

Summary
The ability of injured axons to regenerate declines with age yet the mechanisms that regulate axon regeneration in response to age are not known. Here we show that axon regeneration in aging C. elegans motor neurons is inhibited by the conserved insulin/IGF1 receptor DAF-2. DAF-2’s function in regeneration is mediated by intrinsic neuronal activity of the forkhead transcription factor DAF-16/FOXO. DAF-16 regulates regeneration independently of lifespan, indicating that neuronal aging is an intrinsic, neuron specific, and genetically regulated process. In addition, we found that daf-18/PTEN inhibits regeneration independently of age and FOXO signaling, via the TOR pathway. Finally, DLK-1, a conserved regulator of regeneration, is downregulated by insulin/IGF1 signaling, bound by DAF-16 in neurons, and is required for both DAF-16- and DAF-18-mediated regeneration. Together, our data establish that insulin signaling specifically inhibits regeneration in aging adult neurons, and that this mechanism is independent of PTEN and TOR.
Introduction
Axon regeneration has the potential to repair damaged neurons in response to injury. However, across species, the regenerative potential of injured neurons decreases with age. For example, in the mammalian PNS, where regeneration does occur in adults, regenerative potential is lower in old animals (Pestronk et al., 1980; Tanaka et al., 1992; Verdu et al., 1995; Verdu et al., 2000). In C. elegans, axon regeneration also declines during adulthood (Gabel et al., 2008; Hammarlund et al., 2009; Nix et al., 2011; Wu et al., 2007; Zou et al., 2013). In animals with a mature nervous system, the various effects of age on axon regeneration could be a secondary consequence of aging’s deleterious effects on the organism as a whole, or a result of interactions between neurons and an aging cellular environment. Alternatively, the effects of age could be part of a specific program executed in neurons. Understanding the mechanisms that regulate the decline of regeneration with age could help inform therapies for age-related changes in neuronal function, disease, and recovery. However, the mechanisms that mediate the decline of regeneration in adults are not well understood.
To characterize a neuron’s response to age and injury, we investigated age-related decline in axon regeneration in C. elegans. C. elegans is a leading genetic model of aging (Antebi, 2007; Kenyon, 2010; White et al., 1986). Moreover, C. elegans has a well-characterized, invariant nervous system and has recently emerged as a model for neuronal aging (Pan et al., 2011; Tank et al., 2011; Toth et al., 2012). Finally, C. elegans is a powerful system for studying axon regeneration (Chen and Chisholm, 2011; El Bejjani and Hammarlund, 2012). Individual post-developmental C. elegans axons are readily amenable to pulsed laser axotomy and their regeneration is easily visualized in vivo (Byrne et al., 2011; Gabel et al., 2008; Hammarlund et al., 2009; Rao et al., 2008; Wu et al., 2007; Yanik et al., 2004). Thus, the worm holds the potential to identify mechanisms that regulate how age alters the response of neurons to injury.
Here we analyze how age affects regeneration in the GABA motor neurons of adult C. elegans. We show that as adult animals age, these neurons exhibit decreased retraction, decreased growth response, and decreased extension in response to nerve injury. We show that this decline in regeneration is regulated by the insulin/IGF1 receptor daf-2 and the forkhead transcription factor daf-16/FOXO. daf-16 functions cell-intrinsically in neurons to regulate regeneration, independently of daf-16’s function in lifespan determination. The conserved phosphatase daf-18/PTEN (protein phosphatase and tensin homolog deleted on chromosome ten) also regulates regeneration yet does so in an age- and insulin-independent manner, via TOR (target of rapamycin) signaling. Moreover, regeneration in both daf-2 and daf-18 mutants requires the critical dlk-1/DLK plasticity pathway. Our findings demonstrate that age-related decline in adult neuron regeneration is an intrinsic, neuron-specific, and genetically regulated process, and define separable functions for the INSR/IGF1R-FOXO and the PTEN-TOR pathways in regulating axon regeneration an aged animals.
Results
Insulin/IGF1 signaling inhibits axon regeneration and does so specifically in aged animals
We investigated the relationship between axon regeneration and age using single-neuron pulsed-laser surgery in adult C. elegans GABA (gamma-aminobutyric acid) motor neurons (Figure 1A). To address the effects of adult age (rather than developmental changes) on regeneration, we compared regeneration of severed axons at two adult time points: 1-day adults, aged one day past the final larval stage (young adults) and 5-day adults, aged five days past the final larval stage (aged adults). These adult time points occur well after the GABA neurons complete development by forming functional synaptic connections onto their final post-synaptic targets – a process that is finished by the second of four larval stages that precede adulthood in the C. elegans life cycle (Wood 1988). We found that in young adult wild type animals, 65% of axons regenerated in response to injury (Figure 1B, D), consistent with previous reports (Hammarlund et al., 2009). By contrast, in aged adult wild type animals, few axons (28%) initiated a regeneration response to injury (Figure 1C,D). Therefore, axon regeneration declines in adulthood, in large part due to failure in aged animals of growth cone initiation in response to nerve injury.
Figure 1. Regeneration declines in aged animals.

(A) GABA commissures are severed with a pulsed laser and scored for regeneration after 24 hours in young adult (1-day adult, L4 + 1 day at 25°C) and aged adult (5-day adult, L4 + 5 days at 25°C) animals. (B) GABA commissures 24 hrs after being severed in 1-day adult wild type and (C) 5-day adult wild type animals. (D) Comparison of axon regeneration frequency in young and aged wild type animals. Regeneration in 5-day adult animals is significantly reduced relative to 1-day adult animals (p<0.01, Fisher exact test). (E) Regenerating axons were binned into four categories (depicted in panel A) based on how far they had extended. Regeneration categories: ML-, did not extend past the midline; ML, extended to the midline; ML+, extended ¾ of the distance between the ventral and dorsal cords; Full, full regeneration to the dorsal cord. All animals contain Punc-47∷GFP, which drives GFP expression specifically in the GABA motor neurons. Error bars represent 95% confidence intervals.
Axon regeneration is a complex, multi-step process in which the injured neuron must respond to injury by initiating a growth cone, and then sustain axon extension toward the appropriate target. To characterize the effect of age on regeneration we binned cut axons into four categories based on how far they had regenerated toward their target, the dorsal nerve cord (Figure 1A-D). We found that in aged animals, even axons that did regenerate in response to injury were defective at extension toward the target. Of the severed axons that did regenerate in aged adult wild type animals, only 12% made substantial progress towards the dorsal nerve cord (Figure 1E, ML- and ML vs. ML+ and Full). By contrast, in young adult animals, 31% of regenerating axons made substantial progress towards the dorsal nerve cord after 24 hours (Fig 1E). Thus, of the few axons that do regenerate in aged adult animals, 88% fail to extend towards their targets in the dorsal nerve cord. Therefore, both axon initiation and extension after nerve injury are inhibited in aged adult animals relative to young adult animals. Together, these deficiencies result in an overall failure of axon regeneration in 5-day wild type adults, in stark contrast to the robust axon regeneration observed in wild type adults that are 4 days younger.
We hypothesized that the age-related decline in GABA neuron axon regeneration might be suppressed by manipulations that delay aging and increase lifespan. We tested axon regeneration in several well-characterized models that increase lifespan, including animals that have decreased insulin signaling (daf-2 mutants) (Kenyon et al., 1993; Kimura et al., 1997), animals that overexpress sirtuin (Tissenbaum and Guarente, 2001) (and that have an additional lifespan-increasing mutation (Viswanathan and Guarente, 2011)) (sir-2.1OE), and animals that are calorically-restricted as a result of eating difficulty (eat-2 mutants) (Lakowski and Hekimi, 1998). Of these models, neither sir-2.1 nor eat-2 significantly affected regeneration (p>0.3, Fisher exact test). However, modification of insulin signaling by mutating the insulin/insulin-like growth factor 1 receptor (INSR/IGF1R) DAF-2 enhanced regeneration in aged adults. We found that in contrast to wild type animals, 5-day adult daf-2(-) mutants exhibited no decline in axon regeneration (Figure 2A, B). daf-2 regulates regeneration specifically in aged animals, as the frequency of regeneration in young adult daf-2(-) mutants was indistinguishable from that in young adult wild type animals (Figure 2D). Therefore, daf-2 specifically regulates an age-related decline in axon regeneration.
Figure 2. daf-2 inhibits the number of regenerating axons and limits their extension during early senescence.

(A-C) Regenerating axons were binned into four categories based on how far they had extended. (A) GABA commissures 24 hours after being severed in 5-day adult wild type and 5-day adult daf-2(e1370) animals. (B) Quantification of regeneration in 5-day adult wild type and daf-2(e1370) animals 24 hours after axotomy. Regeneration in 5-day adult daf-2(-) animals is significantly increased relative to 5-day adult wild type animals (p<0.01, Fisher exact test). (C) Regenerating axons were binned into categories according to how far they extended towards the dorsal nerve cord (categories described in Figure Legend 1). More severed axons in aged adult daf-2(-) animals extend significantly (ML+ and Full) compared to severed axons in aged adult wild type animals (p<0.01, Fisher exact test). (D) Axon regeneration frequencies in wild type and daf-2(e1370) animals of increasing age. (E) Lengths of commissural fragments 24 hours after axotomy in 5-day adults. Width represents commissure length prior to axotomy. For precision, axons were severed on the ventral side of the seam cells which are located at the midline. All animals express the Punc-47∷GFP reporter. Error bars represent 95% confidence intervals in B and D, and standard error in E.
We characterized the specific effects of daf-2 on age-related regeneration using the same categorical analysis as described above (Figure 2A, C). In contrast to aged wild type animals, only 30% of axons in aged daf-2(-) mutants failed to respond to injury. Therefore, daf-2 inhibits growth cone initiation in aged axons. Furthermore, in contrast to the 12% of wild type axons that made substantial progress towards the dorsal nerve cord in wild type animals, approximately 48% of regenerating axons made considerable progress towards, or reached, the dorsal nerve cord in daf-2(-) mutants (Figure 2C, ML+ and Full). These results indicate that daf-2 inhibits both growth cone initiation and axon growth in aged animals.
To confirm that the failure of axons in aged wild type animals to regenerate past the midline is due to defects in growth and not inaccurate axon guidance, we measured the length of severed axons. We found that 24 hours after being cut, axons were significantly longer in daf-2(-) mutants than in wild type animals (Figure 2E). Interestingly, retraction after injury was also affected by daf-2. After axotomy in young animals, the injured ends of the axon retract away from the cut site (Hammarlund et al., 2009). We found that this retraction was reduced or absent in aged wild type animals (Figure 2E). The average length of the distal (dorsal) commissural fragments of injured axons in aged wild type animals extended just ventral of the midline, consistent with a lack of retraction away from the cut site (Figures 2A,E). By contrast, we observed axon retraction in the distal (dorsal) commissural fragments of injured axons in aged daf-2(-) animals (Figure 2A,E). Further, the average length of the distal (dorsal) commissural fragments of injured axons in these animals did not reach the midline, consistent with retraction away from the cut site (Figure 2A,C). Together, these results suggest that axon retraction is compromised in aged animals—possibly due to a lack of elasticity—and show that this defect is rescued in daf-2(-) animals. Further, these results show that the increased regeneration we observe in aged daf-2(-) animals is not because the daf-2(-) axons retract less: in fact, daf-2(-) axons retract more, and also regenerate farther. Therefore, daf-2 regulates growth rather than guidance of regenerating axons.
Although lack of daf-2 extended the ability of wild type animals to regenerate, it did not do so indefinitely. The frequency of axon regeneration in daf-2(-) mutants declined by ten days post-development (Figure 2D). However, even in these old animals, some degree of axon regeneration was still present in daf-2(-) animals. By contrast, in wild type animals ten days post-development, regeneration was never observed (Figure 2D). In summary, our results demonstrate that the INS/IGF1 receptor DAF-2 regulates both growth cone initiation and axon extension in aged adult animals after injury.
DAF-2 regulates regeneration in aged animals via DAF-16/FOXO
daf-2/INSR/IGF1R regulates lifespan by activating mechanisms that sequester the forkhead transcription factor daf-16/FOXO in the cytoplasm, thus preventing daf-16/FOXO from translocating to the nucleus and regulating transcription of effector genes (Henderson and Johnson, 2001; Kenyon et al., 1993; Lee et al., 2001; Lin et al., 2001). Mutations that disrupt daf-2/INSR/IGF1R result in a two-fold increase in lifespan that is dependent on daf-16/FOXO (Kenyon et al., 1993). To test whether inhibition of regeneration by daf-2 also functions by inhibiting daf-16, we assessed regeneration in mutants that lacked both daf-2 and daf-16. We found that eliminating daf-16 in daf-2(-) mutants abolished the high regeneration normally observed in aged daf-2(-) mutants (Figure 3A,B). By contrast, axons in aged animals lacking only daf-16 regenerated similarly to wild type axons (Figure 3A,B), consistent with the finding that daf-16/FOXO activity is mostly inhibited in wild type animals (Lin et al., 2001). Therefore, daf-2 inhibits regeneration in aged animals by preventing daf-16 function.
Figure 3. daf-16 mediates age-dependent regeneration downstream of daf-2.

(A) Comparison of axon regeneration frequencies in aged adult insulin pathway mutants, (B) binned into categories described in Figure 1. (C) 5-day adult and 10-day adult time points are indicated on the lifespans of wild type (blue), daf-2(e1370) (purple); daf-16(mu86) (green), and daf-2(e1370); daf-16(mu86) (orange) animals. (D) A simplified representation of the insulin/IGF1 signaling pathway. daf-16 regulates regeneration downstream of daf-2 in aged adults either (i) directly, or (ii) indirectly. Error bars represent 95% confidence intervals.
DAF-16/FOXO regulates regeneration independently of its role in lifespan determination
The effects we observed of daf-2 and daf-16 on axon regeneration correlate with their effects on lifespan (Figure 3A, C). daf-2(-) mutants live longer, and their axons regenerate well when aged; animals lacking both daf-2 and daf-16 do not live longer, and their axons do not regenerate well when aged. One possible model to explain this finding is that increased axon regeneration in 5-day adult daf-2(-) mutants is a secondary consequence of delayed aging (Figure 3D, hatched arrow ii). However, not all manipulations that affect lifespan also affect axon regeneration (see sir-2.1 and eat-2, above), as would be expected if the decline in axon regeneration is secondary to senectitude. Further, the steep decline we observe in regeneration in 5-day aged wild type adults occurs well before significant age-related mortality (Figure 3C). Even at 10 days, when both wild type and daf-2 mutants have lost all or most regenerative ability (Figure 2D), mortality is limited (Figure 3C). Together, these data suggest that senectitude and loss of regenerative ability are separable phenomena, but that both are regulated by daf-2 and daf-16 (Figure 3D, hatched arrow ii).
To test the idea that age-related loss of axon regeneration and lifespan are separately regulated, we attempted to decouple the two phenotypes using daf-16(-) mosaic animals (Figure 4). Although daf-16/FOXO is required for lifespan extension in daf-2/INSR/IGF1R mutants, restoring daf-16 to the intestine of daf-2(-); daf-16(-) double mutants (intestine∷daf-16*) is sufficient to increase their lifespan (Libina et al., 2003). We reasoned that if daf-16 function in neurons--rather than daf-16 function in lifespan extension--is required to increase regeneration in aged animals, the intestine∷daf-16* animals should fail to regenerate when aged, even though they have increased lifespans. We found that intestine∷daf-16* animals did exhibit increased lifespan compared to controls, consistent with previous results (Libina et al., 2003) (Figure 4A, B, +Pges-1:daf-16 and Figure S1). However, 5-day adult intestine∷daf-16* animals showed a low level of axon regeneration that was similar to wild type (Figure 4C,D). Thus, despite increasing the lifespan of daf-2(-); daf-16(-) double mutants, intestinal DAF-16 expression did not affect age-related decline of regeneration (Figure 4B,C, +Pges-1:daf-16). We conclude that daf-16 and insulin/IGF1 signaling can regulate lifespan independently of regeneration, and that increased lifespan is not sufficient to increase axon regeneration and aged animals.
Figure 4. daf-16 mediates age-dependent regeneration independently of lifespan.

(A-E) We assessed lifespan and regeneration in (A) aged daf-2(e1370); daf-16(mu86) worms expressing daf-16 in the intestine (Pges-1∷daf-16) and in the nervous system (Prgef-1∷daf-16). (B) Lifespans of daf-2(e1370); daf-16(mu86) (charcoal), with intestinal (blue) or neuronal (red) daf-16 expression. Duplicates represent separate extrachromosomal array-expressing lines. (C) Regeneration of aged daf-2(e1370); daf-16(mu86) worms expressing daf-16 in the nervous system (Prgef-1∷daf-16) and in the intestine (Pspl-1∷daf-16). (D) Regenerating axons were binned into categories described in Figure 1. Error bars represent 95% confidence intervals. See also Figure S1.
These results suggested that daf-16 might function in neurons to increase regeneration in aged animals. Indeed, the growth potential of injured axons depends in part on intrinsic factors (Neumann et al., 2002; Neumann and Woolf, 1999; Qiu et al., 2002). Thus, FOXO/daf-16 could regulate axon regeneration by acting directly in neurons. If so, we reasoned that neuron-specific daf-16 expression might rescue age-related regeneration in a daf-2(-); daf-16(-) background. We expressed wild type daf-16 under the control of the pan-neuronal promoter Prgef-1 in daf-2(-); daf-16(-) mutants (neuron:daf-16*). In these animals, daf-16 is expressed only in neurons, where it is active due to the lack of daf-2. We found that neuronal-specific expression of daf-16 had no effect on lifespan, consistent with previous results (Libina et al., 2003) (Figure 4A,B, Prgef-1∷daf-16). However, aged neuron:daf-16* animals had improved axon regeneration compared to controls, even though they did not live any longer (Figure 4B,C, Prgef-1∷daf-16). These results show that daf-16 function in neurons is sufficient to regulate axon regeneration.
Although neuron-specific expression of daf-16 improved axon regeneration in aged daf-2(-); daf-16(-) mutants, it was not as robust as axon regeneration in aged daf-2(-) mutants (compare Figure 3B and Figure 4D). This partial rescue may simply be the result of differences in neuronal expression of daf-16 between the endogenous locus and the Prgef-1-driven transgene. Alternatively, it may be an indication that while neuronal daf-16 is necessary and sufficient to promote regeneration, robust regeneration also requires daf-16 function outside the nervous system. Indeed, daf-16 has recently been shown to promote developmental neuronal migration (Christensen et al., 2011; Kennedy et al., 2013). Together, our data demonstrate that age-related decline of regeneration is not a secondary consequence of lifespan-related aging, and define a new function for daf-16 in regulating axon regeneration cell-autonomously in the aging nervous system.
DAF-18/PTEN inhibits regeneration independently of age and DAF-16/FOXO
Like daf-16/FOXO, daf-18/PTEN (protein phosphatase and tensin homolog deleted on chromosome ten) is required for lifespan extension in daf-2/INSR/IGF1R mutants (Dorman et al., 1995; Larsen et al., 1995). However, unlike daf-16, which positively regulates regeneration in daf-2(-) mutants (Figure 3A), PTEN inhibits axon regeneration in mice and flies (Christie et al., 2010; Liu et al., 2010; Park et al., 2008; Song et al., 2012b; Sun et al., 2011). To determine the role of PTEN in axon regeneration in C. elegans, we assessed regeneration in young and aged adult PTEN/daf-18 mutants. We found that like loss of daf-2, loss of PTEN/daf-18 significantly increased regeneration in aged adults (Figures 2B; 5B). However, unlike daf-2 (which had no effect on young animals), loss of PTEN/daf-18 also increased regeneration in young adults (Figures 2D; 5A). The increase in regeneration in aged PTEN/daf-18 mutants was not as large as that seen in aged daf-2 mutants (Figures 5B; 2B), and regenerating axons in aged PTEN/daf-18 mutants often had a less robust growth structure (Figures 5D; Figure 2C). Nonetheless, these data indicate that PTEN’s role as an inhibitor of axon regeneration is conserved in C. elegans.
Figure 5. DAF-18/PTEN does not regulate regeneration via DAF-16/FOXO in aged animals.

(A) Axon regeneration frequency of young and (B) aged animals. Error bars represent 95% confidence intervals. *, p<0.05; **, p<0.01, ***, p<0.001 relative to equivalently aged wild type, Fisher’s exact test. (C) Regenerating axons of young adult and (D) aged adult animals were binned into categories described in Figure 1. (E) Lifespan analysis of various daf-18 mutants. (F) Quantification of axon regeneration in aged animals of the indicated genotypes. (G) Axon regeneration frequency of aged wild type, daf-18(mg198), and daf-18(mg198) animals with extrachromasomal Punc-47∷daf-18cDNA, which is expressed in GABA neurons. *, p<0.05, relative to daf-18(mg198), Fisher exact test. (H) Model of interactions between components of the age-related regeneration response. In contrast to the mechanisms of lifespan determination, daf-18/PTEN regulates regeneration independently of daf-16/FOXO and daf-2. In all panels, daf-2(-), daf-16(-), and daf-18(-) represent daf-2(e1370), daf-16(mu86), and daf-18(mg198), respectively.
Although loss of PTEN/daf-18 and daf-2/INSR/IGF1R both result in increased regeneration in aged adults, the two mutations have different effects on lifespan. Loss of daf-2/INSR/IGF1R increases lifespan, and this increase depends on FOXO/daf-16 activation (Henderson and Johnson, 2001; Kenyon et al., 1993; Lee et al., 2001; Lin et al., 2001). By contrast, loss of PTEN/daf-18 decreases lifespan, and can suppress both extended lifespan and daf-16 activation in daf-2 mutants (Dorman et al., 1995; Larsen et al., 1995; Masse et al., 2005; Ogg and Ruvkun, 1998) (Figure 5E). Since PTEN/daf-18 mutants live shorter than wild type (Dorman et al., 1995; Larsen et al., 1995) (Figure 5E), but retain the ability to regenerate axons when aged (Figure 5B), the loss of PTEN is another way (besides daf-16 mosaics–see Figure 4) to decouple lifespan and axon regeneration in aged animals. Further, the finding that daf-18 and daf-2 both increase regeneration despite having opposite effects on lifespan mutations suggests that they may regulate axon regeneration by different mechanisms.
To determine whether enhanced regeneration in PTEN/daf-18 is due to the same mechanism as enhanced regeneration in daf-2, we determined its dependence on daf-16. In contrast to daf-2 mutants, which increase regeneration (and lifespan) in a daf-16-dependent manner (Kenyon et al., 1993), we found that axon regeneration in PTEN mutants was independent of daf-16. In aged adults, axons in animals that lacked both daf-18 and daf-16 (daf-18(mg150); daf-16(mu86)) regenerated as frequently as axons in daf-18(mg150) animals (Figure 5B). This lack of interaction between daf-18/PTEN and daf-16/FOXO in regulating axon regeneration was also observed in animals that lacked daf-2/INSR: we observed no difference in axon regeneration between aged daf-2(e1370); daf-18(mg198) and aged daf-2(e1370); daf-18(mg198); daf-16(mu86) animals (Figure 5F). In addition, we found that GABA-specific daf-18 expression (driven by the unc-47 (vesicular GABA transporter) promoter) inhibits regeneration in aged daf-18(-) mutants (Figure 5G). Therefore, daf-18 functions cell-intrinsically to inhibit axon regeneration, independently of insulin signaling and FOXO.
Another important output of PTEN activity is inhibition of the TOR (target of rapamycin) pathway (Song et al., 2012a). Mutation of PTEN results in increased AKT activity, leading to increased phosphorylation and reduced activity of the TSC complex (Maehama and Dixon, 1998; Song et al., 2012a; Stambolic et al., 1998; Sun et al., 1999). In turn, reduced TSC activity results in increased signaling by TOR (Manning and Cantley, 2003). We investigated whether increased TOR activity might account for the effect of loss of PTEN on axon regeneration in aged C. elegans GABA neurons. We inhibited TOR with rapamycin and found that had no effect on control animals, suggesting that TOR signaling is not a major component of axon regeneration in aged wild type animals. By contrast, rapamycin treatment abolished the increased axon regeneration phenotype of PTEN mutants (Figure 6). These results suggest that PTEN’s function in regeneration is mediated by TOR in C. elegans. TOR has previously been shown to mediate the effect of PTEN on axon regeneration in mammals (Christie et al., 2010; Liu et al., 2010; Park et al., 2008; Song et al., 2012b; Sun et al., 2011). Thus, the PTEN-TOR axis represents a conserved mechanism that regulates axon regeneration across species.
Figure 6. DAF-18/PTEN, but not insulin, regulates regeneration via TOR signaling.

(A) Representative images of axon regeneration wild type and daf-18(mg198) animals aged on DMSO or rapamycin plates. (B) Axon regeneration frequencies of aged adult animals placed on rapamycin or DMSO as indicated. Error bars represent 95% confidence intervals. (*, p<0.05, relative to daf-18(mg198), Fisher exact test).
Next, we tested whether activated TOR might also mediate the effect of loss of DAF-2 on axon regeneration in aged animals. We found that regeneration in aged daf-2(e1370) mutants is not affected by rapamycin (Figure 6). Therefore daf-2/IGF functions independently of TOR to regulate regeneration, in contrast to daf-18/PTEN. These findings strengthen our conclusion that despite functioning in the same pathway to regulate lifespan, PTEN and insulin signaling function independently of one another to regulate axon regeneration: PTEN via regulation of TOR, and insulin signaling via regulation of DAF-16 activity.
DLK-1 is required for DAF-2- and DAF-18-mediated regeneration
The strong and cell-autonomous regulation of regeneration by the transcription factor FOXO/daf-16 suggests that transcriptional regulation of one or more neuronal genes accounts for the DAF-16-dependent increase in axon regeneration we observe in aged daf-2(-) mutants. One potential target of such regulation is the genes of the dlk-1 MAP kinase pathway – the best-characterized intrinsic regeneration pathway in C. elegans. DLK-1/dlk-1 is a conserved Dual-Leucine zipper Kinase MAPKKK that functions in axon regeneration by activating MAPKK/mkk-4 and MAPK/pmk-3 (Hammarlund et al., 2009; Nakata et al., 2005; Yan et al., 2009). Activated MAPK/pmk-3 promotes regeneration at least in part by post-transcriptional stabilization of the mRNA for the B-Zip protein C/EBP1/cebp-1, likely via MAPKAP2/mak-2 (Yan et al., 2009). dlk-1 is also negatively regulated by the E3 ubiquitin ligase PHR1/rpm-1 (Hammarlund et al., 2009; Nakata et al., 2005; Yan et al., 2009). DLK-1 pathway function is required cell-autonomously to regulate regeneration across species (Hammarlund et al., 2009; Itoh et al., 2009; Shin et al., 2012; Watkins et al., 2013; Xiong et al., 2010). Overexpression of dlk-1 in GABA neurons confers an increase in regenerative potential to both young and aged animals (Hammarlund et al., 2009), without increasing lifespan (Figure S2). Therefore, the dlk-1 MAP kinase pathway is a candidate target for daf-16-mediated regulation of age-related regeneration.
To investigate whether dlk-1 functions with daf-16 to regulate regeneration, we first performed genetic epistasis experiments between dlk-1 and daf-16 (Figure 7A). We found that dlk-1 is absolutely required for regeneration in aged daf-2(-), and aged daf-18(-) mutants (Figure 7A) (Hammarlund et al., 2009). In addition, dlk-1 overexpression was sufficient to rescue regeneration in daf-16 mutants. Since dlk-1 is also absolutely necessary and sufficient for regeneration in aged wild type animals, these results indicate that dlk-1 functions downstream or in parallel to daf-16 and daf-18.
Figure 7. Insulin signaling regulates expression of DLK-1/DLK.
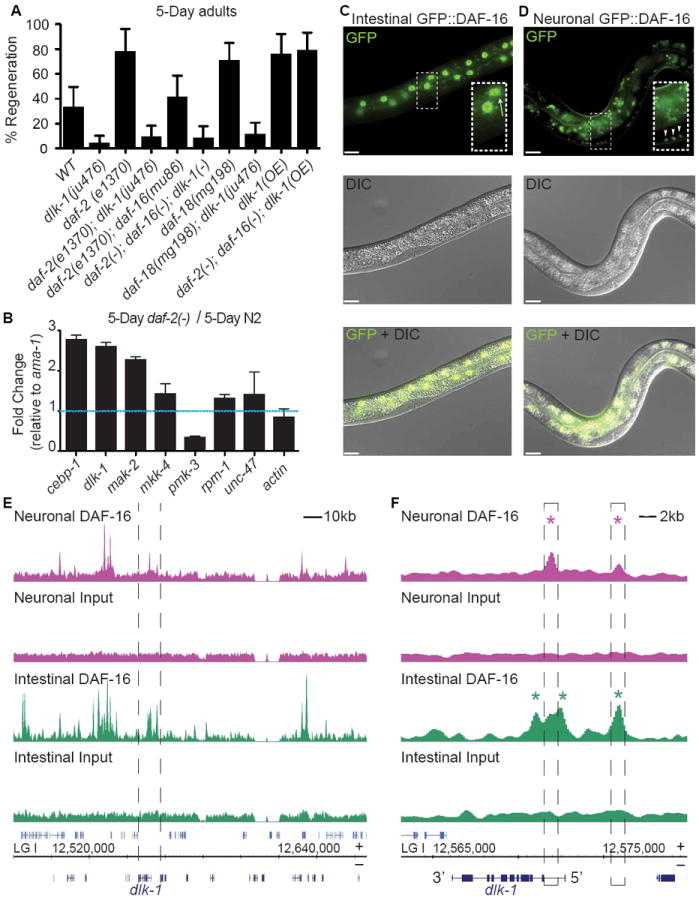
(A) Axon regeneration frequency of aged animals of the indicated genotypes. Due to space restrictions, daf-2(-), daf-16(-), and daf-18(-) represent daf-2(e1370), daf-16(mu86), and daf-18(mg198) respectively. Error bars represent 95% confidence intervals. (B) Quantitative reverse transcription PCR analysis of DLK-1 pathway genes in aged daf-2(e1370) animals. All values were normalized to ama-1 expression levels and to relative mRNA expression in aged wild type animals. Error bars represent 95% confidence intervals. (C) daf-2(e1370); daf-16(mu86) animals expressing GFP-tagged DAF-16A fused to either an intestinal (spl-1) or a (D) neuronal (rgef-1) promoter. GFP∷DAF-16A expression was restricted to the intestinal nuclei (arrow, inset) when fused to the intestinal promoter and was restricted to neuronal nuclei (arrowhead, inset) when fused to the neuronal promoter. Background intestinal autofluorescence was observed in both strains. Scale bars represent 10um. (E) Tissue specific DAF-16A binding profiles of strains presented in C and D. Profiles span the dlk-1 locus (bordered by dashed lines) on chromosome 1 (LG 1). (F) Magnification of DAF-16A binding profiles at the dlk-1 locus. In neurons, two regions in the dlk-1 promoter/locus were significantly enriched for DAF-16A bound sequences (pink asterisks). In the intestine, three regions in the dlk-1 promoter/locus were bound by DAF-16A (green asterisks). See also Figure S2.
To test whether the dlk-1 MAP kinase pathway is regulated in an age-dependent manner by daf-16, we compared the expression levels of dlk-1 pathway genes in aged wild type and daf-2(-) mutants (in which daf-16 function is elevated) using quantitative RT-PCR. We found that in aged daf-2(-) mutants, expression of three dlk-1 pathway genes were specifically increased compared to wild type: dlk-1, mak-2, and cebp-1 (Figure 7B and Figure S2). The increase we observe in cebp-1 expression may be a secondary consequence of dlk-1 upregulation, since dlk-1 activity increases cebp-1 mRNA levels by mak-2 –mediated post-transcriptional stabilization (Yan et al., 2009).
Next, we asked whether daf-16 directly regulates of one or more dlk-1 pathway components. To do so, we examined data from two independent methods--ChipSeq and DamID--that identified physical interactions between DAF-16 and the genome. We found that, uniquely among dlk-1 pathway genes, DAF-16 physically interacts with dlk-1 by both ChipSeq and DamID (Celniker et al., 2009; Schuster et al., 2010). Further, the physical interaction between DAF-16 and the dlk-1 promoter region is partially organized around canonical DAF-16 binding sites. Together, these findings indicate that DAF-16 binds to dlk-1 and may regulate its activity.
With respect to regeneration, DAF-16 functions specifically in the nervous system (Figure 4). We used tissue-specific ChIP-seq data to investigate whether DAF-16 binds directly to dlk-1 in neurons (Figure 7C, D, E, F). Single-copy integration techniques (Frokjaer-Jensen et al., 2012) were used to make transgenes that expressed GFP-tagged DAF-16A under either a neuronal (rgef-1) or an intestinal (spl-1) promoter. Integrated transgenes were placed in a background of daf-2 mutation (to activate nuclear translocation of the GFP-tagged DAF-16) and daf-16 mutation (to avoid competition from untagged DAF-16). An anti-GFP antibody was used to perform ChipSeq from each of the two resulting strains. Two replicates each of neuronal and intestinal DAF-16A-bound DNA were sequenced and analyzed. Overall, the analysis identified 1526 DAF-16 binding sites in neurons and 6525 in intestine (unpublished data and Figure 7E, F). Consistent with previous results, the canonical DAF-16 binding element (TGTTTAC, Murphy et al., 2003) was significantly overrepresented in the 500 most enriched sequences in both tissues (e-values: 1.3e-14, neurons; 1.6e-43 intestine) (Bailey et al., 2009).
Analysis of the Chip-seq data demonstrated that DAF-16 binds the dlk-1 promoter directly in neurons (Figure 7F). DAF-16 also binds dlk-1 in the intestine. However, the binding profiles at the dlk-1 locus differ in neurons and in the intestine (Figure 7F). In neurons, DAF-16 bound two peaks. In the intestine, DAF-16 bound three peaks, one of which was also bound by DAF-16 in neurons. Together with the evidence that daf-16 functions cell-autonomously in neurons to regulate regeneration (Figure 4), and that dlk-1 expression is regulated in an age and daf-16-dependent manner (Figure 7B and Figure S2), these results suggest that one effect of DAF-16 activity in aged animals is to increase expression of dlk-1 in neurons.
Together, our data demonstrate that one notable difference between aged wild type and daf-2(-) animals is expression of dlk-1 pathway genes, and suggest that increased dlk-1 pathway activity in neurons—perhaps due to binding and direct regulation of dlk-1 by neuronal DAF-16—contributes to high regeneration in aged daf-2 animals.
DISCUSSION
INSR/IGFR1 and FOXO function independently of and PTEN and TOR to regulate axon regeneration in aged neurons
Axon regeneration in adult C. elegans declines dramatically with age. In this study, we show that this decline is not a secondary consequence of organismal aging, but is an intrinsic property of aging neurons themselves. We identify two independent neuronal mechanisms that regulate axon regeneration in the GABA neurons of aged animals: activity of the transcription factor FOXO/DAF-16, potentially via the dlk-1 regeneration pathway; and activity of the lipid phosphatase PTEN/DAF-18, via regulation of TOR.
DAF-16 protects an aging nervous system
Our results show that the insulin signaling pathway acts in aged animals to inhibit regeneration via regulation of daf-16 activity in the nervous system. In the absence of insulin signaling, increased daf-16 activity allows the neuron to regenerate in response to injury. Recently, insulin signaling and daf-16 were also shown to regulate the maintenance of neuronal morphology in aging animals. The mechanosensory neurons (as well as some other neuronal types) accumulate aberrant branches and other morphological defects in an age-dependent manner, and accumulation of defects is delayed in daf-2(-) mutant animals, which have increased DAF-16 activity (Pan et al., 2011; Tank et al., 2011; Toth et al., 2012). Together, these data suggest that insulin signaling and DAF-16/FOXO regulate multiple protective processes in aging neurons, including the ability to regenerate in response to injury and the ability to maintain axon morphology.
The functions of DAF-16 in aging neurons are likely complex. Activated daf-16 enhances regenerative growth after laser injury (Figure 3A), but suppresses aberrant growth in neurons that have not received an external injury (Pan et al., 2011; Tank et al., 2011; Toth et al., 2012). Further, regeneration in aged animals is dependent on the dlk-1 MAP kinase pathway (Figure 7), while aberrant growth is independent of the dlk-1 pathway (Tank et al., 2011). Finally, although in aged animals both axon regeneration and maintenance of morphology are both regulated by insulin signaling and daf-16, the effects of aging occur at different rates. We found that insulin signaling acts at an early stage of adult life–between one and five days–to inhibit daf-16 and reduce regeneration of GABA neurons, with regeneration declining 10-fold in this time period (Figure 2D). By contrast, the accumulation of morphological defects in uninjured GABA neurons is much slower: no defects are observed at 8 days (Toth et al., 2012), and even at 10 days of adult life, not all animals have accumulated GABA neuron defects (Tank et al., 2011). Together, these data suggest that DAF-16 has multiple independent protective functions in aging neurons.
Conserved regeneration mechanisms in C. elegans
PTEN and TOR also regulate GABA neuron axon regeneration. In mutant animals that lack daf-18/PTEN, an increase in TOR activity results in increased regeneration (Fig 6). Like daf-16/FOXO, the effect of PTEN and TOR on axon regeneration is intrinsic to the nervous system (Fig 5G). However, the effect of PTEN and TOR on axon regeneration differs from that of daf-16/FOXO in that daf-18/PTEN regulates axon regeneration in both young and aged adult animals (Figure 5A, B). In mice, PTEN disruption promotes axon regeneration of retinal ganglion cells (Park et al., 2008), peripheral sensory (sciatic) neurons (Christie et al., 2010), and corticospinal neurons (Liu et al., 2010). Further, at least in the CNS, the full effect of PTEN deletion on axon regeneration requires mTOR activation (Liu et al., 2010; Park et al., 2008). In Drosophila, disrupting PTEN promotes axon regeneration of the Class IV dendritic arborization (da) neurons (Song et al., 2012b). Thus, PTEN and TOR signaling have emerged as major conserved regulators of axon regeneration, and increasing TOR activity may be useful for improving regeneration, particularly in combination with other pro-regenerative manipulations (Sun et al., 2011).
Our data establish C. elegans as a new model for studying the function of PTEN, TOR, and FOXO in axon regeneration. The conservation of PTEN and TOR’s function in axon regeneration– along with conservation of the DLK-1 pathway (Hammarlund et al., 2009; Itoh et al., 2009; Shin et al., 2012; Watkins et al., 2013; Xiong et al., 2010; Yan et al., 2009), calcium signaling (Ghosh-Roy et al., 2010; Pichichero et al., 1973; Spencer and Filbin, 2004; Ziv and Spira, 1997), and microtubule stability (Ghosh-Roy et al., 2012; Hellal et al., 2011; Sengottuvel et al., 2011) suggests that axon regeneration itself, and the intrinsic molecular mechanisms that regulate it, are of ancient origin and are functionally conserved across highly divergent species.
Both DAF-16/FOXO and TOR have multiple downstream effectors and are global regulators of cellular function. TOR signaling regulates multiple functions via one of two complexes, TORC1 and TORC2. These functions including nutrient sensing, cell growth, actin organization, cytoskeletal dynamics, autophagy, and stress response (Guertin and Sabatini, 2007). DAF-16/FOXO is a transcription factor that, when activated—as in daf-2(-) mutants—regulates the expression of a large number of target genes, (Kenyon et al., 1993; Murphy et al., 2003; Schuster et al., 2010; Wook Oh et al., 2006). DAF-16’s many functions include metabolism, lifespan, synapse maintenance and axon morphology (Murphy, 2006; Pan et al., 2011; Tank et al., 2011; Toth et al., 2012). Our data suggest that one important output of insulin-FOXO signaling is upregulation of the conserved dlk-1 regeneration pathway. Further studies will be necessary to identify other functions of insulin-FOXO signaling in aging neurons, and to determine the downstream effectors of PTEN-TOR.
Lifespan and neuronal healthspan are independent phenotypes
In addition to identifying molecular mechanisms that regulate GABA neuron regeneration in aging animals, our results demonstrate that the regulation of neuronal repair in aging animals is an essential component of healthspan, as opposed to lifespan. In other words, aging and loss of plasticity in neurons is an autonomous process that does not depend on overall lifespan. First, DAF-2/INSR/IGF1R inhibits axon regeneration in older animals by modulating neuronal DAF-16/FOXO function, and this function is independent of lifespan. Second, despite functioning antagonistically to regulate lifespan (Dorman et al., 1995), both loss of daf-18/PTEN and loss of daf-2 result in increased regeneration. Moreover, lifespan and healthspan are distinct at the cellular level, as DAF-16/FOXO functions in intestinal cells to regulate lifespan (Libina et al., 2003) and in neurons to regulate regeneration. Therefore, our results demonstrate that age-related decline in axon regeneration can be decoupled from lifespan, and these two effects of time are functionally distinct. The very early decline of regenerative potential suggests that the mechanisms described here may modulate a critical inflection point on the downward curve of neuronal function.
Experimental Procedures
Axotomy experiments
Axotomy experiments were carried out as previously described (Byrne et al., 2011). Post-axotomy images were acquired with an Olympus DSU mounted on an Olympus BX61® microscope, Andor Neo sCMOS camera, and Lumen light source. Error bars represent 95% confidence intervals. Significance is indicated with an asterisk (p<0.01, Fisher exact test). Lengths of regenerating axons were measured with MetaMorph software.
Lifespan analysis
Lifespans were carried out as previously described (Apfeld and Kenyon, 1999; Hansen et al., 2008; Hansen et al., 2007; Hsin and Kenyon, 1999) on FUDR plates (Kaeberlein et al., 2005). Animals were plated as L4s (t=0) and grown at 25°C. Animals that crawled off of the plate or ruptured were censored. Survival curves were generated with GraphPad Prism. Significance was determined with the Log Rank (Mantel-Cox) test. n values were greater than 55 for each condition tested.
Quantitative RT-PCR
To perform quantitative RT-PCR, total RNA was extracted from 100-worm samples of N2 or CB1370 with TRIzol and Qiagen RNeasy. cDNA was made from 100ng of total RNA with AffinityScript Multiple Temperature cDNA synthesis kit. qPCR was performed with Power SYBR green from Applied Biosystems and cycled on an Applied Biosystems 7500 Fast system. All reactions were repeated multiple times: three technical replicates for each of two biological replicates were performed. No-reverse transcriptase and no-template controls were included in each experiment. Two to three endogeneous controls were included in each experiment and all data was normalized to ama-1. Error bars represent 95% confidence intervals.
Rapamycin
Worms were grown on rapamycin at a previously reported effective concentration for C. elegans culture (Robida-Stubbs et al., 2012).
Chromatin immunoprecipitation-Deep sequencing (ChIP-seq)
ChIP-seq was performed essentially as described in Zhong et al. (2010) for two technical replicates of each strain analyzed, with the following modifications. Synchronized populations of worms were grown on peptone enriched plates seeded with OP50, and were collected at the L4 stage. Worms were fixed in 2% formaldehyde in M9 buffer for 30 minutes at room temperature. Worm pellets were sonicated to prepare protein lysate containing chromatin sheared between 100-1000 bp using a Sonic Dismembrator (Fisher). Immunoprecipitation was completed using 2 mg of protein lysate and 7.5 μg α-GFP (Zhong et al., 2010) at 4°C overnight. 1/10 of a fraction of lysate was used to prepare an input DNA sample.
Library preparation of XE1464 DNA samples for ChIP-seq was completed essentially as described in Zhong et al. (2010). XE1464 input and ChIP DNA samples were size selected to enrich for DNA ranging from 180-400 bp. Libraries prepared from all XE1464 DNA samples were multiplexed as described in Lefrancois et al. (2009), and were sequenced using the Illumina GA2 sequencing platform. Library preparation of XE1593 input and ChIP DNA samples for ChIP-seq was completed using the Ovation Multiplex Library System (NuGEN), in which additional size selection was not performed. Libraries prepared from XE1593 DNA samples were sequenced using the Illumina HiSeq 2000 sequencing platform.
Following sequencing, all samples were uniformly processed using parameters established by the modENCODE consortium for the mapping of transcription factor binding sites (Araya, (to be resubmitted); Trinh et al., 2013). Significantly enriched peaks were called using SPP and IDR analysis tools (Araya, (to be resubmitted); Trinh et al., 2013). Gene annotations for the C. elegans genome were compiled from several sources and mapped to genome build WS220. Coding gene models were collected from Wormbase WS235 and Gerstein et al. (submitted), which includes the analysis of many C. elegans RNA-seq datasets to develop aggregated and improved gene models. Similarly, noncoding RNA gene models were collected from Gerstein et al. (submitted), Lu et al. (2011), and Wormbase WS235 (for tRNAs, snoRNAs, snRNAs, miRNAs, 21U-RNAs, etc). Candidate target gene models were assigned based on closest proximity to the highest point of the binding site, irrespective of whether the binding site is upstream or downstream of the 5’ or 3’ end of the gene (21U-RNAs were excluded from this assignment). In addition, nearby gene models (both coding and noncoding) are listed but not assigned as the candidate target gene.
Strains
Strains (detailed in Supplemental Experimental Procedures) were maintained as previously described at 20°C (Brenner, 1974). The progeny of healthy non-starved animals were transferred to FUDR plates (Sutphin and Kaeberlein, 2009) at the L4 stage and grown at 25°C for the indicated amount of time.
Supplementary Material
Highlights.
Insulin/IGF1 signaling inhibits axon regeneration and does so specifically in aged adult animals.
DAF-16/FOXO regulates regeneration independently of its role in lifespan determination.
DAF-18/PTEN inhibits regeneration independently of age and DAF-16/FOXO, via TOR signaling.
DLK-1 is required for DAF-2- and DAF-18-mediated regeneration.
Acknowledgments
This work was supported by the Christopher and Dana Reeve Foundation to A.B.B., the Ellison Medical Foundation to M.H. and by the National Institute Of General Medical Sciences F32GM101778 to K.E.G.
Footnotes
Supplemental Information
Supplemental Information includes two figures.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Antebi A. Genetics of aging in Caenorhabditis elegans. PLoS genetics. 2007;3:1565–1571. doi: 10.1371/journal.pgen.0030129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apfeld J, Kenyon C. Regulation of lifespan by sensory perception in Caenorhabditis elegans. Nature. 1999;402:804–809. doi: 10.1038/45544. [DOI] [PubMed] [Google Scholar]
- Araya CL, Kawli T, Kundaje A, Cayting P, Jiang L, Wu B, Niu W, Boyle A, Xie D, Ma M, Mace D, Gevirtzman L, Murray J, Reinke V, Waterston RH, Snyder M. A spatiotemporally-resolved cis-regulatory map of the C. elegans genome. to be re-submitted to Nature. [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne AB, Edwards TJ, Hammarlund M. In vivo laser axotomy in C. elegans. J Vis Exp. 2011 doi: 10.3791/2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celniker SE, Dillon LA, Gerstein MB, Gunsalus KC, Henikoff S, Karpen GH, Kellis M, Lai EC, Lieb JD, MacAlpine DM, et al. Unlocking the secrets of the genome. Nature. 2009;459:927–930. doi: 10.1038/459927a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chisholm AD. Axon regeneration mechanisms: insights from C. elegans. Trends in cell biology. 2011;21:577–584. doi: 10.1016/j.tcb.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen R, de la Torre-Ubieta L, Bonni A, Colon-Ramos DA. A conserved PTEN/FOXO pathway regulates neuronal morphology during C. elegans development. Development. 2011;138:5257–5267. doi: 10.1242/dev.069062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie KJ, Webber CA, Martinez JA, Singh B, Zochodne DW. PTEN inhibition to facilitate intrinsic regenerative outgrowth of adult peripheral axons. J Neurosci. 2010;30:9306–9315. doi: 10.1523/JNEUROSCI.6271-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorman JB, Albinder B, Shroyer T, Kenyon C. The age-1 and daf-2 genes function in a common pathway to control the lifespan of Caenorhabditis elegans. Genetics. 1995;141:1399–1406. doi: 10.1093/genetics/141.4.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Bejjani R, Hammarlund M. Neural regeneration in Caenorhabditis elegans. Annual review of genetics. 2012;46:499–513. doi: 10.1146/annurev-genet-110711-155550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frokjaer-Jensen C, Davis MW, Ailion M, Jorgensen EM. Improved Mos1-mediated transgenesis in C. elegans. Nat Methods. 2012;9:117–118. doi: 10.1038/nmeth.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabel CV, Antoine F, Chuang CF, Samuel AD, Chang C. Distinct cellular and molecular mechanisms mediate initial axon development and adult-stage axon regeneration in C. elegans. Development. 2008;135:1129–1136. doi: 10.1242/dev.013995. [DOI] [PubMed] [Google Scholar]
- Gerstein MB, Rozowsky J, Yan K-K, Wang D, Cheng C, Brown JB, Davis CA, Hillier L, Sisu C, Li JJ, Pei B, Harmanci AO, Duff MO, Djebali S, Alexander RP, Alver BH, Auerbach RK, Bell K, Bickel PJ, Boeck ME, Boley NP, Booth BW, Cherbas L, Cherbas P, Di C, Dobin A, Drenkow J, Ewing B, Fang G, Fastuca M, Feingold EA, Frankish A, Gao G, Good PJ, Green P, Guigó R, Hammonds A, Harrow J, Hoskins RA, Howald C, Hu L, Huang H, Hubbard TJP, Huynh C, Jha S, Kasper D, Kato M, Kaufman TC, Kitchen R, Ladewig E, Lagarde J, Lai E, Leng J, Lu Z, MacCoss M, May G, McWhirter R, Merrihew G, Miller DM, Mortazavi A, Murad R, Oliver B, Olson S, Park P, Pazin MJ, Perrimon N, Pervouchine D, Reinke V, Reymond A, Robinson G, Samsonova A, Saunders GI, Schlesinger F, Slack FJ, Spencer WC, Stoiber MH, Strasbourger P, Tanzer A, Thompson OA, Wan KH, Wang G, Wang H, Watkins KL, Wen J, Wen K, Xue C, Yang L, Yip K, Zaleski C, Zhang Y, Zheng H, Brenner SE, Graveley BR, Celniker SE, Gingeras TR, Waterston R. Comparison of 3 metazoan transcriptomes. doi: 10.1038/nature13424. Submitted to Science. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh-Roy A, Goncharov A, Jin Y, Chisholm AD. Kinesin-13 and tubulin posttranslational modifications regulate microtubule growth in axon regeneration. Dev Cell. 2012;23:716–728. doi: 10.1016/j.devcel.2012.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh-Roy A, Wu Z, Goncharov A, Jin Y, Chisholm AD. Calcium and cyclic AMP promote axonal regeneration in Caenorhabditis elegans and require DLK-1 kinase. J Neurosci. 2010;30:3175–3183. doi: 10.1523/JNEUROSCI.5464-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Hammarlund M, Nix P, Hauth L, Jorgensen EM, Bastiani M. Axon regeneration requires a conserved MAP kinase pathway. Science. 2009;323:802–806. doi: 10.1126/science.1165527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M, Chandra A, Mitic LL, Onken B, Driscoll M, Kenyon C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS genetics. 2008;4:e24. doi: 10.1371/journal.pgen.0040024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M, Taubert S, Crawford D, Libina N, Lee SJ, Kenyon C. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell. 2007;6:95–110. doi: 10.1111/j.1474-9726.2006.00267.x. [DOI] [PubMed] [Google Scholar]
- Hellal F, Hurtado A, Ruschel J, Flynn KC, Laskowski CJ, Umlauf M, Kapitein LC, Strikis D, Lemmon V, Bixby J, et al. Microtubule stabilization reduces scarring and causes axon regeneration after spinal cord injury. Science. 2011;331:928–931. doi: 10.1126/science.1201148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson ST, Johnson TE. daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr Biol. 2001;11:1975–1980. doi: 10.1016/s0960-9822(01)00594-2. [DOI] [PubMed] [Google Scholar]
- Hsin H, Kenyon C. Signals from the reproductive system regulate the lifespan of C. elegans. Nature. 1999;399:362–366. doi: 10.1038/20694. [DOI] [PubMed] [Google Scholar]
- Itoh A, Horiuchi M, Bannerman P, Pleasure D, Itoh T. Impaired regenerative response of primary sensory neurons in ZPK/DLK gene-trap mice. Biochem Biophys Res Commun. 2009;383:258–262. doi: 10.1016/j.bbrc.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, Powers RW, 3rd, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310:1193–1196. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- Kennedy LM, Pham SCDL, Grishok A. Nonautonomous Regulation of Neuronal Migration by Insulin Signaling, DAF-16/FOXO, and PAK-1. Cell Reports. 2013 doi: 10.1016/j.celrep.2013.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- Kimura KD, Tissenbaum HA, Liu YX, Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- Lakowski B, Hekimi S. The genetics of caloric restriction in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1998;95:13091–13096. doi: 10.1073/pnas.95.22.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen PL, Albert PS, Riddle DL. Genes that regulate both development and longevity in Caenorhabditis elegans. Genetics. 1995;139:1567–1583. doi: 10.1093/genetics/139.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RY, Hench J, Ruvkun G. Regulation of C. elegans DAF-16 and its human ortholog FKHRL1 by the daf-2 insulin-like signaling pathway. Curr Biol. 2001;11:1950–1957. doi: 10.1016/s0960-9822(01)00595-4. [DOI] [PubMed] [Google Scholar]
- Lefrancois P, Euskirchen GM, Auerbach RK, Rozowsky J, Gibson T, Yellman CM, Gerstein M, Snyder M. Efficient yeast ChIP-Seq using multiplex short-read DNA sequencing. BMC Genomics. 2009;10:37. doi: 10.1186/1471-2164-10-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libina N, Berman JR, Kenyon C. Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell. 2003;115:489–502. doi: 10.1016/s0092-8674(03)00889-4. [DOI] [PubMed] [Google Scholar]
- Lin K, Hsin H, Libina N, Kenyon C. Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat Genet. 2001;28:139–145. doi: 10.1038/88850. [DOI] [PubMed] [Google Scholar]
- Liu K, Lu Y, Lee JK, Samara R, Willenberg R, Sears-Kraxberger I, Tedeschi A, Park KK, Jin D, Cai B, et al. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat Neurosci. 2010;13:1075–1081. doi: 10.1038/nn.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu ZJ, Yip KY, Wang G, Shou C, Hillier LW, Khurana E, Agarwal A, Auerbach R, Rozowsky J, Cheng C, et al. Prediction and characterization of noncoding RNAs in C. elegans by integrating conservation, secondary structure, and high-throughput sequencing and array data. Genome Res. 2011;21:276–285. doi: 10.1101/gr.110189.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. The Journal of biological chemistry. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. Rheb fills a GAP between TSC and TOR. Trends in biochemical sciences. 2003;28:573–576. doi: 10.1016/j.tibs.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Masse I, Molin L, Billaud M, Solari F. Lifespan and dauer regulation by tissue-specific activities of Caenorhabditis elegans DAF-18. Dev Biol. 2005;286:91–101. doi: 10.1016/j.ydbio.2005.07.010. [DOI] [PubMed] [Google Scholar]
- Murphy CT. The search for DAF-16/FOXO transcriptional targets: approaches and discoveries. Exp Gerontol. 2006;41:910–921. doi: 10.1016/j.exger.2006.06.040. [DOI] [PubMed] [Google Scholar]
- Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature. 2003;424:277–284. doi: 10.1038/nature01789. [DOI] [PubMed] [Google Scholar]
- Nakata K, Abrams B, Grill B, Goncharov A, Huang X, Chisholm AD, Jin YS. Regulation of a DLK-1 and p38 MAP kinase pathway by the ubiquitin ligase RPM-1 is required for presynaptic development. Cell. 2005;120:407–420. doi: 10.1016/j.cell.2004.12.017. [DOI] [PubMed] [Google Scholar]
- Neumann S, Bradke F, Tessier-Lavigne M, Basbaum AI. Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron. 2002;34:885–893. doi: 10.1016/s0896-6273(02)00702-x. [DOI] [PubMed] [Google Scholar]
- Neumann S, Woolf CJ. Regeneration of dorsal column fibers into and beyond the lesion site following adult spinal cord injury. Neuron. 1999;23:83–91. doi: 10.1016/s0896-6273(00)80755-2. [DOI] [PubMed] [Google Scholar]
- Nix P, Hisamoto N, Matsumoto K, Bastiani M. Axon regeneration requires coordinate activation of p38 and JNK MAPK pathways. Proc Natl Acad Sci U S A. 2011;108:10738–10743. doi: 10.1073/pnas.1104830108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogg S, Ruvkun G. The C. elegans PTEN homolog, DAF-18, acts in the insulin receptor-like metabolic signaling pathway. Mol Cell. 1998;2:887–893. doi: 10.1016/s1097-2765(00)80303-2. [DOI] [PubMed] [Google Scholar]
- Pan CL, Peng CY, Chen CH, McIntire S. Genetic analysis of age-dependent defects of the Caenorhabditis elegans touch receptor neurons. Proc Natl Acad Sci U S A. 2011;108:9274–9279. doi: 10.1073/pnas.1011711108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, He Z. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestronk A, Drachman DB, Griffin JW. Effects of aging on nerve sprouting and regeneration. Exp Neurol. 1980;70:65–82. doi: 10.1016/0014-4886(80)90006-0. [DOI] [PubMed] [Google Scholar]
- Pichichero M, Beer B, Clody DE. Effects of dibutyryl cyclic AMP on restoration of function of damaged sciatic nerve in rats. Science. 1973;182:724–725. doi: 10.1126/science.182.4113.724. [DOI] [PubMed] [Google Scholar]
- Qiu J, Cai D, Dai H, McAtee M, Hoffman PN, Bregman BS, Filbin MT. Spinal axon regeneration induced by elevation of cyclic AMP. Neuron. 2002;34:895–903. doi: 10.1016/s0896-6273(02)00730-4. [DOI] [PubMed] [Google Scholar]
- Rao GN, Kulkarni SS, Koushika SP, Rau KR. In vivo nanosecond laser axotomy: cavitation dynamics and vesicle transport. Opt Express. 2008;16:9884–9894. doi: 10.1364/oe.16.009884. [DOI] [PubMed] [Google Scholar]
- Robida-Stubbs S, Glover-Cutter K, Lamming DW, Mizunuma M, Narasimhan SD, Neumann-Haefelin E, Sabatini DM, Blackwell TK. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012;15:713–724. doi: 10.1016/j.cmet.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster E, McElwee JJ, Tullet JM, Doonan R, Matthijssens F, Reece-Hoyes JS, Hope IA, Vanfleteren JR, Thornton JM, Gems D. DamID in C. elegans reveals longevity-associated targets of DAF-16/FoxO. Mol Syst Biol. 2010;6:399. doi: 10.1038/msb.2010.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengottuvel V, Leibinger M, Pfreimer M, Andreadaki A, Fischer D. Taxol facilitates axon regeneration in the mature CNS. J Neurosci. 2011;31:2688–2699. doi: 10.1523/JNEUROSCI.4885-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JE, Cho YC, Beirowski B, Milbrandt J, Cavalli V, DiAntonio A. Dual Leucine Zipper Kinase Is Required for Retrograde Injury Signaling and Axonal Regeneration. Neuron. 2012;74:1015–1022. doi: 10.1016/j.neuron.2012.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nature reviews. Molecular cell biology. 2012a;13:283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- Song Y, Ori-McKenney KM, Zheng Y, Han C, Jan LY, Jan YN. Regeneration of Drosophila sensory neuron axons and dendrites is regulated by the Akt pathway involving Pten and microRNA bantam. Genes Dev. 2012b;26:1612–1625. doi: 10.1101/gad.193243.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer T, Filbin MT. A role for cAMP in regeneration of the adult mammalian CNS. Journal of anatomy. 2004;204:49–55. doi: 10.1111/j.1469-7580.2004.00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- Sun F, Park KK, Belin S, Wang D, Lu T, Chen G, Zhang K, Yeung C, Feng G, Yankner BA, He Z. Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature. 2011;480:372–375. doi: 10.1038/nature10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Lesche R, Li DM, Liliental J, Zhang H, Gao J, Gavrilova N, Mueller B, Liu X, Wu H. PTEN modulates cell cycle progression and cell survival by regulating phosphatidylinositol 3,4,5,-trisphosphate and Akt/protein kinase B signaling pathway. Proc Natl Acad Sci U S A. 1999;96:6199–6204. doi: 10.1073/pnas.96.11.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutphin GL, Kaeberlein M. Measuring Caenorhabditis elegans life span on solid media. J Vis Exp. 2009:e1152. doi: 10.3791/1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Zhang QL, Webster HD. Myelinated fiber regeneration after sciatic nerve crush: morphometric observations in young adult and aging mice and the effects of macrophage suppression and conditioning lesions. Exp Neurol. 1992;118:53–61. doi: 10.1016/0014-4886(92)90022-i. [DOI] [PubMed] [Google Scholar]
- Tank EM, Rodgers KE, Kenyon C. Spontaneous age-related neurite branching in Caenorhabditis elegans. J Neurosci. 2011;31:9279–9288. doi: 10.1523/JNEUROSCI.6606-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- Toth ML, Melentijevic I, Shah L, Bhatia A, Lu K, Talwar A, Naji H, Ibanez-Ventoso C, Ghose P, Jevince A, et al. Neurite sprouting and synapse deterioration in the aging Caenorhabditis elegans nervous system. J Neurosci. 2012;32:8778–8790. doi: 10.1523/JNEUROSCI.1494-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh QM, Jen FY, Zhou Z, Chu KM, Perry MD, Kephart ET, Contrino S, Ruzanov P, Stein LD. Cloud-based uniform ChIP-Seq processing tools for modENCODE and ENCODE. BMC Genomics. 2013;14:494. doi: 10.1186/1471-2164-14-494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdu E, Buti M, Navarro X. The effect of aging on efferent nerve fibers regeneration in mice. Brain Res. 1995;696:76–82. doi: 10.1016/0006-8993(95)00762-f. [DOI] [PubMed] [Google Scholar]
- Verdu E, Ceballos D, Vilches JJ, Navarro X. Influence of aging on peripheral nerve function and regeneration. J Peripher Nerv Syst. 2000;5:191–208. doi: 10.1046/j.1529-8027.2000.00026.x. [DOI] [PubMed] [Google Scholar]
- Viswanathan M, Guarente L. Regulation of Caenorhabditis elegans lifespan by sir-2.1 transgenes. Nature. 2011;477:E1–2. doi: 10.1038/nature10440. [DOI] [PubMed] [Google Scholar]
- Watkins TA, Wang B, Huntwork-Rodriguez S, Yang J, Jiang Z, Eastham-Anderson J, Modrusan Z, Kaminker JS, Tessier-Lavigne M, Lewcock JW. DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc Natl Acad Sci U S A. 2013;110:4039–4044. doi: 10.1073/pnas.1211074110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, Brenner S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philosophical transactions of the Royal Society of London. Series B, Biological sciences. 1986;314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- Wook Oh S, Mukhopadhyay A, Dixit BL, Raha T, Green MR, Tissenbaum HA. Identification of direct DAF-16 targets controlling longevity, metabolism and diapause by chromatin immunoprecipitation. Nat Genet. 2006;38:251–257. doi: 10.1038/ng1723. [DOI] [PubMed] [Google Scholar]
- Wu Z, Ghosh-Roy A, Yanik MF, Zhang AZ, Jin YS, Chisholm AD. Caenorhabditis elegans neuronal regeneration is influenced by life stage, ephrin signaling, and synaptic branching. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:15132–15137. doi: 10.1073/pnas.0707001104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Wang X, Ewanek R, Bhat P, Diantonio A, Collins CA. Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J Cell Biol. 2010;191:211–223. doi: 10.1083/jcb.201006039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Wu Z, Chisholm AD, Jin Y. The DLK-1 kinase promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell. 2009;138:1005–1018. doi: 10.1016/j.cell.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanik MF, Cinar H, Cinar HN, Chisholm AD, Jin Y, Ben-Yakar A. Neurosurgery: functional regeneration after laser axotomy. Nature. 2004;432:822. doi: 10.1038/432822a. [DOI] [PubMed] [Google Scholar]
- Zhong M, Niu W, Lu ZJ, Sarov M, Murray JI, Janette J, Raha D, Sheaffer KL, Lam HY, Preston E, et al. Genome-wide identification of binding sites defines distinct functions for Caenorhabditis elegans PHA-4/FOXA in development and environmental response. PLoS genetics. 2010;6:e1000848. doi: 10.1371/journal.pgen.1000848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv NE, Spira ME. Localized and transient elevations of intracellular Ca2+ induce the dedifferentiation of axonal segments into growth cones. J Neurosci. 1997;17:3568–3579. doi: 10.1523/JNEUROSCI.17-10-03568.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Chiu H, Zinovyeva A, Ambros V, Chuang CF, Chang C. Developmental decline in neuronal regeneration by the progressive change of two intrinsic timers. Science. 2013;340:372–376. doi: 10.1126/science.1231321. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.