

Abstract
Ebola viruses (EBOV) can cause severe hemorrhagic disease with high case fatality rates. Currently, no vaccines or therapeutics are approved for use in humans. Ebola virus-like particles (eVLP) comprising of virus protein (VP40), glycoprotein, and nucleoprotein protect rodents and nonhuman primates from lethal EBOV infection, representing as a candidate vaccine for EBOV infection. Previous reports have shown that eVLP stimulate the expression of proinflammatory cytokines in dendritic cells (DCs) and macrophages (MΦs) in vitro. However, the molecular mechanisms and signaling pathways through which eVLP induce innate immune responses remain obscure. In this study, we show that eVLP stimulate not only the expression of proinflammatory cytokines but also the expression of type I interferons (IFNs) and IFN-stimulated genes (ISGs) in murine bone marrow-derived DCs (BMDCs) and MΦs. Our data indicate that eVLP trigger host responses through toll-like receptor (TLR) pathway utilizing 2 distinct adaptors, MyD88 and TRIF. More interestingly, eVLP activated the IFN signaling pathway by inducing a set of potent antiviral ISGs. Last, eVLP and synthetic adjuvants, Poly I:C and CpG DNA, cooperatively increased the expression of cytokines and ISGs. Further supporting this synergy, eVLP when administered together with Poly I:C conferred mice enhanced protection against EBOV infection. These results indicate that eVLP stimulate early innate immune responses through TLR and type I IFN signaling pathways to protect the host from EBOV infection.
Introduction
Ebola viruses (EBOV) are negative-sense RNA viruses of the Filoviridae family that can cause severe and often fatal hemorrhagic fever in humans and nonhuman primates (NHPs) (Feldmann and others 2003; Ascenzi and others 2008). Cell-free Ebola virus-like particles (eVLP) are promising candidates as an EBOV vaccine (Warfield and others 2003, 2007). It has been shown in the rodent and NHP models that animals injected with eVLP before EBOV exposure survive after lethal EBOV infection. In addition, our recent evidence indicates that eVLP can protect mice when given post-EBOV exposure (Bradfute and others, to be submitted, see Discussion). eVLP are composed of matrix protein (VP40), surface glycoprotein (GP), and nucleoprotein (NP) (Bavari and others 2002; Noda and others 2002; Johnson and others 2006). VP40 when coexpressed with GP is responsible for filamentous morphology of eVLP (Noda and others 2002; Yamayoshi and Kawaoka 2007) and NFκB-mediated cytokine induction (Martinez and others 2007; Kaletsky and others 2009). eVLP elicit proinflammatory cytokines, such as TNFα, IL-1β, and IL-6 in dendritic cells (DCs) and macrophages (MΦs), and promote surface expression of costimulatory molecules in vitro, indicating that eVLP facilitate innate immune responses critical for antiviral immunity after EBOV infection (Bosio and others 2003; Warfield and others 2003; Martinez and others 2007). Early innate immune responses stimulated by eVLP may contribute to the timely onset of adaptive immunity, providing antigen-specific protection against the viruses (Warfield and Aman 2011).
Despite active research on eVLP, the molecular pathways and their downstream signaling mechanisms through which eVLP establish the antiviral innate immunity have remained elusive. Toll-like receptor (TLR) signaling is likely to play a major role in eVLP-induced innate immunity. Recently, Okumura and others (2010) showed that eVLP can stimulate proinflammatory cytokines in 293T cells through TLR4. They also showed that GP directly interacts with TLR4 and the inhibition of TLR4 binding prevents eVLP-induced inflammatory responses. Since the analysis was performed with 293T cells, it has remained uncertain whether DCs and MΦs utilize TLR signaling to respond to eVLP. In addition, even though the role of TLRs may be likely, the downstream signaling events that result in cytokine induction have not been fully elucidated. On ligand binding, adaptor proteins, MyD88 and TRIF, that are bound to the TLRs are activated and then recruit downstream signaling molecules to trigger 2 major arms of transcriptional pathways involving NFκB and IRF3/7 (Kabelitz and Medzhitov 2007; Beutler 2009; Kawai and Akira 2010). NFκB activates a number of proinflammatory cytokines, including TNFα, IL-1β, and IL-6 as well as many chemokines, whereas IRF3/IRF7 activates type I interferons (IFNs) (O'Neill and Bowie 2007; Kawai and Akira 2011). MyD88-deficient mice are resistant to LPS-induced endotoxin shock and fail to produce type I IFNs in response to various pathogens (Kawai and others 1999). On the other hand, TRIF controls both TLR3-mediated signaling and TLR4-mediated activation of IRF3/7 (Yamamoto and others 2003; Weighardt and others 2004).
Evidence indicates that type I IFNs are involved in early protection against EBOV infection in mice and NHPs (Jahrling and others 1999; Mahanty and others 2003; Bray and Geisbert 2005). It has been reported that more than 80% of rodents and NHPs injected with eVLP before EBOV infection survive (Warfield and others 2003, 2007). Type I IFNs are induced in DCs by TLRs and other pathogen recognition receptors, which in turn stimulate a large array of IFN-stimulated genes (ISGs) that collectively confer antiviral activities and promote the DC innate immunity (Kumar and others 2011). ISGs are induced through the IFN signaling pathway, distinct from TLR pathway that is activated by the engagement of surface receptors IFNAR1/IFNAR2 leading to the activation of JAK kinases and the STAT1/2/IRF9 complex (Darnell and others 1994; Brierley and Fish 2002; Platanias 2005; Schindler and others 2007).
Despite the potential significance of type I IFNs in anti-EBOV protection, it has not been certain how eVLP can induce innate immune responses in relevant cells. In the present study, we examined the effect of eVLP in murine bone marrow-derived DCs (BMDCs) generated in the presence of Fms-like tyrosine kinase 3 ligand (Flt3L), which supports the development of pDCs, a major source for type I IFN production (Asselin-Paturel and Trinchieri 2005; Tamura and others 2005). Quantitative RT-PCR analysis in DCs generated from MyD88−/− and TRIF−/− mice showed that proinflammatory responses are dependent on MyD88, whereas type I IFNs and ISGs gene expression required TRIF, demonstrating that eVLP engage 2 distinct TLR adaptors to stimulate 2 arms of transcription. ISG induction was a result of a positive feedback loop-dependent activation of IFN signaling pathway since IFNAR−/− DCs failed to induce a set of ISGs on eVLP exposure. Finally, we show that eVLP when added together with the synthetic TLR ligand, Poly I:C, led to enhanced cytokine and ISG induction in vitro. In support of synergy by eVLP and Poly I:C, the 2 agents when administered together in vivo provided enhanced protection in mice against lethal EBOV infection. Together, this study illustrates that eVLP induce type I IFNs, ISGs, and proinflammatory cytokines by mobilizing the TLR signaling and the IFN signaling pathways.
Materials and Methods
Animals
Six- to eight-week-old mice were used for the present study, unless otherwise stated. MyD88−/− mice in C57BL/6 background (provided by M. J. Todd, FDA), TRIF−/− mice (purchased from the Jackson Laboratory), and IFNAR−/− mice in BALB/c background (a gift from J. Durbin, Ohio State University) were maintained in the NICHD animal facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. All experiments were carried out as approved by the NICHD Animal Care and Use Committee (ASP #08-010 and #11-044).
In vitro generation of DCs and MΦs
Bone marrow (BM) mononuclear cells were cultured in the complete RPMI 1640 medium (Invitrogen) in the presence of recombinant Flt3L (100 ng/mL; Peprotech) at 106 cells/mL as described (Tamura and others 2005; Tailor and others 2007). For BM-derived macrophages (BMMΦs), BM cells were cultured in 12-well plate (1×106 cells/mL) with recombinant macrophage–colony-stimulating factor (M-CSF, (20 ng/mL; Invitrogen) for 6 days. A half of culture media was replaced by fresh media containing fresh Flt3L or M-CSF on day 3 or 4. BMMΦs were stimulated with IFN-γ (100 U/mL; Invitrogen) for 24 h before experiments (Hu and others 2008). After 6 days of culture, BMDCs and BMMΦs were stimulated with eVLP (10 μg/mL), Poly I:C (100 μg/mL; Amersham Biosciences), or CpG B (ODN 1826, 1 μg/mL; Operon Biotechnologies) for indicated time period.
Recombinant eVLP
eVLP composed of EBOV GP, NP, and VP40 were prepared as reported (Warfield and others 2005). Briefly, 293T cells were cotransfected with pWRG vectors encoding EBOV VP40, NP, and GP using Lipofectamine 2000 (Invitrogen). Pellets from the supernatants of these cells were separated over a 20%–60% continuous sucrose gradient centrifugation concentrated by a second centrifugation. The gradient fractions containing eVLP were determined, collected, and the total protein concentration of eVLP preparation was determined in the presence of Nonidet P-40 detergent. eVLP preparation was analyzed by immunoblot and electron microscopy to confirm the particle formation and composition as reported by Warfield and others (2007). eVLP preparations used in this study contained <0.03 endotoxin U/mg.
Quantitative real-time PCR
Total RNA was extracted from DCs and MΦ by the TRIzol method (Invitrogen). The quantitative real-time PCR (qRT-PCR) reactions were run in duplicates using the SYBER GREEN master mix as described (Chang and others 2009). The values (ΔCT) were normalized with HPRT as a housekeeping gene to analyze the relative mRNA levels. The results are expressed as fold induction, and the primer sequences used for this study were followed from previous publications (Tamura and others 2005; Tailor and others 2007; Chang and others 2009). Student's t-test was used to evaluate the differences between control (unstimulated) and test samples (eVLP stimulated). Calculations were performed using statistical Excel software. Data are expressed as mean±SEM from 3 independent experiments. P value<0.05 is considered statistically significant.
Flow cytometry
The expression of DC markers and costimulatory molecule MHC class II was detected by the flow cytometry analysis as previously reported (Chang and others 2009). Cells were blocked with anti-mouse FcγR antibody and stained with APC-conjugated anti-CD11c, anti-MHC-II, and PerCP-conjugated anti-B220 antibodies (BD Pharmingen). The percentage of positive cells was determined after collecting 25,000 events and gated based on forward and side scatter parameters for viable cells on the FACSCalibur (Becton Dickinson). Appropriate isotype controls were tested to verify positive staining. Data were analyzed using the FlowJo software.
Vaccination studies
C57Bl/6 mice (8–12 weeks old) were vaccinated intraperitoneally twice, 28 days apart, 0.25 μg of eVLP, with or without 20 μg of poly-I:C, stabilized with Poly lysine and Carboxymethylcellulose (Poly ICLC) (Zhu and other 2007). Twenty-one days after the second vaccination, mice were bled and anti-EBOV antibody titers were determined by enzyme-linked immunosorbent assay (ELISA). Twenty-eight days after the second vaccination, mice were challenged with 1,000 pfu mouse-adapted EBOV and followed for survival. Research performed at the United States Army Medical Research Institute of Infectious Diseases (USAMRIID) was conducted under an IACUC-approved animal protocol in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adheres to principles stated in the Guide for the Care and Use of Laboratory Animals (National Research Council, 1996). USAMRIID is fully accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care International. All virus work was performed, and all infected mice were handled under maximum containment in a biosafety level-4 laboratory at the USAMRIID.
Enzyme-Linked Immunosorbent Assay
Endpoint dilution ELISAs were performed as described previously (Warfield and others 2005) with minor modifications. Briefly, irradiated EBOV was adsorbed onto ELISA plates. Sera was added at 2-fold dilutions, incubated, and washed. EBOV-specific IgG level was detected after the incubation of anti-mouse IgG antibody conjugated to HRP, followed by washing and subsequent incubation with TMB substrate (tetramethylbenzidine) and an acid stop solution. Positive samples were defined as having >2 times the O.D. (at 450 nm) compared to sera from poly ICLC-only injected mice at a given dilution.
Results
eVLP induce type I IFNs, proinflammatory cytokines, and ISGs in DCs and MΦs
We studied whether eVLP stimulate cytokine induction in BMDCs generated in the presence of Flt3L. BMDCs were treated with 10 μg/mL of viral-genome free and endotoxin-free eVLP for up to 12 h, and the expression of mRNAs for type I IFNs (IFNα and IFNβ) and proinflammatory cytokines (TNFα, IL-6, IL-1β, and IL-12p40) were tested by qRT-PCR. BMDC populations generated for this work were composed of more than 95% CD11c+ cells and contained from 25% to 35% plasmacytoid DCs (see Supplementary Fig. S1 for flow cytometry profiles; Supplementary Data are available online at www.liebertpub.com/jir) as expected (Tamura and others 2005; Chang and others 2009). As shown in Fig. 1A, eVLP induced all of the above cytokines with the peak expression at 6 h followed by a decline at 12 h. Induction of these cytokines was reproducibly seen at this concentration of eVLP, although the levels and kinetics varied slightly. These results indicate that eVLP stimulate the induction of both type I IFNs and proinflammatory cytokines. Type I IFNs elicit antiviral activity, whereas TNFα, IL-1β, and IL-6 mediate acute-phase responses and stimulate inflammatory responses in part by mobilizing neutrophils. IL-12p40 together with IL-12p35 activates IFNγ. It should be noted that we used a pan-IFNα primer to detect multiple IFNα subtypes for this study (Tailor and others 2007). Similar to DCs, these cytokines were also induced in BMMΦs after eVLP exposure, although the mRNA expression level was slower in BMMΦs (Supplementary Fig. S2A).
FIG. 1.
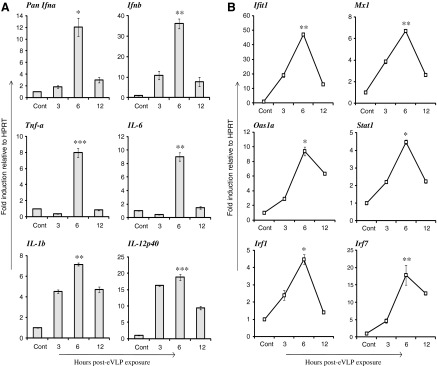
In vitro eVLP stimulation triggers mRNA expression for type I IFNs, proinflammatory cytokines, and ISGs in BMDCs. BMDCs generated by Flt3L were stimulated with 10 μg/mL of eVLP for indicated time period (h). Cont represents unstimulated cells at time 0. mRNA transcripts of indicated genes in (A) (Pan Ifnα, Ifnβ, Tnf-α, IL-6, IL-1β, and IL-12p40) and in (B) (Ifit1, Mx1, Oas1a, Stat1, Irf1, and Irf7) were measured by qRT-PCR, normalized with HPRT, and are expressed as fold induction. Data represent the mean of duplicate samples from 3 independent experiments±SEM. All data were assessed by Student's t-test and are denoted by ***P≤0.001, **P≤0.01 and *P≤0.05. BMDCs, bone marrow-derived DCs; eVLP, Ebola virus-like particles; Flt3L, Fms-like tyrosine kinase 3 ligand; IFNs, interferons; ISGs, IFN-stimulated genes; qRT-PCR, quantitative real-time PCR.
Since eVLP stimulated type I IFN mRNA transcription, we believed that it is important to determine whether eVLP also induce ISGs in DCs. Genes such as Ifit1, Mx1, Oas1a, and Stat1 are classic ISGs that confer antiviral activities (Samuel 2001; Schindler and others 2007). Other ISGs such as Irf1 and Irf7 are transcription factors that enhance the expression of many ISGs and promote the DC innate immunity (Honda and others 2005; Steinman and Banchereau 2007). mRNA expression of ISGs Ifit1, Mx1, Oas1a, Stat1, Irf1, and Irf7 were robustly induced upon eVLP stimulation (Fig. 1B). Mirroring IFN induction kinetics, ISG induction also peaked at 6 h in BMDCs. Likewise, eVLP led to the induction of ISGs in BMMΦs. In keeping with the slower kinetics in BMMΦ, ISG induction also showed a gradual increase with the peak expression at 12 h (Supplementary Fig. S2B).
eVLP trigger inflammatory cytokines through MyD88 and type I IFNs through TRIF
All TLRs, except TLR3, utilize the adaptor MyD88 to initiate signaling cascades to activate NFκB and IRF3/7 (O'Neill and Bowie 2007; Kawai and Akira 2011). TLR3, which recognizes dsRNA, utilizes TRIF to activate NFκB and IRF3/7. TLR4 utilizes both MyD88 and TRIF (Yamamoto and others 2003; Dunne and O'Neill 2005). As eVLP are reported to act through TLR4 in 293T cells (Okumura and others 2010), it was of interest to study the signaling cascade beyond TLR through which eVLP activate cytokine transcription (Okumura and others 2010). To substantiate this, we tested whether eVLP stimulate cytokine mRNA transcription in MyD88−/− mice DC. Data in Fig. 2 show that the induction of IL-6, IL-1β, and IL-12p40 by eVLP stimulation was almost abrogated in MyD88−/− DCs, while MyD88+/+ DCs gave full induction. On the other hand, IFNβ expression was not reduced in MyD88−/− cells, rather levels were somewhat higher than those in MyD88+/+ cells. TNFα expression was only modestly reduced in MyD88−/− cells, indicating that the induction of type I IFN and TNFα requires a MyD88-independent pathway. Consistent with MyD88-independent type I IFN induction, eVLP stimulation of ISGs was not affected in MyD88−/− DCs (Table 1).
FIG. 2.

eVLP induction of some proinflammatory cytokines requires MyD88. BMDCs from MyD88+/+ and MyD88−/− mice were stimulated with eVLP for 6 h, and the induction of indicated cytokines was measured as described previously. MyD88-dependent and MyD88-independent cytokines are shown in this figure and in Table 1, respectively. Data represent the mean of duplicate samples from 3 independent experiments ±SEM. All data were assessed by Student's t test and are denoted by **P≤0.01 and *P≤0.05.
Table 1.
Cytokine Genes Do Not Require MYD88
| Fold induction (eVLP/control) | |||
|---|---|---|---|
| Genes analyzed | MyD88+/+ DCs | MyD88−/−DCs | Significance P value (≤0.05) |
| Pan IFNα | 2.8 | 4.4 | 0.04a |
| IFNβ | 5.6 | 8.4 | 0.03a |
| TNFα | 9.2 | 6.1 | 0.031a |
| Ifit1 | 4.0 | 8.6 | 0.002b |
| Mx1 | 1.8 | 2.2 | 0.06n.s. |
| Oas1a | 1.9 | 2.7 | 0.06n.s. |
| Stat1 | 3.3 | 4.4 | 0.04a |
| Irf1 | 4.2 | 5.8 | 0.01b |
| Irf7 | 6.9 | 8.0 | 0.04a |
P≤0.05; bP≤0.01; n.s., no significance.
BMDCs from MyD88+/+ or MyD88−/− mice were stimulated with eVLP, and mRNA expression of indicated cytokines was measured by qRT-PCR as in Fig. 3. Values represent the average of duplicate samples from 3 independent experiments±SEM.
BMDCs, bone marrow-derived DCs; DCs, dendritic cells; qRT-PCR, quantitative real-time PCR.
Cytokine induction in TRIF−/− DCs was essentially opposite of that of MyD88−/− DCs. As seen in Fig. 3, the induction of IFNβ was almost completely abolished, whereas TNFα induction was reduced by about half in TRIF−/− DCs. Correlating with the inhibition of type I IFN induction, the expression of ISGs was drastically reduced in TRIF−/− cells relative to TRIF+/+ cells. Contrary to these results, mRNA fold induction of IL-6, IL-1β, and IL-12p40 mRNA was unaffected in TRIF−/− DCs or equivalent to TRIF+/+ DCs (Table 2). These results indicate that eVLP utilize 2 different signaling adaptors, MyD88 and TRIF, to induce proinflammatory cytokines and type I IFNs, respectively.
FIG. 3.

eVLP induction of IFNβ and ISGs requires TRIF. BMDCs generated from TRIF+/+ and TRIF−/− mice were stimulated with eVLP for 6 h, and mRNA transcript of indicated cytokines was measured as described previously. TRIF-dependent and TRIF-independent cytokines are shown in this figure and in Table 2, respectively. Data represent the mean of duplicate samples from 3 independent experiments ±SEM. All data were assessed by Student's t test and are denoted by **P≤0.01 and *P≤0.05.
Table 2.
Cytokine Genes Do Not Require TRIF
| Fold induction (eVLP/control) | |||
|---|---|---|---|
| Genes analyzed | TRIF+/+ DCs | TRIF−/−DCs | Significance P value (≤0.05) |
| TNFα | 13.8 | 9.6 | 0.03a |
| IL-6 | 15.1 | 14.3 | 0.06n.s. |
| IL-1β | 5.7 | 5.0 | 0.07n.s. |
| IL-12p40 | 2.8 | 4.0 | 0.04a |
P≤0.05; n.s., no significance.
BMDCs from TRIF+/+ Wt and TIRIF−/− mice were stimulated with eVLP, and mRNA expression of indicated cytokines was measured by qRT-PCR as in Fig. 3. Values represent the mean duplicate samples from 3 independent experiments±SEM.
eVLP activate ISG expression through type I IFN signaling pathway
Type I IFNs play a critical role in anti-EBOV protection, presumably by stimulating multiple ISGs that carry antiviral activities. ISG induction by eVLP may be mediated by type I IFN through the activation of classic IFN signaling pathway that activates JAK/TYK kinases and ISGF3 complex (Darnell and others 1994; Platanias 2005; Schindler and others 2007). However, ISGs can be induced by some dsRNA viruses directly through IRF3 in an IFN-independent manner (Paladino and others 2006). Therefore, it was possible that eVLP induced ISGs directly without relying on prior type I IFN production. To assess how eVLP induce ISGs through type I FIN signaling pathway, we stimulated DCs generated from IFNAR+/+ and IFNAR−/− mice with eVLP for 6 h. The results in Fig. 4 showed that IFNAR−/− DCs were completely defective in expressing ISGs compared to IFNAR+/+ DCs. DCs from both IFNAR+/+ and IFNAR−/− mice, however, expressed Pan IFNα, IFNβ, and TNFα (Fig. 4) in response to eVLP exposure, indicating that the initial activation of NFκB and IRF3/IRF7 was intact in both IFNAR+/+ and IFNAR−/− DCs. It has been shown that IFNβ is strongly expressed even in IFNAR−/− DCs in an IFN signal-independent manner particularly in an early stage, although type I IFN induction is amplified by a positive feedback mechanism later (Barchet and others 2002; Swiecki and Colonna 2010). These results show that eVLP induced ISGs that depends on the IFN signaling pathway activated by the prior induction of type I IFN by eVLP.
FIG. 4.
eVLP induction of ISGs depends on IFNAR-mediated type I IFN signaling. BMDCs generated from type I IFN receptor1 (IFNAR1+/+ and IFNAR1−/−) mice (both BALB/C background) were stimulated with eVLP for 6 h, and the expression of specified cytokines were measured as in Fig. 1. Data represent the mean of duplicate samples from 3 independent experiments ±SEM. All data were assessed by Student's t test and are denoted by **P≤0.01, *P≤0.05 and n.s., no significance.
eVLP and Poly I:C synergistically enhance cytokines and ISG expression
Synthetic TLR ligands such as polyinosinic: polycytidylic acid (Poly I:C) and CpG DNA are shown to boost the efficacy for a wide variety of vaccines (Kwissa and others 2007; Bode and others 2011; Caskey and others 2011). Poly I:C and CpG (ligands for TLR3 and TLR9) can stimulate type I IFNs and proinflammatory cytokines. Poly I:C is also shown to promote type I IFN-induced maturation of DCs (Longhi and others 2009). We asked whether Poly I:C or CpG boost eVLP ability to stimulate the synergistic induction of cytokines in DCs. In Fig. 5A, BMDCs were first treated with eVLP for 1 h followed by an additional treatment with Poly I:C for up to 24 h. Poly I:C alone stimulated the same set of cytokine genes as shown in Fig. 1A at higher levels than eVLP. Importantly, cytokine induction was much higher when cells were treated with both eVLP and Poly I:C. In most cases, the combined treatment led to higher induction than either single treatment, indicating that eVLP and Poly I:C synergistically stimulated these cytokines. Similarly, increased cytokine induction was observed when combined with CpG DNA, although the level of mRNA expression varied in this treatment (Supplementary Fig. S3A). We also found that both combinations (eVLP-Poly I:C and eVLP-CpG) led to higher ISG induction than by a single treatment (Fig. 5B and Supplementary Fig. S3B). These results indicate that the efficacy of eVLP to induce cytokines is amplified by a combined stimulation with Poly I:C or CpG DNA.
FIG. 5.
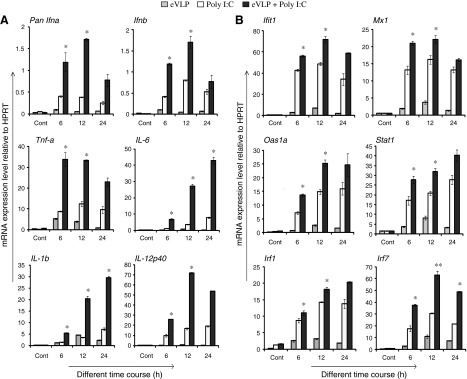
eVLP and Poly I:C synergize to enhance cytokine induction. BMDCs incubated with eVLP (1 μg/mL) for 1 h were stimulated with Poly I:C (100 μg/mL) in the continued presence of eVLP for indicated time course (h) (A). Cytokine induction was measured as in Fig. 1, and data were statistically evaluated. (B) eVLP-induced ISG expression is enhanced by Poly I:C. All data were assessed by Student's t test and are denoted by **P≤0.01 and *P≤0.05.
To ascertain whether Poly I:C could enhance the efficacy of eVLP in vivo, we injected mice with a low dose of eVLP (0.25 μg) alone, Poly ICLC alone, or eVLP plus Poly-ICLC before lethal EBOV infection. Poly-ICLC is a poly I:C stabilized with poly-lysine widely used in vivo (Zhu and others 2007). As shown in Table 3, while only 10% of mice injected with eVLP alone or Poly ICLC alone survived, 100% of mice injected with the combination of eVLP and Poly ICLC survived lethal EBOV infection. In addition, anti-EBOV antibody titer was dramatically enhanced by the addition of Poly-ICLC to eVLP. As expected, Poly ICLC itself did not stimulate the antibody production, whereas eVLP alone led to the production of anti-EBOV antibody at a modest level.
Table 3.
In Vivo Effects of Poly ICLC on eVLP Vaccination Efficacy
| Vaccine | Dose (μg eVLP) | Survival (%) | Anti-EBOV antibody titer |
|---|---|---|---|
| eVLP | 0.25 | 1/10 (10%) | 800±172 |
| eVLP+Poly ICLC | 0.25 | 8/8 (100%)a | 12,800±4,434b |
| Poly ICLC only | N/A | 1/10 (10%) | 0 |
P≤0.05 by log-rank test; bP≤0.05 by 2-tailed Student's t-test.
C57Bl/6 mice were vaccinated i.p. twice, 28 days apart, then challenged with 1,000 pfu mouse-adapted EBOV 28 days after the second vaccination. Poly ICLC was given at 20 μg/dose. Antibody titer is endpoint dilution±standard error.
EBOV, Ebola viruses; eVLP, Ebola virus-like particles; N/A, Not applicable.
Discussion
We show that eVLP activate TLR signaling through 2 different adaptors, MyD88 and TRIF, to induce proinflammatory cytokines and type I IFNs in primary BMDCs (see Fig. 6 for a model). MyD88 was essential for the induction of proinflammatory cytokines, IL-6, IL-1β and IL-12p40, whereas type I IFN and ISGs induction was dependent on TRIF, illustrating an independent adaptor usage by eVLP for inducing the 2 separate groups of cytokines. It may be envisaged that eVLP activation of MyD88 mostly signals through IKKα/β-NFκB pathway, whereas TRIF signals through the cascade of TBK1/IKKɛ-IRF3/7 pathway to induce various cytokines (Fig. 6) (Dunne and O'Neill 2005; O'Neill and Bowie 2007; Kawai and Akira 2010). Given that eVLP GP is shown to bind to TLR4 and stimulate cytokine induction in 293T cells, TRIF activation in DCs may be mediated by TLR4 as well (Okumura and others 2010). However, TRIF may have been activated by TLR3, considering that eVLP preparations used here may have had RNAs copurified along with the intended eVLP components. We found that TNFα induction was only partially reduced in MyD88−/− DCs and unaffected in TRIF−/− DCs, suggesting that pathways other than TLRs such as MAPK pathway, also reported to be activated by eVLP, may modulate the expression of some cytokines (Martinez and others 2007).
FIG. 6.

A 2-step model for eVLP-stimulated cytokine expression. eVLP interact with TLR4 (and presumably other TLRs) and activate the adaptor MyD88 and TRIF bound to the TLRs. MyD88 then activates NFκB (p50/p65) through the TLR signaling cascade involving IRAK, TRAF6, and IKKα/β to trigger proinflammatory cytokine transcription. TRIF activates IRF3/7 through a cascade involving TBK1/IKKi(e) to induce type I IFN genes. Type I IFNs then bind to IFNAR1/2 to activate ISGF3 complex (STAT1/STAT2/IRF9) through JAK1/TYK2 to induce ISGs transcription.
A notable observation in this study is that eVLP stimulated not only proinflammatory cytokines, as previously documented, but also type I IFNs and an array of ISGs, such as Ifit1, Mx1, Oas1a, Stat1, Irf1, and Irf7. The activation of the IFN system by eVLP observed in this study may be in part attributed to the use of BMDCs generated in the presence of Flt3L, which supports the development of plasmacytoid DCs that produce high levels of type I IFNs (Asselin-Paturel and Trinchieri 2005; Tamura and others 2005). Our data show that ISG induction is mediated by the classic IFN signaling pathway since eVLP induction of ISGs was abolished in IFNAR−/− DCs (Fig. 6). Type I IFN signaling results in transcriptional induction of more than 1,000 ISGs (Samuel 2001; Schoggins and others 2011). Many ISGs encode antiviral factors capable of inhibiting various steps of viral growth. Furthermore, some ISGs promote the maturation of DCs and enhance innate and subsequent adaptive immunity (Steinman and Banchereau 2007; Longhi and others 2009). Our data indicate that ISGs are induced rapidly within 6 h after eVLP stimulation, suggesting that the IFN signaling is activated immediately after the induction of type I IFNs by eVLP. Rapid induction of type IFNs and ISGs may be of critical importance in protecting the host, given that type I IFNs are known to play a pivotal role in anti-EBOV innate immunity (Jahrling and others 1999; Mahanty and others 2003; Bray and Geisbert 2005; Bray and Murphy 2007; Chang and others 2009).
It is noteworthy that eVLP induction of cytokines and ISGs observed in vitro are consistent with the function of eVLP in vivo. eVLP has been shown to protect rodents and NHPs from EBOV infection when injected before the viral exposure. Importantly, this eVLP-mediated protection is dependent on Stat1 in mice (Raymond and others 2011). Furthermore, we recently found that eVLP can protect mice when administered 24 h post-EBOV exposures, and this protection depends on IFNAR (Ayithan and others 2013; Bradfute and others 2013, manuscripts in preparation). Thus, the type I IFN system is likely to be a major mechanism of eVLP-based protection against EBOV infection. Type I IFNs may alter host responses broadly by strengthening initial innate immune responses, leading to enhanced adaptive immunity against EBOV.
Finally, eVLP synergized with Poly I:C to enhance the induction of proinflammatory cytokines, type I IFNs, and ISG expression in vitro. Poly I:C acts through TLR3 or the RIG-I pathway and has been proposed as a potential vaccine adjuvant (Beutler 2009; Bode and others 2011; Caskey and others 2011). eVLP also gave increased cytokine induction when combined with CpG DNA, which is known to act through TLR9. CpG similar to Poly I:C has been suggested as a candidate for vaccine adjuvant (Bode and others 2011). Our results are in accordance with synergy documented between different TLRs that result in enhanced innate and adaptive immunity (Napolitani and others 2005; Warger and others 2006; Bagchi and others 2007). The underlying mechanisms of synergy are not completely understood. Different TLR ligands may exert different effects on intermediary factors within the TLR pathways. It is also possible that this synergy is caused by interactions between multiple signaling pathways. For example, eVLP-induced IFNs may result in an alteration of the activity of factors that lie within the TLR pathways. Alternatively, eVLP and TLR ligands may trigger the activity of other signaling pathways, such as MAPK. The ability of Poly I:C and CpG to broadly synergize with other TLR ligands may serve as a basis for their proposed role as vaccine adjuvants. We found that mice injected with a low dose of eVLP mixed with poly ICLC were more effectively protected from EBOV infection than mice injected with eVLP alone or Poly ICLC alone (Table 3). Our results are in agreement with published data showing enhanced efficacy of a subunit EBOV vaccine when mixed with poly I:C (Phoolcharoen and others 2011).
In conclusion, the exposure of naive DCs to eVLP results in early induction of not only proinflammatory cytokines but also type I IFNs, which presumably leads to timely acquisition of antiviral activities mediated by ISGs. The increased capacity to mount innate immunity may contribute to the protection against lethal EBOV infection afforded by eVLP. Efficacy of eVLP may be further augmented by a synthetic vaccine adjuvant, such as Poly I:C and CpG.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Program of NICHD and the Trans-NIH FDA intramural Bio-defense Program, National Institutes of Health, USA. The content of this publication does not necessarily reflect the views or policies of the US Department of Defense or the US Department of the Army.
Author Disclosure Statement
No competing financial interests exist.
References
- Ascenzi P, Bocedi A, Heptonstall J, Capobianchi MR, Di Caro A, Mastrangelo E, Bolognesi M, Ippolito G. 2008. Ebolavirus and Marburgvirus: insight the Filoviridae family. Mol Aspects Med 29(3):151–185 [DOI] [PubMed] [Google Scholar]
- Asselin-Paturel C, Trinchieri G. 2005. Production of type I interferons: plasmacytoid dendritic cells and beyond. J Exp Med 202(4):461–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayithan N, Bradfute SB, Stuthman KS, Anthony SM, Bavari S, Bray M, Ozato K. 2013. VLP post-Ebola infection protects mice via type I IFN signaling mediated negative regulation of inflammatory genes at early time point. (Manuscript to be submitted)
- Bagchi A, Herrup EA, Warren HS, Trigilio J, Shin HS, Valentine C, Hellman J. 2007. MyD88-dependent and MyD88-independent pathways in synergy, priming, and tolerance between TLR agonists. J Immunol 178(2):1164–1171 [DOI] [PubMed] [Google Scholar]
- Barchet W, Cella M, Odermatt B, Asselin-Paturel C, Colonna M, Kalinke U. 2002. Virus-induced interferon alpha production by a dendritic cell subset in the absence of feedback signaling in vivo. J Exp Med 195(4):507–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavari S, Bosio CM, Wiegand E, Ruthel G, Will AB, Geisbert TW, Hevey M, Schmaljohn C, Schmaljohn A, Aman MJ. 2002. Lipid raft microdomains: a gateway compartmentalized trafficking of Ebola and Marburg Viruses. J Exp Med 195(5):593–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutler BA. 2009. TLRs and innate immunity. Blood 113(7):1399–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode C, Zhao G, Steinhagen F, Kinjo T, Klinman DM. 2011. CpG DNA as a vaccine adjuvant. Expert Rev Vaccines 10(4):499–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosio CM, Aman MJ, Grogan C, Hogan R, Ruthel G, Negley D, Mohamadzadeh M, Bavari S, Schmaljohn A. 2003. Ebola and Marburg viruses replicate in monocyte-derived dendritic cells without inducing the production of cytokines and full maturation. J Infect Dis 188(11):1630–1638 [DOI] [PubMed] [Google Scholar]
- Bradfute SB, Anthony SM, Stuthman KS, Ayithan N, Tailor P, Bray M, Ozato K, Bavari S. 2013. Mechanisms of immunity in post-infection vaccination against Ebola virus infection. (Manuscript to be submitted) [DOI] [PMC free article] [PubMed]
- Bray M, Geisbert TW. 2005. Ebola virus: the role of macrophages and dendritic cells in the pathogenesis of Ebola hemorrhagic fever. Int J Biochem Cell Biol 37(8):1560–1566 [DOI] [PubMed] [Google Scholar]
- Bray M, Murphy FA. 2007. Filovirus research: knowledge expands to meet a growing threat. J Infect Dis 196Suppl 2:S438–S443 [DOI] [PubMed] [Google Scholar]
- Brierley MM, Fish EN. 2002. Review: IFN-alpha/beta receptor interactions to biologic outcomes: understanding the circuitry. J Interferon Cytokine Res 22(8):835–845 [DOI] [PubMed] [Google Scholar]
- Caskey M, Lefebvre F, Filali-Mouhim A, Cameron MJ, Goulet JP, Haddad EK, Breton G, Trumpfheller C, Pollak S, Shimeliovich I, Duque-Alarcon A, Pan L, Nelkenbaum A, Salazar AM, Schlesinger SJ, Steinman RM, Sekaly RP. 2011. Synthetic double-stranded RNA induces innate immune responses similar to a live viral vaccine in humans. J Exp Med 208(12):2357–2366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang TH, Kubota T, Matsuoka M, Jones S, Bradfute SB, Bray M, Ozato K. 2009. Ebola Zaire virus blocks type I interferon production by exploiting the host SUMO modification machinery. PLoS Pathog 5(6):e1000493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JE, Jr., Kerr IM, Stark GR. 1994. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264(5164):1415–1421 [DOI] [PubMed] [Google Scholar]
- Dunne A, O'Neill LA. 2005. Adaptor usage and Toll-like receptor signaling specificity. FEBS Lett 579(15):3330–3335 [DOI] [PubMed] [Google Scholar]
- Feldmann H, Jones S, Klenk HD, Schnittler HJ. 2003. Ebola virus: from discovery to vaccine. Nat Rev Immunol 3(8):677–685 [DOI] [PubMed] [Google Scholar]
- Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. 2005. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434(7034):772–777 [DOI] [PubMed] [Google Scholar]
- Hu X, Chakravarty SD, Ivashkiv LB. 2008. Regulation of interferon and Toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol Rev 226:41–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahrling PB, Geisbert TW, Geisbert JB, Swearengen JR, Bray M, Jaax NK, Huggins JW, LeDuc JW, Peters CJ. 1999. Evaluation of immune globulin and recombinant interferon-alpha2b for treatment of experimental Ebola virus infections. J Infect Dis 179Suppl 1:S224–S234 [DOI] [PubMed] [Google Scholar]
- Johnson RF, Bell P, Harty RN. 2006. Effect of Ebola virus proteins GP, NP and VP35 on VP40 VLP morphology. Virol J 3:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabelitz D, Medzhitov R. 2007. Innate immunity—cross-talk with adaptive immunity through pattern recognition receptors and cytokines. Curr Opin Immunol 19(1):1–3 [DOI] [PubMed] [Google Scholar]
- Kaletsky RL, Francica JR, Agrawal-Gamse C, Bates P. 2009. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc Natl Acad Sci U S A 106(8):2886–2891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. 1999. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11(1):115–122 [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. 2010. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11(5):373–384 [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. 2011. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34(5):637–650 [DOI] [PubMed] [Google Scholar]
- Kumar H, Kawai T, Akira S. 2011. Pathogen recognition by the innate immune system. Int Rev Immunol 30(1):16–34 [DOI] [PubMed] [Google Scholar]
- Kwissa M, Kasturi SP, Pulendran B. 2007. The science of adjuvants. Expert Rev Vaccines 6(5):673–684 [DOI] [PubMed] [Google Scholar]
- Longhi MP, Trumpfheller C, Idoyaga J, Caskey M, Matos I, Kluger C, Salazar AM, Colonna M, Steinman RM. 2009. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J Exp Med 206(7):1589–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahanty S, Gupta M, Paragas J, Bray M, Ahmed R, Rollin PE. 2003. Protection from lethal infection is determined by innate immune responses in a mouse model of Ebola virus infection. Virology 312(2):415–424 [DOI] [PubMed] [Google Scholar]
- Martinez O, Valmas C, Basler CF. 2007. Ebola virus-like particle-induced activation of NF-kappaB and Erk signaling in human dendritic cells requires the glycoprotein mucin domain. Virology 364(2):342–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. 2005. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat Immunol 6(8):769–776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda T, Sagara H, Suzuki E, Takada A, Kida H, Kawaoka Y. 2002. Ebola virus VP40 drives the formation of virus-like filamentous particles along with GP. J Virol 76(10):4855–4865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill LA, Bowie AG. 2007. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol 7(5):353–364 [DOI] [PubMed] [Google Scholar]
- Okumura A, Pitha PM, Yoshimura A, Harty RN. 2010. Interaction between Ebola virus glycoprotein and host toll-like receptor 4 leads to induction of proinflammatory cytokines and SOCS1. J Virol 84(1):27–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paladino P, Cummings DT, Noyce RS, Mossman KL. 2006. The IFN-independent response to virus particle entry provides a first line of antiviral defense that is independent of TLRs and retinoic acid-inducible gene I. J Immunol 177(11):8008–8016 [DOI] [PubMed] [Google Scholar]
- Phoolcharoen W, Dye JM, Kilbourne J, Piensook K, Pratt WD, Arntzen CJ, Chen Q, Mason HS, Herbst-Kralovetz MM. 2011. A nonreplicating subunit vaccine protects mice against lethal Ebola virus challenge. Proc Natl Acad Sci U S A 108(51):20695–20700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platanias LC. 2005. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol 5(5):375–386 [DOI] [PubMed] [Google Scholar]
- Raymond J, Bradfute S, Bray M. 2011. Filovirus infection of STAT-1 knockout mice. J Infect Dis 204 (Suppl 3):S986–S990 [DOI] [PubMed] [Google Scholar]
- Samuel CE. 2001. Antiviral actions of interferons. Clin Microbiol Rev 14(4):778–809, table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler C, Levy DE, Decker T. 2007. JAK-STAT signaling: from interferons to cytokines. J Biol Chem 282(28):20059–20063 [DOI] [PubMed] [Google Scholar]
- Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472(7344):481–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman RM, Banchereau J. 2007. Taking dendritic cells into medicine. Nature 449(7161):419–426 [DOI] [PubMed] [Google Scholar]
- Swiecki M, Colonna M. 2010. Unraveling the functions of plasmacytoid dendritic cells during viral infections, autoimmunity, and tolerance. Immunol Rev 234(1):142–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tailor P, Tamura T, Kong HJ, Kubota T, Kubota M, Borghi P, Gabriele L, Ozato K. 2007. The feedback phase of type I interferon induction in dendritic cells requires interferon regulatory factor 8. Immunity 27(2):228–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura T, Tailor P, Yamaoka K, Kong HJ, Tsujimura H, O'Shea JJ, Singh H, Ozato K. 2005. IFN regulatory factor-4 and −8 govern dendritic cell subset development and their functional diversity. J Immunol 174(5):2573–2581 [DOI] [PubMed] [Google Scholar]
- Warfield KL, Aman MJ. 2011. Advances in virus-like particle vaccines for filoviruses. J Infect Dis 204Suppl 3:S1053–S1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warfield KL, Bosio CM, Welcher BC, Deal EM, Mohamadzadeh M, Schmaljohn A, Aman MJ, Bavari S. 2003. Ebola virus-like particles protect from lethal Ebola virus infection. Proc Natl Acad Sci U S A 100(26):15889–15894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warfield KL, Olinger G, Deal EM, Swenson DL, Bailey M, Negley DL, Hart MK, Bavari S. 2005. Induction of humoral and CD8+ T cell responses are required for protection against lethal Ebola virus infection. J Immunol 175(2):1184–1191 [DOI] [PubMed] [Google Scholar]
- Warfield KL, Swenson DL, Olinger GG, Kalina WV, Aman MJ, Bavari S. 2007. Ebola virus-like particle-based vaccine protects nonhuman primates against lethal Ebola virus challenge. J Infect Dis 196Suppl 2:S430–S437 [DOI] [PubMed] [Google Scholar]
- Warger T, Osterloh P, Rechtsteiner G, Fassbender M, Heib V, Schmid B, Schmitt E, Schild H, Radsak MP. 2006. Synergistic activation of dendritic cells by combined Toll-like receptor ligation induces superior CTL responses in vivo. Blood 108(2):544–550 [DOI] [PubMed] [Google Scholar]
- Weighardt H, Jusek G, Mages J, Lang R, Hoebe K, Beutler B, Holzmann B. 2004. Identification of a TLR4- and TRIF-dependent activation program of dendritic cells. Eur J Immunol 34(2):558–564 [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S. 2003. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301(5633):640–643 [DOI] [PubMed] [Google Scholar]
- Yamayoshi S, Kawaoka Y. 2007. Mapping of a region of Ebola virus VP40 that is important in the production of virus-like particles. J Infect Dis 196Suppl 2:S291–S295 [DOI] [PubMed] [Google Scholar]
- Zhu X, Nishimura F, Sasaki K, Fujita M, Dusak JE, Eguchi J, Fellows-Mayle W, Storkus WJ, Walker PR, Salazar AM, Okada H. 2007. Toll like receptor-3 ligand poly-ICLC promotes the efficacy of peripheral vaccinations with tumor antigen-derived peptide epitopes in murine CNS tumor models. J Transl Med 5:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.