

Abstract
Reactive oxygen species (ROS) are deadly weapons used by phagocytes and other cell types, such as lung epithelial cells, against pathogens. ROS can kill pathogens directly by causing oxidative damage to biocompounds or indirectly by stimulating pathogen elimination by various nonoxidative mechanisms, including pattern recognition receptors signaling, autophagy, neutrophil extracellular trap formation, and T-lymphocyte responses. Thus, one should expect that the inhibition of ROS production promote infection. Increasing evidences support that in certain particular infections, antioxidants decrease and prooxidants increase pathogen burden. In this study, we review the classic infections that are controlled by ROS and the cases in which ROS appear as promoters of infection, challenging the paradigm. We discuss the possible mechanisms by which ROS could promote particular infections. These mechanisms are still not completely clear but include the metabolic effects of ROS on pathogen physiology, ROS-induced damage to the immune system, and ROS-induced activation of immune defense mechanisms that are subsequently hijacked by particular pathogens to act against more effective microbicidal mechanisms of the immune system. The effective use of antioxidants as therapeutic agents against certain infections is a realistic possibility that is beginning to be applied against viruses. Antioxid. Redox Signal. 20, 1000–1037.
I. Introduction
Reactive oxygen species (ROS) are used by the immune system as weapons against pathogens; however, antioxidants have long been recognized as protectors of host organism against infections (257). It is plausible that the paradox contained in these statements can be solved by the failure of antioxidants to neutralize the ROS involved in pathogen killing (e.g., by not reaching the appropriate location required to their action) and by the capacity of antioxidants to protect immune cells from the damage caused by ROS. The nature of the microbes and their susceptibility to ROS can also offer a clue to solve the paradox. ROS effectively combat certain microbes, whereas other microbes seem to thrive in oxidative environments. Recognizing the mechanisms by which ROS promote the clearance of microbes and the mechanisms by which particular microbes benefit from ROS generation will help to clarify the paradox.
Phagocytes reside within tissues or are recruited by inflammatory processes. Phagocytes recognize microbes through many molecular patterns displayed by them and try to engulf them. Once a microbe is phagocytosed, the nature of the molecules recognized on microbe's surface dictates the treatment enacted within the phagosome. Respiratory burst, a process by which NADPH oxidase (NADPH oxidase 2 [NOX2]) generates ROS in response to microbe recognition, is a possible outcome of this process and helps to get rid of many microbes. For instance, β-glucan on the surface of the fungi can engage dectin-1 on the surface of phagocyte (69). Once fungi are phagocytosed, NOX2 is promptly assembled at the phagosome membrane and high amounts of superoxide (O2−•) are discharged into the phagosome. The recognition of Escherichia coli generates comparatively smaller amounts of ROS than the recognition of Listeria monocytogenes, and these ROS are less promptly released into the phagosome (347). However, the recognition and phagocytosis of Leishmania spp. is well studied, and, except when recognition is mediated by Fc receptor (FcR), virulent parasites do not usually trigger a severe respiratory burst (325) (see section V[D]). Thus, microbes face different fates after phagocytosis dependent on the molecules presented at their surfaces and how they are targeted by innate immune receptors.
Once a pathogen is phagocytosed, it must subvert the respiratory burst, withstand its oxidative power, or escape the phagosome to survive. Microbe recognition sets the immune system in motion, and ROS are produced not only in the phagocyte respiratory burst but also in other cell compartments, such as mitochondria, as intermediaries in many signal transduction pathways, such as leukocyte pattern recognition receptor (PRR) signaling. The generation of ROS is a prerequisite to the formation of neutrophil extracellular traps (NETs) (28); is actively involved in phagolysosomal formation and enzymatic degradation (281); autophagy (118, 119, 298) and ROS inhibition of mammalian target of rapamycin (mTOR) kinase (171, 323); chemoattraction and inflammation (224, 357); cell death of infection reservoirs (9); antigenic presentation, T-helper (Th) polarization, and lymphocyte proliferation (43, 85, 181, 203, 318, 337); iron redistribution among tissues (198) and cell compartment availability of iron (36, 201, 358); and foam cell formation (1, 316). Many of these mechanisms promote microbe clearance, whereas others can potentially contribute to microbe persistence.
In this review, we first provide a brief summary of antioxidants and their mode of action as a tool to understand their use in infection. Next, we discuss the mechanisms by which ROS directly kill microbes or interfere with the immune response. In addition, we discuss the role of ROS in pathogenic viral, bacterial, and protozoan infections, and we highlight to the cases in which ROS production seems to favor infection instead of combating it. The evidence supporting a role for oxidative stress in fueling certain infections is compelling but remains largely unnoticed in the literature to date.
II. Antioxidants and Their Mode of Action
Antioxidants are molecules that act to deplete ROS. Molecules that inhibit ROS-generating pathways, molecules that directly scavenge ROS, and molecules that interfere with ROS degrading pathways can act as antioxidants. Common antioxidants are ROS-scavengers, NOX2-inhibitors, inhibitors of various ROS-generating pathways, and nuclear factor (erythroid-derived 2)-like 2 (NRF2)-activators, which are a class of compounds that induce the expression of antioxidant enzymes, thus classified as indirect antioxidants. ROS-scavengers, such as N-acetyl-cysteine (NAC), which replenishes glutathione, have been by far the most studied class of antioxidants and became accessible at pharmacies, but recently, NRF2-activators raised much interest. Many NRF2-activators are considered “nutraceuticals,” molecules naturally found in foods to which healthy effects have been ascribed: resveratrol (wine), pterostilbene (blueberry), sulforaphane (broccolis), curcumin (turmeric), cafestol (coffee), quercetin (red onion), epigallocatechin-3-gallate (green tea), and carnosol (rosemary) (13). The food conservation additive tert-butylhydroquinone is also a potent NRF2 activator (13). Cobalt-protoporphyrin (CoPP) is a drug largely used in experimental research that is capable of inducing heme-oxygenase 1 (HO-1) expression through NRF2 activation (295). NOX2 inhibitors, however, have attracted less attention, most likely due to their less specific effects on NOX family proteins. Apocynin, the most studied NOX2 inhibitor, derives from vanillin and is nontoxic but has no current use in clinics (313).
The housekeeping production of ROS is generally neutralized by constitutive antioxidant defenses. Oxidative stress ensues when ROS production overwhelms antioxidant defenses. The oxidative hit then promotes the dissociation of kelch-like ECH-associated protein (Keap) from NRF2, allowing NRF2 to translocate to the nucleus where it activates cytoprotective and antioxidant defenses by turning on the transcription of genes that contain antioxidant response element (ARE) motifs in promoters (335). A subject recently reviewed in the literature is the ability of NRF2 to interact with many other transcription factors (27). The indirect antioxidants that act by activating NRF2-dependent mechanisms are usually oxidants that promote transient surges of ROS production and some of them can even act as pro-oxidants in large concentrations. Most NRF2 activators fit into the general definition of “hormetic” agents: they induce a low level of stress that activates the antioxidant defenses, and the general outcome is beneficial to the organism (27).
The NRF2-target genes include the following phase II enzymes: HO-1, NAD(P)H quinione oxidoreductase 1, glutathione peroxidase, glutamate cysteine ligase, and glutathione S-transferases. However, not all genes under NRF2 control are enzymes of direct antioxidant action. For instance, H-ferritin and ferroportin (FPN)-1, proteins that regulate the labile iron pool, contain ARE motifs in their promoters (112, 186) but are only indirectly linked to redox regulation. NRF2 acts on tissue regeneration, DNA repair, and lipid metabolism genes (335). Some promoters that contain ARE motifs, such as that of CD36, also contain peroxisome proliferator-activated receptor (PPAR)-γ-controlled PPARE motifs and can be operated by both factors at a time (233). Recently, the macrophage phenotype MHem was described to result from a genetic program commanded by activation transcription factor (ATF)-1 and NRF2 simultaneously and to be capable of countering foam cell formation (24). This indicates that an NRF2-dependent event does not necessarily activate antioxidant defenses.
NRF2 activation orchestrates a tolerant response that can protect tissues from damage caused by inflammation, infective, and oxidative insult (335). The immune system is specialized to provide resistance against infection, but body tissues directly deal with pathogens and defend themselves by a myriad of mechanisms. Thus, it follows that when tissue tolerance protects body tissues or the immune system itself against infection-induced damage (192), it can potentially affect resistance.
III. ROS Can Promote Pathogen Elimination by Direct Oxidative Damage or by a Variety of Innate and Adaptive Mechanisms
When a microbe is recognized by phagocytes and engulfed, it triggers a process, named respiratory burst, in which phagocytes elevate their oxygen consumption. The enzyme NADPH-oxidase (NOX2) is pivotal to the respiratory burst and attaches to the phagosomal membrane during phagocytosis (353). Particular stimuli such as phorbol-12-myristate-13-acetate (PMA) stimulation can promote NOX2 attachment to the cell membrane. NOX2-derived ROS promote oxidative (Fig. 1) and nonoxidative (Figs. 2–12) mechanisms of microbe elimination.
FIG. 1.
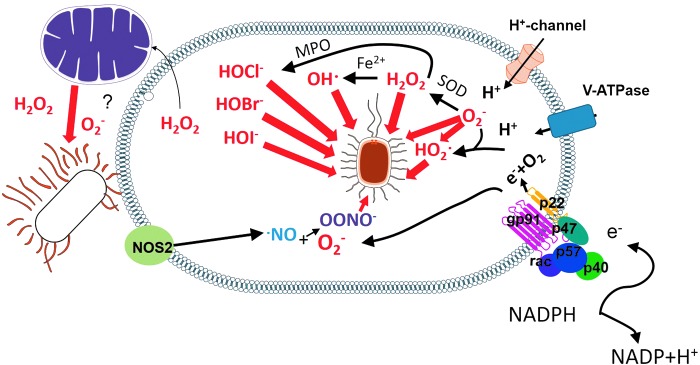
ROS promote pathogen elimination by oxidative mechanisms. Various ROS produced within the phagosome cause oxidative damage (red arrow) to phagocytosed pathogens. ROS from mitochondria also promote pathogen clearance, but whether this effect is caused by oxidative damage is unknown. TLR signaling and NOX2-derived ROS can trigger mitochondrial ROS. NOX2, NADPH oxidase 2; ROS, reactive oxygen species; TLR, toll-like receptor. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 2.
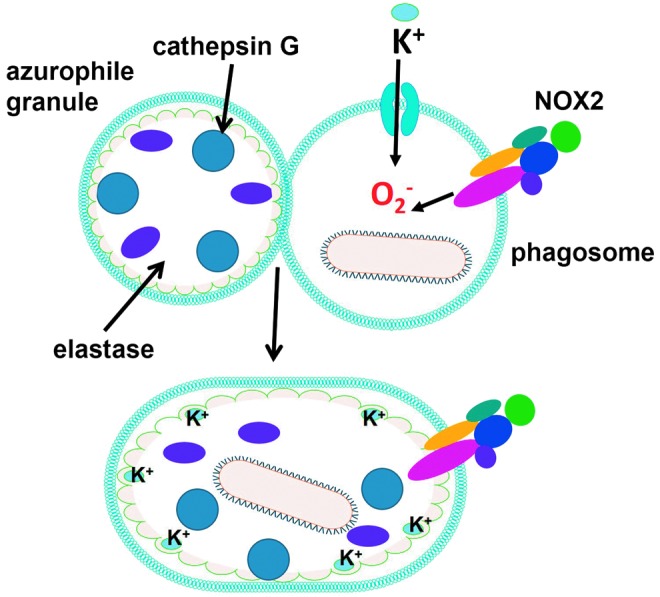
ROS promote pathogen elimination by enzymatic mechanisms. After azurophile granules fuse to the phagosome in neutrophils, superoxide promotes K+ influx to compensate for negative charges. K+ promotes enzyme release from an anionic matrix (green) inside the phagosome, allowing enzymatic assault of pathogens. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 3.
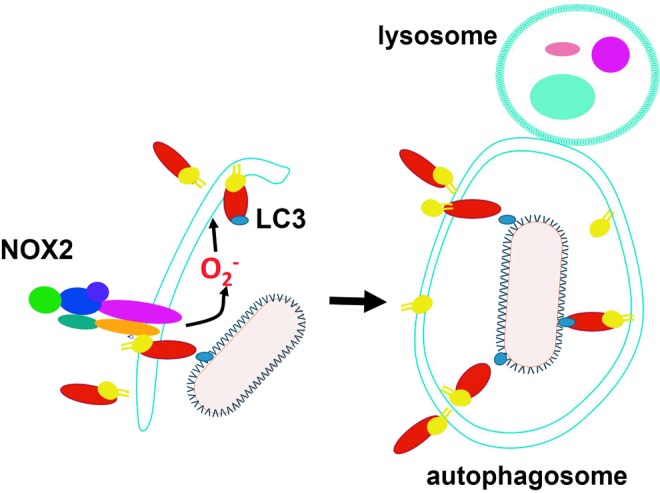
ROS promote pathogen elimination by autophagy. NOX2-derived superoxide promotes LC3 recruitment to the autophagosome. The recognition of microbes in selective autophagy is mediated by proteins that bind to LC3 in a process called xenoautophagy. The autophagosome then fuses with lysosome, where pathogen undergoes an enzymatic assault. LC3, microtubule associated protein 1A/1B light chain 3. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 4.
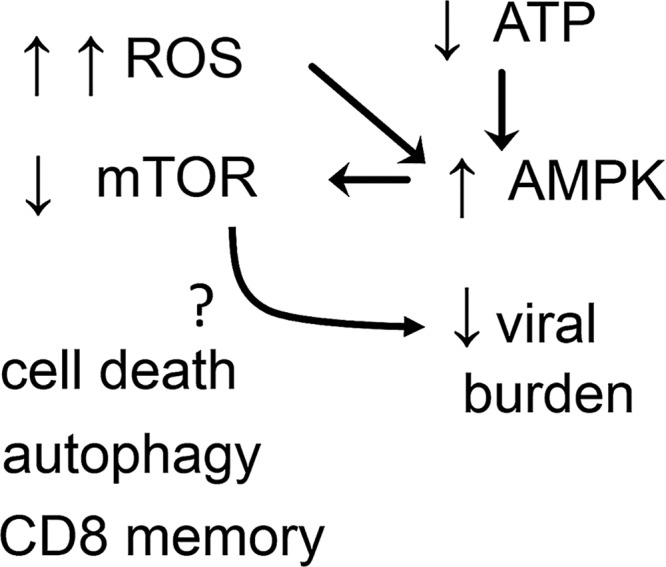
High levels of ROS inhibit mTOR. The nutrient sensor mTOR is kept repressed by AMPK stimulation, a process that can be reinforced by ROS production. The inhibition of mTOR can potentially reduce the viral burden by autophagy, cell death of infected reservoirs, or stimulation of CD8 cell memory. AMPK, AMP-activated kinase; mTOR, mammalian target of rapamycin.
FIG. 5.

ROS production promote ETosis, which traps pathogens in chromatin extracellular traps rich in microbicidal proteins. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 6.
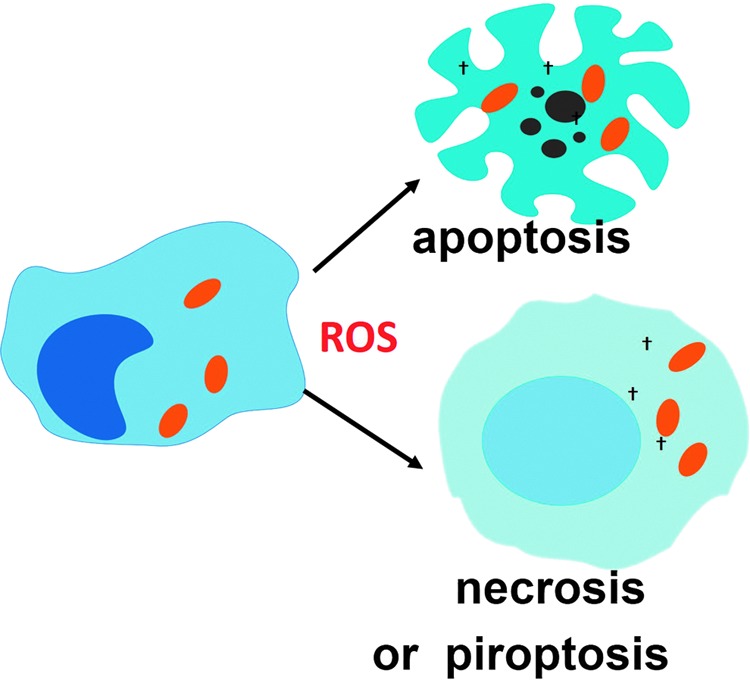
ROS production participates in the various mechanisms of cell death that follows the recognition of intracellular pathogens and contributes to eliminating infection reservoirs. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 7.
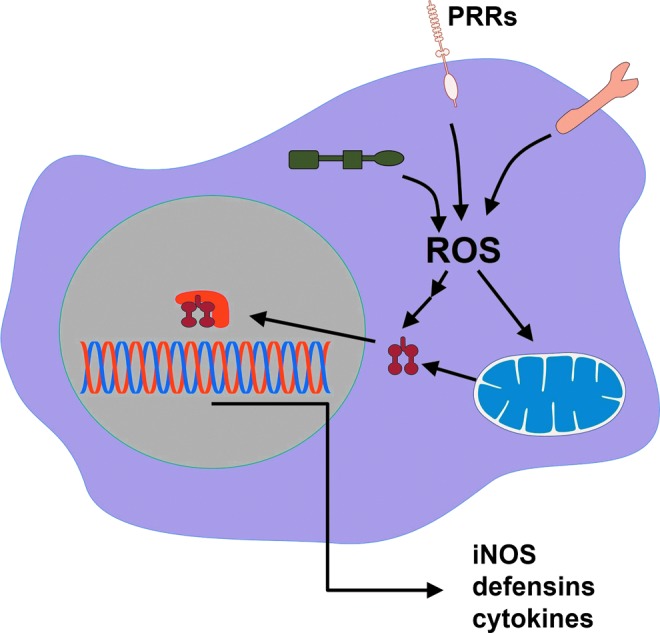
Various PRR signaling pathways use ROS and activate microbicidal mechanisms directly or enhance the expression of proteins involved with microbicidal mechanisms. PRR, pattern recognition receptor. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 8.
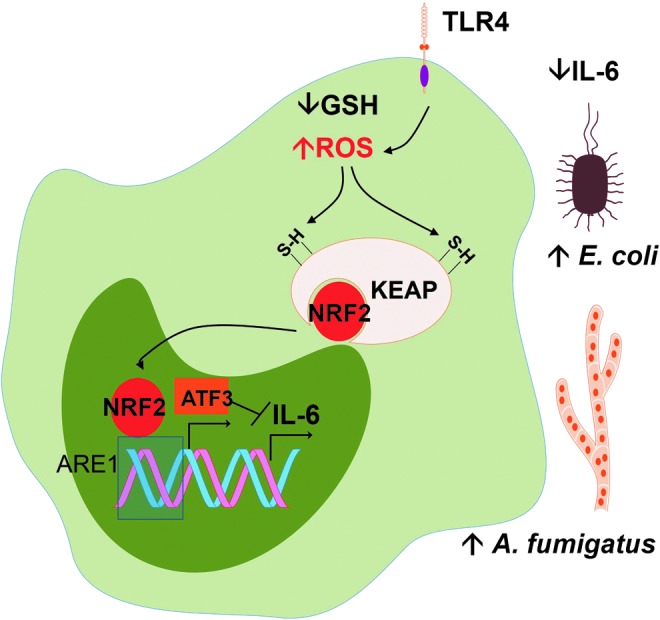
Excessive ROS generated in sepsis reduces defenses against fungi and bacteria. The depletion of endogenous antioxidant GSH, as well as incubation with hydrogen peroxide, increases signaling by TLR4 ligand lipopolysaccharide (LPS), produces NRF2 activation and then increases the expression of ATF3. The two factors act together to reduce transcription of IL-6. Reduction of IL-6 production decreases defenses against Escherichia coli and Aspergillus fumigatus. ATF, activation transcription factor; GSH, reduced glutathione; NRF2, nuclear factor (erythroid-derived 2)-like 2. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 9.
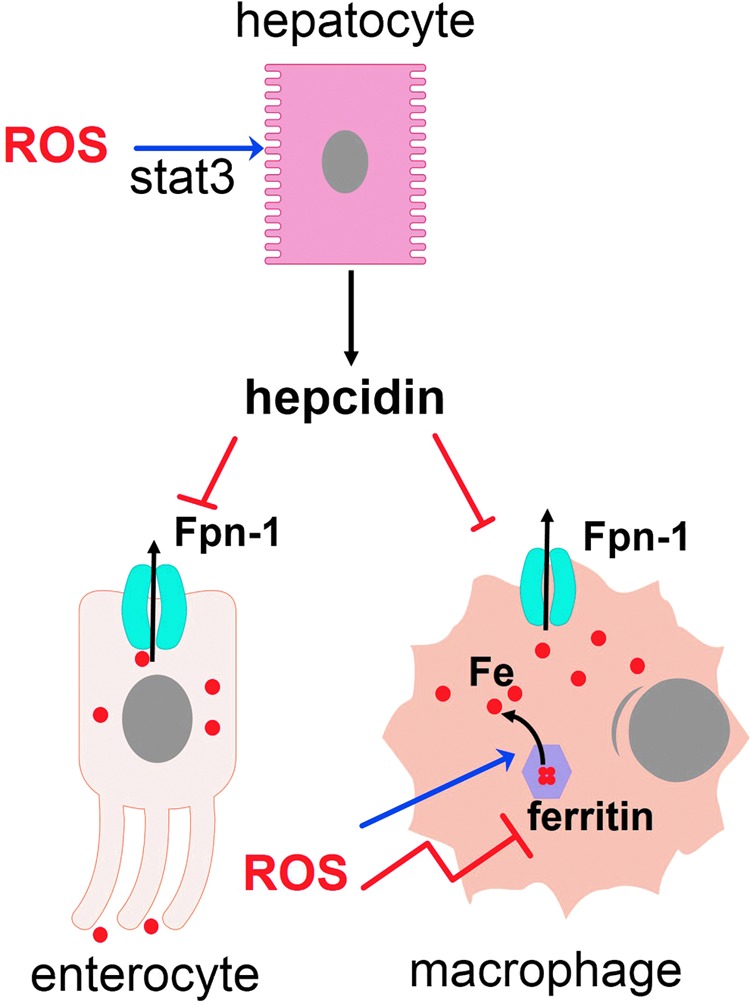
ROS interfere with iron metabolism and iron availability to pathogens residing in different tissues and cell compartments. ROS promote hepcidin secretion by activation of the stat3 pathway in hepatocytes and releases iron from ferritin, increasing the labile iron pool and propagating oxidative stress. The presence of ferroportin-1 (Fpn-1) in cellular membrane is inhibited by hepcidin. Some pathogens that reside in cytosol scavenge ferritin for iron, others degrade heme, but some pathogens feed on the labile iron pool. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 10.

ROS inhibit cholesterol efflux by ABC transporters to high-density lipoprotein (HDL) and contribute to foam cell formation, a macrophage phenotype preferred by various infectious agents, such as HIV, Trypanosoma cruzi, and Mycobacterium tuberculosis. Cholesterol efflux prevents foam cell formation and is associated with iron efflux (through ferroportin-1) in a macrophage phenotype, the so-called MHem. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 11.
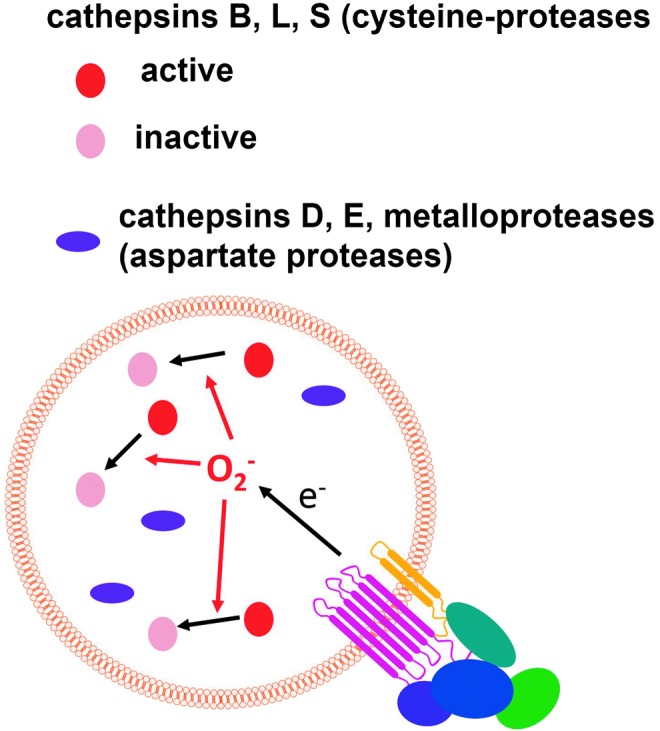
ROS inactivate cathepsins by oxidizing cysteine residues and thus reduce proteolysis inside the phagosome. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 12.
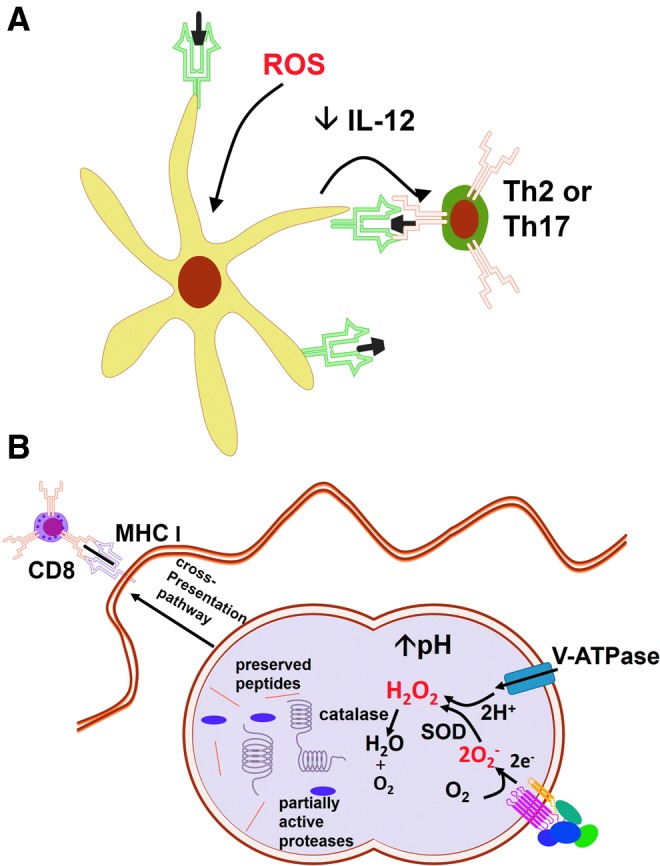
ROS interfere in (A) DC instruction of Th cells and (B) antigen degradation and presentation by DCs. (A) ROS induce physiological changes in DCs, interfering with its antigen presentation capacity, reducing IL-12 secretion, and turning DCs into Th2 or Th17 instructors. (B) ROS enhance antigen cross-presentation in DCs by consuming H+ and increasing phagolysosomal pH, thereby reducing antigenic degradation. DCs, dendritic cells; IL, interleukin. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
NOX2 is a heme-binding protein composed by the subunits gp91phox, p22phox, p67phox, p47phox, p40phox, and Rac1 or 2 (depending on the cell type). The subunit gp91phox forms a transmembrane channel bound to p22phox through which electrons from NADPH are ultimately transferred to oxygen in the phagosome to generate O2−• at micromolar concentrations (353).
Activation of Rac is a critical event for NOX2 assembly at the phagosome membrane. The protein Rac, a member of the Rho family that is usually bound to GDP, is activated by the replacement of GDP with GTP. The GTP-bound Rac and other NOX2 cytosolic components are then recruited to the membrane. The process is further regulated by the following: (i) proteins that help in the replacement of GDP with GTP (guanine nucleotide exchange proteins); (ii) proteins that accelerate the hydrolysis of GTP to GDP and contribute to detaching it from the membrane (GTPase-activating proteins); (iii) proteins that bind to GDP-bound Rac and prevent the interaction between Rac and guanine nucleotide exchange proteins (GDI or GDP dissociation inhibitor family) to prevent Rac activation; (iv) proteins that dissociate the GDI from GDP-bound Rac (GDI dissociation factors) contributing to Rac activation.
Gp91phox and p22phox are stored in intracellular granules, whereas p67phox, p47phox, and p40phox subunits are cytosolic and form a ternary complex (170). On phagocytic stimulation, granules fuse to the phagosome, whereas Rac and the ternary complex bind to the phagosome by independent mechanisms that originate in pathogen recognition. Activated Rac binds to gp91phox. The phosphorylation that p47phox undergoes during phagocytic stimulation allows the interaction of the ternary complex with p22phox-gp91phox. Activated Rac then binds to p67phox leading to NOX2 assembly on the phagosome. The signal pathway from pathogen recognition to Rac activation and its relation to the amount of oxidative attack the microbe will undergo inside the phagosome, if any, are still to be determined.
The host susceptibility to various pathogens in the absence of NOX2-derived ROS production was first observed in chronic granulomatous disease (CGD) patients, which lack a functional NOX2 protein (15, 72). Mice deficient in ROS production have also been shown to be more susceptible to a number of infections. Opportunistic infections such as Salmonella enterica serovar Typhimurium (Salmonella typhimurium), Staphylococcus aureus, Serratia marcescens, and Aspergillus spp. accompany the lack of a functional NOX2 protein. Surprisingly, however, there are scarce demonstrations that the use of pharmacological antioxidants (direct or indirect) can increase pathogen burden, such as in hearts from Trypanosoma cruzi chronically infected mice treated with apocynin (64).In contrast, the antioxidant resveratrol reduced S. marcescens burden in mice (177), and antioxidants NAC and glutathione dramatically increased neutrophil killing of S. aureus in vitro, although they did not kill the bacteria directly (227). The reasons for the scarcity of data indicating that antioxidants can increase pathogen burden and the presence of contradictory data are unknown.
Phagocytosis is tightly coupled to the respiratory burst in phagocytes. As soon as the microbe is engulfed, the delivery of high amounts of ROS inside the phagosome prevents the microbe from escaping to the cytosol. Knowledge of the role of CARD9 in this process is emerging. CARD9-deficient mice are susceptible to L. monocytogenes and fungal infection (90, 106, 117). Wu et al. found that CARD9−/− and wild-type macrophages were able to clear E. coli, but the clearance was faster by wild-type macrophages (347). Nevertheless, CARD9−/− macrophages were unable to clear L. monocytogenes. Knowing that E. coli does not escape the phagosome, whereas L. monocytogenes escapes it 30 min after being engulfed (216), the authors hypothesized that CARD9 was involved in the early ROS surge that kills the bacteria before it can escape from the phagosome. Through further study, researchers found that CARD9 binds to LyGDI, a protein that inhibits the activation of Rac, to unleash Rac to be activated by guanine nucleotide exchange proteins, which leads to the early surge in ROS promoted by NOX2 assembly at the phagosomal membrane (347). Pathogens that take time to escape to the cytosol after phagocytosis most likely undermine CARD9-mediated assembly of NOX2 at the phagosomal membrane or are able to withstand the oxidative stress inside the phagosome.
A. Direct oxidative damage to microbes
Two major lines of evidence support the notion that microbes can be directly killed by ROS. A deficiency in a pathogen's antioxidant machinery can turn a highly virulent pathogen into a ROS-susceptible pathogen, indicating that ROS directly damages microbes (174, 228, 311). Additionally, the oxidative damage caused by ROS, such as lipid peroxidation, DNA strand breakage, base oxidation and deamination, and oxidation of methionine residues (255), can be directly tracked in microbes exposed to respiratory burst. The direct oxidative damage to microbes is shown in Figure 1.
Whether O2−• itself can kill microbes is a question still to be resolved (302). O2−• does not diffuse across membranes efficiently and is rapidly dismutated to hydrogen peroxide (H2O2) by Cu/Zn-superoxide dismutase (SOD) (267). However, H2O2 can diffuse more freely, although it depends on the presence of aquaporin-3 to pass through membranes (196). H2O2 causes direct oxidative damage to many pathogens. It is also a substrate to a metal-catalyzed oxidation that produces hydroxyl radicals (OH·) and a myeloperoxidase (MPO)-catalyzed reaction that produces halogenated acids, such as hypochlorous acid (HOCl−), hypobromous (HOBr−), and hypoiodous (HOI−) acids, which are highly reactive ROS (220). Still, O2−• can also be protonated in the low pH of phagolysosomes to form the highly reactive HO2• species (144).
Although the production of MPO-derived halogenated acids is considered an important oxidative killing mechanism, since MPO-deficient mice and MPO-deficient neutrophils display a decreased capacity to clear a number of pathogens in high doses (8), MPO-deficient individuals do not present particular susceptibilities to infection. A full review on the subject of microbe susceptibility to MPO has been published recently (345).
In the presence of nitric oxide (NO) generated by the enzyme inducible nitric oxide synthase (iNOS), O2−• is consumed to produce peroxynitrite (OONO−) that can be protonated and degraded to form OH· and nitrogen dioxide (·NO2), which are more reactive products (220). However, the expression of iNOS and the ROS/NO ratio that drives the production of a given species depend on the nature of the stimulus recognized by the phagocyte. E. coli, for example, is chlorinated but not nitrated in human neutrophils, suggesting that it is exposed to HOCl− but not to OONO− (277).
Thus, O2−•, although not highly toxic by itself, is a precursor to other ROS, such as HOCl−, OH·, and H2O2, and is combined with NO to form OONO− and ·NO2, which are highly reactive molecules (103).
NOX2-derived ROS production is the main, but not the only, source of oxidative attack on invading organisms (15, 72). Toll-like receptor (TLR) stimulation, for instance, can trigger NOX2 and subsequent mitochondrial-derived ROS production (343). In this case, mitochondria were juxtaposed to the phagocytic vacuoles, and the use of both inhibitors of mitochondrial ROS generation and mitochondrial-targeted catalase expression inhibited the killing of Salmonella typhimurium, demonstrating that mitochondrial ROS can be bactericidal (343). Dual oxidases (DUOXs) are Ca2+-activated NOXs and operate as H2O2-generators in various tissues. ROS production by DUOX2 is required for the killing of L. monocytogenes induced by Nod2 stimulation in intestinal cells (173), indicating that ROS can also promote the elimination of cytoplasmic pathogens, at least in certain circumstances. However, whether mitochondrial and DUOX2-derived ROS kill by direct oxidative damage is still to be fully demonstrated.
Microbes can be eliminated by ROS produced by various nonphagocyte cell types, such as intestinal epithelium (as mentioned above) and lungs, but the subject has not been thoroughly exploited. In airways, DUOX produces H2O2 in response to bacterial infection, and lactoperoxidase acts analogously to phagocyte MPO, converting H2O2 to bactericidal OSCN− and leading to bacterial killing (51).
As the production of ROS is a microbicidal mechanism, it is not surprising that many pathogens downregulate the expression or interfere with the activity of NOX2 or downstream effectors. This subject was reviewed elsewhere (160, 308).
B. O2−• promotes proteolytic elimination of microorganisms indirectly
Although ROS are thought to kill some microbes directly by causing oxidative damage to biocompounds, an alternative mechanism by which the eletrogenic activity of NOX2 is responsible for ROS-promoted elimination of pathogens in neutrophils has been proposed (Fig. 2). O2−• production promotes an increase in K+ concentration in phagosome to compensate for negative charges. Cytoskeleton proteins initially constrain the swelling of the vacuole allowing hypertonicity to develop. The concentration of K+ promotes the release of contents from insoluble granule stores, such as elastase and cathepsin G (bound to an anionic sulphated proteoglycan matrix) enzymes, which in turn kill microbes by proteolytic attack (266). Depending on the cell type, the negative charge of O2−• can be compensated by H+ ions that are pumped by vacuolar-H+-ATPase into the phagosome, indicating that phagosomal acidification and ROS generation can interfere with each other (128, 293). The activation of NOX2 creates a hypertonic, K+ rich, and less acidic environment, which allows the proteases to be activated.
C. ROS promote autophagy
Autophagy is a mechanism of defense against intracellular pathogens such as Mycobacterium tuberculosis (99), Group A Streptococcus (219), Salmonella typhimurium (118), and L. monocytogenes (258). NOX2 has been identified as a factor promoting autophagy [reviewed in (119)] (Fig. 3). In neutrophil infection by Salmonella, NOX2-generated ROS are involved in the recruitment of microtubule associated protein 1A/1B light chain 3 (LC3) to the phagosome, promoting antibacterial autophagy (118). A similar process occurs in intestinal epithelial cells with ROS from other sources. ROS also contribute to the elimination of M. tuberculosis by autophagy (298). The participation of ROS in autophagy is depicted in Figure 3. However, some pathogens sabotage autophagy and take advantage of it to favor infection: Porphyromonas gingivalis (66), Brucella abortus (310), Coxiella burnetii (100), Chlamydia (354), S. aureus (291), Toxoplasma gondii (338), and T. cruzi (275). Whether ROS-promoted autophagy affects pathogen burden in any of these infections remains to be determined.
D. ROS inhibit mTOR kinase, triggering an antiviral response
The activation of the mTOR is a sensor of nutrient status that controls cell growth and metabolism. AMP-activated kinase (AMPK) keeps mTOR repressed, and AMPK is activated by low ATP levels, a phenomenon that signals low nutrient status (340). High amounts of ROS during long timespans inhibit the mTOR pathway (171). Recent experiments demonstrated that ROS inhibit human cytomegalovirus (HCMV) infection by inhibiting mTOR (323) (for more details, see viruses infections combated by ROS in section IV[B]). The inhibition of mTOR can represent an effective mechanism against viral infections (214). The potentially microbicidal mechanisms downstream of mTOR inhibition are autophagy, programmed cell death, and potentiation of CD8 cell memory formation (340) (Fig. 4). Nevertheless, the production of type I interferon (IFN) by plasmacytoid cells depends on mTOR (53). It is not known how mTOR inhibition induced by ROS acts to reduce HCMV infection.
E. ROS promote NETosis
Neutrophils extrude NETs on activation with bacteria, PMA, or interleukin (IL)-8 (95). NETs are formed by chromatin and associated proteins, whereas the neutrophil dies. NETs entrap pathogens and are thought to kill them by delivering bactericidal and fungicidal proteins, such as cathelicidin LL-37, defensins, bactericidal /permeability increasing protein, lactoferrin, MPO, proteinase 3, and elastase. Histones can also kill many pathogens. The ras-related C3 botulinum toxin substrate 2 (Rac2) subunit from NOX2 is required for NET release, and H2O2 can rescue NET release in Rac2-deficient cells (172). Additionally, MPO helps efficient NET formation (240) (Fig. 5). Gene therapy with NOX2 restores the NET formation and controls aspergillosis in cells from CGD patients (18), indicating that NOX2-generated ROS participate in NETs. The NET release mechanism is dependent on a Raf-MEK-ERK pathway of NOX2 activation that is opposed to apoptosis (102). ROS-dependent NET formation was also observed in conditions in which neutrophils remained viable (359). Antioxidants inhibit NET formation (172). Macrophages and monocytes can also release ETs, but their role in the microbicidal activity of macrophages remains unknown.
F. ROS promote cell death of infected reservoirs
Death of infected cells limits the spread of infection by preventing pathogen replication and/or differentiation into a cell-invasive phenotype [reviewed in (9, 159)]. In some particular cases, however, it can aid infection by facilitating the exit of the pathogen from the infected cell or killing phagocytes that could otherwise have acted to eliminate the pathogen. Unbalanced ROS production promotes cell death by various mechanisms including necroptosis, apoptosis, and pyroptosis (38, 42, 78, 161, 329), which can contribute to the demise of infection reservoirs (Fig. 6). Recently, ROS production induced by tumor necrosis factor (TNF) has been shown to participate in both M. tuberculosis elimination and promotion of pathogen infection, depending on the amounts produced by macrophage (269). In this work, low amounts of ROS were shown to be microbicidal and to contribute to controlling M. tuberculosis growth, whereas high amounts induced necroptosis and released bacteria in the surrounding, which favors bacterial extracellular proliferation (exuberant and associated with bacterial cording). The pros and cons of ROS-induced cell death in infection have been reviewed recently (42).
G. PRRs use ROS as signaling intermediaries in inflammation
PRR signaling orchestrates several mechanisms of pathogen elimination, and ROS production can interfere with it. The redox signaling downstream of PRRs, such as toll-like (TLR), nod-like (NLR), C-lectin like, and RIG-like receptors (RLR), controls the expression of genes involved in microbicidal activities (153). In general, it does so by reversible oxidation of cysteine residues in proteins in the NF-κB, AP1, MAPK, and PI3K pathways (321). Thus, it is expected that interference with ROS production can promote pathogen elimination by enhancing redox signaling. TLR-stimulated production of pro-inflammatory cytokines can be enhanced by oxidative stress (74), and TLR-deficient cells do not respond to such stimulation (145, 242, 350). In fact, the literature presents good demonstrations that ROS-enhanced PRR signaling can contribute to pathogen elimination (282, 317, 352).
Inflammasome is a multiprotein complex that senses danger signals and pathogens. The most well-studied inflammasome is the one formed by the NLR NACHT, LRR and PYD domains-containing protein 3 (NALP3). ROS are produced in response to NALP3 activators in Aspergillus fumigatus (284), Candida albicans (91), and influenza A (3) infections, but much controversy surrounds the role of ROS in inflammasomes. There are studies showing that ROS are required for mature IL-1β production induced via P2X7 receptors (206), and antioxidants were shown to prevent pro-IL-1β synthesis or NALP3 expression (67). However, the activation of NRF2, a gene that controls antioxidant defenses (see section III[B]), was shown to be required for cholesterol crystals to activate the inflammasome (80). Rubartelli et al. reviewed the subject and proposed that the activation of antioxidant defenses, which is the usual response to an oxidative hit such as that caused by TLR ligands, is the key to reconcile contradictory studies in favor and against ROS as promoters of inflammasome activation (278). They argue that the greater the activation of Nrf2 by an oxidative insult, the greater the inflammasome response.
H. ROS are chemoattractors to phagocytes
H2O2 from epithelial cells is produced very quickly after tissue injury and appears to be an initial signal that causes neutrophil recruitment to the wound (the region where epithelial tissue was injured) (224). The Src-family kinase Lyn is a sensor of H2O2 and its inhibition impairs neutrophil recruitment to the wound (357). Much is still to be learned about how leukocytes react to oxidative stress through ROS-sensors, but neutrophil recruitment by H2O2 most likely contributes to rapid pathogen clearance after tissue injury.
I. ROS can activate NRF2-target genes, a part of the antioxidant defense response that interferes with innate immunity
In section II, we discussed in detail how ROS activate antioxidant defenses through transcription factor NRF2. For now, we should assume that NRF2 is a transcription factor that orchestrates the antioxidant response to oxidative stress and is the target of many antioxidants, the so-called indirect antioxidants. Oxidative stress in macrophages leads to NRF2-dependent ATF3 (a negative regulator of TLR4) superexpression during sepsis. ATF3 superexpression protects against endotoxic shock but increases susceptibility to fungal and bacterial infections by inhibiting IL-6 production (114) (Fig. 8). In this work, ROS production was increased in reduced glutathione (GSH)-depleted monocytes. The liver load of A. fumigatus and the E. coli bacteremia were increased in response to GSH depletion, challenging the overall notion that antioxidant conditions are detrimental to defenses against fungi and bacteria. Thus, it is possible that oxidative stress can fuel infection in a number of cases by ultimately turning off the production of IL-6.
Another NRF2-target gene that participates in innate immunity is CD36 (58, 123). The molecule CD36 is a scavenger receptor best known for its capacity to bind to oxidized low density lipoprotein (LDL) and mediate its internalization, which is part of the atherosclerotic foam cell formation. CD36 also recognizes some microbes and is able to enhance the phagocytosis of Plasmodium falciparum (233). Nevertheless, the NRF2-dependence of CD36 expression is somewhat controversial, as it seems to depend simultaneously on PPARγ (252) and also on monocyte/macrophage differentiation at least in some instances (149). Still, NRF2 controls genes involved in iron and lipid metabolism, described below.
J. ROS interfere with iron storage and tissue mobilization, influencing iron availability to pathogens
The levels of iron are tightly regulated in the body. Iron is preferentially transported and stored in its redox-inactive form because its free form, the so-called labile iron, can generate cell-damaging ROS (26). Iron is a required nutrient for pathogens and its sequestration away from pathogen reach is part of innate defenses (132). ROS stimulate secretion of hepcidin, a hormone that commands mobilization of iron from the liver to macrophages (198). Ironically, oxidative stress releases iron from intracellular storages (36, 201, 358), thus propagating ROS generation and offering some cytosolic pathogens an opportunity to acquire this nutrient. The outcome of an infection, in certain instances, can presumably depend on the capacity of a given pathogen to withstand oxidative damage, its need for iron, and iron availability in its niche (Fig. 9).
The NRF2-target gene Hmox1 codes for HO-1 (for HO-1 as part of NRF2-orchestrated antioxidant defenses, see section III). HO-1 is an enzyme that has antioxidant capacities, not only because it degrades the prooxidant group heme but also because it leads to biliverdin production, an antioxidant molecule, which is next converted by biliverdin reductase to bilirubin, an even more potent antioxidant (87). Some microorganisms use heme as a source of iron and therefore can be directly affected by the reduced heme availability in the presence of greatly increased HO-1 expression. S. aureus, for instance, takes up hemoglobin and degrades its heme group to produce the free iron it needs (300). HO-1 overexpression produces iron overload in macrophages, but most of this iron is ferritin-bound, due to increased expression of ferritin that comes along with HO-1 overexpression. In fact, HO-1-overexpressing cells have 25% of the free iron usually found in the original cell lineage (162). However, tissue iron is concentrated in splenic and hepatic macrophages in wild-type mice, but in the genetic absence of HO-1 expression, iron leaves these macrophages and can be found in kidneys and hepatocytes (150). The reasons for such redistribution are unknown, but most likely linked to the active heme degrading function in macrophages, since macrophages from HO-1−/− mice die after erythrophagocytosis.
Iron availability to pathogens can greatly influence the outcome of infection. The mononuclear phagocyte system and the liver constitute the main iron reservoirs in the body. A number of hormones and cytokines orchestrate the iron mobilization between those reservoirs.
Hepcidin is mainly secreted by hepatocytes in response to macrophage IL-6 and bone morphogenetic proteins agonists and binds to FPN, a transmembrane protein that exports iron and is expressed in macrophages, hepatocytes, and enterocytes (68). The interaction between hepcidin and FPN produces the degradation of the later. In enterocytes, FPN degradation reduces iron absorption, reducing the flow of iron into the plasma. Plasma iron-binding proteins, which have a general bacteriostatic effect (except for transferrin, targeted by some microorganisms), are upregulated on infection and efficiently sequester iron from extracellular pathogens. These proteins include ferritin (which besides being a major intracellular iron storage, is also present in low amounts in the plasma), lactoferrin, haptoglobin (hemoglobin-binding transporter), and hemopexin (heme-binding transporter). In macrophages, FPN degradation produces iron sequestration inside these cells, so hepcidin provides an opportunity for intracellular microorganisms residing within macrophages (68). The net result of hepcidin action is the mobilization of iron from hepatocytes to macrophages, and the increase in ferritin expression within macrophages. In P. berguei infection, iron mobilization by hepcidin results in decreased liver infection, due to decreased iron availability (253). However, nontyphoidal Salmonella grows within macrophages and its growth is reduced by high levels of FPN expression, whereas hepcidin reverses this effect (48). A similar phenomenon occurs in human immunodeficiency virus (HIV) infection (349). Decreased burden as a consequence of increased FPN expression occurs in M. tuberculosis (131) and also in T. cruzi-infected macrophages (235). Therefore, it should be expected that pathogens that live in the liver and those that live in macrophages exploit the hepcidin-FPN axis to allow maximal iron availability.
In addition, depending on the cell compartment in which the pathogen lives, iron availability can vary. Intracellular pathogens that live inside a phagosome (such as Leishmania, Mycobacteria, and Salmonella) can be subjected to iron starvation mediated by natural resistance-associated macrophage protein (NRAMP)-1, a protein that pumps iron out of the phagosome (132). However, pathogens that live in the cytosol can directly scavenge iron-laden ferritin, as is performed by Neisseria meningiditis (164), or feed on the labile iron pool (non-bound to ferritin), as is performed by Leishmania donovani (61). L. amazonensis expresses an iron transporter called LIT1 and depends greatly on iron availability inside the phagolysosome to express the iron superoxide dismutase (FeSOD), to differentiate into amastigotes, and to grow efficiently inside macrophages (200). Contrary to expectations, L. major can be eliminated in certain conditions of systemic iron delivery, apparently due to increased NF-κB activation caused by increased ROS (19).
M2 macrophages release iron, whereas M1 macrophages exhibit iron retention (34). The generation of NO by macrophages is dependent on the labile iron pool. The activity of iNOS has previously been associated with iron content inside macrophages (341). This finding was confirmed in Mycobacterium-infected macrophages because the upregulation of FPN reduced bacterial burden in its early stages and also decreased NO production in response to macrophage stimulation by lipopolysaccharide (LPS) (131).
Oxidative stress and iron metabolism are closely related. The intricate relations between ROS and iron had been the subject of recent studies. It is well known that excess free iron generates oxidative stress, whereas iron sequestration by ferritin precludes it (26). ROS are in fact able to release iron from iron-containing proteins (201), inhibit ferritin synthesis, increase iron uptake (36), and propagate ROS formation. Inside the cell, nonferritin bound iron complexes with small molecules in different cell compartments that comprise the labile iron pool. In the cytosol, most labile iron forms a complex with glutathione, a molecule involved in antioxidant defenses of the cell (111), and high levels of glutathione are limiting for iron metabolism (154). A short exposure to redox-active agents produces parallel increases in ROS and the labile iron pool (25), an effect that seems to be the net result of a number of phenomena. H2O2 increases the degradation of l-ferritin by oxidative modification and produces iron release from ferritin, increasing oxidative stress (358). In hepatocytes, H2O2 upregulates hepcidin secretion through STAT3 induction (198), most likely deviating iron from hepatocytes to ferritin-bound macrophage storages. NAC reverses this effect. Together, these data demonstrate that ROS can mobilize the labile iron pool and produce the propagation of ROS production, while also contributing to the redistribution of iron among tissues (136).
Loading of macrophages with saturated Fe-ferritin produces lysosomal membrane permeabilization (a condition associated with cell damage) when cells are subsequently exposed to H2O2. Incubation of macrophages with apo-ferritin prevented lysosomal membrane permeabilization, suggesting that the progressive chelation of free iron by ferritin represents a defense against oxidative stress by preventing lysosomal membrane permeabilization and cell damage (157). In fact, certain microorganisms exploit the redox balance of the cell by inducing the expression of ferritin, as is observed with Chlamydia trichomatis, which reduces ROS-induced apoptosis to preserve the host cell (330). Moreover, NRF2 activators, the so-called indirect antioxidants, increase ferritin and FPN expression (112, 186) since both genes have ARE consensus in their promoters, in agreement with the general role of NRF2 in tissue protection against damage. The outcome of oxidative stress actions on iron availability to each microorganism will depend on the tissue the microorganism infects and/or the intracellular compartment it lives in, and the iron availability can greatly impact the infection. The case for T. cruzi, a cytosolic infection in which oxidative stress acts to increase the labile iron pool in macrophages and to favor parasite growth (235), is discussed in detail in a section below (V[D]).
K. ROS interfere with lipid metabolism and foam cell formation
Atherosclerosis and oxidative stress are closely associated. Macrophage accumulation in the vascular wall occurs in atherosclerotic lesions and contributes to plaque formation. On uptake of oxidized lipids through scavenger receptors, macrophages turn into foam cells, a phenotype specialized in lipid accumulation within cytosolic droplets (1, 316). Several pathogens induce foam cell formation, and some pathogens seem to benefit from this formation to maintain their optimal growth, whereas it appears to be counterproductive to others. Some of these pathogens, such as Actinobacillus actionomycetemcomitans, P. gingivalis, Chlamydia pneumonia, HIV, have even been observed in clinical specimens taken from atherosclerotic plaques (151, 155, 202, 209). Foam cell formation can be stimulated by the inhibition of cholesterol efflux through ABCA1 and ABCG1 to high-density lipoprotein (HDL) (360), and recent studies have demonstrated that ROS is a major factor inhibiting cholesterol efflux through ABC transporters, favoring foam cell formation (46, 76, 319). In fact, the oxidative stress and lipid peroxidation produced by iron ascorbate in macrophages (through Fenton reaction) inhibit cholesterol efflux through ABC transporters (183). Therefore, it is highly likely that ROS can influence the outcome of macrophage infections through the inhibition of cholesterol efflux and the generation of the seven cell phenotype.
Several pathogens (both those that are harbored inside macrophages and not) induce foam cell formation: HIV (209), Streptococcus sanguinis (232), Porphyromonas gingivalis (259), M. leprae (55), M. tuberculosis (299), C. pneumoniae (37), T. cruzi (60), L. amazonensis (249), T. gondii (45), P. falciparum (124), and P. berghei (273), to name a few. HIV (209) and C. pneumonia (155) have been observed infecting foam cells in atherosclerotic plaques, indicating that infection and foam cell formation co-exist in the highly oxidant environment of atherosclerotic plaque. HIV Nef is responsible for foam cell formation (209), whereas C. pneumoniae LPS is capable of inhibiting cholesterol efflux and turning macrophages into foam cells (137). In fact, bacterial LPS can induce foam cell formation (40), and other TLR ligands from many different bacteria also appear capable of inducing foam cells (223). M. tuberculosis diverts the cell metabolism toward ketone body synthesis, contributing to the formation of foam cells (299). Studies on L. major-infected macrophages have revealed that the microbe downregulates the expression of ABCA1, producing lipid accumulation inside the cell (262). In some of these infections, a role for foam cell generation in pathogen survival and growth has been proposed. In other infections, the opposite holds true.
Some pathogens reduce cholesterol efflux to HDL and alter cholesterol homeostasis in favor of foam cell formation, such as C. pneumoniae (via the JNK-PPARγ-ABCA1 pathway) (176), HIV (the HIV protein Nef downregulates ABCA1 expression) (209), and M. tuberculosis (299). In HIV infection, there is evidence that the inhibition of cholesterol efflux contributes to virus persistence at least in CD4 T cells because liver X receptors (LXRs) agonists, which promote ABCA1 expression, reduce viral load, an effect that can be reversed by cholesterol replenishment (130). In macrophages, the stimulation of cholesterol efflux reduces the productivity of the virions, reducing infectivity (209), which suggests that foam cell formation is associated with HIV growth. In patients, a decreased HDL level is observed along with increased ABCA1 and paralleling HIV load, a finding interpreted by authors as being compensatory to the dysfunctional cholesterol efflux (73). Thus, in HIV infection, the evidence points to foam cell formation as a factor contributing to enhancing infection. In C. pneumoniae infection, however, the opposite situation occurs: established foam cells can normally uptake bacteria, but this gives rise to smaller bacterial burdens (21). In agreement with these data, in the closely related C. trachomatis, the growth is also impaired along with the inhibition of cholesterol efflux through ABCA1 (54), indicating that the generation of foam cells represents an innate mechanism of defense against this pathogen. M. tuberculosis, however, depends on the generation of foam cells for its survival (244, 299). In T. cruzi-infected cells, the formation of foam cells is associated with the prostaglandin E2 synthesis that favors macrophage deactivation of microbicidal actions and parasite growth (59).
L. monocytogenes growth is increased in LXRα−/− (a nuclear receptor that controls ABCA1 expression) macrophages, but this phenomenon does not appear to be related to cholesterol efflux, as authors of one study examining the issue claim that both LXRα and LXRβ participate redundantly in cholesterol efflux, and they found a role for the former but not for the latter in affecting the bacterial burden (134). Nevertheless, LXRα−/−LXRβ−/− macrophages presented additional susceptibility that demanded further explanation. The recent finding that ABCA1−/− macrophages have decreased L. monocytogenes burden (364) suggests instead a role of foam cell formation as an innate defense mechanism. It appears that foam cells can be either more susceptible or more resistant than macrophages, depending on the pathogen considered. ROS promotes foam cell formation, and it is possible that ROS promotes infection when a pathogen grows better in foam cells and vice versa.
The interplay between lipid accumulation within macrophage foam cells, oxidative stress, and labile iron pool has been the subject of some recent works. In general, oxidative stress reduces the cholesterol efflux through ABCA1 and ABCG1 transporters (46), contributing to the preservation of the foam cell functional phenotype, whereas antioxidants increase cholesterol efflux and prevent foam cell formation (31, 239, 363). In agreement with these data, the exposure of macrophages to haptoglobin:hemoglobin complexes in vitro for 7 days increases FPN expression, decreases labile iron, and increases ABC cholesterol efflux transporters (ABCA1 and ABCG1) expression by reducing ROS generation and turns macrophages into a phenotype resistant to cholesterol loading. The degradation of FPN by hepcidin reverses the foam cell-resistant phenotype, reduces cholesterol efflux, and increases labile iron and ROS generation. The incubation with SOD significantly increased ABCA1 and ABCG1 expression, which is in perfect agreement with the idea that oxidative stress inhibits cholesterol efflux to HDL (76). In addition, the pharmacological suppression of hepcidin (see section III[J]) reduces the labile iron pool in macrophages, increases FPN expression, enhances macrophage cholesterol efflux, increases ABCA1 expression, decreases H2O2 generation, decreases foam cell formation, and atherosclerotic fatty streaks (283). Paralleling these results, in one study, incubation with heme induced a macrophage phenotype resistant to cholesterol loading due to LXRα and ABCA1 expression (24). This new macrophage phenotype is apparently the same in three different works, and J. J. Boyle proposed calling it MHem (23). Together, these data indicate that cholesterol accumulation in lipid droplets, foam cell formation, and atherosclerosis are in fact related to a macrophage phenotype that is characterized by iron retention and high ROS generation. Whether this phenotype MHem is present in foam cell-inducing infections and plays a role in their infectivity or pathogenicity remains unclear.
As shown in this section and in the previous section, HIV, T. cruzi, and M. tuberculosis benefit from foam cell formation (59, 209, 299) and grow less actively when FPN expression increases iron efflux (131, 235, 349) (Fig. 10). Whether this correlation is related to the MHem versus foam cell phenotype remains to be investigated.
The activation of the Nrf2 gene results in a plethora of mechanisms specialized in lipid metabolism, as detected by proteomic studies (143), and although the effects of Nrf2 on atherosclerosis have been the subject of much debate (279), it appears that NRF2-activators increase cholesterol efflux and prevent foam cell formation (152, 239). Whether ROS determine the outcome of macrophage infections through cholesterol efflux and foam cell formation remains to be established.
L. ROS influence phagosomal proteolysis through cathepsin inactivation
Phagosomal proteolysis is controlled by NOX2-derived ROS, which produce the inactivation of specific cathepsins by oxidation of the catalytic cysteine residues inside macrophage phagosomes (Fig. 11), whereas antioxidants such as resveratrol, quercetin, and diphenylene iodonium (DPI) increase the bulk proteolysis within phagosomes (281). The cathepsin-specific inactivation produced by NOX2 can potentially produce a particular pattern of proteolysis with the emphasis on some antigenic properties during infection.
M. ROS interfere with protein immunogenicity, antigenic presentation, Th polarization, and co-stimulation by dendritic cells
Dendritic cells (DCs) are responsible for the antigen capture, processing, and presentation to T cells that promote T-cell activation and influence the achievement of particular T-cell effector functions. NOX2-derived ROS is required for antigen processing by DCs and can interfere with adaptive immunity (181), increasing immunogenicity of proteins, inducing physiologic changes in DC, or influencing polarization of lymphocytes during antigen-specific responses [reviewed by (148)]. In general, antioxidants produce Th1-polarized responses, whereas the oxidative stress induced by exposure to prooxidants, oxidized proteins, or exhaustion of antioxidant defenses, generates Th2 (203, 243) or Th17 responses (336, 337) (Fig. 12A). An interesting picture has emerged from a work in which responses to oxidative stress generated by diesel particles interfered with the ability of TLR agonists to induce the expression of maturation receptors in DC, thus programming DCs to instruct Th2 responses (43). Oxidative stress increases influenza virus titers in association with increased polarization of Th2 responses (85). The generation of ROS stimulated by papain was shown to orchestrate Th2 responses by generating TLR4-induced lymphopoietin secretion, directly inhibiting IL-12 secretion by DCs, and promoting DC–basophil interaction in lymph nodes (318). It is possible that oxidative stress acts through the activation of NRF2, as Nrf2−/− DC fail to react to diesel particles by programming cells to Th2 responses (43), and NRF2 activation is known to directly induce Th2 polarization (271).
ROS interference in adaptive immunity can potentially alter resistance to pathogens, and antigen processing and presentation are subject to ROS regulation. ROS and pH are critical to antigen degradation within DC phagosomes, as reviewed elsewhere (148). The consumption of V-ATPase-pumped H+ by NOX2-derived O2− increases pH in DC phagosomes, reducing proteolysis and allowing antigen preservation for presentation (Fig. 12B). In fact, ovalbumin presentation to CD4+ cells is impaired by DPI, an inhibitor of flavoproteins that decreases NOX2-derived ROS (189). The MHC class I presentation may be compromised under oxidative stress, as demonstrated in macrophage T. cruzi infection, in which ROS inhibit protein tyrosine phosphatase activity, thereby inhibiting IFNγ-mediated immunoproteasome synthesis and causing MHC class I downregulation (17). In various models, ROS have been demonstrated to interfere with antigenic cross-presentation to CD8 cells (148). Recently, NOX2-inhibitors were shown to decrease the antigenic cross-presentation by DC to CD8 T cells due to defective autophagy and alkalization of phagosomes, in a model in which NOX2 controls autophagy of Aspergillus conidia (178). It is possible that the susceptibility of NOX2-deficient mice as well as of CGD patients to some of the pathogens depend on the adaptive CD8 response instead of on the microbicidal phagocyte response.
IV. Pathogens That ROS Contribute to Eliminating
A. Bacterial infections combated by ROS
Most bacterial infections are at least partially susceptible to ROS produced by phagocytes. In CGD, there is susceptibility to fungal (Aspergillus, Candida) and to particular bacterial infections, indicating that the immune system relies on NOX2-derived ROS production to clear these pathogens. The main bacteria that prosper in the absence of NOX2 are S. aureus, L. monocytogenes, Francisella tularensis, S. typhimurium, and S. marcescens (15, 303). It has been previously proposed that CGD patients are susceptible to catalase-positive microbes but exhibit normal killing of noncatalase microbes through the production of HOCl− using the H2O2 produced by microbes themselves in the phagosomes (168, 339).
The most frequently isolated bacterial pathogen from CGD patients is S. aureus (303). S. aureus activates oxidative and nonoxidative microbicidal mechanisms in neutrophils (110). NOX2 (gp91phox) hemizygous mice infected with S. aureus develop abscesses in the abdominal cavity, different from wild-type mice, and have impaired bacterial clearance in peritoneum but can ultimately clear the infection (251). NOX2 (p47phox)−/− mice kill 10-fold fewer S. aureus than wild-type mice (125). These results indicate that ROS production is effective against S. aureus infection, but in its absence, other less effective immune mechanisms undertake clearance and ultimately eliminate the infection.
L. monocytogenes grow unrestricted in NOX2 (gp91phox)−/− mice, and the mice develop high splenic and hepatic bacterial burdens, along with an increased number of microabscesses (65). The enhanced susceptibility of gp91phox−/− mice can be detected from 2 days on, the time when lesions are caused by neutrophils in the liver (65). Nevertheless, by day 6 postinfection, the mice are able to control the infection, most likely due to the establishment of an adaptive response. NOX2 (gp91phox)−/− mice have been shown by another group to resist L. monocytogenes infection with a splenic bacterial load equal to that of wild-type mice, even though their macrophages displayed no ability to kill L. monocytogenes in vitro (297). Another NOX2-deficient murine model, p47phox−/− mice, is relatively resistant to infection (70). The differences between these two CGD murine models were exploited recently (356). The authors observed that the macrophages from the p47phox−/− mice have an increased STAT6 phosphorylation in response to IL-4 compared with gp91phox-/- or wild-type mice, producing the alternatively activated macrophage phenotype (high expression of Ym1 and FIZZ1). How precisely the alternatively activated macrophage phenotype produces resistance to L. monocytogenes remains to be elucidated, although it is known that the cells hypersecrete IL-1α, and this secretion is capable of increasing resistance to the bacteria.
Activated macrophages retain L. monocytogenes in vacuoles for microbicidal purposes. In nonactivated macrophages, L. monocytogenes escapes from macrophages vacuoles within 30 min and incubation with the iNOS inhibitor NG-Monomethyl-l-arginine acetate enhances this escape; in contrast, SOD produces only a mild enhancement of the escape (216), indicating that NO is more important for the prevention of cytosolic escape of L. monocytogenes. However, both macrophages from NOX2 (gp91phox)−/− and iNOS (NOS2)−/− mice allowed easier L. monocytogenes escape from the phagosome in activated and nonactivated cells, with a greater enhancement of escape from phagosomes from NOX2 (gp91phox)−/− mice (216). The discovery of CARD9, a molecule that mediates the initial respiratory burst that eliminates certain bacteria before they have the chance to escape from the phagosome, shed light on the role of gp91phox in the phagosomal escape (347) (see section I).
F. tularensis is a facultative intracellular pathogen that causes intense oxidative stress (250). NOX2 (gp91phox)−/− mice are more susceptible to infection with F. tularensis, developing significantly higher bacterial burdens in lungs and spleens and dying 1 day sooner than wild-type mice (156). Nevertheless, the differences observed in existing studies were small, and the authors have proposed that NOX2 plays a minor role in the host defense to F. tularensis. Neutrophils are recruited to the lungs as soon as mice are infected, but the depletion of neutrophils with monoclonal antibodies did not alter bacterial burden (156). A live vaccine strain opsonized by serum was phagocytosed by neutrophils, but not destroyed (191). In this work, F. tularensis was observed to impair neutrophil activation and disrupt the assembly of NOX2 in the phagosome membrane, preventing ROS production. The genes involved in this process have been characterized (292). In another work, the oxidative stress induced by the infection of macrophages with F. tularensis was dissected. The genes leading to the depletion of glutathione were upregulated, but the incubation with NAC did not affect the bacterial burden (7). Taken together, the evidence supports rather limited importance of NOX2-mediated respiratory burst in the killing of F. tularensis.
S. marcescens is one of the most prevalent pathogens found as an opportunistic infection in CGD (15), but no studies concerning how human phagocyte ROS kill S. marcescens or mouse CGD models infected with S. marcescens have been published.
In one study, M. avis, which is pathogenic to humans and mice, was lethal to 40% of NOX2 (gp91phox)−/− mice by 4–6 weeks postinfection (81). These mice displayed a much higher bacterial load in the lungs and their macrophages developed increased loads when infected in vitro with M. avis. These data are consistent with a case of human CGD that was first manifested by the presentation of M. avis infection (231), indicating that NOX2-derived ROS are an important mechanism of elimination in this case.
One of the most striking cases of bacterial susceptibility to NOX2-derived ROS is Acinetobacter baumannii infection in mice. These bacteria are closely related to Pseudomonas aeruginosa and cause a nosocomial pulmonary infection. Multiple drug resistance contributes to its emerging status. Intranasal infection with A. baumannii kills 100% of NOX2 (gp91phox)−/− mice in 48 h but does not kill NOS2−/− or wild-type mice (261). The bacterial counts are 1000-fold increased in the lungs and 10-fold in the spleens of infected gp91phox−/− mice compared with wild-type mice. To our knowledge, no cases of A. baumannii infections have been reported in CGD patients.
B. Viral infections combated by ROS
There is a paucity of data concerning the role of phagocyte ROS in mediating virucidal activity, but in general, findings support a role for ROS in apoptosis induction as a first line of defense against infection reservoirs (301). However, in betanodavirus (nervous necrosis virus) infection, ROS have been shown to induce apoptosis of the host cells, but catalase overexpression still reduced viral titers at the early stages of infection, most likely because apoptosis helps in the spread of infection in early stages (44).
In 1982, Rager-Zisman et al. used a variant of the J774 monocyte cell line defective in oxidative metabolism to evaluate the contribution of respiratory burst to vesicular stomatitis virus (VSV) clearance (263). PMA-induced oxidative burst significantly reduced viral burden when small amounts of viral particles were used to infect cells. No reduction of viral burden was observed on stimulation of oxidative metabolism-deficient cells and catalase was able to inhibit PMA effects on parental cells, indicating that respiratory burst was indeed responsible for the reduction of viral burden. Free virus suspended in H2O2 solutions were killed only when a limited amount of viral particles were used, suggesting that there is an amount of H2O2 required to inactivate each virus and leading the authors to speculate that it most likely acts on the small extracellular input virus rather than on intracellular viral progeny. In 1992, VSV-infected cells were transfected with CuZnSOD to evaluate whether their sensitivity to type IFN I-antiviral state depended on ROS generation. In fact, the more gene copies of CuZnSOD used in transfection, the more cells became resistant to induction of antiviral state and susceptible to infection, indicating that O2−• participates in determining the sensitivity to type I IFN (120). In this study, paraquat, a drug that induces oxidative stress, reduced viral burden, confirming the role of ROS in reducing viral burden. VSV-infected VERO cells were spared from cell death by antioxidants, which offer a clue to explain the decreased viral burden (268). Epithelial cells deficient in autophagy (Atg5−/−) were observed to be resistant to VSV infection, apparently due to increased RLR signaling (317). Such increased RLR signaling was produced by the deficient autophagy of mitochondria, the main sources of ROS in nonphagocytic cells, generating increased ROS signaling and increasing viral clearance via effector mechanisms downstream of RLR signaling (see section III[G]).
HCMV induces ROS production as soon as it invades a cell (307). These ROS would normally contribute to the effective activation of NF-κB, but the virus has ways to overcome this barrier. Recent experiments have shed light on the subject. HCMV induces non-NRF2-mediated induction of the antioxidant enzymes, SOD, GLCG, and GPX-1 (323). The addition of buthionine sulfoximine (BSO) to HCMV-infected cells, a drug capable of depleting antioxidant GSH, produced unbalanced ROS production that inhibited mTOR and decreased viral growth. Thus, as a result of antioxidant enzymes induction, the levels of ROS are reduced, and H2O2 does not reach the levels necessary to inhibit mTOR. The inhibition of mTOR, a nutrient sensor, halts cell growth and represents an important effector mechanism against viruses (see section III[D]).
Chikungunya virus (CHIKV) is an arbovirus transmitted by mosquitoes. Autophagosome induction in infected fibroblasts is reduced in infected NAC-treated cells, indicating the mediation by ROS (135). Moreover, the oxidative stress caused by CHIKV infection inhibits mTOR (mTORC1, linked to autophagy). In this model, autophagy has been observed to be prosurvival, inhibiting the apoptosis of the host cell, whereas the blockage of autophagy results in massive apoptosis (Z-vad-sensitive). The inhibition of apoptosis decreases the numbers of infected cells, apparently due to the apoptosis-mediated propagation of infection in cell culture, most likely achieved by the enhancement of virus release from apoptotic cells. Although presumably ROS production should inhibit the viral burden as a result of inhibition of both apoptosis and mTOR activation, in the study, the authors focused on the linkage of autophagy, apoptosis and viral propagation and did not directly study how ROS affected viral burden.
In another study, the growth of herpes simplex virus 1 (HSV1) in corneal cells was inhibited by granulocytes, which released H2O2, and the amounts of H2O2 equivalent to that measured were sufficient to cause viral inactivation (109). HSV1 was observed to be somewhat resistant to inactivation by H2O2, and this resistance was mediated by the presence of its viral catalase since catalase inhibition increases its susceptibility (222). These data indicate that the virus is adapted to the oxidative environment where it grows. However, treatment with sulforaphane, an efficient NRF2 activator, did not alter HSV1 burden, despite causing great a reduction in the production of ROS by neutrophils and macrophages infiltrating the central nervous system (288).
Porcine reproductive and respiratory syndrome arterivirus stimulates ROS generation in infected cells, promoting the activation of NF-κB (166). Despite the possible role in pathology, in this case, ROS production and the activation of NF-κB do not seem to be related to viral reproduction, as treatment with antioxidants greatly reduces NF-κB activation but does not alter viral titers.
V. When the Paradigm Fails Us: Pathogens That Thrive on Oxidative Stress
Intriguingly, NRF2 activation results in the reduction of viral burden in respiratory syncytial virus (RSV) infection (49), whereas Nrf2−/− mice are more susceptible to P. aeruginosa (265). The expression of HO-1, a NRF2-activated heme-degrading enzyme that has many antioxidant properties, has also been shown to reduce pathogen burden in hepatitis B (256), hepatitis C (167), enterovirus-71 (324), and HIV (62) infections, to mediate macrophage resistance to S. typhimurium (361) and T. cruzi (235), and to enhance the bacterial clearance of Enterococcus faecalis (50). Whether NRF2-target genes participate directly in innate immunity against pathogens or act by protecting tissues from infection-induced damage and allowing them to respond to pathogens remains to be elucidated. However, ROS scavengers or NOX2 inhibitors also contribute to the control of the burden of some pathogens, and NOX2-deficiency is associated with decreased burden in some cases (84, 188, 235), indicating that the production of ROS is counterproductive against some pathogens.
In the following sections, we dissect infection cases in which oxidative stress appears to enhance a pathogen's growth and antioxidants inhibit infection. Cell death can be a drawback in studies with prooxidants, as they tend to increase the number of dead cells, and the uptake of pathogens released from dead cells by live cells can potentially distort the results (44, 135), and therefore, most of these studies are based only on antioxidants. In addition, one must keep in mind that prooxidants may later turn into functional antioxidants, as they stimulate NRF2-target genes. Heme, for example, is capable of acting as both a prooxidant, as it can induce oxidative stress, and an antioxidant, as it induces HO-1 expression with delayed kinetics.
A. Bacterial infections in which the participation of ROS in clearance is dubious, controversial, or irrelevant
CGD patients have increased susceptibility to M. tuberculosis and also to environmental mycobacteria (32, 158, 165). Human neutrophils are rather inefficient at eliminating M. tuberculosis (52), and neutrophil respiratory burst, besides apparently being dispensable as a mycobactericidal mechanism (133), produces neutrophil necrotic death and allows bacterial escape (52). However, the respiratory burst contributes to the elimination of M. tuberculosis in macrophages, as shown recently in patients with a NOX2 (gp91phox)-deficiency (CGD) that only affects macrophages (33). In mice, the inhibition of TNF-induced macrophage apoptosis is achieved by M. tuberculosis through the inhibition of NOX2-derived ROS by its type I NADH dehydrogenase (197), a source of ROS that seems to be involved in the redox signaling pathway to apoptosis downstream of TNF. These data indicates that ROS in this case participate in a second line of defense beyond the initial respiratory burst, which is the induction of host cell apoptosis that favors the elimination of infection reservoirs, but the bacteria manages to escape this innate mechanism of control.
M. tuberculosis is relatively resistant to ROS, but NOX2-derived ROS mediates the TLR2 inflammatory responses that contribute to clearing M. tuberculosis through the enhancement of vitamin-D receptor-induced cathelicidin expression in macrophages (352). However, the M. tuberculosis gene Eis reduces ROS production, downstream autophagy and pro-inflammatory cytokine production, but still, does not alter bacterial burden (298). Treating M. tuberculosis-infected guinea pigs with NAC resulted in decreased splenic bacterial counts (236), indicating that the role of oxidative stress in tuberculosis deserves further consideration. In fact, GSH is toxic to M. tuberculosis and GSH depletion increases the bacterial numbers inside macrophages (332, 333), but it remains unknown whether GSH disturbs the redox balance of the bacteria. At least some effects of GSH appear to be related to the stimulation of the NK activity (92). The effects of GSH on M. tuberculosis infection are reviewed elsewhere (207).
Streptococcus pneumoniae is not an opportunistic infection in CGD (169). In 2003, Schaper et al. observed that NOX2 (p47phox)−/− mice have an increased bacterial burden in S. pneumonia-induced meningitis, but NOX2 (gp91phox)−/− mice did not differ from wild-type mice (289). Confirming these findings in gp91phox−/− mice, the administration of a low inoculum resulted in bacterial clearance similar to wild-type controls (184). In 2008, examining gp91phox-/- mice subjected to high inoculum pneumonitis, Marriot et al. observed a surprisingly decreased mortality and decreased bacterial burden in the lungs and blood (185). The enhanced recruitment and activation of neutrophils were observed in gp91phox−/− mice, and the authors concluded that gp91phox is fundamental for control inflammation in this infection but not for microbial clearance. The role of ROS in clearance of S. pneumoniae remains to be fully unraveled.
The killing of P. aeruginosa by neutrophils is normal in CGD patients (306). In this work, the authors found evidence that CGD is associated with P. cepacia, but not with P. aeruginosa infection. These results contrast with NOX2 (p47phox)-deficient mice, which present deficient P. aeruginosa clearance in the lungs and by macrophages, due to decreased TLR4-induced NF-κB (282). In fact, p47phox−/− mice have additional macrophage deficiencies (175), and it is important to note that these results have been confirmed in a NOX2 (gp91phox)-deficient model. Accordingly, mice deficient in Vav, a guanine nucleotide exchange factor involved in Rac activation, display increased mortality, deficient oxidative burst, and deficient clearance of P. aeruginosa by neutrophils, but the authors observed that this protein has additional signaling functions to those predicted by its Rac-activating role, and the mechanism by which Vav deficiency promotes infection requires further research to clarify (88). In addition, when studying the response of mice deficient in vitamin D3-upregulated protein-1 to P. aeruginosa bacteremic shock, the authors observed the mice to be resistant, to have increased bacterial clearance, and to produce increased amounts of ROS (248). Treatment with NAC reversed ROS production and resistance, demonstrating that, in this case, ROS was responsible for increased bacterial clearance. Moreover, in cystic fibrosis conductance regulator molecule-deficient mice, the reduced clearance of P. aeruginosa by alveolar macrophages has been associated with the failure to cluster gp91phox in ceramide-enriched membrane platforms, to release ROS, and to acidify vesicles. The inhibition of ROS production by the NOX2 inhibitor apocynin or incubation with ROS-degrading enzymes (SOD and catalase) of P. aeruginosa-infected macrophages results in increased infection (362). Taken together, these data indicate that ROS contribute to the elimination of P. aeruginosa, at least in mice.
Some studies have not found a role for ROS in P. aeruginosa clearance, or even supported a role for ROS in enhancing infection. Pyocyanin, a redox toxin from P. aeruginosa that induces ROS and depletes GSH in neutrophils, does not alter killing by neutrophils (210). A recent study analyzed the role of MUNC13-4 in P. aeruginosa infection. The molecule is necessary for the regulation of p22phox trafficking to the plasma membrane and extracellular ROS production by neutrophils. Phagosomal maturation after phagocytosis of P. aeruginosa was dramatically impaired in MUNC13-4−/− neutrophils, a stimulation that triggers exclusively intracellular ROS production, but the assembly of p22phox in phagosomes did not depend on MUNC13-4. However, the fusion of phagosomes to azurophilic granules depended on MUNC13-4, as well as the killing of P. aeruginosa. To determine which of these effects was required for MUNC13-4 killing of P. aeruginosa, the authors used gp91phox−/− neutrophils in killing assays and observed that NOX2 is totally dispensable for the neutrophil killing of P. aeruginosa (204).
In a different study, treatment with the NRF2-activator sulforaphane produced the enhanced clearance of bacteria (P. aeruginosa and Haemophilus influenza) by the macrophages from chronic obstructive pulmonary disease patients, an effect that could be reversed by siRNA against NRF2 (107). Sulforaphane also enhanced bacterial clearance in the lungs of wild-type mice but failed to do so in Nrf2−/− mice. NAC failed to enhance bacterial clearance, indicating that this is an NRF2-mediated effect rather than a general antioxidant effect, but the failure of NAC to perform appropriately in vivo is a rather common finding and should not be taken as a conclusive evidence of a nonantioxidant effect. Nrf2−/− mice displayed reduced bacterial clearance on exposure to cigarette smoke, an effect that could be credited to the reduced phagocytosis by alveolar macrophages ex vivo under oxidative conditions. In another work, Nrf2−/− mice succumbed to P. aeruginosa infection provoked after hyperoxia insult and had increased bacterial burden compared with wild-type mice (265). The reduced capacity to clear bacteria could be verified as early as 4 h after infection in Nrf2−/− mice. After 48 h, hyperoxia-exposed infected Nrf2−/− mice presented increases in IL-6 and IL-1 production and dramatically reduced the expression of phagocytosis receptors in alveolar macrophages compared with wild-type cells. Supplementation with GSH restored the ability of Nrf2−/− cells to clear the bacterial burden and suppressed the increases in cytokine expression, indicating that in this case, the antioxidant effects of NRF2 mediate the protective effect against P. aeruginosa. Contradictory evidence favoring or refuting a role for ROS in bacterial clearance are most likely due the various cell mechanisms controlled by ROS or due to the differences between mice and humans. These differences need to be dissected and reconciled.
Salmonella is the second most prevalent bacterial infection in CGD (208). Nontyphoidal Salmonella is a common infection causing bacteremia in CGD patients (86), and Salmonella typhimurium has been observed, as an initial manifestation of CGD, to cause bilateral pneumonia and sepsis in association with Pneumocystis jiroveci (303). S. typhimurium is believed to be easily killed by NOX2-derived ROS, despite avoiding NOX2 assembly and remaining in phagocyte vacuoles that do not co-localize with gp91phox (82) since NOX2 (gp91phox−/−)-deficient mice presented increased mortality (297), increased bacterial loads in spleens, livers, and macrophages (187), and their macrophages present less efficient killing of S. typhimurium in vitro (297). These experiments were performed with gp91phox−/− mice in an Nramp1s background to allow for susceptibility to Salmonella and relying on the exclusive and noninteractive effects of the two phenotypes. The susceptibility of Salmonella to killing by ROS was confirmed by the increase in bacterial load observed in mice treated with SOD (326). The survival of S. typhimurium inside peritoneal macrophages from gp91phox−/− mice was greatly increased, and the main killing effects of gp91phox-dependent respiratory burst lasted for less than 5 h since wild-type mice and even NOS2−/− mice cleared a percentage of the bacteria within this interval, which was different from gp91phox−/− and gp91phox−/− NOS2−/− mice (331). Salmonella variants susceptible to oxidative burst (29) or with low capacity to live inside macrophages (75) were identified as avirulent in vivo. In NOX2 (p47phox)−/− mice, an oxidative burst-sensitive mutant regained virulence (328). The restoration of the virulence of a Salmonella variant unable to repair damaged DNA (thereby sensitive to oxidative damage) in gp91phox−/− and gp91phox−/−NOS2−/− mice suggests a role for direct oxidative damage as a mechanism of bacterial killing (297). Taken together, these data appear to draw a clear picture in which Salmonella depends greatly on respiratory burst evasion mechanisms to establish infection, and its growth is favored by the lack of NOX2. Salmonella susceptibility to ROS is reviewed elsewhere (72).
However, Salmonella resists direct oxidative damage, as it is capable of rapidly fixing ROS-induced DNA damage and can adapt to grow normally in highly oxidative environment (250 μM H2O2) within 24 h (182). Surprisingly, Salmonella has been shown to benefit from neutrophil transmigration and ROS production in inflamed guts (312) due to the generation of a luminal oxidized electron acceptor that confers a metabolic advantage and allows Salmonella to outcompete anaerobic microbiota (344). The contrast between Salmonella resistance and enhanced growth in the oxidative environment and its susceptibility to NOX2 activity in macrophages demand further explanation. It is possible that a percentage of Salmonella resists oxidative damage while NOX2-derived ROS promotes Salmonella elimination by nonoxidative mechanisms, such as autophagy (118).
B. Viral infections that thrive in oxidative environments
It is widely accepted that the ROS produced by phagocytes during respiratory burst contribute to the elimination of pathogens. Whereas NOX2-generated ROS can promote direct elimination of microbes engulfed by phagocytes, such as bacteria, fungi, and protozoa, the virus case is more complicated. Viral infections are more frequently inhibited by antioxidants (Table 1), a subject previously examined by Fraternale et al. (79).
Table 1.
Viral Infections Fueled by ROS
| Pathogen | Observed effects of ROS/antioxidants on pathogen's burden | Model | Associated mechanisms of resistance | Suspected mechanisms underlying results | References |
|---|---|---|---|---|---|
| KSHV | H2O2 promotes viral reactivation and replication; NAC, catalase, and gluthatione inhibit replication in culture; NAC reduces viral load in vivo | In vitro infection of BCBL1 cells; KSHV-induced lymphoma in mice | H2O2 promotes viral reactivation by activating ERK1/2 quinase, JNK and p38 | Iron exposure probably acts through ROS generation to reativate KSHV | (355) |
| HIV | PMA promoted viral replication and NAC inhibited it | In vitro infection of human peripheral blood or T-cell lines | Activation of NF-κB was responsible for PMA-driven infection; NAC inhibited such activation | Activation of NF-κB was associated with decreased thiols and NAC increased thiols | (274, 309) |
| HO-1 induction by hemin reduced burden in monocytes and in a humanized mice model | In vitro infection of human monocytes; in vivo infection of Hu-Nod-Scid mice | Inhibition of Tat-dependent activation of LTR promoter | Tat-induced ROS is involved in promotion of infection, and antioxidants possibly act by its inhibition | (62) | |
| Influenza A | Antioxidant NAC reduces viral burden in vitro and in vivo. | In vitro infection of human lung carcinoma and vero cells | NAC inhibited: caspases 8, 9, 3–7 and nuclear export of the virus; production of CCL5, CXCL8, CXCL10, IL-6 and monocyte migration; p38 and NF-κB activation by infection | Casual findings which do not demonstrate why antioxidants reduce influenza A viral burden | (84, 188) |
| NOX2-deficiency reduces viral burden; NOX2-inhibition with apocynin reduces viral burden | Infection of NOX2−/− mice | Preservation of CD8+ producing IL-2, IFNγ, and IL-12; higher levels of IL-1β were found in lungs from NOX2−/− mice | None of the observed effects could offer a clue to how inhibition of NOX2 activity contributes to reduce viral burden | (122, 334) | |
| Activation of NRF2 with sulforaphane and EGCG reduces viral burden; knockdown of NRF2 (lentivirus containing shRNA NRF2) increases viral burden | Infection of nasal epithelial cells | NRF2-activators induced type I IFN, RIG1, and MxA, molecules involved in virucidal mechanisms | Whether the virucidal effects of type I IFN, RIG1, and MxA are involved in the reduction of viral burden induced by NRF2 activation remains to be demonstrated | (140) | |
| RSV | Virus induces oxidative stress; antioxidant NAC reduces viral burden. | In vitro infected human epithelial pulmonary cell A549 | NAC reduced expression of IL-6, IL-8, TNF, MUC5AC, and blocked NF-κB and p38 activation | The possible relations between viral burden and ROS production were considered elsewhere (83) | (41, 188) |
| Nrf2−/− mice has increased viral replication and wild-type, but not Nrf2−/− mice, react to NRF2-activator sulforaphane with decreased viral burden. Oxidative stress in Nrf2−/− mice associated with decreased expression of NRF2-target genes (HO-1, NQO1, GCLC,GST-P1, GPx2). | Infection of wild-type and Nrf2−/− mice and treatment with sulforaphane | Nrf2−/− had increased IL-6, IL-18 and IL-13, eosinophil and neutrophil infiltration, and decreased IFNγ pulmonary levels | Increased burden possibly related to decreased IFNγ pulmonary levels | (49) | |
| PCV2 | Viral replication is inhibited by NAC; GSH depletion restores growth | In vitro infection of PK15 (porcine kidney) cells | Blockage of ROS-induced NF-κB activation inhibits GSH-promoted viral replication | Inhibition of NF-κB activation is known to reduce viral replication and was achieved by antioxidants | (47) |
| Enterovirus 71 | Virus induced ROS generation through integrin β1/Rac1/NOX2 activation. Treatment of infected cell cultures with antioxidants, siRNA to NOX2 p47, or Ad-HO-1 reduced burden. HO-1 by-product CO reduced ROS generation | Infected Vero and SK-N-SH cells | CO generated by HO-1 did not interfere with integrin β1-Rac pathway, but inhibited ROS and viral replication dependent on increased cGMP and PKG activation | No clue to mechanisms by which ROS promoted infection or cGMP/PKG inhibited ROS generation | (324) |
| HCV | HO-1 activity decreases viral burden (CoPP, hemin, transfection) | Human hepatoma cell lines | Inhibition of HCV protein NS3/4A by biliverdin or heme was shown to reverse the blockade on TLR3 and RIG-1 signaling caused by infection and to restore the type I IFN production in infected hepatocytes | Results suggest that HO-1 acts through rescue of type I IFN production | (98, 116, 296, 366) |
| H2O2 increases HCV replication in hepatocytes | Human hepatoma cell lines | Low doses of H2O2 (100 μM) increases HCV replication in hepatocytes, while high doses (1 mM) inhibit it | No mechanism was shown that could explain why H2O2 increases HCV replication | (167) | |
| HBV | Induction of HO-1 (CoPP) reduced viral burden in liver or in hepatocytes (treatment with CoPP was reversed by HO-1-specific siRNA knockdown) | HBV-transfected hepatomas and HBV-transgenic mice were used in the study | Induction of HO-1 reduced the levels of HBV core protein | Post-transcriptionally inhibition of HBV core protein mRNA by HO-1 was indicated by authors as a possible mechanism of action for CoPP | (256) |
CoPP, cobalt-protoporphyrin; GCLC, glutamate cysteine ligase catalytic subunit; GSH, reduced glutathione; GST, glutathione S-transferase; H2O2, hydrogen peroxide; HBV, human hepatitis B virus; HCV, human hepatitis C virus; HIV, human immunodeficiency virus; HO-1, heme oxygenase 1; IFN, interferon; IL, interleukin; KSHV, Kaposi's sarcoma-associated herpesvirus; NAC, N-acetyl-cysteine; NOX2, NADPH oxidase 2; NRF2, nuclear factor (erythroid-derived 2)-like 2; pCV2, porcine circovirus type 2; PMA, phorbol-12-myristate-13-acetate; ROS, reactive oxygen species; RSV, respiratory syncytial virus.
Viruses are usually targeted by autophagy but are barely detected by PRRs on the surface of phagocytes for engulfment and subsequent exposure to ROS and reactive nitrogen species (RNS). Rather, dying virus-infected cells (2) or Ig-opsonized viruses are engulfed (104). Endosomal TLRs, sensors of viral infections, do not augment ROS production, which is different from cell surface TLRs (343). Nevertheless, other sensors of viral infections, such as cytosolic NLRP and RLR, employ ROS as part of cell signaling. ROS can thus potentially influence the elimination of viruses by nonoxidative routes, such as by increasing PRR signaling. Virus detection by intracellular PRRs trigger the production of inflammatory cytokines, type I IFN, autophagy, unfolded protein response, apoptosis, necroptosis, and the onset of an adaptive response that targets virus reservoirs. ROS can also potentially interfere with all these processes. Moreover, many viruses have developed an evasion strategy that depends on NF-κB to allow their growth, such as influenza (225) and HIV (217). In this case, ROS enhance NF-κB activation and thus increase viral burden.
Kaposi's sarcoma-associated herpesvirus (KSHV) enters cells by binding to the NRF2-target cysteine glutathione exchanger (xCT), a cystine/glutamate exchanger that contributes to reducing oxidative stress by importing cysteine to replenish glutathione. Viral miRNAs upregulate the macrophage production of RNS, but RNS-induced apoptosis is prevented by concomitant upregulation of xCT, which is also induced by viral miRNAs in infected cells (260). In this case, the inhibition of RNS generation reduces viral burden, revealing the pathogen's stratagem. Incubation with H2O2 produces KSHV reactivation from latency, whereas ROS-scavenging NAC, glutathione, and peroxide-degrading catalase inhibit KSHV replication in culture, and treatment with NAC in vivo prolongs mice lifespan and reduces their viral load (355). It remains to be demonstrated how ROS and RNS generation contribute to promoting KSHV infection.
When acquired immunodeficiency syndrome (AIDS) became an epidemic, it was soon discovered that NAC could reduce viral burden in vitro (274), and Stanford Hospital in San Francisco observed that ROS-scavenging NAC could prolong patient survival (126, 127). Evidence was found in favor of NAC-induced reduction of viral burden, and the effects of oxidative stress in promoting HIV infection soon became clear (14, 274, 309). It was also observed that PMA promoted viral replication, which could be blocked with NAC (309). In fact, HIV infection leads to chronic oxidative stress, and the induction of HO-1 by heme can produce >90% reduction in the viral DNA found in infected human monocytes, a process reversed by the inhibitor of HO-1 activity tin-protoporphyrin (SnPP) (62). In a humanized mouse model of HIV infection using Hu-NOD-Scid mice, heme was capable of reducing viral burden. The inhibition of NF-κB activation is a suspected mechanism in this case, as hemin-induced HO-1 inhibits NF-κB, and NF-κB activation is a known mechanism promoting infection. Although HIV replication is enhanced by NF-κB activators, treatment with LPS, a TLR4 ligand that activates NF-κB, is capable of inhibiting infection through the induction of HO-1 (63).
Influenza A virus causes oxidative stress, and its inhibition by antioxidants effectively reduces viral replication. ROS-scavenging NAC reduces infection by influenza A (H3N2, H5N1, but less effective against H1N1) (84, 188) and B viruses (188) and is used in clinics to inhibit mucin secretion and reduce viral load. The inhibition of NOX2 by apocynin has been shown to reduce viral titers of influenza A H3N2 (X31) and H1N1 (PR8) in the lungs (334), despite the participation of ROS in RIG-I-elicited antiviral effects (304). Mice deficient in the NOX2 subunit gp91phox present reduced viral titers of influenza A H3N2 (X31) compared with wild-type mice and increased amounts of IL-1β in the lungs (334), whereas mice deficient in NOX2 p47phox present reduced H5N1 burden (122). The effects of NOX2 inhibition include the preservation of CD8+ producing IL-2, IFNγ, and IL-12 (334), but it still remains to be demonstrated how NOX2 inhibition acts to inhibit influenza A infection. In addition, the induction of NRF2 activation (using sulforaphane and epigallocatechin gallate [EGCG]) can reduce influenza A H3N2 (Bangkok/1/79) titer, whereas NRF2 inhibition (via lentivirus-containing shRNA NRF2) has been shown to increase epithelial viral burden (140). EGCG, the NRF2-activator, found in green tea, has been shown to be effective against H1N1, H3N2, and B virus. In these cases, the NRF2-activators induced type I IFN, RIG-I, and MxA molecules involved in virucidal mechanisms and potential mediators of their effects. However, NRF2 overexpression induced by an adenovirus containing Nrf2 gene (AdNRF2) infection reduced viral burden in epithelial and alveolar macrophages but did not increase IFNλ levels (147). However, in another study, HO-1 overexpression resulting from AdHO-1 infection did not reduce H1N1 PR8 titers (108).
RSV infection generates oxidative stress (41). Treatment with NAC reduces viral burden in vivo (188). The administration of aerosols containing SOD reduced the pulmonary RSV load in rats (348), whereas synthetic catalytic scavengers that possess SOD, catalase, and glutathione-peroxidase activity reduced viral replication in human epithelial pulmonary cell (115). Similarly, Nrf2−/− mice present increased RSV replication compared with wild-type mice, which can be verified as soon as 1 day after infection (49). Moreover, wild-type mice, but not Nrf2−/− mice, reacted to treatment with NRF2-activator sulforaphane with reduced viral burden in lungs. Taken together, these results indicate that ROS promote RSV infection. The mechanisms by which ROS can do so are discussed in detail elsewhere (83).
Porcine circovirus type 2 (PCV2) replication inside the kidney cells is inhibited by treatment with NAC, whereas GSH depletion with BSO increases PCV2 replication (47). ROS-induced NF-κB activation appears to be involved in promoting this infection.
Enterovirus-71 induces oxidative stress by increasing NOX2-mediated ROS production. Antioxidants such as the NRF2 activators EGCG and GCG have been shown to decrease the viral burden of infected VERO cells (113). Treatment of cell cultures with NAC, apocynin, or siRNA to the p47phox subunit of NOX2 reduces viral burden, and the overexpression of HO-1 (by prior infection with Ad-HO-1) suppresses ROS production and also enterovirus-71 replication (324). The inhibitory effect of Ad-HO-1 on virus replication can be reversed by the inhibitor of HO-1 activity SnPP. The HO-1 metabolite CO is able to inhibit ROS generation and to mimic the effects of HO-1 on the inhibition of virus replication. These results are highly indicative that oxidative stress promotes infection with enterovirus 71.
Chronic viral hepatitis C (human hepatitis C virus [HCV]) is associated with high oxidative stress and reduced expression of NRF2-target genes (39, 342). However, in one study, no positive effects were observed when NAC was added to the conventional IFNα treatment (89). The treatment of hepatocytes with inducers of HO-1 expression such as CoPP or heme decreases HCV replication, as wells as genetically induces HO-1 overexpression (366) or repression of Bach with miRNA (116, 296). A similar situation was observed in human hepatitis B virus (HBV)-infected mice, in which CoPP-induced HO-1 expression reduced viral replication in livers, which could be verified as early as 5 days after infection (256). In hepatocytes, the inhibition of HBV replication with CoPP was reversed by HO-1-specific siRNA and treatment with CoPP promoted HBC core protein degradation in hepatocytes within 6 h (256). As the product of heme degradation billiverdin induces the upregulation of type I IFN and proteins involved in the antiviral state, it is possible that the induction of HO-1 reduces viral burden by increasing type I IFN production. Recently, the inhibition of HCV protein NS3/4A by billiverdin or heme was shown to reverse the blockade of TLR3 and RIGI signaling caused by infection and to restore the type I IFN production in infected hepatocytes (98, 167, 365). The role of HO-1 and heme in viral hepatitis was recently reviewed elsewhere (290), and much of their effects appear to be directly antiviral and unrelated to antioxidant capacities. Nevertheless, it has been shown that H2O2 also increases HCV replication in hepatocytes (10–100 μM), except when incubated at a high and nonphysiological concentration (1 mM) (167). Silybin, the active principle of silymarin, an antioxidant and hepatoprotective plant used since the ancient Greeks, dramatically reduces the viral load in HCV patients (280), indicating that antioxidants in general act on HCV replication.
Lymphocytic choriomeningitis virus (LCMV) is eliminated in mice by perforin and IFNγ-producing CD8 cells. In NOX2 (p47phox)-deficient mice, disease development is attenuated, a phenomenon associated with reduced viral burden, increased numbers of antigen-specific CD8 cells, and increased IFNγ production, starting 8 days after the onset of infection (163). The evidence indicate that CD8 cells and their IFNγ production are sensitive to ROS, as depletion of GSH using BSO is capable of preventing the increase in virus-specific T-cell frequencies and the fraction of these cells that produces IFNγ. In fact, the binding and growth of LCMV in peritoneal macrophages also depends on the ROS generated upon infection (195), indicating that multiple effects of ROS contributes to favoring LCMV infection.
C. Bacterial infections that thrive in oxidative environments
Certain bacterial infections can be promoted by oxidative stress (Table 2). Studying P. gingivalis mutants deficient in rubrerythrin, a molecule involved in oxidative burst defense, Mydel et al. observed that rubrerythrin ensured bacteria proliferation. Surprisingly, NOX2 (gp91phox)-deficient mice were observed to be highly resistant to both wild-type and rubrerythrin-deficient bacteria (215). The authors concluded that the host's oxidative burst enhanced the survival of P. gingivalis and observed evidence that the compensatory increase in NO production was responsible for the resistance of NOX2-deficient mice to infection.
Table 2.
Bacterial Infections Fueled by ROS
| Pathogen | Observed effects of ROS/antioxidants on pathogen's burden | Model | Associated mechanisms | Suspected mechanisms underlying results | References |
|---|---|---|---|---|---|
| Porphyromonas gingivalis | Decreased burden in mice in the absence of oxidative burst; rubrerythrin promotes growth in the presence of gp91phox | gp91phox−/− mice are highly resistant to infection | No mechanism was found that could be responsible for gp91phox−/− mice resistance to bacteria | Possible compensatory NO production. Pathogen thrives on autophagy-, a suspect process, as it is usually regulated by ROS | (215) |
| Mycobacterium abscessus | Increased growth in macrophages under oxidative conditions; antioxidants NAC and MnTE-2-PyP reduced bacterial viability in macrophages | Infected THP-1 macrophages were stimulated to respiratory burst with PMA, incubated with H2O2, NAC, or MnTE-2-PyP (later also in primary human monocytes) | Infection reduces antioxidant defenses; MnTE-2-PyP promotes phagosome–lysosome fusion and prevents apoptosis | Antioxidant MnTE-2-PyP does not act directly on bacteria, as demonstrated by authors. Possibly acts by preventing phagosome–lysosome fusion. No functional demonstration | (229, 230) |
| Helicobacter pylori | Usual growth is in oxidative environment; decreased colonization upon sulforaphane (NRF2-activator) or UDCA (ROS-scavenger) administration | Brocollis administered to wild-type or Nrf2−/− mice; UDCA administered to mice | No mechanism assigned | Sulforaphane, NAC, and UDCA have direct bactericidal effects, but at higher doses | (351, 322) |
| Increased bacterial elimination when NAC is administered along with antibiotics to nonsmoker patients | NAC administered along with antibiotics to smokers and nonsmokers | (97) | |||
| Bacillus anthracis | Endospores germinate in oxidative environment; they germinate readily upon uptake by macrophages, where they generate huge oxidative stress as a consequence of LeTX toxin effects | Germination of spores in vitro under O2− flux; infection of macrophages; exposure of macrophages to LeTX | Direct effect of ROS exposure on endospore's physiology | ROS is probably a signal for endospores to germinate | (12, 94, 105, 287) |
UDCA, ursodeoxycholic acid.
The first clear demonstration that ROS could directly promote a bacterial infection was made in M. abscessus-infected macrophages (230). The incubation of infected THP-1 macrophages with H2O2 or PMA, a respiratory burst inducer, was capable of increasing the growth of M. abscessus, whereas ROS-scavenger antioxidants inhibited the pathogen growth during 4–7 days of infection. The authors had previously demonstrated that the antioxidant MnTE-2-PyP reduces burden in infected macrophages, a phenomenon associated with rescue from infection-induced cell death and promotion of phagosome–lysosome fusion (229). To date, there are no clues to the mechanism by which oxidative stress promotes M. abscessus growth, but it is possible that it acts by preventing phagosome–lysosome fusion, as preventing fusion appears to be an important step in establishing infection.
Helicobacter pylori is known to induce severe oxidative stress and to feed on damaged tissue. Anecdotal evidence led researchers to conduct studies with sulforaphane-rich broccoli sprouts in H. pylori-infected mice and humans. Sulforaphane is a well-known NRF2 activator. Broccoli reduced bacterial colonization, an effect that could not be obtained in Nrf2−/− mice (351), indicating that sulforaphane acts through NRF2 activation. In fact, the transfection of gastric epithelial cells with the HspB protein from H. pylori increased Keap expression and reduced the expression of the phase II detoxifying enzymes HO-1, SOD1, and NQO1, a mechanism that appears to be involved in the decrease in antioxidant defenses that follows infection (30). The mechanisms by which NRF2 activation reduces H. pylori colonization remain to be clarified. In addition, ROS-scavenging ursodeoxycholic acid (UDCA) reduced H. pylori bacterial colonization (322), whereas NAC helped to efficiently eradicate infection when associated with antibiotics, an effect that could not be attained in oxidative stress-exposed smokers (97). However, those studies are difficult to interpret, as sulforaphane, UDCA and NAC have direct bactericidal effects, although at higher doses (241, 351). The bacterial load of NOS2−/− gp91phox−/− or gp91phox−/− mice infected with H. pylori was far smaller than that of wild-type or NOS2−/− mice, indicating that indeed ROS are counterproductive against these bacteria, and gp91phox−/− mice also had greatly decreased H. felis burden (20). An increase in gastric mononuclear inflammation was associated with the absence of gp91phox expression, but whether it is related to the reduced bacterial load remains unknown. In another work, no significant decrease in H. pylori burden was observed in NOX2 (gp91phox)−/− mice, although the results were suggestive of a moderate decrease (139).
Bacillus anthracis infect macrophages, a critical step for their propagation, as spores germinate within the cell (94). B. anthracis is highly cytolytic to macrophages in vivo. The cytolytic toxin has been identified previously and is known to be responsible for the sudden shock that occurs during infection. Within macrophages, the toxin induces a lethal oxidative burst that kills the cells in hours (105, 287). Therefore, B. anthracis spores must germinate in the oxidative environment induced in macrophages. In fact, it appears that spores from B. anthracis are highly protected against oxidative stress by various SODs (57) and are stimulated to germinate by O2− exposure (12). B. anthracis is therefore able not only to cope with oxidative stress but also is able to thrive on its generation.
Transgenic mice overexpressing HO-1 present increased survival in the cecal ligation and puncture (CLP) model of polymicrobial sepsis and also decreased bacterial burden, whereas Hmox1−/− mice do not survive CLP and have higher bacterial burdens than wild-type mice (50). When instead of CLP, a bacterial fibrin clot was placed in their abdominal cavities, HO-1-overexpressing mice presented improved survival and phagocytosis of E. faecalis but not E. coli. These results indicate that HO-1 enhances clearance of particular bacterial species. In addition, when CLP was performed in Nrf2−/− mice, bacteremia was increased, whereas in Keap1−/− mice, which present increased activation of NRF2, bacteremia was reduced, a phenomenon associated with altered phagocytosis of bacteria (146).
Infection with the protozoan T. gondii increases enterococci and E. coli counts in the ileum, whereas treatment with NRF2 activators such as curcumin and resveratrol reverse this increase in ileum counts (16). No direct bactericidal activity has been observed that could be ascribed to these compounds, but inflammation is greatly reduced and the intestinal tissue is well preserved after treatment, suggesting that changes in intestinal flora are caused by their interactions with tissues protected from oxidative and inflammatory damage by the NRF2 activators (192).
Recently, HO-1-inducer CoPP was observed to be capable of reducing the numbers of live Salmonella in the lamina propria, mesenteric lymph note, spleen, and liver, whereas mice deficient in the expression of HO-1 in macrophages presented impaired clearance of E. fecalis, E. coli, and S. typhimurium (234). The by-product of HO-1 activity CO mimicked the effects of HO-1, suggesting that in this case, the effects of HO-1 were dependent on CO production. Antioxidant studies are still required to test whether the inhibition of ROS production is involved in the phenomenon. No differences in intestinal permeability were observed among nontreated CO-releasing molecule or CoPP-treated mice, indicating that the effects of HO-1/CO are not due to the protection of the epithelial intestinal barrier against damage.
D. The role of ROS in clearance or promotion of infection by protozoan parasites of medical importance
Whereas in bacterial and viral infections, it is clear that certain pathogens thrive in oxidative environment and others are eliminated by ROS, the situation with protozoans is a bit more complex. Parasites have complex genomes, life cycles, and evasion strategies from the mammalian immune system, and ROS may have different effects depending on the infected cell type or compartment the parasite occupies. The evidence that some protozoan infections can be fueled by oxidative stress is summarized in Table 3.
Table 3.
Role of ROS in Protozoan Infections of Medical Importance
| Pathogen | Observed effects of ROS/antioxidants on pathogen's burden | Model | Associated mechanisms of resistance | Suspected mechanisms underlying results | References |
|---|---|---|---|---|---|
| Leishmania major | NAC treatment of BALB/c mice reduced burden, whereas BSO (a drug that depletes gluthatione) increased burden in C57BL/6 footpads | Mice were treated with NAC | Increase in the frequency of IFN-γ-secreting lymphocytes in draining popliteal lymph nodes from NAC-treated mice | It remains unknown whether reduction of burden is dependent on IFN-γ-secreting lymphocytes | (56, 270, 276) |
| Treatment of infected macrophages with GSH-depleting BSO before IFN-γ/LPS activation increased burden | Elicited peritoneal macrophages(C57BL/6 mice) treated with BSO for 24 h, activated with IFN-γ/LPS | GSH depletion was associated with a decrease in NO production | It remains unknown whether GSH inhibited L. major growth in macrophages | (276) | |
| NRF2-activator resveratrol reduced parasite burden of infected macrophages | Elicited peritoneal macrophages or J774 murine cell line treated with resveratrol | Resveratrol acts to reduce parasite burden in macrophages at lower doses than directly | The mechanisms activated by resveratrol remain unknown | (138) | |
| Leishmania amazonensis | NAC treatment reduced parasite burden in footpads from infected BALB/c mice | NAC treatment of BALB/c mice | No clues to how NAC reduced footpad parasite burden | No suspected mechanism | (205) |
| DETC, an inhibitor of SOD activity, promoted parasite destruction in infected macrophages or reduced parasite burden in vivo, NAC reversed this effect | Peritoneal macrophages from BALB/c mice | Superoxide production seems to be a killing mechanism. No controls treated only with NAC | IFN β increases parasite burden through induction of SOD and decrease of superoxide | (141) | |
| Trypanosoma cruzi | ROS exposure of epimastigotes increased their growth | Epimastigotes grow more actively in LIT medium containing H2O2 | ROS affects cell signaling. | No similar mechanisms are known in amastigotes | (77, 226) |
| Increased infection under oxidative conditions and vice versa, in macrophages and in vivo. Burden reduced in infected macrophages incubated with antioxidants, transfected with Nrf2 or HO-1, and vice versa in H2O2 or PMA. Treatment with NRF2-activators reduce parasitemia, also reduced in gp91phox−/−.mice | Elicited peritoneal macrophages, BMMs or THP-1 cells; C57BL/6 and NOX2(gp91phox)−/− mice | Reduction of labile iron pool associated with reduction in parasite growth by antioxidants. NO, type I IFN, and apoptosis of infected cells not involved | Direct effects of ROS on amastigote proliferation were not ruled out, nor were mechanisms indirectly associated with reduction of labile iron pool | (93, 210, 218, 235, 272, 285) |
BSO, buthionine sulfoximine; LPS, lipopolysaccharide; NO, nitric oxide; SOD, superoxide dismutase.
Treatment with NAC reduces the parasitism in footpads of BALB/c mice infected with L. major (270, 276), whereas treatment with BSO, a drug that inhibits the synthesis of glutathione, increases the burden in the footpads of C57BL/6 mice infected with L. major (56). None of these works established whether innate or adaptive immunity was involved in the effect or when treatment started to result in altered burden, but BSO effects were verified at the second week postinfection and an increase in the frequency of IFNγ-secreting lymphocytes was observed in the draining popliteal lymph nodes of NAC-treated mice (56). In another work, however, macrophages were treated with BSO for 24 h, infected with L. major and then activated with LPS/IFNγ (276). Prior BSO treatment resulted in increased macrophage parasite burden. The authors credited the result to the decrease in NO production in BSO-treated macrophages. The NRF2-activator resveratrol, although directly leishmanicidal, reduces the macrophage L. major burden at much lower doses than it does acting directly on the parasite (138), indicating that the antioxidant effects stimulate leishmanicidal macrophage mechanisms. In different study, NOX2 (gp91phox)−/− female homozygous mice had smaller acute infection lesions than wild-type controls, although in macrophages and neutrophils, no role for gp91phox was observed in the killing of L. major (22). Later in the course of infection (from 127 dpi on), L. major burden did not decrease in skin, popliteal lymph nodes, and spleens from gp91phox−/− mice as it did in wild-type mice, and the burden was greatly elevated in spleen, indicating visceralization of leishmaniasis for unknown reasons (22). Taken together, these results suggest that NOX2-derived ROS does not contribute to killing L. major inside macrophages but does interfere with the establishment of later immune responses. A full review on the subject of Leishmania susceptibility to ROS has recently been published elsewhere (327).
Not all infections with Leishmania respond to antioxidants with reduced burden. In L. amazonensis-infected macrophages, diethyldithiocarbamate, an inhibitor of SOD activity, promotes parasite destruction, and NAC reverses this effect (141). However, these findings contrast with the reduced parasite burden observed in the footpads from L. amazonensis-infected mice treated with NAC (205). It is possible, however, that NAC does not act on macrophages to reduce footpad parasite loads but instead on adaptive immunity. Recently, studying the iron uptake by L. amazonensis, Mittra et al. observed iron uptake to be increased after promastigotes were subjected to the iron-deficient medium, a compensatory mechanism dependent on the upregulation of iron transporter LIT1 and which promoted differentiation into amastigotes (200). The increased iron uptake allowed the synthesis of FeSOD, an amastigote antioxidant enzyme that depends greatly on iron availability. When macrophages were infected with L. amazonensis promastigotes subjected to iron depletion or to O2−• induction by menadione, the parasite displayed increased cell invasion and growth. A similar situation occurred in vivo: O2−• induction or iron stress increased the parasite burden in footpads. The ROS-enhancing effect on L. amazonensis infection appears to be related to the differentiation into amastigotes, as controls prepared with axenic amastigotes produced similar results.
Leishmania spp. have evolved a complex lipophosphoglycan that allows them to carefully avoid binding to mannose receptors, which triggers respiratory burst and other microbicidal mechanisms in phagocytes (325). Binding to FcR induces intense respiratory burst upon phagocytosis (325), but L. major amastigotes are coated with IgG1 in the footpads of infected mice and bind to macrophages through IgG1 (101). How L. major uses FcR to enter the macrophages and still survive respiratory burst remains unknown. In fact, L. major parasites trigger intense ROS production upon macrophage infection, different from L. amazonensis, which proceeds through a relatively silent cell invasion (4). FcR most likely represent an entry portal to the macrophages at least in L. amazonensis or L. pifanoi, as FcR-deficient mice present smaller cutaneous lesions (142).
L. guyanensis and L. brasiliensis do not cause large lesions in BALB/c mice. J. Souza-Franco et al. observed that the infection of BALB/c macrophages with L. guyanensis triggers an intense respiratory burst able to kill parasites (305).
Human infection with L. chagasi induces the upregulation of HO-1, and treatment with antileishmanial downregulates it (179). L. chagasi apparently benefits from HO-1 upregulation, as induction of HO-1 by treatment with CoPP increases parasite burden in macrophages, and macrophages from HO-1 knockouts infected in vitro have decreased burden and do not respond to treatment with CoPP. Antioxidants such as NAC were not tested in this infection and the role of NOX2 was also not assessed, but L. chagasi has enzymes that protects it against RNS and enhances its survival within macrophages.
Mice deficient in NOX2 (gp91phox) and infected with L. donovani presented increased peak parasite burden and delayed control of infection but were still, ultimately, able to control the infection (213). L. pifanoi interferes with NOX2 recruitment to phagosomes, and only a premature 65 kDa form of gp91phox is found in vacuoles containing amastigotes, which is different from 91 and 65 kDa-containing zymosan vacuoles (245). The maturation of gp91phox to its full-weight form of 95 kDa is known to depend on the availability of heme. L. pifanoi also upregulates HO-1, and the authors speculated that HO-1 overexpression could interfere with NOX2, as it promotes heme degradation. In fact, the HO-1 inducer CoPP reduces O2−• production (245). Nevertheless, they did not demonstrate the outcome of infection in macrophages treated with the HO-1 inducer CoPP or the parasite burden in gp91phox-knockout macrophages. Therefore, we can only speculate on the role of ROS on L. pifanoi infection.
NOX2 (gp91phox)-deficient mice do not display any differences in parasite burden when infected with P. berghei ANKA, P. yoelli, or P. chabaudi (254), indicating that phagocyte respiratory burst does not participate in Plasmodium clearance. Nevertheless, in P. berghei ANKA infection, although HO-1−/− mice did not differ from controls in parasite burden, the NRF2/HO-1-inducer CoPP did reduce parasitemia in an early stage of infection, an effect that was lost after the drug was discontinued (237). The authors considered that perhaps CoPP interfered with the ability of the parasites to polymerize heme, decreasing parasitemia, but no functional data support this assumption. In the liver, HO-1 expression promoted infection by P. berghei and yoelii, whereas NAC did not alter parasitism, indicating that HO-1 effects are not related to its antioxidant effects (71).
CD36, a receptor for the uptake of oxidized LDL, is a NRF2-target gene that also mediates phagocytosis and clearance of Plasmodium by macrophages (199, 233, 294). CD36 does not have known antioxidant actions, although its expression can be an NRF2-dependent event triggered by an oxidative hit or other kind of stimulus. Thus, although some results might be a bit misleading, we consider that there is no strong evidence in the literature to support a role for ROS in promoting Plasmodium infection.
P. vivax induces oxidative stress in the gut of Anopheles aquasalis, a mosquito vector of malaria. Gene silencing of catalase in the A. aquasalis midgut epithelium increases the percentage of infected insects. A decrease has been observed in the load of potential bacterial competitors, which could be responsible for the association between increased ROS and increased P. vivax infection in the midgut (11). The role of ROS in mammalian infection by P. vivax awaits investigation.
T. cruzi induces oxidative stress (193), which causes much of the cardiac damage associated with Chagas disease (342). T. cruzi infection activates macrophages to respiratory burst, as shown by the decreased production of ROS by macrophages from NOX2 knockout mice (235), whereas in cardiomyocytes, T. cruzi induces mitochondrial ROS (342). Details about the molecular pathway that elicits ROS production in these cases remain unknown. The parasite is well equipped to withstand oxidative insults (246, 247), and the epimastigote forms even proliferate more actively in the presence of H2O2 (77, 226) and heme (226), which probably mimic the highly oxidative environment the epimastigotes face within the insect gut. Nevertheless, a role for ROS in T. cruzi killing was established by early studies. The first evidence came from studies of exhausted respiratory burst in macrophages, in which T. cruzi growth increased, suggesting that ROS are protective against infection (212). This finding was supported by the association between macrophage trypanocidal activity and the production of H2O2 (221), even though others claimed that they observed no evidence that macrophage-derived ROS contribute to T. cruzi killing (190, 211). Arguing against a role for ROS in T. cruzi elimination, another study demonstrated that the amastigote burden was significantly decreased when preinfected macrophages were treated with SOD and catalase during stimulation with IFNγ (194), although the decrease was small. These authors interpreted this finding as a possible consequence of the increased NO production, as ROS inhibition decreases OONO− formation and increases NO availability. They also observed that SOD and catalase did not affect parasite burden when the macrophages were activated before infection and enzymatic treatment. In another work, the percentage of Y-strain-infected cells appeared to be reduced when nonstimulated inflammatory macrophages were infected and then incubated with SOD or catalase, whereas increased percentages were demonstrated when they were incubated with PMA 1.5–3 h before infection (190). An even more clear picture emerged when the percentages of infected cells were compared among PMA (15%) and SOD (1%) treated cells, but the finding was most likely overlooked due to the bad sensitivity of the assay (190).
Recently, we have demonstrated that in elicited macrophages infected in vitro with T. cruzi, incubation with antioxidants (NAC, apocynin, billiverdin), permeant ROS-degrading enzymes (SOD-polyethylene glycol [PEG], catalase-PEG), and transfection of NRF2 or HO-1, reduced parasite burden, whereas incubation with SnPP, an inhibitor of HO-1 activity, or prooxidants (H2O2 or respiratory-burst activator PMA) increased parasitism (235). Consistently, we observed that infected NOX2 (gp91phox)-deficient mice have a decreased macrophage parasite burden and that elicited macrophages from gp91phox−/−-mice subjected to in vitro infection have reduced parasitism. In elicited macrophages infected in vitro before treatment, NRF2 activators, including resveratrol, sulforaphane, pterostilbene, and oltipraz, reduced parasitism (235), whereas in another work, the NRF2 activator curcumin reduced parasitism of infected fibroblasts (218). The mechanisms by which NRF2 activators operate remain to be fully elucidated. The classic mechanisms of parasite elimination such as NO production are not involved in CoPP-mediated reduction of parasitemia or parasitism, as well as the production of type I IFN, or adaptive immunity (235). The labile iron pool is reduced by treatment of infected cells with antioxidants, and the reduction of labile iron pool achieved by various strategies can reduce parasitism (235), indicating that iron sequestration is most likely involved in the burden-reducing effects of antioxidants.
In another work, however, performed with the T. cruzi CL-Brenner clone, apocynin did not alter parasite burden when incubated with infected nonstimulated macrophages and even blocked the killing of T. cruzi in IFNγ/LPS prestimulated macrophages (5). A role for NOX2-derived ROS in TLR4 signaling was not tested in this model. Evidence for OONO− as a mediator of T. cruzi killing has been observed in IFNγ/LPS preactivated macrophages (5, 6). Protein oxidation in parasites has been detected soon after phagocytosis mainly in IFNγ/LPS stimulated macrophages, and NOX2 assembly was confirmed in amastigote-containing vacuoles (5). Parasites overexpressing T. cruzi cytosolic tryparedoxin peroxidase (TcCPX), an enzyme that degrades OONO−, presented increased survival in short-term phagocytosis assays (5), but the parasite burden in nonstimulated macrophages did not differ between the wild-type and TcCPX-overexpressing parasites (247). These findings indicate that OONO− participates in T. cruzi killing. The reasons for the contradictory results concerning the role of ROS in T. cruzi infection await further studies.
The main capacity to induce ROS production in macrophages has been ascribed to T. cruzi cruzipain (96). Cruzipain enhances the oxidative burst by inducing the expression of p47phox and gp91 and enhancing the co-localization of NADPH oxidase enzyme subunits. As cruzipain is known to increase susceptibility of macrophages to T. cruzi infection (314, 315), it remains to be demonstrated if it does so by increasing ROS production by macrophages.
Slamf1-deficient mice were recently described as resistant to T. cruzi Y strain infection (35). As Slamf1 is a molecule expressed in myeloid cells that promote NOX2 activation (180), it is possible that the promotion of T. cruzi infection by ROS underlies these effects.
The effects of antioxidants in vivo are a bit more complex. The HO-1 inducer CoPP administered to T. cruzi Y-strain C57BL/6-infected mice reduces parasitism, whereas the inhibitor of HO-1 activity SnPP increases parasitism (235). The prooxidant paraquat also increases peak parasitemia and mortality in T. cruzi acutely infected mice, indicating that oxidative stress fuels T. cruzi infection in vivo (235). However, we failed to reduce parasitemia by treating mice with NAC in vivo, and only slightly decreased parasitemia by treating with apocynin (our unpublished results), whereas other researchers have succeeded by treating mice with NAC or glutathione (93). In mice infected with the Sylvio strain and treated with the antioxidant phenyl-alpha-tert-butyl-nitrone at the acute or chronic stages, heart parasite burden did not change (342), but conversely, in mice similarly infected and treated with apocynin, parasite burden was observed to be increased in heart and blood (64), indicating that NOX2 is required to control parasitemia in this model. Treatment with NRF2 activators, such as resveratrol, pterostilbene (235), melatonin (285), 15ΔPGJ2 (272), and curcumin (218) reduced T. cruzi parasite burden in mice. The reversal of CoPP's effects on parasitemia by peroral iron sulfate indicated that iron mobilization is involved in the effects of antioxidants on T. cruzi infection (235).
NOX2 (gp91phox−/−)-deficient mice infected with T. cruzi Y strain were examined by us and by Santiago et al. (286). Although we observed a significant decrease in parasitemia (235), they showed a nonsignificant trend toward decreased parasite burden in the gp91phox−/− mice. Similar to Santiago et al., we observed increased parasite burden at some timepoints postinfection in hearts and spleens by quantitative polymerase chain reaction (our unpublished results), and we believe that the lack of gp91phox expression causes distinct effects depending on the tissue and interval postinfection. Both groups observed that T. cruzi-infected gp91phox−/− mice die during acute infection, despite the early control of parasitemia. Their deaths were credited by Santiago et al. for the arterial pressure drop caused by NO overexpression, but an iNOS inhibitor, despite being capable of reversing the drop in arterial pressure, did not prevent the deaths of the gp91phox−/− mice (286). However, we observed that the treatment of gp91phox−/− mice with the HO-1-inducer CoPP produced a further decrease in their parasitemia and totally prevented their deaths. It is not clear to us whether treatment with CoPP spared the lives of infected gp91phox−/− by reducing parasitism in critical tissues or by reducing non-NOX2 generated ROS, and the phenomenon awaits a mechanistic explanation. There are various obscure points in these studies and the role of NOX2 deserves further consideration in T. cruzi infection.
VI. What Do Pathogens That Thrive on Oxidative Stress Have in Common?
Pathogens differ in their escape strategy within phagocytes (121). Some bacteria persist in the phagosome but inhibit their maturation to a fully microbicidal organelle, such as Legionella pneumophila and C. burnetii. M. tuberculosis and L. monocytogenes, not only arrest phagosomal maturation, but also ultimately escape to the cytosol. Listeria can alternatively live within large modified vacuoles (spacious Listeria-containing phagosomes). T. cruzi trypomastigotes induce their phagocytosis, escape phagosomes and change into amastigotes to grow in the cytoplasm. Most viruses invade cells and go directly to their cytoplasm, although some of them stimulate phagocytosis and use the phagosomal vacuole to enter the cell. Whether a pathogen will survive ROS production, live with it, or thrive on it, will probably depend on the cell type that harbors them, the location they reach within the cell, and their susceptibility to particular ROS-promoted mechanisms of microbe elimination.
It is noteworthy that none of the pathogens shown to thrive on oxidative stress spends most of their lifespan inside a mature phagosome, and it is probably an indication that pathogens that benefit from ROS production are not fully exposed to direct oxidative damage.
The literature is full of examples of immune mechanisms sabotaged by pathogens. Most intracellular pathogens succumb to type I IFN-elicited mechanisms of microbe elimination. L. monocytogenes, however, benefits from type I IFN secretion (10), most likely because it desensitizes the cell to the bactericidal mechanisms induced by IFNγ, which are effective against the bacteria (264). In this case, the innate mechanism of protection mediated by IFNγ has been somehow victimized by the pathogen, which turns type I IFN production against it, using the strength of the opponent against himself. It is like an “aikido” blow (Japanese martial art performed by redirecting the force of the attacker rather than opposing it head-on), to get rid of the much more efficient microbicidal mechanism commanded by IFNγ. The production of type I IFN was also stimulated by infection with M. leprae, turning on IL-10 production and downregulation of IFNγ, which in turn inhibits vitamin D-CAMP (cathelicidin)/DEFB4 (beta-defensin) antimicrobial pathway (320). Similarly, in M. tuberculosis infection, PKR activation, part of a microbicidal mechanism that engenders the activation of eIF2α, turns against the host, sensitizing macrophages to enhanced IFNγ-activation and subsequent apoptosis (346). It is possible that an analogous situation occurs with ROS-induced mechanisms of pathogen elimination, such that some pathogens, which are shielded by their own antioxidants, benefit from ROS production as it turns against effective killing mechanisms (Fig. 5). Iron sequestration could be one of these mechanisms, as oxidative stress determines the release of iron from ferritin in T. cruzi infection (235).
Alternatively, it is possible that some pathogens indulge ROS as an indirect metabolic supplement. T. cruzi epimastigotes multiply more actively in axenic cultures supplied with H2O2 (77, 226), for unknown reasons. Salmonella typhimurium grows more actively in inflamed intestinal lumen when ROS is actively produced (312). In this case, intestinal ROS converts luminal thiosulfate to tetrathionate, an electron acceptor that provides a metabolic advantage to Salmonella (344), as demonstrated by the outgrowth of wild-type bacteria over mutants unable to use tetrathionate in normal but not in gp91phox−/− mice.
In Caenorhabditis elegans, the worm NRF2-homolog SKN-1 regulates the responses to xenobiotics and oxidative stress (129). Recently, SKN-1-deficient C. elegans was shown to be susceptible to P. aeruginosa and E. faecalis (238), in a case reminiscent of the role of NRF2-target genes in enhancing immunity against certain bacteria (50, 107, 265). The conserved participation of NRF2-target genes in innate immunity appears to indicate a common microbicidal mechanism underlying most of these cases described here that has yet to be identified.
VII. Concluding Remarks
Oxidative attack is a major weapon used by phagocytes and other cell types against invading pathogens and ROS production can interfere with microbe elimination through a myriad of mechanisms. Although the current paradigm predicts that ROS production contributes to elimination of pathogens, it is becoming clear that for viruses, bacteria, and protozoans, ROS production can contribute to increasing pathogen burden. These new findings open an avenue to the use of antioxidants against particular infections.
Abbreviations Used
- AMPK
AMP-activated kinase
- ARE
antioxidant response element
- ATF
activation transcription factor
- BSO
buthionine sulfoximine
- CGD
chronic granulomatous disease
- CHIKV
chikungunya virus
- CLP
cecal ligation and puncture
- CoPP
cobalt-protoporphyrin
- DC
dendritic cell
- DPI
diphenylene iodonium
- DUOXs
dual oxidases
- EGCG
epigallocatechin gallate
- FcR
Fc receptor
- FeSOD
iron superoxide dismutase
- FPN-1
ferroportin-1
- GCLC
glutamate cysteine ligase catalytic subunit
- GSH
reduced glutathione
- GST
glutathione S-transferase
- HBV
human hepatitis B virus
- HCMV
human cytomegalovirus
- HCV
human hepatitis C virus
- HIV
human immunodeficiency virus
- H2O2
hydrogen peroxide
- HO-1
heme oxygenase 1
- HOBr−
hypobromous acid
- HOCl−
hypochlorous acid
- HOI−
hypoiodous acid
- HSV1
herpes simplex virus 1
- IFN
interferon
- IL
interleukin
- Keap-1
kelch-like ECH-associated protein 1
- KSHV
Kaposi's sarcoma-associated herpesvirus
- LC3
microtubule associated protein 1A/1B light chain 3
- LCMV
lymphocytic choriomeningitis virus
- LXR
liver X receptors
- MPO
myeloperoxidase
- mTOR
mammalian target of rapamycin
- NAC
N-acetyl-cysteine
- NALP3
NACHT, LRR and PYD domains-containing protein 3
- NETs
neutrophil extracellular traps
- NLR
nod like receptor
- NO
nitric oxide
- ·NO2
nitrogen dioxide radical
- NOX2
NADPH oxidase 2
- NRAMP
natural resistance-associated macrophage protein
- NRF2
nuclear factor (erythroid-derived 2)-like 2
- Nrf2
nuclear factor erythroid derived 2
- O2−•
superoxide
- OH·
hydroxyl radical
- ONOO−
peroxynitrite anion
- OSCN−
hypothiocyanite
- pCV2
porcine circovirus type 2
- PMA
phorbol-12-myristate-13-acetate
- PPAR
peroxisome proliferator activated receptor
- PPR
pattern recognition receptor
- Rac2
ras-related C3 botulinum toxin substrate 2
- RLR
RIG like receptor
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- RSV
respiratory syncytial virus
- SnPP
tin-protoporphyrin
- SOD
superoxide dismutase
- TcCPX
T. cruzi cytosolic tryparedoxin peroxidase
- Th
T helper
- TLR
toll-like receptor
- TNF
tumor necrosis factor
- UDCA
ursodeoxycholic acid
- VSV
vesicular stomatitis virus
- xCT
cysteine glutathione exchanger
Acknowledgments
The authors received financial support from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Pesquisa (CNPq), Fundaçao de Amparo à Pesquisa do Rio de Janeiro (FAPERJ) and INCTDengue. “Figures were made with the help of Motifolio (www.motifolio.com)”.
References
- 1.Adamson S. and Leitinger N. Phenotypic modulation of macrophages in response to plaque lipids. Curr Opin Lipidol 22: 335–342, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Albert ML, Sauter B, and Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature 392: 86–89, 1998 [DOI] [PubMed] [Google Scholar]
- 3.Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ, and Ting JP. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 30: 556–565, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Almeida TF, Palma LC, Mendez LC, Noronha-Dutra AA, and Veras PS. Leishmania amazonensis fails to induce the release of reactive oxygen intermediates by CBA macrophages. Parasite Immunol 34: 492–498, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alvarez MN, Peluffo G, Piacenza L, and Radi R. Intraphagosomal peroxynitrite as a macrophage-derived cytotoxin against internalized Trypanosoma cruzi: consequences for oxidative killing and role of microbial peroxiredoxins in infectivity. J Biol Chem 286: 6627–6640, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alvarez MN, Piacenza L, Irigoin F, Peluffo G, and Radi R. Macrophage-derived peroxynitrite diffusion and toxicity to Trypanosoma cruzi. Arch Biochem Biophys 432: 222–232, 2004 [DOI] [PubMed] [Google Scholar]
- 7.Andersson H, Hartmanova B, Ryden P, Noppa L, Naslund L, and Sjostedt A. A microarray analysis of the murine macrophage response to infection with Francisella tularensis LVS. J Med Microbiol 55: 1023–1033, 2006 [DOI] [PubMed] [Google Scholar]
- 8.Aratani Y, Kura F, Watanabe H, Akagawa H, Takano Y, Suzuki K, Dinauer MC, Maeda N, and Koyama H. In vivo role of myeloperoxidase for the host defense. Jpn J Infect Dis 57: S15, 2004 [PubMed] [Google Scholar]
- 9.Ashida H, Mimuro H, Ogawa M, Kobayashi T, Sanada T, Kim M, and Sasakawa C. Cell death and infection: a double-edged sword for host and pathogen survival. J Cell Biol 195: 931–942, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Auerbuch V, Brockstedt DG, Meyer-Morse N, O'Riordan M, and Portnoy DA. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med 200: 527–533, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bahia AC, Oliveira JH, Kubota MS, Araujo HR, Lima JB, Rios-Velasquez CM, Lacerda MV, Oliveira PL, Traub-Cseko YM, and Pimenta PF. The role of reactive oxygen species in Anopheles aquasalis response to Plasmodium vivax infection. PLoS One 8: e57014, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baillie L, Hibbs S, Tsai P, Cao GL, and Rosen GM. Role of superoxide in the germination of Bacillus anthracis endospores. FEMS Microbiol Lett 245: 33–38, 2005 [DOI] [PubMed] [Google Scholar]
- 13.Balstad TR, Carlsen H, Myhrstad MC, Kolberg M, Reiersen H, Gilen L, Ebihara K, Paur I, and Blomhoff R. Coffee, broccoli and spices are strong inducers of electrophile response element-dependent transcription in vitro and in vivo - studies in electrophile response element transgenic mice. Mol Nutr Food Res 55: 185–197, 2011 [DOI] [PubMed] [Google Scholar]
- 14.Baruchel S. and Wainberg MA. The role of oxidative stress in disease progression in individuals infected by the human immunodeficiency virus. J Leukoc Biol 52: 111–114, 1992 [DOI] [PubMed] [Google Scholar]
- 15.Ben-Ari J, Wolach O, Gavrieli R, and Wolach B. Infections associated with chronic granulomatous disease: linking genetics to phenotypic expression. Expert Rev Anti Infect Ther 10: 881–894, 2012 [DOI] [PubMed] [Google Scholar]
- 16.Bereswill S, Munoz M, Fischer A, Plickert R, Haag LM, Otto B, Kuhl AA, Loddenkemper C, Gobel UB, and Heimesaat MM. Anti-inflammatory effects of resveratrol, curcumin and simvastatin in acute small intestinal inflammation. PloS One 5: e15099, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bergeron M, Blanchette J, Rouleau P, and Olivier M. Abnormal IFN-gamma-dependent immunoproteasome modulation by Trypanosoma cruzi-infected macrophages. Parasite Immunol 30: 280–292, 2008 [DOI] [PubMed] [Google Scholar]
- 18.Bianchi M, Hakkim A, Brinkmann V, Siler U, Seger RA, Zychlinsky A, and Reichenbach J. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood 114: 2619–2622, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bisti S, Konidou G, Boelaert J, Lebastard M, and Soteriadou K. The prevention of the growth of Leishmania major progeny in BALB/c iron-loaded mice: a process coupled to increased oxidative burst, the amplitude and duration of which depend on initial parasite developmental stage and dose. Microbes Infect 8: 1464–1472, 2006 [DOI] [PubMed] [Google Scholar]
- 20.Blanchard TG, Yu F, Hsieh CL, and Redline RW. Severe inflammation and reduced bacteria load in murine helicobacter infection caused by lack of phagocyte oxidase activity. J Infect Dis 187: 1609–1615, 2003 [DOI] [PubMed] [Google Scholar]
- 21.Blessing E, Kuo CC, Lin TM, Campbell LA, Bea F, Chesebro B, and Rosenfeld ME. Foam cell formation inhibits growth of Chlamydia pneumoniae but does not attenuate Chlamydia pneumoniae-induced secretion of proinflammatory cytokines. Circulation 105: 1976–1982, 2002 [DOI] [PubMed] [Google Scholar]
- 22.Blos M, Schleicher U, Soares Rocha FJ, Meissner U, Rollinghoff M, and Bogdan C. Organ-specific and stage-dependent control of Leishmania major infection by inducible nitric oxide synthase and phagocyte NADPH oxidase. Eur J Immunol 33: 1224–1234, 2003 [DOI] [PubMed] [Google Scholar]
- 23.Boyle JJ. Heme and haemoglobin direct macrophage Mhem phenotype and counter foam cell formation in areas of intraplaque haemorrhage. Curr Opin Lipidol 23: 453–461, 2012 [DOI] [PubMed] [Google Scholar]
- 24.Boyle JJ, Johns M, Kampfer T, Nguyen AT, Game L, Schaer DJ, Mason JC, and Haskard DO. Activating transcription factor 1 directs Mhem atheroprotective macrophages through coordinated iron handling and foam cell protection. Circ Res 110: 20–33, 2012 [DOI] [PubMed] [Google Scholar]
- 25.Breuer W, Epsztejn S, and Cabantchik ZI. Dynamics of the cytosolic chelatable iron pool of K562 cells. FEBS Lett 382: 304–308, 1996 [DOI] [PubMed] [Google Scholar]
- 26.Breuer W, Shvartsman M, and Cabantchik ZI. Intracellular labile iron. Int J Biochem Cell Biol 40: 350–354, 2008 [DOI] [PubMed] [Google Scholar]
- 27.Brigelius-Flohe R. and Flohe L. Basic principles and emerging concepts in the redox control of transcription factors. Antioxid Redox Signal 15: 2335–2381, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, and Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science 303: 1532–1535, 2004 [DOI] [PubMed] [Google Scholar]
- 29.Buchmeier NA, Lipps CJ, So MY, and Heffron F. Recombination-deficient mutants of Salmonella typhimurium are avirulent and sensitive to the oxidative burst of macrophages. Mol Microbiol 7: 933–936, 1993 [DOI] [PubMed] [Google Scholar]
- 30.Buommino E, Donnarumma G, Manente L, De Filippis A, Silvestri F, Iaquinto S, Tufano MA, and De Luca A. The Helicobacter pylori protein HspB interferes with Nrf2/Keap1 pathway altering the antioxidant response of Ags cells. Helicobacter 17: 417–425, 2012 [DOI] [PubMed] [Google Scholar]
- 31.Burke MF, Khera AV, and Rader DJ. Polyphenols and cholesterol efflux: is coffee the next red wine? Circ Res 106: 627–629, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bustamante J, Aksu G, Vogt G, de Beaucoudrey L, Genel F, Chapgier A, Filipe-Santos O, Feinberg J, Emile JF, Kutukculer N, and Casanova JL. BCG-osis and tuberculosis in a child with chronic granulomatous disease. J Allergy Clin Immunol 120: 32–38, 2007 [DOI] [PubMed] [Google Scholar]
- 33.Bustamante J, Arias AA, Vogt G, Picard C, Galicia LB, Prando C, Grant AV, Marchal CC, Hubeau M, Chapgier A, de Beaucoudrey L, Puel A, Feinberg J, Valinetz E, Janniere L, Besse C, Boland A, Brisseau JM, Blanche S, Lortholary O, Fieschi C, Emile JF, Boisson-Dupuis S, Al-Muhsen S, Woda B, Newburger PE, Condino-Neto A, Dinauer MC, Abel L, and Casanova JL. Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat Immunol 12: 213–221, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cairo G, Recalcati S, Mantovani A, and Locati M. Iron trafficking and metabolism in macrophages: contribution to the polarized phenotype. Trends Immunol 32: 241–247, 2011 [DOI] [PubMed] [Google Scholar]
- 35.Calderon J, Maganto-Garcia E, Punzon C, Carrion J, Terhorst C, and Fresno M. The receptor Slamf1 on the surface of myeloid lineage cells controls susceptibility to infection by Trypanosoma cruzi. PLoS Pathog 8: e1002799, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Caltagirone A, Weiss G, and Pantopoulos K. Modulation of cellular iron metabolism by hydrogen peroxide. Effects of H2O2 on the expression and function of iron-responsive element-containing mRNAs in B6 fibroblasts. J Biol Chem 276: 19738–19745, 2001 [DOI] [PubMed] [Google Scholar]
- 37.Cao F, Castrillo A, Tontonoz P, Re F, and Byrne GI. Chlamydia pneumoniae—induced macrophage foam cell formation is mediated by Toll-like receptor 2. Infect Immun 75: 753–759, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carneiro LA, Travassos LH, Soares F, Tattoli I, Magalhaes JG, Bozza MT, Plotkowski MC, Sansonetti PJ, Molkentin JD, Philpott DJ, and Girardin SE. Shigella induces mitochondrial dysfunction and cell death in nonmyleoid cells. Cell Host Microbe 5: 123–136, 2009 [DOI] [PubMed] [Google Scholar]
- 39.Carvajal-Yepes M, Himmelsbach K, Schaedler S, Ploen D, Krause J, Ludwig L, Weiss T, Klingel K, and Hildt E. Hepatitis C virus impairs the induction of cytoprotective Nrf2 target genes by delocalization of small Maf proteins. J Biol Chem 286: 8941–8951, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Castrillo A, Joseph SB, Vaidya SA, Haberland M, Fogelman AM, Cheng G, and Tontonoz P. Crosstalk between LXR and toll-like receptor signaling mediates bacterial and viral antagonism of cholesterol metabolism. Mol Cell 12: 805–816, 2003 [DOI] [PubMed] [Google Scholar]
- 41.Castro SM, Guerrero-Plata A, Suarez-Real G, Adegboyega PA, Colasurdo GN, Khan AM, Garofalo RP, and Casola A. Antioxidant treatment ameliorates respiratory syncytial virus-induced disease and lung inflammation. Am J Respir Crit Care Med 174: 1361–1369, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Challa S. and Chan FK. Going up in flames: necrotic cell injury and inflammatory diseases. Cell Mol Life Sci 67: 3241–3253, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chan RC, Wang M, Li N, Yanagawa Y, Onoe K, Lee JJ, and Nel AE. Pro-oxidative diesel exhaust particle chemicals inhibit LPS-induced dendritic cell responses involved in T-helper differentiation. J Allergy Clin Immunol 118: 455–465, 2006 [DOI] [PubMed] [Google Scholar]
- 44.Chang CW, Su YC, Her GM, Ken CF, and Hong JR. Betanodavirus induces oxidative stress-mediated cell death that prevented by anti-oxidants and zfcatalase in fish cells. PLoS One 6: e25853, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Charron AJ. and Sibley LD. Host cells: mobilizable lipid resources for the intracellular parasite Toxoplasma gondii. J Cell Sci 115: 3049–3059, 2002 [DOI] [PubMed] [Google Scholar]
- 46.Chen M, Li W, Wang N, Zhu Y, and Wang X. ROS and NF-kappaB but not LXR mediate IL-1beta signaling for the downregulation of ATP-binding cassette transporter A1. Am J Physiol Cell Physiol 292: C1493–C1501, 2007 [DOI] [PubMed] [Google Scholar]
- 47.Chen X, Ren F, Hesketh J, Shi X, Li J, Gan F, and Huang K. Reactive oxygen species regulate the replication of porcine circovirus type 2 via NF-kappaB pathway. Virology 426: 66–72, 2012 [DOI] [PubMed] [Google Scholar]
- 48.Chlosta S, Fishman DS, Harrington L, Johnson EE, Knutson MD, Wessling-Resnick M, and Cherayil BJ. The iron efflux protein ferroportin regulates the intracellular growth of Salmonella enterica. Infect Immun 74: 3065–3067, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cho HY, Imani F, Miller-DeGraff L, Walters D, Melendi GA, Yamamoto M, Polack FP, and Kleeberger SR. Antiviral activity of Nrf2 in a murine model of respiratory syncytial virus disease. Am J Respir Crit Care Med 179: 138–150, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chung SW, Liu X, Macias AA, Baron RM, and Perrella MA. Heme oxygenase-1-derived carbon monoxide enhances the host defense response to microbial sepsis in mice. J Clin Invest 118: 239–247, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Conner GE, Salathe M, and Forteza R. Lactoperoxidase and hydrogen peroxide metabolism in the airway. Am J Respir Crit Care Med 166: S57–S61, 2002 [DOI] [PubMed] [Google Scholar]
- 52.Corleis B, Korbel D, Wilson R, Bylund J, Chee R, and Schaible UE. Escape of Mycobacterium tuberculosis from oxidative killing by neutrophils. Cell Microbiol 14: 1109–1121, 2012 [DOI] [PubMed] [Google Scholar]
- 53.Costa-Mattioli M. and Sonenberg N. RAPping production of type I interferon in pDCs through mTOR. Nat Immunol 9: 1097–1099, 2008 [DOI] [PubMed] [Google Scholar]
- 54.Cox JV, Naher N, Abdelrahman YM, and Belland RJ. Host HDL biogenesis machinery is recruited to the inclusion of Chlamydia trachomatis-infected cells and regulates chlamydial growth. Cell Microbiol 14: 1497–1512, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cruz D, Watson AD, Miller CS, Montoya D, Ochoa MT, Sieling PA, Gutierrez MA, Navab M, Reddy ST, Witztum JL, Fogelman AM, Rea TH, Eisenberg D, Berliner J, and Modlin RL. Host-derived oxidized phospholipids and HDL regulate innate immunity in human leprosy. J Clin Invest 118: 2917–2928, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cruz KK, Fonseca SG, Monteiro MC, Silva OS, Andrade VM, Cunha FQ, and Romao PR. The influence of glutathione modulators on the course of Leishmania major infection in susceptible and resistant mice. Parasite Immunol 30: 171–174, 2008 [DOI] [PubMed] [Google Scholar]
- 57.Cybulski RJ, Jr., Sanz P, Alem F, Stibitz S, Bull RL, and O'Brien AD. Four superoxide dismutases contribute to Bacillus anthracis virulence and provide spores with redundant protection from oxidative stress. Infect Immun 77: 274–285, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.D'Archivio M, Scazzocchio B, Filesi C, Vari R, Maggiorella MT, Sernicola L, Santangelo C, Giovannini C, and Masella R. Oxidised LDL up-regulate CD36 expression by the Nrf2 pathway in 3T3-L1 preadipocytes. FEBS Lett 582: 2291–2298, 2008 [DOI] [PubMed] [Google Scholar]
- 59.D'Avila H, Freire-de-Lima CG, Roque NR, Teixeira L, Barja-Fidalgo C, Silva AR, Melo RC, Dosreis GA, Castro-Faria-Neto HC, and Bozza PT. Host cell lipid bodies triggered by Trypanosoma cruzi infection and enhanced by the uptake of apoptotic cells are associated with prostaglandin E(2) generation and increased parasite growth. J Infect Dis 204: 951–961, 2011 [DOI] [PubMed] [Google Scholar]
- 60.D'Avila H, Maya-Monteiro CM, and Bozza PT. Lipid bodies in innate immune response to bacterial and parasite infections. Int Immunopharmacol 8: 1308–1315, 2008 [DOI] [PubMed] [Google Scholar]
- 61.Das NK, Biswas S, Solanki S, and Mukhopadhyay CK. Leishmania donovani depletes labile iron pool to exploit iron uptake capacity of macrophage for its intracellular growth. Cell Microbiol 11: 83–94, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Devadas K. and Dhawan S. Hemin activation ameliorates HIV-1 infection via heme oxygenase-1 induction. J Immunol 176: 4252–4257, 2006 [DOI] [PubMed] [Google Scholar]
- 63.Devadas K, Hewlett IK, and Dhawan S. Lipopolysaccharide suppresses HIV-1 replication in human monocytes by protein kinase C-dependent heme oxygenase-1 induction. J Leukoc Biol 87: 915–924, 2010 [DOI] [PubMed] [Google Scholar]
- 64.Dhiman M. and Garg NJ. NADPH oxidase inhibition ameliorates Trypanosoma cruzi-induced myocarditis during Chagas disease. J Pathol 225: 583–596, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dinauer MC, Deck MB, and Unanue ER. Mice lacking reduced nicotinamide adenine dinucleotide phosphate oxidase activity show increased susceptibility to early infection with Listeria monocytogenes. J Immunol 158: 5581–5583, 1997 [PubMed] [Google Scholar]
- 66.Dorn BR, Dunn WA, Jr., and Progulske-Fox A. Porphyromonas gingivalis traffics to autophagosomes in human coronary artery endothelial cells. Infect Immun 69: 5698–5708, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, and Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 320: 674–677, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Drakesmith H. and Prentice AM. Hepcidin and the iron-infection axis. Science 338: 768–772, 2012 [DOI] [PubMed] [Google Scholar]
- 69.Drummond RA. and Brown GD. The role of Dectin-1 in the host defence against fungal infections. Curr Opin Microbiol 14: 392–399, 2011 [DOI] [PubMed] [Google Scholar]
- 70.Endres R, Luz A, Schulze H, Neubauer H, Futterer A, Holland SM, Wagner H, and Pfeffer K. Listeriosis in p47(phox−/−) and TRp55−/− mice: protection despite absence of ROI and susceptibility despite presence of RNI. Immunity 7: 419–432, 1997 [DOI] [PubMed] [Google Scholar]
- 71.Epiphanio S, Mikolajczak SA, Goncalves LA, Pamplona A, Portugal S, Albuquerque S, Goldberg M, Rebelo S, Anderson DG, Akinc A, Vornlocher HP, Kappe SH, Soares MP, and Mota MM. Heme oxygenase-1 is an anti-inflammatory host factor that promotes murine plasmodium liver infection. Cell Host Microbe 3: 331–338, 2008 [DOI] [PubMed] [Google Scholar]
- 72.Fang FC. Antimicrobial actions of reactive oxygen species. mBio 2:pii: e00141–e00111, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Feeney ER, McAuley N, O'Halloran JA, Rock C, Low J, Satchell CS, Lambert JS, Sheehan GJ, and Mallon PW. The expression of cholesterol metabolism genes in monocytes from HIV-infected subjects suggests intracellular cholesterol accumulation. J Infect Dis 207: 628–637, 2013 [DOI] [PubMed] [Google Scholar]
- 74.Fernandez PL, Dutra FF, Alves L, Figueiredo RT, Mourao-Sa D, Fortes GB, Bergstrand S, Lonn D, Cevallos RR, Pereira RM, Lopes UG, Travassos LH, Paiva CN, and Bozza MT. Heme amplifies the innate immune response to microbial molecules through spleen tyrosine kinase (Syk)-dependent reactive oxygen species generation. J Biol Chem 285: 32844–32851, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fields PI, Swanson RV, Haidaris CG, and Heffron F. Mutants of Salmonella typhimurium that cannot survive within the macrophage are avirulent. Proc Natl Acad Sci U S A 83: 5189–5193, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Finn AV, Nakano M, Polavarapu R, Karmali V, Saeed O, Zhao X, Yazdani S, Otsuka F, Davis T, Habib A, Narula J, Kolodgie FD, and Virmani R. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. J Am Coll Cardiol 59: 166–177, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Finzi JK, Chiavegatto CW, Corat KF, Lopez JA, Cabrera OG, Mielniczki-Pereira AA, Colli W, Alves MJ, and Gadelha FR. Trypanosoma cruzi response to the oxidative stress generated by hydrogen peroxide. Mol Biochem Parasitol 133: 37–43, 2004 [DOI] [PubMed] [Google Scholar]
- 78.Fortes GB, Alves LS, de Oliveira R, Dutra FF, Rodrigues D, Fernandez PL, Souto-Padron T, De Rosa MJ, Kelliher M, Golenbock D, Chan FK, and Bozza MT. Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood 119: 2368–2375, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fraternale A, Paoletti MF, Casabianca A, Nencioni L, Garaci E, Palamara AT, and Magnani M. GSH and analogs in antiviral therapy. Mol Aspects Med 30: 99–110, 2009 [DOI] [PubMed] [Google Scholar]
- 80.Freigang S, Ampenberger F, Spohn G, Heer S, Shamshiev AT, Kisielow J, Hersberger M, Yamamoto M, Bachmann MF, and Kopf M. Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. Eur J Immunol 41: 2040–2051, 2011 [DOI] [PubMed] [Google Scholar]
- 81.Fujita M, Harada E, Matsumoto T, Mizuta Y, Ikegame S, Ouchi H, Inoshima I, Yoshida S, Watanabe K, and Nakanishi Y. Impaired host defence against Mycobacterium avium in mice with chronic granulomatous disease. Clin Exp Immunol 160: 457–460, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gallois A, Klein JR, Allen LA, Jones BD, and Nauseef WM. Salmonella pathogenicity island 2-encoded type III secretion system mediates exclusion of NADPH oxidase assembly from the phagosomal membrane. J Immunol 166: 5741–5748, 2001 [DOI] [PubMed] [Google Scholar]
- 83.Garofalo RP, Kolli D, and Casola A. Respiratory syncytial virus infection: mechanisms of redox control and novel therapeutic opportunities. Antioxid Redox Signal 18: 186–217, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Geiler J, Michaelis M, Naczk P, Leutz A, Langer K, Doerr HW, and Cinatl J., Jr.N-acetyl-L-cysteine (NAC) inhibits virus replication and expression of pro-inflammatory molecules in A549 cells infected with highly pathogenic H5N1 influenza A virus. Biochem Pharmacol 79: 413–420, 2010 [DOI] [PubMed] [Google Scholar]
- 85.Gilmour MI. Influence of air pollutants on allergic sensitization: the paradox of increased allergies and decreased resistance to infection. Toxicol Pathol 40: 312–314, 2012 [DOI] [PubMed] [Google Scholar]
- 86.Gordon MA. Salmonella infections in immunocompromised adults. J Infect 56: 413–422, 2008 [DOI] [PubMed] [Google Scholar]
- 87.Gozzelino R, Jeney V, and Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol 50: 323–354, 2010 [DOI] [PubMed] [Google Scholar]
- 88.Graham DB, Robertson CM, Bautista J, Mascarenhas F, Diacovo MJ, Montgrain V, Lam SK, Cremasco V, Dunne WM, Faccio R, Coopersmith CM, and Swat W. Neutrophil-mediated oxidative burst and host defense are controlled by a Vav-PLCgamma2 signaling axis in mice. J Clin Invest 117: 3445–3452, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Grant PR, Black A, Garcia N, Prieto J, and Garson JA. Combination therapy with interferon-alpha plus N-acetyl cysteine for chronic hepatitis C: a placebo controlled double-blind multicentre study. J Med Virol 61: 439–442, 2000 [DOI] [PubMed] [Google Scholar]
- 90.Gross O, Gewies A, Finger K, Schafer M, Sparwasser T, Peschel C, Forster I, and Ruland J. Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature 442: 651–656, 2006 [DOI] [PubMed] [Google Scholar]
- 91.Gross O, Poeck H, Bscheider M, Dostert C, Hannesschlager N, Endres S, Hartmann G, Tardivel A, Schweighoffer E, Tybulewicz V, Mocsai A, Tschopp J, and Ruland J. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature 459: 433–436, 2009 [DOI] [PubMed] [Google Scholar]
- 92.Guerra C, Johal K, Morris D, Moreno S, Alvarado O, Gray D, Tanzil M, Pearce D, and Venketaraman V. Control of Mycobacterium tuberculosis growth by activated natural killer cells. Clin Exp Immunol 168: 142–152, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Guevara AG, Guilvard E, Borges MM, Cordeiro da Silva A, and Ouaissi A. N-acetylcysteine and glutathione modulate the behaviour of Trypanosoma cruzi experimental infection. Immunol Lett 71: 79–83, 2000 [DOI] [PubMed] [Google Scholar]
- 94.Guidi-Rontani C, Weber-Levy M, Labruyere E, and Mock M. Germination of Bacillus anthracis spores within alveolar macrophages. Mol Microbiol 31: 9–17, 1999 [DOI] [PubMed] [Google Scholar]
- 95.Guimaraes-Costa AB, Nascimento MT, Wardini AB, Pinto-da-Silva LH, and Saraiva EM. ETosis: a microbicidal mechanism beyond cell death. J Parasitol Res 2012: 929743, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Guinazu N, Carrera-Silva EA, Becerra MC, Pellegrini A, Albesa I, and Gea S. Induction of NADPH oxidase activity and reactive oxygen species production by a single Trypanosoma cruzi antigen. Int J Parasitol 40: 1531–1538, 2010 [DOI] [PubMed] [Google Scholar]
- 97.Gurbuz AK, Ozel AM, Ozturk R, Yildirim S, Yazgan Y, and Demirturk L. Effect of N-acetyl cysteine on Helicobacter pylori. South Med J 98: 1095–1097, 2005 [DOI] [PubMed] [Google Scholar]
- 98.Gutierrez-Grobe Y, Vitek L, Tiribelli C, Kobashi-Margain RA, Uribe M, and Mendez-Sanchez N. Biliverdin and heme oxygenase antiviral activity against hepatitis C virus. Ann Hepatol 10: 105–107, 2011 [PubMed] [Google Scholar]
- 99.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, and Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119: 753–766, 2004 [DOI] [PubMed] [Google Scholar]
- 100.Gutierrez MG, Vazquez CL, Munafo DB, Zoppino FC, Beron W, Rabinovitch M, and Colombo MI. Autophagy induction favours the generation and maturation of the Coxiella-replicative vacuoles. Cell Microbiol 7: 981–993, 2005 [DOI] [PubMed] [Google Scholar]
- 101.Guy RA. and Belosevic M. Comparison of receptors required for entry of Leishmania major amastigotes into macrophages. Infect Immun 61: 1553–1558, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, and Waldmann H. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol 7: 75–77, 2011 [DOI] [PubMed] [Google Scholar]
- 103.Halliwell B. Phagocyte-derived reactive species: salvation or suicide? Trends Biochem Sci 31: 509–515, 2006 [DOI] [PubMed] [Google Scholar]
- 104.Halstead SB, Mahalingam S, Marovich MA, Ubol S, and Mosser DM. Intrinsic antibody-dependent enhancement of microbial infection in macrophages: disease regulation by immune complexes. Lancet Infect Dis 10: 712–722, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hanna PC, Kruskal BA, Ezekowitz RA, Bloom BR, and Collier RJ. Role of macrophage oxidative burst in the action of anthrax lethal toxin. Mol Med 1: 7–18, 1994 [PMC free article] [PubMed] [Google Scholar]
- 106.Hara H, Ishihara C, Takeuchi A, Imanishi T, Xue L, Morris SW, Inui M, Takai T, Shibuya A, Saijo S, Iwakura Y, Ohno N, Koseki H, Yoshida H, Penninger JM, and Saito T. The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nat Immunol 8: 619–629, 2007 [DOI] [PubMed] [Google Scholar]
- 107.Harvey CJ, Thimmulappa RK, Sethi S, Kong X, Yarmus L, Brown RH, Feller-Kopman D, Wise R, and Biswal S. Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci Transl Med 3: 78ra32, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hashiba T, Suzuki M, Nagashima Y, Suzuki S, Inoue S, Tsuburai T, Matsuse T, and Ishigatubo Y. Adenovirus-mediated transfer of heme oxygenase-1 cDNA attenuates severe lung injury induced by the influenza virus in mice. Gene Ther 8: 1499–1507, 2001 [DOI] [PubMed] [Google Scholar]
- 109.Hayashi K, Hooper LC, Okuno T, Takada Y, and Hooks JJ. Inhibition of HSV-1 by chemoattracted neutrophils: supernatants of corneal epithelial cells (HCE) and macrophages (THP-1) treated with virus components chemoattract neutrophils (PMN), and supernatants of PMN treated with these conditioned media inhibit viral growth. Arch Virol 157: 1377–1381, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hermann M, Jaconi ME, Dahlgren C, Waldvogel FA, Stendahl O, and Lew DP. Neutrophil bactericidal activity against Staphylococcus aureus adherent on biological surfaces. Surface-bound extracellular matrix proteins activate intracellular killing by oxygen-dependent and -independent mechanisms. J Clin Invest 86: 942–951, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hider RC. and Kong XL. Glutathione: a key component of the cytoplasmic labile iron pool. Biometals 24: 1179–1187, 2011 [DOI] [PubMed] [Google Scholar]
- 112.Hintze KJ. and Theil EC. DNA and mRNA elements with complementary responses to hemin, antioxidant inducers, and iron control ferritin-L expression. Proc Natl Acad Sci U S A 102: 15048–15052, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ho HY, Cheng ML, Weng SF, Leu YL, and Chiu DT. Antiviral effect of epigallocatechin gallate on enterovirus 71. J Agric Food Chem 57: 6140–6147, 2009 [DOI] [PubMed] [Google Scholar]
- 114.Hoetzenecker W, Echtenacher B, Guenova E, Hoetzenecker K, Woelbing F, Bruck J, Teske A, Valtcheva N, Fuchs K, Kneilling M, Park JH, Kim KH, Kim KW, Hoffmann P, Krenn C, Hai T, Ghoreschi K, Biedermann T, and Rocken M. ROS-induced ATF3 causes susceptibility to secondary infections during sepsis-associated immunosuppression. Nat Med 18: 128–134, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hosakote YM, Komaravelli N, Mautemps N, Liu T, Garofalo RP, and Casola A. Antioxidant mimetics modulate oxidative stress and cellular signaling in airway epithelial cells infected with respiratory syncytial virus. Am J Physiol Lung Cell Mol Physiol 303: L991–L1000, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hou W, Tian Q, Zheng J, and Bonkovsky HL. MicroRNA-196 represses Bach1 protein and hepatitis C virus gene expression in human hepatoma cells expressing hepatitis C viral proteins. Hepatology 51: 1494–1504, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hsu YM, Zhang Y, You Y, Wang D, Li H, Duramad O, Qin XF, Dong C, and Lin X. The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat Immunol 8: 198–205, 2007 [DOI] [PubMed] [Google Scholar]
- 118.Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MA, Glogauer M, Grinstein S, and Brumell JH. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci U S A 106: 6226–6231, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Huang J, Lam GY, and Brumell JH. Autophagy signaling through reactive oxygen species. Antioxid Redox Signal 14: 2215–2231, 2011 [DOI] [PubMed] [Google Scholar]
- 120.Huang TT, Carlson EJ, Epstein LB, and Epstein CJ. The role of superoxide anions in the establishment of an interferon-alpha-mediated antiviral state. Free Radic Res Commun 17: 59–72, 1992 [DOI] [PubMed] [Google Scholar]
- 121.Hybiske K. and Stephens RS. Exit strategies of intracellular pathogens. Nat Rev Microbiol 6: 99–110, 2008 [DOI] [PubMed] [Google Scholar]
- 122.Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen R, Leung YH, Wang H, Liu H, Sun Y, Pasparakis M, Kopf M, Mech C, Bavari S, Peiris JS, Slutsky AS, Akira S, Hultqvist M, Holmdahl R, Nicholls J, Jiang C, Binder CJ, and Penninger JM. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 133: 235–249, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ishii T, Itoh K, Ruiz E, Leake DS, Unoki H, Yamamoto M, and Mann GE. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: activation by oxidatively modified LDL and 4-hydroxynonenal. Circ Res 94: 609–616, 2004 [DOI] [PubMed] [Google Scholar]
- 124.Jackson KE, Klonis N, Ferguson DJ, Adisa A, Dogovski C, and Tilley L. Food vacuole-associated lipid bodies and heterogeneous lipid environments in the malaria parasite, Plasmodium falciparum. Mol Microbiol 54: 109–122, 2004 [DOI] [PubMed] [Google Scholar]
- 125.Jackson SH, Gallin JI, and Holland SM. The p47phox mouse knock-out model of chronic granulomatous disease. J Exp Med 182: 751–758, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.James JS. Stanford NAC study: glutathione level predicts survival. AIDS Treat News 1–5, 1997 [PubMed] [Google Scholar]
- 127.James JS. NAC: Stanford San Francisco study report shows blood glutathione improvement, possible survival benefit. AIDS Treat News 2–5, 2000 [PubMed] [Google Scholar]
- 128.Jankowski A. and Grinstein S. Modulation of the cytosolic and phagosomal pH by the NADPH oxidase. Antioxid Redox Signal 4: 61–68, 2002 [DOI] [PubMed] [Google Scholar]
- 129.Jasper H. SKNy worms and long life. Cell 132: 915–916, 2008 [DOI] [PubMed] [Google Scholar]
- 130.Jiang H, Badralmaa Y, Yang J, Lempicki R, Hazen A, and Natarajan V. Retinoic acid and liver X receptor agonist synergistically inhibit HIV infection in CD4+ T cells by up-regulating ABCA1-mediated cholesterol efflux. Lipids Health Dis 11: 69, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Johnson EE, Sandgren A, Cherayil BJ, Murray M, and Wessling-Resnick M. Role of ferroportin in macrophage-mediated immunity. Infect Immun 78: 5099–5106, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Johnson EE. and Wessling-Resnick M. Iron metabolism and the innate immune response to infection. Microbes Infect 14: 207–216, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jones GS, Amirault HJ, and Andersen BR. Killing of Mycobacterium tuberculosis by neutrophils: a nonoxidative process. J Infect Dis 162: 700–704, 1990 [DOI] [PubMed] [Google Scholar]
- 134.Joseph SB, Bradley MN, Castrillo A, Bruhn KW, Mak PA, Pei L, Hogenesch J, O'connell RM, Cheng G, Saez E, Miller JF, and Tontonoz P. LXR-dependent gene expression is important for macrophage survival and the innate immune response. Cell 119: 299–309, 2004 [DOI] [PubMed] [Google Scholar]
- 135.Joubert PE, Werneke S, de la Calle C, Guivel-Benhassine F, Giodini A, Peduto L, Levine B, Schwartz O, Lenschow D, and Albert ML. Chikungunya-induced cell death is limited by ER and oxidative stress-induced autophagy. Autophagy 8: 1261–1263, 2012 [DOI] [PubMed] [Google Scholar]
- 136.Kakhlon O. and Cabantchik ZI. The labile iron pool: characterization, measurement, and participation in cellular processes(1). Free Radic Biol Med 33: 1037–1046, 2002 [DOI] [PubMed] [Google Scholar]
- 137.Kalayoglu MV. and Byrne GI. A Chlamydia pneumoniae component that induces macrophage foam cell formation is chlamydial lipopolysaccharide. Infect Immun 66: 5067–5072, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kedzierski L, Curtis JM, Kaminska M, Jodynis-Liebert J, and Murias M. In vitro antileishmanial activity of resveratrol and its hydroxylated analogues against Leishmania major promastigotes and amastigotes. Parasitol Res 102: 91–97, 2007 [DOI] [PubMed] [Google Scholar]
- 139.Keenan JI, Peterson RA, 2nd, and Hampton MB. NADPH oxidase involvement in the pathology of Helicobacter pylori infection. Free Radic Biol Med 38: 1188–1196, 2005 [DOI] [PubMed] [Google Scholar]
- 140.Kesic MJ, Simmons SO, Bauer R, and Jaspers I. Nrf2 expression modifies influenza A entry and replication in nasal epithelial cells. Free Radic Biol Med 51: 444–453, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Khouri R, Bafica A, Silva Mda P, Noronha A, Kolb JP, Wietzerbin J, Barral A, Barral-Netto M, and Van Weyenbergh J. IFN-beta impairs superoxide-dependent parasite killing in human macrophages: evidence for a deleterious role of SOD1 in cutaneous leishmaniasis. J Immunol 182: 2525–2531, 2009 [DOI] [PubMed] [Google Scholar]
- 142.Kima PE, Constant SL, Hannum L, Colmenares M, Lee KS, Haberman AM, Shlomchik MJ, and McMahon-Pratt D. Internalization of Leishmania mexicana complex amastigotes via the Fc receptor is required to sustain infection in murine cutaneous leishmaniasis. J Exp Med 191: 1063–1068, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kinter CS, Lundie JM, Patel H, Rindler PM, Szweda LI, and Kinter M. A quantitative proteomic profile of the Nrf2-mediated antioxidant response of macrophages to oxidized LDL determined by multiplexed selected reaction monitoring. PLoS One 7: e50016, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol 77: 598–625, 2005 [DOI] [PubMed] [Google Scholar]
- 145.Koarai A, Sugiura H, Yanagisawa S, Ichikawa T, Minakata Y, Matsunaga K, Hirano T, Akamatsu K, and Ichinose M. Oxidative stress enhances toll-like receptor 3 response to double-stranded RNA in airway epithelial cells. Am J Respir Cell Mol Biol 42: 651–660, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kong X, Thimmulappa R, Craciun F, Harvey C, Singh A, Kombairaju P, Reddy SP, Remick D, and Biswal S. Enhancing Nrf2 pathway by disruption of Keap1 in myeloid leukocytes protects against sepsis. Am J Respir Crit Care Med 184: 928–938, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kosmider B, Messier EM, Janssen WJ, Nahreini P, Wang J, Hartshorn KL, and Mason RJ. Nrf2 protects human alveolar epithelial cells against injury induced by influenza A virus. Respir Res 13: 43, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kotsias F, Hoffmann E, Amigorena S, and Savina A. Reactive oxygen species production in the phagosome: impact on antigen presentation in dendritic cells. Antioxid Redox Signal 18: 714–729, 2013 [DOI] [PubMed] [Google Scholar]
- 149.Kou MC, Chiou SY, Weng CY, Wang L, Ho CT, and Wu MJ. Curcuminoids distinctly exhibit antioxidant activities and regulate expression of scavenger receptors and heme oxygenase-1. Mol Nutr Food Res 57: 1598–1610, 2013 [DOI] [PubMed] [Google Scholar]
- 150.Kovtunovych G, Eckhaus MA, Ghosh MC, Ollivierre-Wilson H, and Rouault TA. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: effects on macrophage viability and tissue iron distribution. Blood 116: 6054–6062, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kozarov EV, Dorn BR, Shelburne CE, Dunn WA, Jr., and Progulske-Fox A. Human atherosclerotic plaque contains viable invasive Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis. Arterioscler Thromb Vasc Biol 25: e17–e18, 2005 [DOI] [PubMed] [Google Scholar]
- 152.Kuhn AM, Tzieply N, Schmidt MV, von Knethen A, Namgaladze D, Yamamoto M, and Brune B. Antioxidant signaling via Nrf2 counteracts lipopolysaccharide-mediated inflammatory responses in foam cell macrophages. Free Radic Biol Med 50: 1382–1391, 2011 [DOI] [PubMed] [Google Scholar]
- 153.Kumagai Y. and Akira S. Identification and functions of pattern-recognition receptors. J Allergy Clin Immunol 125: 985–992, 2010 [DOI] [PubMed] [Google Scholar]
- 154.Kumar C, Igbaria A, D'Autreaux B, Planson AG, Junot C, Godat E, Bachhawat AK, Delaunay-Moisan A, and Toledano MB. Glutathione revisited: a vital function in iron metabolism and ancillary role in thiol-redox control. EMBO J 30: 2044–2056, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kuo CC, Shor A, Campbell LA, Fukushi H, Patton DL, and Grayston JT. Demonstration of Chlamydia pneumoniae in atherosclerotic lesions of coronary arteries. J Infect Dis 167: 841–849, 1993 [DOI] [PubMed] [Google Scholar]
- 156.KuoLee R, Harris G, Conlan JW, and Chen W. Role of neutrophils and NADPH phagocyte oxidase in host defense against respiratory infection with virulent Francisella tularensis in mice. Microbes Infect 13: 447–456, 2011 [DOI] [PubMed] [Google Scholar]
- 157.Kurz T, Gustafsson B, and Brunk UT. Cell sensitivity to oxidative stress is influenced by ferritin autophagy. Free Radic Biol Med 50: 1647–1658, 2011 [DOI] [PubMed] [Google Scholar]
- 158.Kusuhara K, Ohga S, Hoshina T, Saito M, Sasaki Y, Ishimura M, Takada H, Fujita M, and Hara T. Disseminated Bacillus Calmette-Guerin lymphadenitis in a patient with gp91phox- chronic granulomatous disease 25 years after vaccination. Eur J Pediatr 168: 745–747, 2009 [DOI] [PubMed] [Google Scholar]
- 159.Labbe K. and Saleh M. Cell death in the host response to infection. Cell Death Differ 15: 1339–1349, 2008 [DOI] [PubMed] [Google Scholar]
- 160.Lam GY, Huang J, and Brumell JH. The many roles of NOX2 NADPH oxidase-derived ROS in immunity. Semin Immunopathol 32: 415–430, 2010 [DOI] [PubMed] [Google Scholar]
- 161.Lamkanfi M. and Dixit VM. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe 8: 44–54, 2010 [DOI] [PubMed] [Google Scholar]
- 162.Lanceta L, Li C, Choi AM, and Eaton JW. Haem oxygenase-1 overexpression alters intracellular iron distribution. Biochem J 449: 189–194, 2013 [DOI] [PubMed] [Google Scholar]
- 163.Lang PA, Xu HC, Grusdat M, McIlwain DR, Pandyra AA, Harris IS, Shaabani N, Honke N, Kumar Maney S, Lang E, Pozdeev VI, Recher M, Odermatt B, Brenner D, Haussinger D, Ohashi PS, Hengartner H, Zinkernagel RM, Mak TW, and Lang KS. Reactive oxygen species delay control of lymphocytic choriomeningitis virus. Cell Death Differ 20: 649–658, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Larson JA, Howie HL, and So M. Neisseria meningitidis accelerates ferritin degradation in host epithelial cells to yield an essential iron source. Mol Microbiol 53: 807–820, 2004 [DOI] [PubMed] [Google Scholar]
- 165.Lee PP, Chan KW, Jiang L, Chen T, Li C, Lee TL, Mak PH, Fok SF, Yang X, and Lau YL. Susceptibility to mycobacterial infections in children with X-linked chronic granulomatous disease: a review of 17 patients living in a region endemic for tuberculosis. Pediatr Infect Dis J 27: 224–230, 2008 [DOI] [PubMed] [Google Scholar]
- 166.Lee SM. and Kleiboeker SB. Porcine arterivirus activates the NF-kappaB pathway through IkappaB degradation. Virology 342: 47–59, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lehmann E, El-Tantawy WH, Ocker M, Bartenschlager R, Lohmann V, Hashemolhosseini S, Tiegs G, and Sass G. The heme oxygenase 1 product biliverdin interferes with hepatitis C virus replication by increasing antiviral interferon response. Hepatology 51: 398–404, 2010 [DOI] [PubMed] [Google Scholar]
- 168.Lehrer RI. The fungicidal mechanisms of human monocytes. I. Evidence for myeloperoxidase-linked and myeloperoxidase-independent candidacidal mechanisms. J Clin Invest 55: 338–346, 1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lekstrom-Himes JA. and Gallin JI. Immunodeficiency diseases caused by defects in phagocytes. N Engl J Med 343: 1703–1714, 2000 [DOI] [PubMed] [Google Scholar]
- 170.Leto TL, Morand S, Hurt D, and Ueyama T. Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid Redox Signal 11: 2607–2619, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Li M, Zhao L, Liu J, Liu A, Jia C, Ma D, Jiang Y, and Bai X. Multi-mechanisms are involved in reactive oxygen species regulation of mTORC1 signaling. Cell Signal 22: 1469–1476, 2010 [DOI] [PubMed] [Google Scholar]
- 172.Lim MB, Kuiper JW, Katchky A, Goldberg H, and Glogauer M. Rac2 is required for the formation of neutrophil extracellular traps. J Leukoc Biol 90: 771–776, 2011 [DOI] [PubMed] [Google Scholar]
- 173.Lipinski S, Till A, Sina C, Arlt A, Grasberger H, Schreiber S, and Rosenstiel P. DUOX2-derived reactive oxygen species are effectors of NOD2-mediated antibacterial responses. J Cell Sci 122: 3522–3530, 2009 [DOI] [PubMed] [Google Scholar]
- 174.Liu GY, Essex A, Buchanan JT, Datta V, Hoffman HM, Bastian JF, Fierer J, and Nizet V. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J Exp Med 202: 209–215, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Liu Q, Cheng LI, Yi L, Zhu N, Wood A, Changpriroa CM, Ward JM, and Jackson SH. p47phox deficiency induces macrophage dysfunction resulting in progressive crystalline macrophage pneumonia. Am J Pathol 174: 153–163, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Liu W, He P, Cheng B, Mei CL, Wang YF, and Wan JJ. Chlamydia pneumoniae disturbs cholesterol homeostasis in human THP-1 macrophages via JNK-PPARgamma dependent signal transduction pathways. Microbes Infect 12: 1226–1235, 2010 [DOI] [PubMed] [Google Scholar]
- 177.Lu CC, Lai HC, Hsieh SC, and Chen JK. Resveratrol ameliorates Serratia marcescens-induced acute pneumonia in rats. J Leukoc Biol 83: 1028–1037, 2008 [DOI] [PubMed] [Google Scholar]
- 178.Luca AD, Iannitti RG, Bozza S, Beau R, Casagrande A, D'Angelo C, Moretti S, Cunha C, Giovannini G, Massi-Benedetti C, Carvalho A, Boon L, Latge JP, and Romani L. CD4+ T cell vaccination overcomes defective cross-presentation of fungal antigens in a mouse model of chronic granulomatous disease. J Clin Invest 122: 1816–1831, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Luz NF, Andrade BB, Feijo DF, Araujo-Santos T, Carvalho GQ, Andrade D, Abanades DR, Melo EV, Silva AM, Brodskyn CI, Barral-Netto M, Barral A, Soares RP, Almeida RP, Bozza MT, and Borges VM. Heme oxygenase-1 promotes the persistence of Leishmania chagasi infection. J Immunol 188: 4460–4467, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ma C, Wang N, Detre C, Wang G, O'Keeffe M, and Terhorst C. Receptor signaling lymphocyte-activation molecule family 1 (Slamf1) regulates membrane fusion and NADPH oxidase 2 (NOX2) activity by recruiting a Beclin-1/Vps34/ultraviolet radiation resistance-associated gene (UVRAG) complex. J Biol Chem 287: 18359–18365, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Mantegazza AR, Savina A, Vermeulen M, Perez L, Geffner J, Hermine O, Rosenzweig SD, Faure F, and Amigorena S. NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood 112: 4712–4722, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Mantena RK, Wijburg OL, Vindurampulle C, Bennett-Wood VR, Walduck A, Drummond GR, Davies JK, Robins-Browne RM, and Strugnell RA. Reactive oxygen species are the major antibacterials against Salmonella typhimurium purine auxotrophs in the phagosome of RAW 264.7 cells. Cell Microbiol 10: 1058–1073, 2008 [DOI] [PubMed] [Google Scholar]
- 183.Marcil V, Delvin E, Sane AT, Tremblay A, and Levy E. Oxidative stress influences cholesterol efflux in THP-1 macrophages: role of ATP-binding cassette A1 and nuclear factors. Cardiovasc Res 72: 473–482, 2006 [DOI] [PubMed] [Google Scholar]
- 184.Marriott HM, Hellewell PG, Whyte MK, and Dockrell DH. Contrasting roles for reactive oxygen species and nitric oxide in the innate response to pulmonary infection with Streptococcus pneumoniae. Vaccine 25: 2485–2490, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Marriott HM, Jackson LE, Wilkinson TS, Simpson AJ, Mitchell TJ, Buttle DJ, Cross SS, Ince PG, Hellewell PG, Whyte MK, and Dockrell DH. Reactive oxygen species regulate neutrophil recruitment and survival in pneumococcal pneumonia. Am J Respir Crit Care Med 177: 887–895, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Marro S, Chiabrando D, Messana E, Stolte J, Turco E, Tolosano E, and Muckenthaler MU. Heme controls ferroportin1 (FPN1) transcription involving Bach1, Nrf2 and a MARE/ARE sequence motif at position −7007 of the FPN1 promoter. Haematologica 95: 1261–1268, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Mastroeni P, Vazquez-Torres A, Fang FC, Xu Y, Khan S, Hormaeche CE, and Dougan G. Antimicrobial actions of the NADPH phagocyte oxidase and inducible nitric oxide synthase in experimental salmonellosis. II. Effects on microbial proliferation and host survival in vivo. J Exp Med 192: 237–248, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Mata M, Morcillo E, Gimeno C, and Cortijo J. N-acetyl-L-cysteine (NAC) inhibit mucin synthesis and pro-inflammatory mediators in alveolar type II epithelial cells infected with influenza virus A and B and with respiratory syncytial virus (RSV). Biochem Pharmacol 82: 548–555, 2011 [DOI] [PubMed] [Google Scholar]
- 189.Matsue H, Edelbaum D, Shalhevet D, Mizumoto N, Yang C, Mummert ME, Oeda J, Masayasu H, and Takashima A. Generation and function of reactive oxygen species in dendritic cells during antigen presentation. J Immunol 171: 3010–3018, 2003 [DOI] [PubMed] [Google Scholar]
- 190.McCabe RE. and Mullins BT. Failure of Trypanosoma cruzi to trigger the respiratory burst of activated macrophages. Mechanism for immune evasion and importance of oxygen-independent killing. J Immunol 144: 2384–2388, 1990 [PubMed] [Google Scholar]
- 191.McCaffrey RL. and Allen LA. Francisella tularensis LVS evades killing by human neutrophils via inhibition of the respiratory burst and phagosome escape. J Leukoc Biol 80: 1224–1230, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Medzhitov R, Schneider DS, and Soares MP. Disease tolerance as a defense strategy. Science 335: 936–941, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Melo RC, Fabrino DL, D'Avila H, Teixeira HC, and Ferreira AP. Production of hydrogen peroxide by peripheral blood monocytes and specific macrophages during experimental infection with Trypanosoma cruzi in vivo. Cell Biol Int 27: 853–861, 2003 [DOI] [PubMed] [Google Scholar]
- 194.Metz G, Carlier Y, and Vray B. Trypanosoma cruzi upregulates nitric oxide release by IFN-gamma-preactivated macrophages, limiting cell infection independently of the respiratory burst. Parasite Immunol 15: 693–699, 1993 [DOI] [PubMed] [Google Scholar]
- 195.Michalek RD, Pellom ST, Holbrook BC, and Grayson JM. The requirement of reactive oxygen intermediates for lymphocytic choriomeningitis virus binding and growth. Virology 379: 205–212, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Miller EW, Dickinson BC, and Chang CJ. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proc Natl Acad Sci U S A 107: 15681–15686, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Miller JL, Velmurugan K, Cowan MJ, and Briken V. The type I NADH dehydrogenase of Mycobacterium tuberculosis counters phagosomal NOX2 activity to inhibit TNF-alpha-mediated host cell apoptosis. PLoS Pathog 6: e1000864, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Millonig G, Ganzleben I, Peccerella T, Casanovas G, Brodziak-Jarosz L, Breitkopf-Heinlein K, Dick TP, Seitz HK, Muckenthaler MU, and Mueller S. Sustained submicromolar H2O2 levels induce hepcidin via signal transducer and activator of transcription 3 (STAT3). J Biol Chem 287: 37472–37482, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Mimche PN, Thompson E, Taramelli D, and Vivas L. Curcumin enhances non-opsonic phagocytosis of Plasmodium falciparum through up-regulation of CD36 surface expression on monocytes/macrophages. J Antimicrob Chemother 67: 1895–1904, 2012 [DOI] [PubMed] [Google Scholar]
- 200.Mittra B, Cortez M, Haydock A, Ramasamy G, Myler PJ, and Andrews NW. Iron uptake controls the generation of Leishmania infective forms through regulation of ROS levels. J Exp Med 210: 401–416, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Mladenka P, Simunek T, Hubl M, and Hrdina R. The role of reactive oxygen and nitrogen species in cellular iron metabolism. Free Radic Res 40: 263–272, 2006 [DOI] [PubMed] [Google Scholar]
- 202.Moazed TC, Kuo C, Grayston JT, and Campbell LA. Murine models of Chlamydia pneumoniae infection and atherosclerosis. J Infect Dis 175: 883–890, 1997 [DOI] [PubMed] [Google Scholar]
- 203.Moghaddam AE, Gartlan KH, Kong L, and Sattentau QJ. Reactive carbonyls are a major Th2-inducing damage-associated molecular pattern generated by oxidative stress. J Immunol 187: 1626–1633, 2011 [DOI] [PubMed] [Google Scholar]
- 204.Monfregola J, Johnson JL, Meijler MM, Napolitano G, and Catz SD. MUNC13-4 protein regulates the oxidative response and is essential for phagosomal maturation and bacterial killing in neutrophils. J Biol Chem 287: 44603–44618, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Monteiro MC, Marques FC, Blazius RD, Santos da Silva O, de Queiroz Cunha F, Bento DB, and Torres Romao PR. N-acetyl-L: -cysteine reduces the parasitism of BALB/c mice infected with Leishmania amazonensis. Parasitol Res 102: 801–803, 2008 [DOI] [PubMed] [Google Scholar]
- 206.Moore SF. and MacKenzie AB. NADPH oxidase NOX2 mediates rapid cellular oxidation following ATP stimulation of endotoxin-primed macrophages. J Immunol 183: 3302–3308, 2009 [DOI] [PubMed] [Google Scholar]
- 207.Morris D, Khurasany M, Nguyen T, Kim J, Guilford F, Mehta R, Gray D, Saviola B, and Venketaraman V. Glutathione and infection. Biochim Biophys Acta 1830: 3329–3349, 2013 [DOI] [PubMed] [Google Scholar]
- 208.Mouy R, Fischer A, Vilmer E, Seger R, and Griscelli C. Incidence, severity, and prevention of infections in chronic granulomatous disease. J Pediatr 114: 555–560, 1989 [DOI] [PubMed] [Google Scholar]
- 209.Mujawar Z, Rose H, Morrow MP, Pushkarsky T, Dubrovsky L, Mukhamedova N, Fu Y, Dart A, Orenstein JM, Bobryshev YV, Bukrinsky M, and Sviridov D. Human immunodeficiency virus impairs reverse cholesterol transport from macrophages. PLoS Biol 4: e365, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Muller PK, Krohn K, and Muhlradt PF. Effects of pyocyanine, a phenazine dye from Pseudomonas aeruginosa, on oxidative burst and bacterial killing in human neutrophils. Infect Immun 57: 2591–2596, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Munoz-Fernandez MA, Fernandez MA, and Fresno M. Activation of human macrophages for the killing of intracellular Trypanosoma cruzi by TNF-alpha and IFN-gamma through a nitric oxide-dependent mechanism. Immunol Lett 33: 35–40, 1992 [DOI] [PubMed] [Google Scholar]
- 212.Murray HW. Pretreatment with phorbol myristate acetate inhibits macrophage activity against intracellular protozoa. J Reticuloendothel Soc 31: 479–487, 1982 [PubMed] [Google Scholar]
- 213.Murray HW. and Nathan CF. Macrophage microbicidal mechanisms in vivo: reactive nitrogen versus oxygen intermediates in the killing of intracellular visceral Leishmania donovani. J Exp Med 189: 741–746, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Murray JL, McDonald NJ, Sheng J, Shaw MW, Hodge TW, Rubin DH, O'Brien WA, and Smee DF. Inhibition of influenza A virus replication by antagonism of a PI3K-AKT-mTOR pathway member identified by gene-trap insertional mutagenesis. Antivir Chem Chemother 22: 205–215, 2012 [DOI] [PubMed] [Google Scholar]
- 215.Mydel P, Takahashi Y, Yumoto H, Sztukowska M, Kubica M, Gibson FC, 3rd, Kurtz DM, Jr., Travis J, Collins LV, Nguyen KA, Genco CA, and Potempa J. Roles of the host oxidative immune response and bacterial antioxidant rubrerythrin during Porphyromonas gingivalis infection. PLoS Pathog 2: e76, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Myers JT, Tsang AW, and Swanson JA. Localized reactive oxygen and nitrogen intermediates inhibit escape of Listeria monocytogenes from vacuoles in activated macrophages. J Immunol 171: 5447–5453, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Nabel G. and Baltimore D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 326: 711–713, 1987 [DOI] [PubMed] [Google Scholar]
- 218.Nagajyothi F, Zhao D, Weiss LM, and Tanowitz HB. Curcumin treatment provides protection against Trypanosoma cruzi infection. Parasitol Res 110″ 2491–2499, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, Hamada S, and Yoshimori T. Autophagy defends cells against invading group A Streptococcus. Science 306: 1037–1040, 2004 [DOI] [PubMed] [Google Scholar]
- 220.Nathan C. and Ding A. SnapShot: reactive oxygen intermediates (ROI). Cell 140: 951–951 e2, 2010 [DOI] [PubMed] [Google Scholar]
- 221.Nathan C, Nogueira N, Juangbhanich C, Ellis J, and Cohn Z. Activation of macrophages in vivo and in vitro. Correlation between hydrogen peroxide release and killing of Trypanosoma cruzi. J Exp Med 149: 1056–1068, 1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Newcomb WW. and Brown JC. Internal catalase protects herpes simplex virus from inactivation by hydrogen peroxide. J Virol 86: 11931–11934, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Nicolaou G, Goodall AH, and Erridge C. Diverse bacteria promote macrophage foam cell formation via Toll-like receptor-dependent lipid body biosynthesis. J Atheroscler Thromb 19: 137–148, 2012 [DOI] [PubMed] [Google Scholar]
- 224.Niethammer P, Grabher C, Look AT, and Mitchison TJ. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 459: 996–999, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Nimmerjahn F, Dudziak D, Dirmeier U, Hobom G, Riedel A, Schlee M, Staudt LM, Rosenwald A, Behrends U, Bornkamm GW, and Mautner J. Active NF-kappaB signalling is a prerequisite for influenza virus infection. J Gen Virol 85: 2347–2356, 2004 [DOI] [PubMed] [Google Scholar]
- 226.Nogueira NP, de Souza CF, Saraiva FM, Sultano PE, Dalmau SR, Bruno RE, Goncalves Rde L, Laranja GA, Leal LH, Coelho MG, Masuda CA, Oliveira MF, and Paes MC. Heme-induced ROS in Trypanosoma cruzi activates CaMKII-like that triggers epimastigote proliferation. One helpful effect of ROS. PloS One 6: e25935, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Nygren H, Malmberg P, and Sahlin H. Development of a wound dressing targeting neutrophil function. World J Surg 28: 337–342, 2004 [DOI] [PubMed] [Google Scholar]
- 228.O'Rourke EJ, Chevalier C, Pinto AV, Thiberge JM, Ielpi L, Labigne A, and Radicella JP. Pathogen DNA as target for host-generated oxidative stress: role for repair of bacterial DNA damage in Helicobacter pylori colonization. Proc Natl Acad Sci U S A 100: 2789–2794, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Oberley-Deegan RE, Lee YM, Morey GE, Cook DM, Chan ED, and Crapo JD. The antioxidant mimetic, MnTE-2-PyP, reduces intracellular growth of Mycobacterium abscessus. Am J Respir Cell Mol Biol 41: 170–178, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Oberley-Deegan RE, Rebits BW, Weaver MR, Tollefson AK, Bai X, McGibney M, Ovrutsky AR, Chan ED, and Crapo JD. An oxidative environment promotes growth of Mycobacterium abscessus. Free Radic Biol Med 49: 1666–1673, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Ohga S, Ikeuchi K, Kadoya R, Okada K, Miyazaki C, Suita S, and Ueda K. Intrapulmonary Mycobacterium avium infection as the first manifestation of chronic granulomatous disease. J Infect 34: 147–150, 1997 [DOI] [PubMed] [Google Scholar]
- 232.Okahashi N, Okinaga T, Sakurai A, Terao Y, Nakata M, Nakashima K, Shintani S, Kawabata S, Ooshima T, and Nishihara T. Streptococcus sanguinis induces foam cell formation and cell death of macrophages in association with production of reactive oxygen species. FEMS Microbiol Lett 323: 164–170, 2011 [DOI] [PubMed] [Google Scholar]
- 233.Olagnier D, Lavergne RA, Meunier E, Lefevre L, Dardenne C, Aubouy A, Benoit-Vical F, Ryffel B, Coste A, Berry A, and Pipy B. Nrf2, a PPARgamma alternative pathway to promote CD36 expression on inflammatory macrophages: implication for malaria. PLoS Pathog 7: e1002254, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Onyiah JC, Sheikh SZ, Maharshak N, Steinbach EC, Russo SM, Kobayashi T, Mackey LC, Hansen JJ, Moeser AJ, Rawls JF, Borst LB, Otterbein LE, and Plevy SE. Carbon monoxide and heme oxygenase-1 prevent intestinal inflammation in mice by promoting bacterial clearance. Gastroenterology 144: 789–798, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Paiva CN, Feijo DF, Dutra FF, Carneiro VC, Freitas GB, Alves LS, Mesquita J, Fortes GB, Figueiredo RT, Souza HS, Fantappie MR, Lannes-Vieira J, and Bozza MT. Oxidative stress fuels Trypanosoma cruzi infection in mice. J Clin Invest 122: 2531–2542, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Palanisamy GS, Kirk NM, Ackart DF, Shanley CA, Orme IM, and Basaraba RJ. Evidence for oxidative stress and defective antioxidant response in guinea pigs with tuberculosis. PloS One 6: e26254, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Pamplona A, Ferreira A, Balla J, Jeney V, Balla G, Epiphanio S, Chora A, Rodrigues CD, Gregoire IP, Cunha-Rodrigues M, Portugal S, Soares MP, and Mota MM. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat Med 13: 703–710, 2007 [DOI] [PubMed] [Google Scholar]
- 238.Papp D, Csermely P, and Soti C. A Role for SKN-1/Nrf in pathogen resistance and immunosenescence in Caenorhabditis elegans. PLoS Pathog 8: e1002673, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Park DW, Baek K, Kim JR, Lee JJ, Ryu SH, Chin BR, and Baek SH. Resveratrol inhibits foam cell formation via NADPH oxidase 1-mediated reactive oxygen species and monocyte chemotactic protein-1. Exp Mol Med 41: 171–179, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Parker H, Albrett AM, Kettle AJ, and Winterbourn CC. Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. J Leukoc Biol 91: 369–376, 2012 [DOI] [PubMed] [Google Scholar]
- 241.Parry MF. and Neu HC. Effect of N-acetylcysteine on antibiotic activity and bacterial growth in vitro. J Clin Microbiol 5: 58–61, 1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Paul-Clark MJ, McMaster SK, Sorrentino R, Sriskandan S, Bailey LK, Moreno L, Ryffel B, Quesniaux VF, and Mitchell JA. Toll-like receptor 2 is essential for the sensing of oxidants during inflammation. Am J Respir Crit Care Med 179: 299–306, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Peterson JD, Herzenberg LA, Vasquez K, and Waltenbaugh C. Glutathione levels in antigen-presenting cells modulate Th1 versus Th2 response patterns. Proc Natl Acad Sci U S A 95: 3071–3076, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Peyron P, Vaubourgeix J, Poquet Y, Levillain F, Botanch C, Bardou F, Daffe M, Emile JF, Marchou B, Cardona PJ, de Chastellier C, and Altare F. Foamy macrophages from tuberculous patients' granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog 4: e1000204, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Pham NK, Mouriz J, and Kima PE. Leishmania pifanoi amastigotes avoid macrophage production of superoxide by inducing heme degradation. Infect Immun 73: 8322–8333, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Piacenza L, Alvarez MN, Peluffo G, and Radi R. Fighting the oxidative assault: the Trypanosoma cruzi journey to infection. Curr Opin Microbiol 12: 415–421, 2009 [DOI] [PubMed] [Google Scholar]
- 247.Piacenza L, Peluffo G, Alvarez MN, Martinez A, and Radi R. Trypanosoma cruzi antioxidant enzymes as virulence factors in Chagas disease. Antioxid Redox Signal 19: 723–734, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Piao ZH, Kim MS, Jeong M, Yun S, Lee SH, Sun HN, Song HY, Suh HW, Jung H, Yoon SR, Kim TD, Lee YH, and Choi I. VDUP1 exacerbates bacteremic shock in mice infected with Pseudomonas aeruginosa. Cell Immunol 280: 1–9, 2012 [DOI] [PubMed] [Google Scholar]
- 249.Pinheiro RO, Nunes MP, Pinheiro CS, D'Avila H, Bozza PT, Takiya CM, Corte-Real S, Freire-de-Lima CG, and DosReis GA. Induction of autophagy correlates with increased parasite load of Leishmania amazonensis in BALB/c but not C57BL/6 macrophages. Microbes Infect 11: 181–190, 2009 [DOI] [PubMed] [Google Scholar]
- 250.Pohanka M, Pavlis O, Ruttkay-Nedecky B, Sochor J, Sobotka J, Pikula J, Adam V, and Kizek R. Tularemia progression accompanied with oxidative stress and antioxidant alteration in spleen and liver of BALB/c mice. J Microbiol 50: 401–408, 2012 [DOI] [PubMed] [Google Scholar]
- 251.Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, Orkin SH, Doerschuk CM, and Dinauer MC. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet 9: 202–209, 1995 [DOI] [PubMed] [Google Scholar]
- 252.Polvani S, Tarocchi M, and Galli A. PPARgamma and oxidative stress: con(beta) catenating NRF2 and FOXO. PPAR Res 2012: 641087, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Portugal S, Carret C, Recker M, Armitage AE, Goncalves LA, Epiphanio S, Sullivan D, Roy C, Newbold CI, Drakesmith H, and Mota MM. Host-mediated regulation of superinfection in malaria. Nat Med 17: 732–737, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Potter SM, Mitchell AJ, Cowden WB, Sanni LA, Dinauer M, de Haan JB, and Hunt NH. Phagocyte-derived reactive oxygen species do not influence the progression of murine blood-stage malaria infections. Infect Immun 73: 4941–4947, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Pratico D. In vivo measurement of the redox state. Lipids 36Suppl: S45–S47, 2001 [DOI] [PubMed] [Google Scholar]
- 256.Protzer U, Seyfried S, Quasdorff M, Sass G, Svorcova M, Webb D, Bohne F, Hosel M, Schirmacher P, and Tiegs G. Antiviral activity and hepatoprotection by heme oxygenase-1 in hepatitis B virus infection. Gastroenterology 133: 1156–1165, 2007 [DOI] [PubMed] [Google Scholar]
- 257.Puertollano MA, Puertollano E, de Cienfuegos GA, and de Pablo MA. Dietary antioxidants: immunity and host defense. Curr Top Med Chem 11: 1752–1766, 2011 [DOI] [PubMed] [Google Scholar]
- 258.Py BF, Lipinski MM, and Yuan J. Autophagy limits Listeria monocytogenes intracellular growth in the early phase of primary infection. Autophagy 3: 117–125, 2007 [DOI] [PubMed] [Google Scholar]
- 259.Qi M, Miyakawa H, and Kuramitsu HK. Porphyromonas gingivalis induces murine macrophage foam cell formation. Microb Pathog 35: 259–267, 2003 [DOI] [PubMed] [Google Scholar]
- 260.Qin Z, Freitas E, Sullivan R, Mohan S, Bacelieri R, Branch D, Romano M, Kearney P, Oates J, Plaisance K, Renne R, Kaleeba J, and Parsons C. Upregulation of xCT by KSHV-encoded microRNAs facilitates KSHV dissemination and persistence in an environment of oxidative stress. PLoS Pathog 6: e1000742, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Qiu H, Kuolee R, Harris G, and Chen W. Role of NADPH phagocyte oxidase in host defense against acute respiratory Acinetobacter baumannii infection in mice. Infect Immun 77: 1015–1021, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Rabhi I, Rabhi S, Ben-Othman R, Rasche A, Daskalaki A, Trentin B, Piquemal D, Regnault B, Descoteaux A, Guizani-Tabbane L, and Sysco C. Transcriptomic signature of Leishmania infected mice macrophages: a metabolic point of view. PLoS Negl Trop Dis 6: e1763, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Rager-Zisman B, Kunkel M, Tanaka Y, and Bloom BR. Role of macrophage oxidative metabolism in resistance to vesicular stomatitis virus infection. Infect Immun 36: 1229–1237, 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Rayamajhi M, Humann J, Penheiter K, Andreasen K, and Lenz LL. Induction of IFN-alphabeta enables Listeria monocytogenes to suppress macrophage activation by IFN-gamma. J Exp Med 207: 327–337, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Reddy NM, Suryanarayana V, Kalvakolanu DV, Yamamoto M, Kensler TW, Hassoun PM, Kleeberger SR, and Reddy SP. Innate immunity against bacterial infection following hyperoxia exposure is impaired in NRF2-deficient mice. J Immunol 183: 4601–4608, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, Potma EO, Warley A, Roes J, and Segal AW. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 416: 291–297, 2002 [DOI] [PubMed] [Google Scholar]
- 267.Reeves EP, Nagl M, Godovac-Zimmermann J, and Segal AW. Reassessment of the microbicidal activity of reactive oxygen species and hypochlorous acid with reference to the phagocytic vacuole of the neutrophil granulocyte. J Med Microbiol 52: 643–651, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Riva DA, de Molina MC, Rocchetta I, Gerhardt E, Coulombie FC, and Mersich SE. Oxidative stress in vero cells infected with vesicular stomatitis virus. Intervirology 49: 294–298, 2006 [DOI] [PubMed] [Google Scholar]
- 269.Roca FJ. and Ramakrishnan L. TNF dually mediates resistance and susceptibility to Mycobacteria via mitochondrial reactive oxygen species. Cell 153: 521–534, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Rocha-Vieira E, Ferreira E, Vianna P, De Faria DR, Gaze ST, Dutra WO, and Gollob KJ. Histopathological outcome of Leishmania major-infected BALB/c mice is improved by oral treatment with N-acetyl-l-cysteine. Immunology 108: 401–408, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Rockwell CE, Zhang M, Fields PE, and Klaassen CD. Th2 skewing by activation of Nrf2 in CD4(+) T cells. J Immunol 188: 1630–1637, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Rodrigues WF, Miguel CB, Chica JE, and Napimoga MH. 15d-PGJ(2) modulates acute immune responses to Trypanosoma cruzi infection. Mem Inst Oswaldo Cruz 105: 137–143, 2010 [DOI] [PubMed] [Google Scholar]
- 273.Rodriguez-Acosta A, Finol HJ, Pulido-Mendez M, Marquez A, Andrade G, Gonzalez N, Aguilar I, Giron ME, and Pinto A. Liver ultrastructural pathology in mice infected with Plasmodium berghei. J Submicrosc Cytol Pathol 30: 299–307, 1998 [PubMed] [Google Scholar]
- 274.Roederer M, Staal FJ, Raju PA, Ela SW, and Herzenberg LA. Cytokine-stimulated human immunodeficiency virus replication is inhibited by N-acetyl-L-cysteine. Proc Natl Acad Sci U S A 87: 4884–4888, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Romano PS, Arboit MA, Vazquez CL, and Colombo MI. The autophagic pathway is a key component in the lysosomal dependent entry of Trypanosoma cruzi into the host cell. Autophagy 5: 6–18, 2009 [DOI] [PubMed] [Google Scholar]
- 276.Romao PR, Fonseca SG, Hothersall JS, Noronha-Dutra AA, Ferreira SH, and Cunha FQ. Glutathione protects macrophages and Leishmania major against nitric oxide-mediated cytotoxicity. Parasitology 118 (Pt 6): 559–566, 1999 [DOI] [PubMed] [Google Scholar]
- 277.Rosen H, Crowley JR, and Heinecke JW. Human neutrophils use the myeloperoxidase-hydrogen peroxide-chloride system to chlorinate but not nitrate bacterial proteins during phagocytosis. J Biol Chem 277: 30463–30468, 2002 [DOI] [PubMed] [Google Scholar]
- 278.Rubartelli A, Gattorno M, Netea MG, and Dinarello CA. Interplay between redox status and inflammasome activation. Trends Immunol 32: 559–566, 2011 [DOI] [PubMed] [Google Scholar]
- 279.Ruotsalainen AK, Inkala M, Partanen ME, Lappalainen JP, Kansanen E, Makinen PI, Heinonen SE, Laitinen HM, Heikkila J, Vatanen T, Horkko S, Yamamoto M, Yla-Herttuala S, Jauhiainen M, and Levonen AL. The absence of macrophage Nrf2 promotes early atherogenesis. Cardiovasc Res 98: 107–115, 2013 [DOI] [PubMed] [Google Scholar]
- 280.Rutter K, Scherzer TM, Beinhardt S, Kerschner H, Stattermayer AF, Hofer H, Popow-Kraupp T, Steindl-Munda P, and Ferenci P. Intravenous silibinin as ‘rescue treatment’ for on-treatment non-responders to pegylated interferon/ribavirin combination therapy. Antiviral Ther 16: 1327–1333, 2011 [DOI] [PubMed] [Google Scholar]
- 281.Rybicka JM, Balce DR, Khan MF, Krohn RM, and Yates RM. NADPH oxidase activity controls phagosomal proteolysis in macrophages through modulation of the lumenal redox environment of phagosomes. Proc Natl Acad Sci U S A 107: 10496–10501, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Sadikot RT, Zeng H, Yull FE, Li B, Cheng DS, Kernodle DS, Jansen ED, Contag CH, Segal BH, Holland SM, Blackwell TS, and Christman JW. p47phox deficiency impairs NF-kappa B activation and host defense in Pseudomonas pneumonia. J Immunol 172: 1801–1808, 2004 [DOI] [PubMed] [Google Scholar]
- 283.Saeed O, Otsuka F, Polavarapu R, Karmali V, Weiss D, Davis T, Rostad B, Pachura K, Adams L, Elliott J, Taylor WR, Narula J, Kolodgie F, Virmani R, Hong CC, and Finn AV. Pharmacological suppression of hepcidin increases macrophage cholesterol efflux and reduces foam cell formation and atherosclerosis. Arterioscler Thromb Vasc Biol 32: 299–307, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Said-Sadier N, Padilla E, Langsley G, and Ojcius DM. Aspergillus fumigatus stimulates the NLRP3 inflammasome through a pathway requiring ROS production and the Syk tyrosine kinase. PloS One 5: e10008, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Santello FH, Frare EO, dos Santos CD, Toldo MP, Kawasse LM, Zucoloto S, and do Prado JC., Jr.Melatonin treatment reduces the severity of experimental Trypanosoma cruzi infection. J Pineal Res 42: 359–363, 2007 [DOI] [PubMed] [Google Scholar]
- 286.Santiago HC, Gonzalez Lombana CZ, Macedo JP, Utsch L, Tafuri WL, Campagnole-Santos MJ, Alves RO, Alves-Filho JC, Romanha AJ, Cunha FQ, Teixeira MM, Radi R, and Vieira LQ. NADPH phagocyte oxidase knockout mice control Trypanosoma cruzi proliferation, but develop circulatory collapse and succumb to infection. PLoS Negl Trop Dis 6: e1492, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Sapra R, Gaucher SP, Lachmann JS, Buffleben GM, Chirica GS, Comer JE, Peterson JW, Chopra AK, and Singh AK. Proteomic analyses of murine macrophages treated with Bacillus anthracis lethal toxin. Microb Pathog 41: 157–167, 2006 [DOI] [PubMed] [Google Scholar]
- 288.Schachtele SJ, Hu S, and Lokensgard JR. Modulation of experimental herpes encephalitis-associated neurotoxicity through sulforaphane treatment. PLoS One 7: e36216, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Schaper M, Leib SL, Meli DN, Brandes RP, Tauber MG, and Christen S. Differential effect of p47phox and gp91phox deficiency on the course of pneumococcal meningitis. Infect Immun 71: 4087–4092, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Schmidt WN, Mathahs MM, and Zhu Z. Heme and HO-1 inhibition of HCV, HBV, and HIV. Front Pharmacol 3: 129, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Schnaith A, Kashkar H, Leggio SA, Addicks K, Kronke M, and Krut O. Staphylococcus aureus subvert autophagy for induction of caspase-independent host cell death. J Biol Chem 282: 2695–2706, 2007 [DOI] [PubMed] [Google Scholar]
- 292.Schulert GS, McCaffrey RL, Buchan BW, Lindemann SR, Hollenback C, Jones BD, and Allen LA. Francisella tularensis genes required for inhibition of the neutrophil respiratory burst and intramacrophage growth identified by random transposon mutagenesis of strain LVS. Infect Immun 77: 1324–1336, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Segal AW. How neutrophils kill microbes. Annu Rev Immunol 23: 197–223, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Serghides L. and Kain KC. Peroxisome proliferator-activated receptor gamma-retinoid X receptor agonists increase CD36-dependent phagocytosis of Plasmodium falciparum-parasitized erythrocytes and decrease malaria-induced TNF-alpha secretion by monocytes/macrophages. J Immunol 166: 6742–6748, 2001 [DOI] [PubMed] [Google Scholar]
- 295.Shan Y, Lambrecht RW, Donohue SE, and Bonkovsky HL. Role of Bach1 and Nrf2 in up-regulation of the heme oxygenase-1 gene by cobalt protoporphyrin. FASEB J 20: 2651–2653, 2006 [DOI] [PubMed] [Google Scholar]
- 296.Shan Y, Zheng J, Lambrecht RW, and Bonkovsky HL. Reciprocal effects of micro-RNA-122 on expression of heme oxygenase-1 and hepatitis C virus genes in human hepatocytes. Gastroenterology 133: 1166–1174, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Shiloh MU, MacMicking JD, Nicholson S, Brause JE, Potter S, Marino M, Fang F, Dinauer M, and Nathan C. Phenotype of mice and macrophages deficient in both phagocyte oxidase and inducible nitric oxide synthase. Immunity 10: 29–38, 1999 [DOI] [PubMed] [Google Scholar]
- 298.Shin DM, Jeon BY, Lee HM, Jin HS, Yuk JM, Song CH, Lee SH, Lee ZW, Cho SN, Kim JM, Friedman RL, and Jo EK. Mycobacterium tuberculosis eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PLoS Pathog 6: e1001230, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Singh V, Jamwal S, Jain R, Verma P, Gokhale R, and Rao KV. Mycobacterium tuberculosis-driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell Host Microbe 12: 669–681, 2012 [DOI] [PubMed] [Google Scholar]
- 300.Skaar EP, Humayun M, Bae T, DeBord KL, and Schneewind O. Iron-source preference of Staphylococcus aureus infections. Science 305: 1626–1628, 2004 [DOI] [PubMed] [Google Scholar]
- 301.Skulachev VP. Possible role of reactive oxygen species in antiviral defense. Biochemistry (Mosc) 63: 1438–1440, 1998 [PubMed] [Google Scholar]
- 302.Slauch JM. How does the oxidative burst of macrophages kill bacteria? Still an open question. Mol Microbiol 80: 580–583, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Soler-Palacin P, Margareto C, Llobet P, Asensio O, Hernandez M, Caragol I, and Espanol T. Chronic granulomatous disease in pediatric patients: 25 years of experience. Allergol Immunopathol (Madr) 35: 83–89, 2007 [DOI] [PubMed] [Google Scholar]
- 304.Soucy-Faulkner A, Mukawera E, Fink K, Martel A, Jouan L, Nzengue Y, Lamarre D, Vande Velde C, and Grandvaux N. Requirement of NOX2 and reactive oxygen species for efficient RIG-I-mediated antiviral response through regulation of MAVS expression. PLoS Pathog 6: e1000930, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Sousa-Franco J, Araujo-Mendes E, Silva-Jardim I, J LS, Faria DR, Dutra WO, and Horta MF. Infection-induced respiratory burst in BALB/c macrophages kills Leishmania guyanensis amastigotes through apoptosis: possible involvement in resistance to cutaneous leishmaniasis. Microbes Infect 8: 390–400, 2006 [DOI] [PubMed] [Google Scholar]
- 306.Speert DP, Bond M, Woodman RC, and Curnutte JT. Infection with Pseudomonas cepacia in chronic granulomatous disease: role of nonoxidative killing by neutrophils in host defense. J Infect Dis 170: 1524–1531, 1994 [DOI] [PubMed] [Google Scholar]
- 307.Speir E, Shibutani T, Yu ZX, Ferrans V, and Epstein SE. Role of reactive oxygen intermediates in cytomegalovirus gene expression and in the response of human smooth muscle cells to viral infection. Circ Res 79: 1143–1152, 1996 [DOI] [PubMed] [Google Scholar]
- 308.Spooner R. and Yilmaz O. The role of reactive-oxygen-species in microbial persistence and inflammation. Int J Mol Sci 12: 334–352, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Staal FJ, Roederer M, and Herzenberg LA. Intracellular thiols regulate activation of nuclear factor kappa B and transcription of human immunodeficiency virus. Proc Natl Acad Sci U S A 87: 9943–9947, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Starr T, Child R, Wehrly TD, Hansen B, Hwang S, Lopez-Otin C, Virgin HW, and Celli J. Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe 11: 33–45, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Staudinger BJ, Oberdoerster MA, Lewis PJ, and Rosen H. mRNA expression profiles for Escherichia coli ingested by normal and phagocyte oxidase-deficient human neutrophils. J Clin Invest 110: 1151–1163, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Stecher B, Robbiani R, Walker AW, Westendorf AM, Barthel M, Kremer M, Chaffron S, Macpherson AJ, Buer J, Parkhill J, Dougan G, von Mering C, and Hardt WD. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol 5: 2177–2189, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Stefanska J. and Pawliczak R. Apocynin: molecular aptitudes. Mediat Inflamm 2008: 106507, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Stempin CC, Garrido VV, Dulgerian LR, and Cerban FM. Cruzipain and SP600125 induce p38 activation, alter NO/arginase balance and favor the survival of Trypanosoma cruzi in macrophages. Acta Tropica 106: 119–127, 2008 [DOI] [PubMed] [Google Scholar]
- 315.Stempin CC, Tanos TB, Coso OA, and Cerban FM. Arginase induction promotes Trypanosoma cruzi intracellular replication in Cruzipain-treated J774 cells through the activation of multiple signaling pathways. European journal of immunology 34: 200–209, 2004 [DOI] [PubMed] [Google Scholar]
- 316.Swirski FK. and Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science 339: 161–166, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, and Iwasaki A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci U S A 106: 2770–2775, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Tang H, Cao W, Kasturi SP, Ravindran R, Nakaya HI, Kundu K, Murthy N, Kepler TB, Malissen B, and Pulendran B. The T helper type 2 response to cysteine proteases requires dendritic cell-basophil cooperation via ROS-mediated signaling. Nat Immunol 11: 608–617, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Tavakoli S. and Asmis R. Reactive oxygen species and thiol redox signaling in the macrophage biology of atherosclerosis. Antioxid Redox Signal 17: 1785–1795, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Teles RM, Graeber TG, Krutzik SR, Montoya D, Schenk M, Lee DJ, Komisopoulou E, Kelly-Scumpia K, Chun R, Iyer SS, Sarno EN, Rea TH, Hewison M, Adams JS, Popper SJ, Relman DA, Stenger S, Bloom BR, Cheng G, and Modlin RL. Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science 339: 1448–1453, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Thannickal VJ. and Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol 279: L1005–L1028, 2000 [DOI] [PubMed] [Google Scholar]
- 322.Thao TD, Ryu HC, Yoo SH, and Rhee DK. Antibacterial and anti-atrophic effects of a highly soluble, acid stable UDCA formula in Helicobacter pylori-induced gastritis. Biochem Pharmacol 75: 2135–2146, 2008 [DOI] [PubMed] [Google Scholar]
- 323.Tilton C, Clippinger AJ, Maguire T, and Alwine JC. Human cytomegalovirus induces multiple means to combat reactive oxygen species. J Virol 85: 12585–12593, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Tung WH, Hsieh HL, Lee IT, and Yang CM. Enterovirus 71 induces integrin beta1/EGFR-Rac1-dependent oxidative stress in SK-N-SH cells: role of HO-1/CO in viral replication. J Cell Physiol 226: 3316–3329, 2011 [DOI] [PubMed] [Google Scholar]
- 325.Ueno N. and Wilson ME. Receptor-mediated phagocytosis of Leishmania: implications for intracellular survival. Trends Parasitol 28: 335–344, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Umezawa K, Ohnishi N, Tanaka K, Kamiya S, Koga Y, Nakazawa H, and Ozawa A. Granulation in livers of mice infected with Salmonella typhimurium is caused by superoxide released from host phagocytes. Infect Immun 63: 4402–4408, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Van Assche T, Deschacht M, da Luz RA, Maes L, and Cos P. Leishmania-macrophage interactions: insights into the redox biology. Free Radic Biol Med 51: 337–351, 2011 [DOI] [PubMed] [Google Scholar]
- 328.van Diepen A, van der Straaten T, Holland SM, Janssen R, and van Dissel JT. A superoxide-hypersusceptible Salmonella enterica serovar typhimurium mutant is attenuated but regains virulence in p47(phox−/−) mice. Infect Immun 70: 2614–2621, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Vandenabeele P, Galluzzi L, Vanden Berghe T, and Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11: 700–714, 2010 [DOI] [PubMed] [Google Scholar]
- 330.Vardhan H, Bhengraj AR, Jha R, Srivastava P, Jha HC, and Mittal A. Higher expression of ferritin protects Chlamydia trachomatis infected HeLa 229 cells from reactive oxygen species mediated cell death. Biochem Cell Biol 88: 835–842, 2010 [DOI] [PubMed] [Google Scholar]
- 331.Vazquez-Torres A, Xu Y, Jones-Carson J, Holden DW, Lucia SM, Dinauer MC, Mastroeni P, and Fang FC. Salmonella pathogenicity island 2-dependent evasion of the phagocyte NADPH oxidase. Science 287: 1655–1658, 2000 [DOI] [PubMed] [Google Scholar]
- 332.Venketaraman V, Dayaram YK, Amin AG, Ngo R, Green RM, Talaue MT, Mann J, and Connell ND. Role of glutathione in macrophage control of mycobacteria. Infect Immun 71: 1864–1871, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Venketaraman V, Dayaram YK, Talaue MT, and Connell ND. Glutathione and nitrosoglutathione in macrophage defense against Mycobacterium tuberculosis. Infect Immun 73: 1886–1889, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Vlahos R, Stambas J, Bozinovski S, Broughton BR, Drummond GR, and Selemidis S. Inhibition of nox2 oxidase activity ameliorates influenza a virus-induced lung inflammation. PLoS Pathog 7: e1001271, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Wakabayashi N, Slocum SL, Skoko JJ, Shin S, and Kensler TW. When NRF2 talks, who's listening? Antioxid Redox Signal 13: 1649–1663, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Wang G, Wang J, Fan X, Ansari GA, and Khan MF. Protein adducts of malondialdehyde and 4-hydroxynonenal contribute to trichloroethene-mediated autoimmunity via activating Th17 cells: dose- and time-response studies in female MRL+/+ mice. Toxicology 292: 113–122, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Wang J, Ren Z, Xu Y, Xiao S, Meydani SN, and Wu D. Epigallocatechin-3-gallate ameliorates experimental autoimmune encephalomyelitis by altering balance among CD4+ T-cell subsets. Am J Pathol 180: 221–234, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Wang Y, Weiss LM, and Orlofsky A. Host cell autophagy is induced by Toxoplasma gondii and contributes to parasite growth. J Biol Chem 284: 1694–1701, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Washburn RG, Gallin JI, and Bennett JE. Oxidative killing of Aspergillus fumigatus proceeds by parallel myeloperoxidase-dependent and -independent pathways. Infect Immun 55: 2088–2092, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Weichhart T. and Saemann MD. The multiple facets of mTOR in immunity. Trends Immunol 30: 218–226, 2009 [DOI] [PubMed] [Google Scholar]
- 341.Weiss G, Werner-Felmayer G, Werner ER, Grunewald K, Wachter H, and Hentze MW. Iron regulates nitric oxide synthase activity by controlling nuclear transcription. J Exp Med 180: 969–976, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Wen JJ, Gupta S, Guan Z, Dhiman M, Condon D, Lui C, and Garg NJ. Phenyl-alpha-tert-butyl-nitrone and benzonidazole treatment controlled the mitochondrial oxidative stress and evolution of cardiomyopathy in chronic chagasic Rats. J Am Coll Cardiol 55: 2499–2508, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, and Ghosh S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472: 476–480, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, Russell JM, Bevins CL, Adams LG, Tsolis RM, Roth JR, and Baumler AJ. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467: 426–429, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Winterbourn CC. and Kettle AJ. Redox reactions and microbial killing in the neutrophil phagosome. Antioxid Redox Signal 18: 642–660, 2013 [DOI] [PubMed] [Google Scholar]
- 346.Wu K, Koo J, Jiang X, Chen R, Cohen SN, and Nathan C. Improved control of tuberculosis and activation of macrophages in mice lacking protein kinase R. PLoS One 7: e30512, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Wu W, Hsu Y-MS, Bi L, Songyang Z, and Lin X. CARD9 facilitates microbe-elicited production of reactive oxygen species by regulating the LyGDI-Rac1 complex. Nat Immunol 10: 1208–1214, 2009 [DOI] [PubMed] [Google Scholar]
- 348.Wyde PR, Moore DK, Pimentel DM, Gilbert BE, Nimrod R, and Panet A. Recombinant superoxide dismutase (SOD) administered by aerosol inhibits respiratory syncytial virus infection in cotton rats. Antivir Res 31: 173–184, 1996 [DOI] [PubMed] [Google Scholar]
- 349.Xu M, Kashanchi F, Foster A, Rotimi J, Turner W, Gordeuk VR, and Nekhai S. Hepcidin induces HIV-1 transcription inhibited by ferroportin. Retrovirology 7: 104, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Yanagisawa S, Koarai A, Sugiura H, Ichikawa T, Kanda M, Tanaka R, Akamatsu K, Hirano T, Matsunaga K, Minakata Y, and Ichinose M. Oxidative stress augments toll-like receptor 8 mediated neutrophilic responses in healthy subjects. Respir Res 10: 50, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Yanaka A, Fahey JW, Fukumoto A, Nakayama M, Inoue S, Zhang S, Tauchi M, Suzuki H, Hyodo I, and Yamamoto M. Dietary sulforaphane-rich broccoli sprouts reduce colonization and attenuate gastritis in Helicobacter pylori-infected mice and humans. Cancer Prev Res 2: 353–360, 2009 [DOI] [PubMed] [Google Scholar]
- 352.Yang CS, Shin DM, Kim KH, Lee ZW, Lee CH, Park SG, Bae YS, and Jo EK. NADPH oxidase 2 interaction with TLR2 is required for efficient innate immune responses to mycobacteria via cathelicidin expression. J Immunol 182: 3696–3705, 2009 [DOI] [PubMed] [Google Scholar]
- 353.Yang HC, Cheng ML, Ho HY, and Chiu DT. The microbicidal and cytoregulatory roles of NADPH oxidases. Microb Infect 13: 109–120, 2011 [DOI] [PubMed] [Google Scholar]
- 354.Yasir M, Pachikara ND, Bao X, Pan Z, and Fan H. Regulation of chlamydial infection by host autophagy and vacuolar ATPase-bearing organelles. Infect Immun 79: 4019–4028, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Ye F, Zhou F, Bedolla RG, Jones T, Lei X, Kang T, Guadalupe M, and Gao SJ. Reactive oxygen species hydrogen peroxide mediates Kaposi's sarcoma-associated herpesvirus reactivation from latency. PLoS Pathog 7: e1002054, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Yi L, Liu Q, Orandle MS, Sadiq-Ali S, Koontz SM, Choi U, Torres-Velez FJ, and Jackson SH. p47(phox) directs murine macrophage cell fate decisions. Am J Pathol 180: 1049–1058, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Yoo SK, Starnes TW, Deng Q, and Huttenlocher A. Lyn is a redox sensor that mediates leukocyte wound attraction in vivo. Nature 480: 109–112, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Yoon JH, An SH, Kyeong IG, Lee MS, Kwon SC, and Kang JH. Oxidative modification of ferritin induced by hydrogen peroxide. BMB Rep 44: 165–169, 2011 [DOI] [PubMed] [Google Scholar]
- 359.Yousefi S, Mihalache C, Kozlowski E, Schmid I, and Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ 16: 1438–1444, 2009 [DOI] [PubMed] [Google Scholar]
- 360.Yvan-Charvet L, Ranalletta M, Wang N, Han S, Terasaka N, Li R, Welch C, and Tall AR. Combined deficiency of ABCA1 and ABCG1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J Clin Invest 117: 3900–3908, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Zaki MH, Fujii S, Okamoto T, Islam S, Khan S, Ahmed KA, Sawa T, and Akaike T. Cytoprotective function of heme oxygenase 1 induced by a nitrated cyclic nucleotide formed during murine salmonellosis. J Immunol 182: 3746–3756, 2009 [DOI] [PubMed] [Google Scholar]
- 362.Zhang Q, Pi J, Woods CG, and Andersen ME. A systems biology perspective on Nrf2-mediated antioxidant response. Toxicol Appl Pharmacol 244: 84–97, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Zhao JF, Ching LC, Huang YC, Chen CY, Chiang AN, Kou YR, Shyue SK, and Lee TS. Molecular mechanism of curcumin on the suppression of cholesterol accumulation in macrophage foam cells and atherosclerosis. Mol Nutr Food Res 56: 691–701, 2012 [DOI] [PubMed] [Google Scholar]
- 364.Zhu X, Westcott MM, Bi X, Liu M, Gowdy KM, Seo J, Cao Q, Gebre AK, Fessler MB, Hiltbold EM, and Parks JS. Myeloid cell-specific ABCA1 deletion protects mice from bacterial infection. Circ Res 111: 1398–1409, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Zhu Z, Wilson AT, Luxon BA, Brown KE, Mathahs MM, Bandyopadhyay S, McCaffrey AP, and Schmidt WN. Biliverdin inhibits hepatitis C virus nonstructural 3/4A protease activity: mechanism for the antiviral effects of heme oxygenase? Hepatology 52: 1897–1905, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Zhu Z, Wilson AT, Mathahs MM, Wen F, Brown KE, Luxon BA, and Schmidt WN. Heme oxygenase-1 suppresses hepatitis C virus replication and increases resistance of hepatocytes to oxidant injury. Hepatology 48: 1430–1439, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]