (Gong et al., 2007). Additionally, a recent study demonstrated that in rat hippocampal neurons, the scaffolding molecule guanylate kinase-associated protein (GKAP) plays a key role in both scaling up synaptic activity (after TTX application) and scaling down of activity (after bicuculline) (Shin et al., 2012). It was found that blockade of Calcium/Calmodulin-dependent Kinase II (CaMKII) activity by the drug KN-93 impairs both the recruitment and the removal of GKAP at synapses. Further examination showed that the source of calcium that results in the activation of CaMKII is important. NMDAR blockade by 2-amino-5-phosphonopentanoic acid (APV) stops bicuculline-driven GKAP removal from synapses; by contrast, CaV1 impairment by nimodipine stops TTX-induced GKAP enhancement at synapses (Shin et al., 2012).
Chronic neuronal silencing not only enhances the abundance of excitatory neurotransmitter receptors in a homeostatic fashion, but it can also decrease the abundance of inhibitory receptors at GABAergic synapses—consistent with network stability working through a balance of excitatory and inhibitory connections (Kilman et al., 2002; Swanwick et al., 2006; Saliba et al., 2007). Electrophysiological experiments have revealed a role for CaV1 in this homeostatic process too. For example, in hippocampal cultures, 24 h of nifedipine application reduces GABAergic synaptic transmission, likely due to an increased turnover and a decreased insertion of GABAA receptors (Saliba et al., 2009).
CaV1 integrates responses to chronic activation
If loss of calcium influx through postsynaptic CaV1 channels mediates homeostatic forms of potentiation (Figure 1A), could increased calcium influx through CaV1 mediate homeostatic forms of depression (Figure 1B)? Yes—one elegant analysis showed that this is likely true and went further to demonstrate that homeostatic signaling events governed by CaV1 occur not just on the level of entire networks, but also on the level of single cells (Goold and Nicoll, 2010). By taking advantage of optogenetics, the authors of this study excited individual Channelrhodopsin 2 (ChR2)-expressing CA1 pyramidal neurons for 24 h. In response to this excitatory stimulation, there is a homeostatic downregulation of both NMDAR and AMPAR-mediated responses postsynaptically. This compensatory, homeostatic depression works via L-type calcium channels, as nifedipine occludes the effect (Goold and Nicoll, 2010).
Cellular mechanisms after chronic activation
What are the cellular mechanisms that drive homeostatic downscaling after chronic activation? This question has recently been reviewed (Siddoway et al., 2014). Here, we cover data integrating CaV1 with homeostatic downscaling. In the ChR2-expressing CA1 neurons, this process appears to require Calcium/Calmodulin-dependent protein kinase kinase (CaMKK)—the CaMKK inhibitor STO-609 blocks homeostatic depression (Goold and Nicoll, 2010). This process also requires CaM kinase 4 (CaMK4), as a CaMK4 dominant negative construct also occludes depression (Goold and Nicoll, 2010).
Similar manipulations have uncovered additional calcium-responsive integrators of HSP. In an analysis employing cultured mouse cortical neurons, it was found that chronic bicuculline application causes phosphorylation of the protein phosphatase 1 (PP1) inhibitor I-2. In turn, I-2 phosphorylation is required for appropriate AMPAR trafficking and homeostatic downregulation of synaptic function (Siddoway et al., 2013). L-type calcium channels and calmodulin activity are required for I-2 phosphorylation. Addition of nimodipine or the calmodulin antagonist W7 both block increases in I-2 phosphorylation provoked by bicuculline (Siddoway et al., 2013).
Another example crops up in neocortical neurons, where prolonged hyperactivity induced by gabazine induces increases of production of vesicular glutamate transporter (VGLUT2) and neuronal activity-regulated pentraxin (Narp; Doyle et al., 2010). These increases are hypothesized to be homeostatic in nature because they may lead to increased activation of GABAergic inhibitory feedback neurons (Rutherford et al., 1997; Turrigiano and Nelson, 2004). This form of excitation-transcription coupling is dependent upon CaV1 channels—VGLUT2/Narp induction by gabazine is blocked by nifedipine and verapamil application (Doyle et al., 2010). What acts downstream of CaV1? More classical calcium signaling molecules: VGLUT2/Narp induction is blocked by CaMK antagonists KN-62 and KN-93, as well as mitogen-activated protein kinase (MAPK) antagonists PD98059 and U0126 (Doyle et al., 2010).
CaV channels and the homeostatic growth and pruning of synapses
If the end goal of homeostatic plasticity is to keep synapses and circuits functioning within normal physiological ranges, then a logical way to accomplish this task is to add or prune synaptic connections and couple development with activity. The relationship between synaptic growth, developmental plasticity, neurotransmission, and HSP is not entirely understood. The coupling of these processes appears to depend upon the particular system or manipulation examined. With Hebbian forms of plasticity like LTP or LTD, one observes a clear correlation between the growth (potentiation) or shrinkage (depression) of dendritic spines in central synapses (Matsuzaki et al., 2004; Zhou et al., 2004; Tada and Sheng, 2006; Lisman et al., 2012). This is not always the case with homeostatic plasticity. Above we have considered several examples of long-lasting homeostatic mechanisms gated by VGCCs. Many of these involve changes to presynaptic release probability or to postsynaptic neurotransmitter receptor composition, yet do not involve gross morphological changes to synaptic architecture.
Nevertheless, it would be wrong to claim that synapse/circuit function is completely divorced from developmental forms of homeostatic plasticity. The visual systems of mammals and fruit flies offer elegant examples in which developmental homeostatic programs compensate for visual or activity depravation (Mrsic-Flogel et al., 2007; Yuan et al., 2011; Whitt et al., 2014). In the realm of VGCCs and downstream calcium signaling, there are data that suggest direct or indirect control synaptic growth processes that could influence homeostasis. We consider a few examples from both invertebrate and vertebrate systems.
Drosophila neuromuscular junction (NMJ)
Do long-lasting disruptions of neurotransmission cause synapses to assume alternate, homeostatic developmental programs? Conversely, do developmental alterations result in aberrant neurophysiology? At the Drosophila NMJ, much data suggest that developmental changes are not necessarily coupled to alterations of neurotransmission. A recent study examining evolutionarily diverged species of Drosophila showed that wide variations of “wild-type” synaptic growth—including marked differences in bouton number and branching at the NMJ—redound to indistinguishable physiology (Campbell and Ganetzky, 2012, 2013). These data and the data of others suggest that the properties of synapse growth and function can be uncoupled at insect NMJs.
Concerning CaV channels, two studies reported that partial cac loss-of-function mutations affect synaptic growth by causing a mild decrease in the number of synaptic boutons that are formed (Rieckhof et al., 2003; Xing et al., 2005). Another has shown that null mutations in the α2δ subunit gene of CaV2 fail to develop NMJ boutons in embryos (Kurshan et al., 2009). Finally, in a study examining synaptic overgrowth caused by potassium channel mutations, it was found that both postsynaptic L-type CaV1 channels (α1 subunit encoded by Drosophila Dmca1D) and presynaptic CaV2/Cac channels participate in enabling the overgrowth, at the respective stages of bouton budding (DmCa1D) and maturation (Cac) (Lee and Wu, 2010). It is unclear whether the roles of VGCCs during overgrowth are mechanistically the same as during normal developmental growth.
In all, the aggregate data show that the activity of Drosophila CaV channels can positively influence NMJ maturation and growth (Figure 2A). In the sense that synapse growth is developmentally coincident with larval growth, this could be considered to be a homeostatic function. However, NMJ bouton developmental phenotypes can clearly be uncoupled from neurotransmission phenotypes. This point highlights the importance of examining both development and electrophysiological responses in this model synapse.
Caenorhabditis elegans synapse development
CaV2 and CaV1 channels govern a number of behaviors studied in nematode worms, such as coordination of normal movement and egg laying (Brenner, 1974; Trent et al., 1983; Schafer and Kenyon, 1995; Lee et al., 1997). In C. elegans, the CaV2 α1 homolog is called UNC-2 (Schafer and Kenyon, 1995), and the CaV1 α1 homolog is called EGL-19 (Lee et al., 1997). C. elegans is an excellent system to study CaV functions because there are strong loss-of-function mutations in the genes encoding these pore-forming subunits, as well as mutations in the genes encoding auxiliary subunits, like the α2δ homolog, UNC-36 (Figure 2B). Interestingly, UNC-36 has been shown to be important not only for UNC-2/CaV2 localization and function as would be expected for α2δ (Saheki and Bargmann, 2009), but it also modulates the activation and conductance of EGL-19/CaV1-mediated calcium currents (Frøkjaer-Jensen et al., 2006; Lainé et al., 2011).
Throughout C. elegans larval development, the density of synapses (the number of synapses per unit length) containing GLR-1 glutamate receptors in the ventral nerve cord (VNC) remains at a set point level, even though absolute synapse number increases dramatically over time (Rongo and Kaplan, 1999). The tight coupling between GLR-1 synapse formation and VNC growth is likely homeostatic, and it is reminiscent of the coupling between organism growth and synapse formation seen at the Drosophila and mammalian NMJs. In C. elegans, mutations in the unc-43 gene—which encodes CaMKII—significantly reduce synaptic density of transgenic GLR-1::GFP protein in the VNCs of adult worms (Rongo and Kaplan, 1999). This indicates an uncoupling between growth and synapse development. This result prompted an investigation into the source of calcium upstream of UNC-43/CaMKII; it was found that unc-2 and egl-19 mutations also significantly reduce synapse density (Rongo and Kaplan, 1999). However, in addition to synapse density, GLR-1::GFP puncta intensity was examined in a follow-up study—and here it was found that unc-2 loss results in a compensatory increase in glutamate receptor intensity (Grunwald et al., 2004). Taken together with data from other systems, these C. elegans data are consistent with an ancient role in synapse development for both P/Q- and L-type channels.
The importance of CaMKII and CaV function carries over to the NMJs of C. elegans. A recent study demonstrated that mutations in unc-43, unc-2, or unc-36 all have altered NMJ morphology (Caylor et al., 2013). A close examination of the NMJs during the L4 larval stage revealed that wild-type NMJs add new boutons in a dynamic process, evidenced by enlarged or elongated puncta of a GFP-tagged Synaptobrevin marker. By contrast, in unc-2 mutants, this process is muted (Figure 2B; Caylor et al., 2013).
Striatal medium spiny neurons
Striatal medium spiny neurons (MSNs) offer a vertebrate model of homeostatic control of synaptic growth gated by VGCCs. In MSNs, increased striatal dopamine causes an increase in MSN spine density (Kim et al., 2009). By contrast, decreased dopamine levels (as in Parkinson’s Disease) result in a marked pruning of the MSN spine density, but also a decrease of glutamatergic synapses onto D2 dopamine receptor (D2R)-expressing MSNs (Day et al., 2006; Deutch et al., 2007). Elimination of D2R synapses enhances MSN excitability (Shen et al., 2008). Thus the pruning of D2R-containing synapses likely represents a homeostatic response to the lack of dopamine. Follow-up work employing a combination of potassium-induced membrane depolarization and nimodipine blockage of L-type calcium channels demonstrates that MSN synaptic reduction is dependent upon CaV1.2 function. By contrast, the L-type channel agonist Bay K8644 enhances the effects of membrane depolarization (Tian et al., 2010). Downstream of calcium entry through CaV1.2, there is Calcineurin-mediated activation of Mef2 transcription factor activity (Tian et al., 2010). Consistently, Mef2 has been reported to regulate developmental synaptic remodeling by controlling the expression of a variety of target genes (Flavell et al., 2006, 2008; Fiore et al., 2009; Ye et al., 2013).
Possible links between CaV channelopathies and homeostatic plasticity
Ion channel disorders (channelopathies) can have debilitating manifestations (Kullmann, 2010; Ryan and Ptacek, 2010). The fact that stable synapse function depends on homeostatic signaling leads to a logical question: do some channelopathies result from impaired homeostatic systems? The answer is not 100% clear for any disorder. However, several channelopathies—such as forms of epilepsy and migraine—display a possible hallmark of impaired HSP: long periods of neuronal stability followed by sudden, episodic attacks. A series of reviews recently surveyed many calcium channelopathies, including those caused by mutations in subunit genes for CaV1-, CaV2-, and CaV3-type channels (Liao and Soong, 2010; Pietrobon, 2010a; Striessnig et al., 2010; Zamponi et al., 2010; Cain and Snutch, 2011). Another recent review examined compelling connections between HSP and neurological disorders (Wondolowski and Dickman, 2013). Knowing what we know about VGCCs and HSP, we may draw speculative links between some VGCC channelopathies and homeostatic plasticity. Here we consider a subset of them, including episodic forms of migraine, ataxia, myasthenia, epilepsy, and paralysis.
CaV2.1 channelopathies: migraine, ataxia, and myasthenia
Two CaV2.1 channelopathies—familial hemiplegic migraine type 1 (FHM1) and episodic ataxia type 2 (EA2)—result from mutations (gain- and loss-of-function, respectively) in human CACNA1A, which encodes the α1 subunit of presynaptic CaV2.1-type calcium channels (Ophoff et al., 1996; Pietrobon, 2010a). A third disorder—spinocerebellar ataxia type 6 (SCA6)—results from poly-glutamine (polyQ) expansion in the CACNA1A gene product (Zhuchenko et al., 1997). There is some question about whether SCA6 is more properly classified as polyQ expansion disorder rather than a channelopathy. Here we focus on FHM1 and EA2.
Migraine is the most common neurological disorder—about 12% of the population suffers from it, with a high lifetime incidence rate for women (Barrett et al., 2008; Stewart et al., 2008; NINDS/NIH, 2009). FHM1 is a rare subtype; it is an inherited CaV2.1 channelopathy that causes migraine with an accompanying aura (Ophoff et al., 1996; Pietrobon, 2010a,b). While some gain-of-function CACNA1A mutations cause only FHM1, others can lead to additional maladies, such as epileptic attacks (Kors et al., 2001; Pietrobon, 2010a,b; van den Maagdenberg et al., 2010). Cortical Spreading Depression (CSD) is associated with aura and FHM1 migraines. CSD is defined as a wave of depolarization of neural cells along the cerebral cortex, followed a prolonged period of inactivity (Charles and Baca, 2013). CSD can be monitored in real time with Blood-Oxygen-Level Dependent functional Magnetic Resonance Imaging (BOLD-fMRI). It is unclear whether CSD is coincident with types of migraine other than FHM. However, the fact that a sudden wave of cortical depolarization is tightly coupled to a form of headache is consistent with the idea that homeostatic mechanisms keep neuronal functions within normal physiological ranges—and more importantly, that these mechanisms could be impaired in FHM1 migraine sufferers. There exist knock-in mouse models of FHM1 (van den Maagdenberg et al., 2004, 2010). These knock-in models have been a wonderful resource to deduce P/Q-channel properties of FHM1-inducing amino acid substitutions and to establish that the substitutions do represent gains of channel function. Animal models may also prove to be valuable in examining whether forms of migraine correlate with an underlying disruption in HSP.
In contrast to FHM1, EA2 is caused by loss-of-function mutations in CACNA1A (Ophoff et al., 1996; Pietrobon, 2010a). Most of the known EA2 mutations are dominant missense mutations that affect the trafficking of channel subunits, but some cause CACNA1A splicing defects (Pietrobon, 2010a). Like most forms of ataxia, EA2 is marked by sudden attacks of uncoordinated movement. The episodic nature of these attacks again suggests a possible impairment in homeostatic plasticity—speculation made all the more intriguing given that Drosophila cac loss-of-function mutations impair homeostatic plasticity at the NMJ (Frank et al., 2006, 2009; Müller and Davis, 2012). Again, several rodent EA2 models exist, including spontaneous loss-of-function CACNA1A mutations in tottering, leaner, rolling Nagoya, and rocker mice—as well as mice lacking functional P/Q channels altogether (Pietrobon, 2005, 2010a; Miki et al., 2008; Plomp et al., 2009). Homozygous CACNA1A –/– loss-of-function mutant mice display severe forms of ataxia. It may be fruitful to probe these mice (or neuronal cultures derived from them) to check if homeostatic plasticity is disrupted.
Forms of myasthenia cause muscle weakness. For example, myasthenia gravis is an autoimmune disorder that directly affects the NMJ. Antibodies formed against acetylcholine receptors impair muscle function. This occurs despite the NMJ’s apparent attempts to correct the problem via homeostatic increases in presynaptic quantal content—an observation from both human myasthenic muscle and rodent models of myasthenia gravis (Cull-Candy et al., 1980; Plomp et al., 1992, 1994, 1995). A separate, rare form of myasthenia is Lambert-Eaton myasthenic syndrome (LEMS; Marion et al., 1984; Pascuzzi, 2002; Mareska and Gutmann, 2004). Like myasthenia gravis, LEMS is an autoimmune disorder, but it is also a channelopathy because autoantibodies are formed against CaV2.1-type calcium channels (Motomura et al., 2000). Mammalian NMJs appear to be endowed with homeostatic coping mechanisms, much like insect NMJs. However, in the case of LEMS, interfering with the NMJ’s ability to generate sufficient presynaptic calcium influx may cause sufficient stress to cause myasthenic symptoms.
CaV mutations and epilepsy
The term epilepsy encompasses a range of seizure-associated maladies. Epilepsy is broadly defined by multiple seizure events in a single individual (NINDS/NIH, 2004). It is likely the most frequently invoked neurological disorder that is hypothesized to be triggered or facilitated by defects in neuronal homeostatic signaling. This is logical because epilepsies are marked by an underlying instability of neuronal function.
In addition to ataxia, CaV2.1-deficient “EA2” mice experience absence seizures—i.e., brief events that are marked by an abrupt arrest in activity, followed by a return to normal activity (Noebels and Sidman, 1979; Jun et al., 1999). Mice that have loss-of-function mutations in genes encoding α2δ and γ2 calcium channel subunits and a reduction in CaV2.1 channel activity also display absence seizures (Letts et al., 1998; Barclay et al., 2001; Letts et al., 2003). In humans with heterozygous EA2-causing mutations, absence seizures are not common, but there are instances in which they are present (Zamponi et al., 2010).
CaV3.1 and CaV3.2 T-type calcium channels contribute to a spectrum of polygenic epilepsies. Mutations in the CaV3.1 α1 subunit gene CACNA1G have been implicated in idiopathic generalized epilepsy (IGE) susceptibility (Singh et al., 2007). Likewise, mutations in the CaV3.2 α1 subunit gene CACNA1H have been implicated in childhood absence epilepsy (CAE) susceptibility (Chen et al., 2003; Vitko et al., 2005). Canonically, T-type calcium channels are responsible for low-threshold spiking activity in thalamic neurons (Llinas, 1988). As T-type calcium channels open, there is a significant increase in local intracellular calcium levels (Errington et al., 2010); thus, one can easily hypothesize that proper regulation of T-type currents is critical to neuronal stability. In several rodent models of absence epilepsy, there is a robust increase in T-type calcium currents (Tsakiridou et al., 1995; Talley et al., 2000; Broicher et al., 2008). This is true not only for rodent models in which CaV3.1 currents themselves are high (Ernst et al., 2009), but also for cases in which a loss of P/Q-channel activity results in an increase in T-channel activity (Zhang et al., 2002). The latter example may illustrate a hierarchy of homeostatic signals, in which overall calcium currents are maintained at the expense of neuronal stability. A similar type of model has been proposed for potassium currents and homeostasis at the Drosophila NMJ (Bergquist et al., 2010).
CaV1.1 and hypokalemic periodic paralysis
Starting around adolescence, individuals who suffer from hypokalemic periodic paralysis (HPP) experience intermittent episodes of muscle weakness and paralysis that last on the order of 3–24 h in length. The attacks often occur upon waking or after high levels of carbohydrate consumption (USNLM/NIH, 2011). The episodic nature of these attacks and the mild challenges that trigger them could suggest defective homeostasis in muscle cells. Familial HPP type 1 is caused by missense mutations in CACNA1S, which encodes the α1 subunit of CaV1.1, and most of the mutations alter arginine residues in the S4 voltage sensor (Sipos et al., 1995; Morrill and Cannon, 1999). Further experiments exploiting muscle fibers from HPP1 patients have suggested that these arginine amino acid substitutions induce cation gating pore currents and that HPP1 muscle has a higher concentration of sodium (Jurkat-Rott et al., 2009).
Concluding comments
Understanding how VGCCs help to execute homeostatic forms of neuroplasticity represents a new area of research. It offers a fresh way to consider why VGCCs are regulated in the ways that they are. In healthy neurons, it is logical that calcium channels are co-opted to gate homeostatic signaling processes. Presynaptically, neurotransmitter release is dependent upon calcium influx or local domains of high calcium concentration. Postsynaptically, many signaling processes are dependent upon calcium that enters the cell through receptor complexes and L-type calcium channels. Neurons, synapses, and circuits across metazoan nervous systems are endowed with powerful homeostatic set points of activity, and small adjustments to calcium channel activity parameters could help fine tune synaptic outputs in a variety of ways.
An important frontier for research will be to understand how CaV channels interact with intracellular signaling mechanisms and with the activities of other ion channels to control homeostatic set points of excitable cells. What signaling processes directly modulate CaV2 during homeostatic modulations of neurotransmitter release? What postsynaptic cascades are activated or suppressed downstream of calcium influx through CaV1? We have excellent clues at this date from diverse experimental systems reviewed here. Concerning HSP, it will be interesting to see which modes of regulation prove to be universal and which ones prove to be synapse- or organism-specific. With that knowledge in hand, it may be possible to better understand and address numerous phenomena, including CaV channelopathies.
Conflict of interest statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Many thanks to members of the Frank laboratory for helpful discussions. This work was supported in part by NIH grant NS062738, as well as funds from the Department of Anatomy and Cell Biology at the University of Iowa Carver College of Medicine, and a Carver Trust Medical Research Initiative Grant.
References
- Barclay J., Balaguero N., Mione M., Ackerman S. L., Letts V. A., Brodbeck J., et al. (2001). Ducky mouse phenotype of epilepsy and ataxia is associated with mutations in the Cacna2d2 gene and decreased calcium channel current in cerebellar Purkinje cells. J. Neurosci. 21, 6095–6104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergquist S., Dickman D. K., Davis G. W. (2010). A hierarchy of cell intrinsic and target-derived homeostatic signaling. Neuron 66, 220–234 10.1016/j.neuron.2010.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett C. F., van den Maagdenberg A. M., Frants R. R., Ferrari M. D. (2008). Familial hemiplegic migraine. Adv. genet. 63, 57–83 10.1016/S0065-2660(08)01003-1 [DOI] [PubMed] [Google Scholar]
- Brenner S. (1974). The genetics of Caenorhabditis elegans. Genetics 77, 71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broicher T., Kanyshkova T., Meuth P., Pape H. C., Budde T. (2008). Correlation of T-channel coding gene expression, IT and the low threshold Ca2+ spike in the thalamus of a rat model of absence epilepsy. Mol. Cell. Neurosci. 39, 384–399 10.1016/j.mcn.2008.07.012 [DOI] [PubMed] [Google Scholar]
- Brooks I. M., Felling R., Kawasaki F., Ordway R. W. (2003). Genetic analysis of a synaptic calcium channel in Drosophila: intragenic modifiers of a temperature-sensitive paralytic mutant of cacophony. Genetics 164, 163–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosenitsch T. A., Katz D. M. (2001). Physiological patterns of electrical stimulation can induce neuronal gene expression by activating N-type calcium channels. J. Neurosci. 21, 2571–2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrone J., O’Byrne M., Murthy V. N. (2002). Multiple forms of synaptic plasticity triggered by selective suppression of activity in individual neurons. Nature 420, 414–418 10.1038/nature01242 [DOI] [PubMed] [Google Scholar]
- Cain S. M., Snutch T. P. (2011). Voltage-gated calcium channels and disease. Biofactors 37, 197–205 10.1002/biof.158 [DOI] [PubMed] [Google Scholar]
- Campbell M., Ganetzky B. (2012). Extensive morphological divergence and rapid evolution of the larval neuromuscular junction in Drosophila. Proc. Natl. Acad. Sci. U S A 109, E648–E655 10.1073/pnas.1201176109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell M., Ganetzky B. (2013). Identification of Mob2, a novel regulator of larval neuromuscular junction morphology, in natural populations of drosophila melanogaster. Genetics 195, 915–926 10.1534/genetics.113.156562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao G., Platisa J., Pieribone V. A., Raccuglia D., Kunst M., Nitabach M. N. (2013). Genetically targeted optical electrophysiology in intact neural circuits. Cell 154, 904–913 10.1016/j.cell.2013.07.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall W. A. (2000). Structure and regulation of voltage-gated Ca2+ channels. Annu. Rev. Cell Dev. Biol. 16, 521–555 10.1146/annurev.cellbio.16.1.521 [DOI] [PubMed] [Google Scholar]
- Catterall W. A., Few A. P. (2008). Calcium channel regulation and presynaptic plasticity. Neuron 59, 882–901 10.1016/j.neuron.2008.09.005 [DOI] [PubMed] [Google Scholar]
- Catterall W. A., Perez-Reyes E., Snutch T. P., Striessnig J. (2005). International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol. Rev. 57, 411–425 10.1124/pr.57.4.5 [DOI] [PubMed] [Google Scholar]
- Caylor R. C., Jin Y., Ackley B. D. (2013). The Caenorhabditis elegans voltage-gated calcium channel subunits UNC-2 and UNC-36 and the calcium-dependent kinase UNC-43/CaMKII regulate neuromuscular junction morphology. Neural Dev. 8:10 10.1186/1749-8104-8-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles A. C., Baca S. M. (2013). Cortical spreading depression and migraine. Nat. Rev. Neurol. 9, 637–644 10.1038/nrneurol.2013.192 [DOI] [PubMed] [Google Scholar]
- Chen L., Lau A. G., Sarti F. (2014). Synaptic retinoic acid signaling and homeostatic synaptic plasticity. Neuropharmacology 78, 3–12 10.1016/j.neuropharm.2012.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Lu J., Pan H., Zhang Y., Wu H., Xu K., et al. (2003). Association between genetic variation of CACNA1H and childhood absence epilepsy. Ann. Neurol. 54, 239–243 10.1002/ana.10607 [DOI] [PubMed] [Google Scholar]
- Christel C., Lee A. (2012). Ca2+-dependent modulation of voltage-gated Ca2+ channels. Biochim. Biophys. Acta 1820, 1243–1252 10.1016/j.bbagen.2011.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin T., Marty A., Llano I. (2005). Presynaptic calcium stores and synaptic transmission. Curr. Opin. Neurobiol. 15, 275–281 10.1016/j.conb.2005.05.003 [DOI] [PubMed] [Google Scholar]
- Cull-Candy S. G., Miledi R., Uchitel O. D. (1980). Diffusion of acetylcholine in the synaptic cleft of normal and myasthenia gravis human endplates. Nature 286, 500–502 10.1038/286500a0 [DOI] [PubMed] [Google Scholar]
- Currie K. P. (2010). G protein modulation of CaV2 voltage-gated calcium channels. Channels (Austin) 4, 497–509 10.4161/chan.4.6.12871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis D. R., Duggan A. W., Felix D., Johnston G. A. (1970). GABA, bicuculline and central inhibition. Nature 226, 1222–1224 10.1038/2261222a0 [DOI] [PubMed] [Google Scholar]
- Davis G. W. (2006). Homeostatic control of neural activity: from phenomenology to molecular design. Annu. Rev. Neurosci. 29, 307–323 10.1146/annurev.neuro.28.061604.135751 [DOI] [PubMed] [Google Scholar]
- Davis G. W. (2013). Homeostatic signaling and the stabilization of neural function. Neuron 80, 718–728 10.1016/j.neuron.2013.09.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis G. W., DiAntonio A., Petersen S. A., Goodman C. S. (1998). Postsynaptic PKA controls quantal size and reveals a retrograde signal that regulates presynaptic transmitter release in Drosophila. Neuron 20, 305–315 10.1016/s0896-6273(00)80458-4 [DOI] [PubMed] [Google Scholar]
- Day M., Wang Z., Ding J., An X., Ingham C. A., Shering A. F., et al. (2006). Selective elimination of glutamatergic synapses on striatopallidal neurons in Parkinson disease models. Nat. Neurosci. 9, 251–259 10.1038/nn1632 [DOI] [PubMed] [Google Scholar]
- Deutch A. Y., Colbran R. J., Winder D. J. (2007). Striatal plasticity and medium spiny neuron dendritic remodeling in parkinsonism. Parkinsonism Relat. Disord. 13(Suppl. 3), S251–S258 10.1016/s1353-8020(08)70012-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiAntonio A., Petersen S. A., Heckmann M., Goodman C. S. (1999). Glutamate receptor expression regulates quantal size and quantal content at the Drosophila neuromuscular junction. J. Neurosci. 19, 3023–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickman D. K., Davis G. W. (2009). The schizophrenia susceptibility gene dysbindin controls synaptic homeostasis. Science 326, 1127–1130 10.1126/science.1179685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickman D. K., Tong A., Davis G. W. (2012). Snapin is critical for presynaptic homeostatic plasticity. J. Neurosci. 32, 8716–8724 10.1523/jneurosci.5465-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch R. E., Pajvani U., Fife K., Spotts J. M., Greenberg M. E. (2001). Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science 294, 333–339 10.1126/science.1063395 [DOI] [PubMed] [Google Scholar]
- Dolphin A. C. (2009). Calcium channel diversity: multiple roles of calcium channel subunits. Curr. Opin. Neurobiol. 19, 237–244 10.1016/j.conb.2009.06.006 [DOI] [PubMed] [Google Scholar]
- Doyle S., Pyndiah S., De Gois S., Erickson J. D. (2010). Excitation-transcription coupling via calcium/calmodulin-dependent protein kinase/ERK1/2 signaling mediates the coordinate induction of VGLUT2 and Narp triggered by a prolonged increase in glutamatergic synaptic activity. J. Biol. Chem. 285, 14366–14376 10.1074/jbc.m109.080069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlap K. (2007). Calcium channels are models of self-control. J. Gen. Physiol. 129, 379–383 10.1085/jgp.200709786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst W. L., Zhang Y., Yoo J. W., Ernst S. J., Noebels J. L. (2009). Genetic enhancement of thalamocortical network activity by elevating alpha 1g-mediated low-voltage-activated calcium current induces pure absence epilepsy. J. Neurosci. 29, 1615–1625 10.1523/jneurosci.2081-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errington A. C., Renger J. J., Uebele V. N., Crunelli V. (2010). State-dependent firing determines intrinsic dendritic Ca2+ signaling in thalamocortical neurons. J. Neurosci. 30, 14843–14853 10.1523/jneurosci.2968-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiore R., Khudayberdiev S., Christensen M., Siegel G., Flavell S. W., Kim T. K., et al. (2009). Mef2-mediated transcription of the mi R379-410 cluster regulates activity-dependent dendritogenesis by fine-tuning Pumilio2 protein levels. EMBO J. 28, 697–710 10.1038/emboj.2009.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavell S. W., Cowan C. W., Kim T. K., Greer P. L., Lin Y., Paradis S., et al. (2006). Activity-dependent regulation of MEF2 transcription factors suppresses excitatory synapse number. Science 311, 1008–1012 10.1126/science.1122511 [DOI] [PubMed] [Google Scholar]
- Flavell S. W., Kim T. K., Gray J. M., Harmin D. A., Hemberg M., Hong E. J., et al. (2008). Genome-wide analysis of MEF2 transcriptional program reveals synaptic target genes and neuronal activity-dependent polyadenylation site selection. Neuron 60, 1022–1038 10.1016/j.neuron.2008.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouquet W., Owald D., Wichmann C., Mertel S., Depner H., Dyba M., et al. (2009). Maturation of active zone assembly by drosophila bruchpilot. J. Cell. Biol. 186, 129–145 10.1083/jcb.200812150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank C. A. (2014). Homeostatic plasticity at the Drosophila neuromuscular junction. Neuropharmacology 78, 63–74 10.1016/j.neuropharm.2013.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank C. A., Kennedy M. J., Goold C. P., Marek K. W., Davis G. W. (2006). Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron 52, 663–677 10.1016/j.neuron.2006.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank C. A., Pielage J., Davis G. W. (2009). A presynaptic homeostatic signaling system composed of the Eph receptor, ephexin, Cdc42 and CaV2.1 calcium channels. Neuron 61, 556–569 10.1016/j.neuron.2008.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frøkjaer-Jensen C., Kindt K. S., Kerr R. A., Suzuki H., Melnik-Martinez K., Gerstbreih B., et al. (2006). Effects of voltage-gated calcium channel subunit genes on calcium influx in cultured C. elegans mechanosensory neurons. J. Neurobiol. 66, 1125–1139 10.1002/neu.20261 [DOI] [PubMed] [Google Scholar]
- Giagtzoglou N., Lin Y. Q., Haueter C., Bellen H. J. (2009). Importin 13 regulates neurotransmitter release at the Drosophila neuromuscular junction. J. Neurosci. 29, 5628–5639 10.1523/jneurosci.0794-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraudo C. G., Eng W. S., Melia T. J., Rothman J. E. (2006). A clamping mechanism involved in SNARE-dependent exocytosis. Science 313, 676–680 10.1126/science.1129450 [DOI] [PubMed] [Google Scholar]
- Gong B., Wang H., Gu S., Heximer S. P., Zhuo M. (2007). Genetic evidence for the requirement of adenylyl cyclase 1 in synaptic scaling of forebrain cortical neurons. Eur. J. Neurosci. 26, 275–288 10.1111/j.1460-9568.2007.05669.x [DOI] [PubMed] [Google Scholar]
- Goold C. P., Davis G. W. (2007). The BMP ligand Gbb gates the expression of synaptic homeostasis independent of synaptic growth control. Neuron 56, 109–123 10.1016/j.neuron.2007.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goold C. P., Nicoll R. A. (2010). Single-cell optogenetic excitation drives homeostatic synaptic depression. Neuron 68, 512–528 10.1016/j.neuron.2010.09.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf E. R., Valakh V., Wright C. M., Wu C., Liu Z., Zhang Y. Q., et al. (2012). RIM promotes calcium channel accumulation at active zones of the drosophila neuromuscular junction. J. Neurosci. 32, 16586–16596 10.1523/jneurosci.0965-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunwald M. E., Mellem J. E., Strutz N., Maricq A. V., Kaplan J. M. (2004). Clathrin-mediated endocytosis is required for compensatory regulation of GLR-1 glutamate receptors after activity blockade. Proc. Natl. Acad. Sci. U S A 101, 3190–3195 10.1073/pnas.0306156101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halling D. B., Aracena-Parks P., Hamilton S. L. (2006). Regulation of voltage-gated Ca2+ channels by calmodulin. Sci. STKE 2006:er1 10.1126/stke.3152005re15 [DOI] [PubMed] [Google Scholar]
- Hardingham G. E., Cruzalegui F. H., Chawla S., Bading H. (1998). Mechanisms controlling gene expression by nuclear calcium signals. Cell Calcium 23, 131–134 10.1016/s0143-4160(98)90111-7 [DOI] [PubMed] [Google Scholar]
- Hell J. W., Westenbroek R. E., Warner C., Ahlijanian M. K., Prystay W., Gilbert M. M., et al. (1993). Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J. Cell Biol. 123, 949–962 10.1083/jcb.123.4.949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helton T. D., Xu W., Lipscombe D. (2005). Neuronal L-type calcium channels open quickly and are inhibited slowly. J. Neurosci. 25, 10247–10251 10.1523/jneurosci.1089-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry F. E., McCartney A. J., Neely R., Perez A. S., Carruthers C. J., Stuenkel E. L., et al. (2012). Retrograde changes in presynaptic function driven by dendritic mTORC1. J. Neurosci. 32, 17128–17142 10.1523/jneurosci.2149-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlitze S., Xie M., Han J., Hummer A., Melnik-Martinez K. V., Moreno R. L., et al. (2003). Targeting mechanisms of high voltage-activated Ca2+ channels. J. Bioenerg. Biomembr. 35, 621–637 10.1023/b:jobb.0000008027.19384.c0 [DOI] [PubMed] [Google Scholar]
- Honore T., Davies S. N., Drejer J., Fletcher E. J., Jacobsen P., Lodge D., et al. (1988). Quinoxalinediones: potent competitive non-NMDA glutamate receptor antagonists. Science 241, 701–703 10.1126/science.2899909 [DOI] [PubMed] [Google Scholar]
- Jakawich S. K., Nasser H. B., Strong M. J., McCartney A. J., Perez A. S., Rakesh N., et al. (2010). Local presynaptic activity gates homeostatic changes in presynaptic function driven by dendritic BDNF synthesis. Neuron 68, 1143–1158 10.1016/j.neuron.2010.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun K., Piedras-Renteria E. S., Smith S. M., Wheeler D. B., Lee S. B., Lee T. G., et al. (1999). Ablation of P/Q-type Ca(2+) channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the alpha(1A)-subunit. Proc. Natl. Acad. Sci. U S A 96, 15245–15250 10.1073/pnas.96.26.15245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkat-Rott K., Weber M. A., Fauler M., Guo X. H., Holzherr B. D., Paczulla A., et al. (2009). K+-dependent paradoxical membrane depolarization and Na+ overload, major and reversible contributors to weakness by ion channel leaks. Proc. Natl. Acad. Sci. U S A 106, 4036–4041 10.1073/pnas.0811277106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B., Miledi R. (1970). Further study of the role of calcium in synaptic transmission. J. Physiol. 207, 789–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki F., Felling R., Ordway R. W. (2000). A temperature-sensitive paralytic mutant defines a primary synaptic calcium channel in Drosophila. J. Neurosci. 20, 4885–4889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki F., Zou B., Xu X., Ordway R. W. (2004). Active zone localization of presynaptic calcium channels encoded by the cacophony locus of Drosophila. J. Neurosci. 24, 282–285 10.1523/jneurosci.3553-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilman V., van Rossum M. C., Turrigiano G. G. (2002). Activity deprivation reduces miniature IPSC amplitude by decreasing the number of postsynaptic GABA(A) receptors clustered at neocortical synapses. J. Neurosci. 22, 1328–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. H., Ryan T. A. (2010). CDK5 serves as a major control point in neurotransmitter release. Neuron 67, 797–809 10.1016/j.neuron.2010.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. H., Ryan T. A. (2013). Balance of calcineurin Aalpha and CDK5 activities sets release probability at nerve terminals. J. Neurosci. 33, 8937–8950 10.1523/jneurosci.4288-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y., Teylan M. A., Baron M., Sands A., Nairn A. C., Greengard P. (2009). Methylphenidate-induced dendritic spine formation and DeltaFosB expression in nucleus accumbens. Proc. Natl. Acad. Sci. U S A 106, 2915–2920 10.1073/pnas.0813179106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kors E. E., Terwindt G. M., Vermeulen F. L., Fitzsimons R. B., Jardine P. E., Heywood P., et al. (2001). Delayed cerebral edema and fatal coma after minor head trauma: role of the CACNA1A calcium channel subunit gene and relationship with familial hemiplegic migraine. Ann. Neurol. 49, 753–760 10.1002/ana.1031 [DOI] [PubMed] [Google Scholar]
- Kulkarni S. J., Hall J. C. (1987). Behavioral and cytogenetic analysis of the cacophony courtship song mutant and interacting genetic variants in Drosophila melanogaster. Genetics 115, 461–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann D. M. (2010). Neurological channelopathies. Annu. Rev. Neurosci. 33, 151–172 10.1146/annurev-neuro-060909-153122 [DOI] [PubMed] [Google Scholar]
- Kurshan P. T., Oztan A., Schwarz T. L. (2009). Presynaptic alpha2delta-3 is required for synaptic morphogenesis independent of its Ca2+-channel functions. Nat. Neurosci. 12, 1415–1423 10.1038/nn.2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lainé V., Frøkjaer-Jensen C., Couchoux H., Jospin M. (2011). The alpha1 subunit EGL-19, the alpha2/delta subunit UNC-36 and the beta subunit CCB-1 underlie voltage-dependent calcium currents in Caenorhabditis elegans striated muscle. J. Biol. Chem. 286, 36180–36187 10.1074/jbc.m111.256149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarevic V., Pothula S., Andres-Alonso M., Fejtova A. (2013). Molecular mechanisms driving homeostatic plasticity of neurotransmitter release. Front. Cell. Neurosci. 7:244 10.3389/fncel.2013.00244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarevic V., Schone C., Heine M., Gundelfinger E. D., Fejtova A. (2011). Extensive remodeling of the presynaptic cytomatrix upon homeostatic adaptation to network activity silencing. J. Neurosci. 31, 10189–10200 10.1523/jneurosci.2088-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Wu C. F. (2010). Orchestration of stepwise synaptic growth by K+ and Ca2+ channels in Drosophila. J. Neurosci. 30, 15821–15833 10.1523/jneurosci.3448-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. F., Soares C., Béique J. C. (2014). Tuning into diversity of homeostatic synaptic plasticity. Neuropharmacology 78, 31–37 10.1016/j.neuropharm.2013.03.016 [DOI] [PubMed] [Google Scholar]
- Lee R. Y., Lobel L., Hengartner M., Horvitz H. R., Avery L. (1997). Mutations in the alpha1 subunit of an L-type voltage-activated Ca2+ channel cause myotonia in Caenorhabditis elegans. EMBO J. 16, 6066–6076 10.1093/emboj/16.20.6066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei S., Pelkey K. A., Topolnik L., Congar P., Lacaille J. C., McBain C. J. (2003). Depolarization-induced long-term depression at hippocampal mossy fiber-CA3 pyramidal neuron synapses. J. Neurosci. 23, 9786–9795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letts V. A., Felix R., Biddlecome G. H., Arikkath J., Mahaffey C. L., Valenzuela A., et al. (1998). The mouse stargazer gene encodes a neuronal Ca2+-channel gamma subunit. Nat. Genet. 19, 340–347 10.1038/1228 [DOI] [PubMed] [Google Scholar]
- Letts V. A., Kang M. G., Mahaffey C. L., Beyer B., Tenbrink H., Campbell K. P., et al. (2003). Phenotypic heterogeneity in the stargazin allelic series. Mamm. Genome 14, 506–513 10.1007/s00335-003-2268-x [DOI] [PubMed] [Google Scholar]
- Liao P., Soong T. W. (2010). CaV1.2 channelopathies: from arrhythmias to autism, bipolar disorder and immunodeficiency. Pflugers Arch. 460, 353–359 10.1007/s00424-009-0753-0 [DOI] [PubMed] [Google Scholar]
- Lipscombe D., Allen S. E., Toro C. P. (2013). Control of neuronal voltage-gated calcium ion channels from RNA to protein. Trends Neurosci. 36, 598–609 10.1016/j.tins.2013.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscombe D., Helton T. D., Xu W. (2004). L-type calcium channels: the low down. J. Neurophysiol. 92, 2633–2641 10.1152/jn.00486.2004 [DOI] [PubMed] [Google Scholar]
- Lisman J., Yasuda R., Raghavachari S. (2012). Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 13, 169–182 10.1038/nrn3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Chen B., Ge Q., Wang Z. W. (2007). Presynaptic Ca2+/calmodulin-dependent protein kinase II modulates neurotransmitter release by activating BK channels at Caenorhabditis elegans neuromuscular junction. J. Neurosci. 27, 10404–10413 10.1523/jneurosci.5634-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q., Chen B., Yankova M., Morest D. K., Maryon E., Hand A. R., et al. (2005). Presynaptic ryanodine receptors are required for normal quantal size at the Caenorhabditis elegans neuromuscular junction. J. Neurosci. 25, 6745–6754 10.1523/jneurosci.1730-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R. R. (1988). The intrinsic electrophysiological properties of mammalian neurons: insights into central nervous system function. Science 242, 1654–1664 10.1126/science.3059497 [DOI] [PubMed] [Google Scholar]
- Maffei A., Fontanini A. (2009). Network homeostasis: a matter of coordination. Curr. Opin. Neurobiol. 19, 168–173 10.1016/j.conb.2009.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee J. C., Johnston D. (1997). A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science 275, 209–213 10.1126/science.275.5297.209 [DOI] [PubMed] [Google Scholar]
- Marder E. (2012). Neuromodulation of neuronal circuits: back to the future. Neuron 76, 1–11 10.1016/j.neuron.2012.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E., Bucher D. (2007). Understanding circuit dynamics using the stomatogastric nervous system of lobsters and crabs. Annu. Rev. Physiol. 69, 291–316 10.1146/annurev.physiol.69.031905.161516 [DOI] [PubMed] [Google Scholar]
- Marder E., Goaillard J. M. (2006). Variability, compensation and homeostasis in neuron and network function. Nat. Rev. Neurosci. 7, 563–574 10.1038/nrn1949 [DOI] [PubMed] [Google Scholar]
- Mareska M., Gutmann L. (2004). Lambert-Eaton myasthenic syndrome. Semin. Neurol. 24, 149–153 10.1055/s-2004-830900 [DOI] [PubMed] [Google Scholar]
- Marie B., Pym E., Bergquist S., Davis G. W. (2010). Synaptic homeostasis is consolidated by the cell fate gene gooseberry, a Drosophila pax3/7 homolog. J. Neurosci. 30, 8071–8082 10.1523/jneurosci.5467-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion J. L., Pennec Y., Mottier D., Le Menn G. (1984). Lambert-Eaton syndrome and myasthenia. Nosologic discussion. Sem. Hop. 60, 43–45 [PubMed] [Google Scholar]
- Matsuzaki M., Honkura N., Ellis-Davies G. C., Kasai H. (2004). Structural basis of long-term potentiation in single dendritic spines. Nature 429, 761–766 10.1038/nature02617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCobb D. P., Beam K. G. (1991). Action potential waveform voltage-clamp commands reveal striking differences in calcium entry via low and high voltage-activated calcium channels. Neuron 7, 119–127 10.1016/0896-6273(91)90080-j [DOI] [PubMed] [Google Scholar]
- Mermelstein P. G., Bito H., Deisseroth K., Tsien R. W. (2000). Critical dependence of cAMP response element-binding protein phosphorylation on L-type calcium channels supports a selective response to EPSPs in preference to action potentials. J. Neurosci. 20, 266–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T., Zwingman T. A., Wakamori M., Lutz C. M., Cook S. A., Hosford D. A., et al. (2008). Two novel alleles of tottering with distinct Ca(v)2.1 calcium channel neuropathologies. Neuroscience 155, 31–44 10.1016/j.neuroscience.2008.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor D. L., Jr., Findeisen F. (2010). Progress in the structural understanding of voltage-gated calcium channel (CaV) function and modulation. Channels (Austin) 4, 459–474 10.4161/chan.4.6.12867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrill J. A., Cannon S. C. (1999). Effects of mutations causing hypokalaemic periodic paralysis on the skeletal muscle L-type Ca2+ channel expressed in Xenopus laevis oocytes. J. Physiol. 520, 321–336 10.1111/j.1469-7793.1999.00321.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motomura M., Hamasaki S., Nakane S., Fukuda T., Nakao Y. K. (2000). Apheresis treatment in Lambert-Eaton myasthenic syndrome. Ther. Apher. 4, 287–290 10.1046/j.1526-0968.2000.004004287.x [DOI] [PubMed] [Google Scholar]
- Mrsic-Flogel T. D., Hofer S. B., Ohki K., Reid R. C., Bonhoeffer T., Hubener M. (2007). Homeostatic regulation of eye-specific responses in visual cortex during ocular dominance plasticity. Neuron 54, 961–972 10.1016/j.neuron.2007.05.028 [DOI] [PubMed] [Google Scholar]
- Müller M., Davis G. W. (2012). Transsynaptic control of presynaptic Ca(2)(+) influx achieves homeostatic potentiation of neurotransmitter release. Curr. Biol. 22, 1102–1108 10.1016/j.cub.2012.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M., Liu K. S., Sigrist S. J., Davis G. W. (2012). RIM controls homeostatic plasticity through modulation of the readily-releasable vesicle pool. J. Neurosci. 32, 16574–16585 10.1523/jneurosci.0981-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M., Pym E. C., Tong A., Davis G. W. (2011). Rab3-GAP controls the progression of synaptic homeostasis at a late stage of vesicle release. Neuron 69, 749–762 10.1016/j.neuron.2011.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy T. H., Worley P. F., Baraban J. M. (1991). L-type voltage-sensitive calcium channels mediate synaptic activation of immediate early genes. Neuron 7, 625–635 10.1016/0896-6273(91)90375-a [DOI] [PubMed] [Google Scholar]
- Murthy V. N., Schikorski T., Stevens C. F., Zhu Y. (2001). Inactivity produces increases in neurotransmitter release and synapse size. Neuron 32, 673–682 10.1016/s0896-6273(01)00500-1 [DOI] [PubMed] [Google Scholar]
- Narahashi T. (2008). Tetrodotoxin: a brief history. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 84, 147–154 10.2183/pjab.84.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- NINDS/NIH (2004). Seizures and Epilepsy: Hope Through Research, ed NINDS/NIH (Department of Health and Human Services, Bethesda, MD: Office of Communications and Public Liason, NINDS/NIH, Publication; ), 04–156 [Google Scholar]
- NINDS/NIH (2009). Headache: Hope Through Research, ed NINDS/NIH (Department of Health and Human Services, Bethesda, MD: Office of Communications and Public Liason, NINDS/NIH, Publication; ), 09–158 [Google Scholar]
- Noebels J. L., Sidman R. L. (1979). Inherited epilepsy: spike-wave and focal motor seizures in the mutant mouse tottering. Science 204, 1334–1336 10.1126/science.572084 [DOI] [PubMed] [Google Scholar]
- Norton R. S., McDonough S. I. (2008). Peptides targeting voltage-gated calcium channels. Curr. Pharm. Des. 14, 2480–2491 10.2174/138161208785777478 [DOI] [PubMed] [Google Scholar]
- Obermair G. J., Szabo Z., Bourinet E., Flucher B. E. (2004). Differential targeting of the L-type Ca2+ channel alpha 1C (CaV1.2) to synaptic and extrasynaptic compartments in hippocampal neurons. Eur. J. Neurosci. 19, 2109–2122 10.1111/j.0953-816x.2004.03272.x [DOI] [PubMed] [Google Scholar]
- O’Brien R. J., Kamboj S., Ehlers M. D., Rosen K. R., Fischbach G. D., Huganir R. L. (1998). Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron 21, 1067–1078 10.1016/s0896-6273(00)80624-8 [DOI] [PubMed] [Google Scholar]
- Ophoff R. A., Terwindt G. M., Vergouwe M. N., van Eijk R., Oefner P. J., Hoffman S. M., et al. (1996). Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell 87, 543–552 10.1016/s0092-8674(00)81373-2 [DOI] [PubMed] [Google Scholar]
- Pak D. T., Sheng M. (2003). Targeted protein degradation and synapse remodeling by an inducible protein kinase. Science 302, 1368–1373 10.1126/science.1082475 [DOI] [PubMed] [Google Scholar]
- Paradis S., Sweeney S. T., Davis G. W. (2001). Homeostatic control of presynaptic release is triggered by postsynaptic membrane depolarization. Neuron 30, 737–749 10.1016/s0896-6273(01)00326-9 [DOI] [PubMed] [Google Scholar]
- Pascuzzi R. M. (2002). Myasthenia gravis and Lambert-Eaton syndrome. Ther. Apher. 6, 57–68 10.1046/j.1526-0968.2002.00403.x [DOI] [PubMed] [Google Scholar]
- Penney J., Tsurudome K., Liao E. H., Elazzouzi F., Livingstone M., Gonzalez M., et al. (2012). TOR is required for the retrograde regulation of synaptic homeostasis at the Drosophila neuromuscular junction. Neuron 74, 166–178 10.1016/j.neuron.2012.01.030 [DOI] [PubMed] [Google Scholar]
- Pérez-Otaño I., Ehlers M. D. (2005). Homeostatic plasticity and NMDA receptor trafficking. Trends Neurosci. 28, 229–238 10.1016/j.tins.2005.03.004 [DOI] [PubMed] [Google Scholar]
- Perrier J. F., Alaburda A., Hounsgaard J. (2002). Spinal plasticity mediated by postsynaptic L-type Ca2+ channels. Brain Res. Brain Res. Rev. 40, 223–229 10.1016/s0165-0173(02)00204-7 [DOI] [PubMed] [Google Scholar]
- Petersen S. A., Fetter R. D., Noordermeer J. N., Goodman C. S., DiAntonio A. (1997). Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron 19, 1237–1248 10.1016/s0896-6273(00)80415-8 [DOI] [PubMed] [Google Scholar]
- Pietrobon D. (2005). Function and dysfunction of synaptic calcium channels: insights from mouse models. Curr. Opin. Neurobiol. 15, 257–265 10.1016/j.conb.2005.05.010 [DOI] [PubMed] [Google Scholar]
- Pietrobon D. (2010a). CaV2.1 channelopathies. Pflugers Arch. 460, 375–393 10.1007/s00424-010-0802-8 [DOI] [PubMed] [Google Scholar]
- Pietrobon D. (2010b). Insights into migraine mechanisms and CaV2.1 calcium channel function from mouse models of familial hemiplegic migraine. J. Physiol. 588, 1871–1878 10.1113/jphysiol.2010.188003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilgram G. S., Potikanond S., Baines R. A., Fradkin L. G., Noordermeer J. N. (2010). The roles of the dystrophin-associated glycoprotein complex at the synapse. Mol. Neurobiol. 41, 1–21 10.1007/s12035-009-8089-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilgram G. S., Potikanond S., van der Plas M. C., Fradkin L. G., Noordermeer J. N. (2011). The RhoGAP crossveinless-c interacts with Dystrophin and is required for synaptic homeostasis at the Drosophila neuromuscular junction. J. Neurosci. 31, 492–500 10.1523/jneurosci.4732-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomp J. J., van den Maagdenberg A. M., Kaja S. (2009). The ataxic Cacna1a-mutant mouse rolling nagoya: an overview of neuromorphological and electrophysiological findings. Cerebellum 8, 222–230 10.1007/s12311-009-0117-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomp J. J., Van Kempen G. T., De Baets M. B., Graus Y. M., Kuks J. B., Molenaar P. C. (1995). Acetylcholine release in myasthenia gravis: regulation at single end-plate level. Ann. Neurol. 37, 627–636 10.1002/ana.410370513 [DOI] [PubMed] [Google Scholar]
- Plomp J. J., van Kempen G. T., Molenaar P. C. (1992). Adaptation of quantal content to decreased postsynaptic sensitivity at single endplates in alpha-bungarotoxin-treated rats. J. Physiol. 458, 487–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plomp J. J., van Kempen G. T., Molenaar P. C. (1994). The upregulation of acetylcholine release at endplates of alpha-bungarotoxin-treated rats: its dependency on calcium. J. Physiol. 478(Pt. 1), 125–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozo K., Goda Y. (2010). Unraveling mechanisms of homeostatic synaptic plasticity. Neuron 66, 337–351 10.1016/j.neuron.2010.04.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribiag H., Stellwagen D. (2014). Neuroimmune regulation of homeostatic synaptic plasticity. Neuropharmacology 78, 13–22 10.1016/j.neuropharm.2013.06.008 [DOI] [PubMed] [Google Scholar]
- Rieckhof G. E., Yoshihara M., Guan Z., Littleton J. T. (2003). Presynaptic N-type calcium channels regulate synaptic growth. J. Biol. Chem. 278, 41099–41108 10.1074/jbc.m306417200 [DOI] [PubMed] [Google Scholar]
- Rongo C., Kaplan J. M. (1999). CaMKII regulates the density of central glutamatergic synapses in vivo. Nature 402, 195–199 [DOI] [PubMed] [Google Scholar]
- Rutherford L. C., DeWan A., Lauer H. M., Turrigiano G. G. (1997). Brain-derived neurotrophic factor mediates the activity-dependent regulation of inhibition in neocortical cultures. J. Neurosci. 17, 4527–4535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan D. P., Ptacek L. J. (2010). Episodic neurological channelopathies. Neuron 68, 282–292 10.1016/j.neuron.2010.10.008 [DOI] [PubMed] [Google Scholar]
- Saheki Y., Bargmann C. I. (2009). Presynaptic CaV2 calcium channel traffic requires CALF-1 and the alpha(2)delta subunit UNC-36. Nat. Neurosci. 12, 1257–1265 10.1038/nn.2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliba R. S., Gu Z., Yan Z., Moss S. J. (2009). Blocking L-type voltage-gated Ca2+ channels with dihydropyridines reduces gamma-aminobutyric acid type A receptor expression and synaptic inhibition. J. Biol. Chem. 284, 32544–32550 10.1074/jbc.m109.040071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliba R. S., Michels G., Jacob T. C., Pangalos M. N., Moss S. J. (2007). Activity-dependent ubiquitination of GABA(A) receptors regulates their accumulation at synaptic sites. J. Neurosci. 27, 13341–13351 10.1523/jneurosci.3277-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer W. R., Kenyon C. J. (1995). A calcium-channel homologue required for adaptation to dopamine and serotonin in Caenorhabditis elegans. Nature 375, 73–78 10.1038/375073a0 [DOI] [PubMed] [Google Scholar]
- Schaub J. R., Lu X., Doneske B., Shin Y. K., McNew J. A. (2006). Hemifusion arrest by complexin is relieved by Ca2+-synaptotagmin I. Nat. Struct. Mol. Biol. 13, 748–750 10.1038/nsmb1124 [DOI] [PubMed] [Google Scholar]
- Schild L. (2010). The epithelial sodium channel and the control of sodium balance. Biochim. Biophys. Acta 1802, 1159–1165 10.1016/j.bbadis.2010.06.014 [DOI] [PubMed] [Google Scholar]
- Sheardown M. J. (1993). The pharmacology of AMPA receptors and their antagonists. Stroke 24, I146–147; discussion I153–I154 [PubMed] [Google Scholar]
- Shen W., Flajolet M., Greengard P., Surmeier D. J. (2008). Dichotomous dopaminergic control of striatal synaptic plasticity. Science 321, 848–851 10.1126/science.1160575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S. M., Zhang N., Hansen J., Gerges N. Z., Pak D. T., Sheng M., et al. (2012). GKAP orchestrates activity-dependent postsynaptic protein remodeling and homeostatic scaling. Nat. Neurosci. 15, 1655–1666 10.1038/nn.3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddoway B., Hou H., Xia H. (2014). Molecular mechanisms of homeostatic synaptic downscaling. Neuropharmacology 78, 38–44 10.1016/j.neuropharm.2013.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddoway B. A., Altimimi H. F., Hou H., Petralia R. S., Xu B., Stellwagen D., et al. (2013). An essential role for inhibitor-2 regulation of protein phosphatase-1 in synaptic scaling. J. Neurosci. 33, 11206–11211 10.1523/jneurosci.5241-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B., Monteil A., Bidaud I., Sugimoto Y., Suzuki T., Hamano S., et al. (2007). Mutational analysis of CACNA1G in idiopathic generalized epilepsy. Mutation in brief #962. Online. Hum. Mutat. 28, 524–525 10.1002/humu.9491 [DOI] [PubMed] [Google Scholar]
- Sipos I., Jurkat-Rott K., Harasztosi C., Fontaine B., Kovacs L., Melzer W., et al. (1995). Skeletal muscle DHP receptor mutations alter calcium currents in human hypokalaemic periodic paralysis myotubes. J. Physiol. 483(Pt. 2), 299–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith L. A., Peixoto A. A., Kramer E. M., Villella A., Hall J. C. (1998). Courtship and visual defects of cacophony mutants reveal functional complexity of a calcium-channel alpha1 subunit in Drosophila. Genetics 149, 1407–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith L. A., Wang X., Peixoto A. A., Neumann E. K., Hall L. M., Hall J. C. (1996). A Drosophila calcium channel alpha1 subunit gene maps to a genetic locus associated with behavioral and visual defects. J. Neurosci. 16, 7868–7879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolova I. V., Mody I. (2008). Silencing-induced metaplasticity in hippocampal cultured neurons. J. Neurophysiol. 100, 690–697 10.1152/jn.90378.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart W. F., Wood C., Reed M. L., Roy J., Lipton R. B., Group A. A. (2008). Cumulative lifetime migraine incidence in women and men. Cephalalgia 28, 1170–1178 10.1111/j.1468-2982.2008.01666.x [DOI] [PubMed] [Google Scholar]
- Striessnig J., Bolz H. J., Koschak A. (2010). Channelopathies in Cav1.1, Cav1.3 and Cav1.4 voltage-gated L-type Ca2+ channels. Pflugers Arch. 460, 361–374 10.1007/s00424-010-0800-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton M. A., Ito H. T., Cressy P., Kempf C., Woo J. C., Schuman E. M. (2006). Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell 125, 785–799 10.1016/j.cell.2006.03.040 [DOI] [PubMed] [Google Scholar]
- Swanwick C. C., Murthy N. R., Kapur J. (2006). Activity-dependent scaling of GABAergic synapse strength is regulated by brain-derived neurotrophic factor. Mol. Cell. Neurosci. 31, 481–492 10.1016/j.mcn.2005.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada T., Sheng M. (2006). Molecular mechanisms of dendritic spine morphogenesis. Curr. Opin. Neurobiol. 16, 95–101 10.1016/j.conb.2005.12.001 [DOI] [PubMed] [Google Scholar]
- Talley E. M., Solorzano G., Depaulis A., Perez-Reyes E., Bayliss D. A. (2000). Low-voltage-activated calcium channel subunit expression in a genetic model of absence epilepsy in the rat. Brain Res. Mol. Brain Res. 75, 159–165 10.1016/s0169-328x(99)00307-1 [DOI] [PubMed] [Google Scholar]
- Tang J., Maximov A., Shin O. H., Dai H., Rizo J., Sudhof T. C. (2006). A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell 126, 1175–1187 10.1016/j.cell.2006.08.030 [DOI] [PubMed] [Google Scholar]
- Tedford H. W., Zamponi G. W. (2006). Direct G protein modulation of Cav2 calcium channels. Pharmacol. Rev. 58, 837–862 10.1124/pr.58.4.11 [DOI] [PubMed] [Google Scholar]
- Thalhammer A., Cingolani L. A. (2014). Cell adhesion and homeostatic synaptic plasticity. Neuropharmacology 78, 23–30 10.1016/j.neuropharm.2013.03.015 [DOI] [PubMed] [Google Scholar]
- Thiagarajan T. C., Lindskog M., Tsien R. W. (2005). Adaptation to synaptic inactivity in hippocampal neurons. Neuron 47, 725–737 10.1016/j.neuron.2005.06.037 [DOI] [PubMed] [Google Scholar]
- Tian X., Kai L., Hockberger P. E., Wokosin D. L., Surmeier D. J. (2010). MEF-2 regulates activity-dependent spine loss in striatopallidal medium spiny neurons. Mol. Cell. Neurosci. 44, 94–108 10.1016/j.mcn.2010.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trent C., Tsuing N., Horvitz H. R. (1983). Egg-laying defective mutants of the nematode Caenorhabditis elegans. Genetics 104, 619–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsakiridou E., Bertollini L., de Curtis M., Avanzini G., Pape H. C. (1995). Selective increase in T-type calcium conductance of reticular thalamic neurons in a rat model of absence epilepsy. J. Neurosci. 15, 3110–3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsurudome K., Tsang K., Liao E. H., Ball R., Penney J., Yang J. S., et al. (2010). The Drosophila miR-310 cluster negatively regulates synaptic strength at the neuromuscular junction. Neuron 68, 879–893 10.1016/j.neuron.2010.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. (2012). Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb. Perspect. Biol. 4:a005736 10.1101/cshperspect.a005736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. G. (2008). The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135, 422–435 10.1016/j.cell.2008.10.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. G., Nelson S. B. (2004). Homeostatic plasticity in the developing nervous system. Nat. Rev. Neurosci. 5, 97–107 10.1038/nrn1327 [DOI] [PubMed] [Google Scholar]
- Turrigiano G. G., Leslie K. R., Desai N. S., Rutherford L. C., Nelson S. B. (1998). Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391, 892–896 10.1038/36103 [DOI] [PubMed] [Google Scholar]
- Uchida I., Cestari I. N., Yang J. (1996). The differential antagonism by bicuculline and SR95531 of pentobarbitone-induced currents in cultured hippocampal neurons. Eur. J. Pharmacol. 307, 89–96 10.1016/0014-2999(96)00156-2 [DOI] [PubMed] [Google Scholar]
- USNLM/NIH (2011). Hypokalemic Periodic Paralysis (MedlinePlus Medical Encyclopedia: USNLM/NIH http://www.nlm.nih.gov/medlineplus/ency/article/000312.htm). [Google Scholar]
- van den Maagdenberg A. M., Pietrobon D., Pizzorusso T., Kaja S., Broos L. A., Cesetti T., et al. (2004). A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron 41, 701–710 10.1016/s0896-6273(04)00085-6 [DOI] [PubMed] [Google Scholar]
- van den Maagdenberg A. M., Pizzorusso T., Kaja S., Terpolilli N., Shapovalova M., Hoebeek F. E., et al. (2010). High cortical spreading depression susceptibility and migraine-associated symptoms in Ca(v)2.1 S218L mice. Ann. Neurol. 67, 85–98 10.1002/ana.21815 [DOI] [PubMed] [Google Scholar]
- Verkhratsky A. J., Petersen O. H. (1998). Neuronal calcium stores. Cell Calcium 24, 333–343 10.1016/s0143-4160(98)90057-4 [DOI] [PubMed] [Google Scholar]
- Vitko I., Chen Y., Arias J. M., Shen Y., Wu X. R., Perez-Reyes E. (2005). Functional characterization and neuronal modeling of the effects of childhood absence epilepsy variants of CACNA1H, a T-type calcium channel. J. Neurosci 25, 4844–4855 10.1523/jneurosci.0847-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt A. J., Desai N. S. (2010). Homeostatic plasticity and STDP: keeping a neuron’s cool in a fluctuating world. Front. Synaptic Neurosci. 2:5 10.3389/fnsyn.2010.00005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisskopf M. G., Bauer E. P., LeDoux J. E. (1999). L-type voltage-gated calcium channels mediate NMDA-independent associative long-term potentiation at thalamic input synapses to the amygdala J. Neurosci. 19, 10512–10519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenner P. (2014). Homeostatic synaptic plasticity in developing spinal networks driven by excitatory GABAergic currents. Neuropharmacology 78, 55–62 10.1016/j.neuropharm.2013.04.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyhersmüller A., Hallermann S., Wagner N., Eilers J. (2011). Rapid active zone remodeling during synaptic plasticity. J. Neurosci. 31, 6041–6052 10.1523/jneurosci.6698-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitt J. L., Petrus E., Lee H. K. (2014). Experience-dependent homeostatic synaptic plasticity in neocortex. Neuropharmacology 78, 45–54 10.1016/j.neuropharm.2013.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga C. J., Ibata K., Turrigiano G. G. (2005). Postsynaptic expression of homeostatic plasticity at neocortical synapses. J. Neurosci. 25, 2895–2905 10.1523/jneurosci.5217-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams G. S., Boyman L., Chikando A. C., Khairallah R. J., Lederer W. J. (2013). Mitochondrial calcium uptake. Proc. Natl. Acad. Sci. U S A 110, 10479–10486 10.1073/pnas.1300410110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wondolowski J., Dickman D. (2013). Emerging links between homeostatic synaptic plasticity and neurological disease. Front. Cell. Neurosci. 7:223 10.3389/fncel.2013.00223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing B., Ashleigh Long A., Harrison D. A., Cooper R. L. (2005). Developmental consequences of neuromuscular junctions with reduced presynaptic calcium channel function. Synapse 57, 132–147 10.1002/syn.20165 [DOI] [PubMed] [Google Scholar]
- Ye J., Llorian M., Cardona M., Rongvaux A., Moubarak R. S., Comella J. X., et al. (2013). A pathway involving HDAC5, cFLIP and caspases regulates expression of the splicing regulator polypyrimidine tract binding protein in the heart. J. Cell Sci. 126, 1682–1691 10.1242/jcs.121384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younger M. A., Müller M., Tong A., Pym E. C., Davis G. W. (2013). A presynaptic ENaC channel drives homeostatic plasticity. Neuron 79, 1183–1196 10.1016/j.neuron.2013.06.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Q., Xiang Y., Yan Z., Han C., Jan L. Y., Jan Y. N. (2011). Light-induced structural and functional plasticity in Drosophila larval visual system. Science 333, 1458–1462 10.1126/science.1207121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi G. W. (2005). Voltage-Gated Calcium Channels. Georgetown, Tex. New York, NY: Landes Bioscience/Eurekah.com; Kluwer Academic/Plenum Publishers [Google Scholar]
- Zamponi G. W., Currie K. P. (2013). Regulation of Ca(V)2 calcium channels by G protein coupled receptors. Biochim. Biophys. Acta 1828, 1629–1643 10.1016/j.bbamem.2012.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi G. W., Lory P., Perez-Reyes E. (2010). Role of voltage-gated calcium channels in epilepsy. Pflugers Arch. 460, 395–403 10.1007/s00424-009-0772-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Mori M., Burgess D. L., Noebels J. L. (2002). Mutations in high-voltage-activated calcium channel genes stimulate low-voltage-activated currents in mouse thalamic relay neurons. J. Neurosci. 22, 6362–6371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C., Dreosti E., Lagnado L. (2011). Homeostatic synaptic plasticity through changes in presynaptic calcium influx. J. Neurosci. 31, 7492–7496 10.1523/jneurosci.6636-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q., Homma K. J., Poo M. M. (2004). Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 44, 749–757 10.1016/j.neuron.2004.11.011 [DOI] [PubMed] [Google Scholar]
- Zhuchenko O., Bailey J., Bonnen P., Ashizawa T., Stockton D. W., Amos C., et al. (1997). Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat. Genet. 15, 62–69 10.1038/ng0197-62 [DOI] [PubMed] [Google Scholar]