

Abstract
Bromodomain-containing protein 4 (Brd4) and hexamethylene bisacetamide (HMBA) inducible protein 1 (HEXIM1) are two opposing regulators of the positive transcription elongation factor b (P-TEFb), which is the master modulator of RNA polymerase II during transcriptional elongation. While Brd4 recruits P-TEFb to promoter-proximal chromatins to activate transcription, HEXIM1 sequesters P-TEFb into an inactive complex containing the 7SK small nuclear RNA. Besides regulating P-TEFb's transcriptional activity, recent evidence demonstrates that both Brd4 and HEXIM1 also play novel roles in cell cycle progression and tumorigenesis. Here we will discuss the current knowledge on Brd4 and HEXIM1 and their implication as novel therapeutic options against cancer.
1. Introduction
Transcriptional regulation is a fundamental process for converting the genetic codes into RNA synthesis for proper cellular functions in an organism. The main cellular machinery for transcribing all protein-coding genes is the RNA polymerase II (RNAPII). Due to the enormous numbers of different genes to be coordinately expressed at any given time, the transcriptional activity of RNAPII is subjected to meticulous modulation by a myriad of regulators. One of the most significant RNAPII regulators identified to date is the positive transcriptional elongation factor b (P-TEFb). P-TEFb controls the transition of RNAPII from promoter proximal pausing to productive elongation for efficient full-length mRNA synthesis [1, 2]. At the early phase of mRNA synthesis shortly after the initiation of transcription, the progression of RNAPII is paused near the transcriptional start site by the concerted actions of the negative elongation factor (NELF) [3], DRB (5,6-dichloro-1-b-D-ribofuranosyl-benzimidazole) sensitivity-inducing factor (DSIF) [4], and Gdown1 [5]. In order to release the paused RNAPII, P-TEFb is recruited to the promoter, where its kinase activity phosphorylates the C-terminal domain (CTD) of RNAPII and the negative elongation factors NELF and DSIF [6, 7]. These phosphorylation events are believed to induce conformational changes that allow RNAPII to escape promoter proximal pausing and enter the productive phase of transcriptional elongation. More importantly, the pausing of RNAPII serves as a rate-limiting step for transcriptional regulation, especially for inducible genes, such as those induced by stress and inflammatory responses [8, 9]. Cells treated with flavopiridol, the most potent and selective P-TEFb inhibiting compound, result in inhibition of 60–70% of RNAPII transcription [10, 11]. Furthermore, genome-wide studies find that RNAPII occupies the promoters of most protein-coding genes in Drosophila and human embryonic stem cells without entering into productive elongation [12–16]. Therefore, P-TEFb plays a critical role in RNAPII regulation and has a broad impact on global gene expression patterns that affect almost every aspect of cellular functions.
Besides controlling the expression of cellular genes, P-TEFb is also required for transcriptional regulation of a number of viruses. The best-known example is the human immunodeficiency virus (HIV). Mathews and coworkers detected two classes of HIVpromoted cytoplasmic RNAs, a fulllength transcript, and a short transcript ending 55–59 nucleotides from the start site of transcription [17]. This observation clearly implies that transcription of HIV may also be regulated at the elongation stage. Later studies demonstrate that the HIV Tat protein recruits P-TEFb to the viral promoter and triggers the transition of RNAPII into productive elongation, resulting in the generation of full-length viral transcripts [18–20].
P-TEFb is composed of cyclin-dependent kinase 9 (CDK9) and its regulatory cyclin partner cyclin T1 [21, 22]. The Ser2 of the RNA Polymerase II CTD repeat (i.e., YSPTSPS) has been identified as the P-TEFb phosphorylation site during elongation [23, 24]. CDK12 is also a CTD Ser2 kinase; however, it is not within the scope of this review [25]. CDK9 exists as two isoforms, a major 42 kDa form and a minor 55 kDa form [26]. Besides cyclin T1, minor CDK9-associated cyclins, such as T2a, T2b, and K, are also present but at much lower levels in many cell types [27, 28]. Given the important roles of P-TEFb in RNAPII-dependent transcription, P-TEFb's activity is constantly being modulated through dynamic association with positive and negative regulators. Among these factors, hexamethylene bisacetamide (HMBA) inducible protein 1 (HEXIM1) and bromodomain-containing protein 4 (Brd4) are the two major regulators of P-TEFb. In log-phase HeLa cells, roughly half of the P-TEFb is sequestered into an inactive complex containing the kinase inhibitor HEXIM1 [29, 30] and other auxiliary proteins, MePCE [31], and LARP7 [32] held together by the non-coding 7SK small nuclear RNA (snRNA) (Figure 1). HEXIM1 exerts its inhibitory function on P-TEFb only when associated with the 7SK snRNA, while neither 7SK nor HEXIM1 alone instigates any effects [29, 33]. It has been proposed that association of the 7SK snRNA with HEXIM1 leads to a conformational change that renders the cyclin T1-binding domain of HEXIM1 accessible for P-TEFb binding [34]. Besides the inactive pool of P-TEFb, the remaining half of P-TEFb is transcriptionally active and bound to Brd4. Brd4 functions to recruit P-TEFb to active promoter through its affinity to acetylated histones [35, 36] (Figure 1). The distribution of P-TEFb between the active and inactive complexes is a dynamic process that can be rapidly changed in response to cellular stress and transcriptional demand. The regulation of P-TEFb by Brd4 and HEXIM1 has been extensively reviewed elsewhere [37, 38]. In this review, we will focus on the recent findings on Brd4/HEXIM1 with respect to their newly discovered roles in cell cycle progression and cancer.
Figure 1.
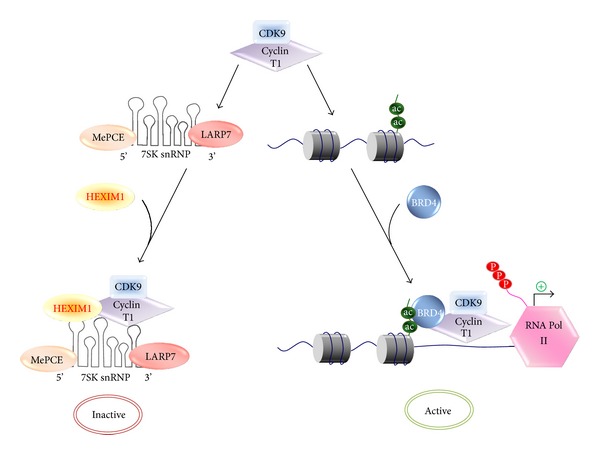
Regulation of P-TEFb activity by its positive regulator HEXIM1 and negative regulator Brd4.
2. Brd4
During eukaryotic transcription, the epigenetic information packaged in chromatin is interpreted by “reader” proteins. These proteins contain specialized domains for recognizing the vocabularies of histone codes that are composed of combinatorial histone modifications [39]. Histone acetylation, for instance, is specifically recognized by protein module termed bromodomain [40]. In human, there are a total of 61 different bromodomains. These bromodomains are evolutionarily conserved and present in 46 distinct proteins, which belong to eight families: histone acetyltransferases, ATP-dependent chromatin-remodeling complexes, helicases, methyltransferases, transcriptional coactivators, transcriptional mediators, nuclear-scaffolding proteins, and the BET family proteins [41, 42].
Brd4 is a ubiquitously expressed nuclear protein that belongs to the BET protein family. Brd4 contains two N-terminal tandem bromodomains (BDI and BDII) and an extraterminal domain (ET) [43]. The ET domain functions as a motif that interacts with WHSC1L1 (Wolf-Hirschhorn syndrome candidate 1-like 1 or NSD3) for the regulation of H3K36 methylation [44]. Early reports indicated that the bromodomains of Brd4 are essential for its binding to chromatin via acetylated histones [45, 46]. A recent study with systematic peptide array screening combined with crystallography further revealed that the first bromodomain BD1 of Brd4 contributes mostly for its association with acetylated chromatin by simultaneous binding and recognition of diverse diacetyl-containing histones [41].
2.1. The Dual Faces of Brd4: Cell Cycle Control and Transcriptional Regulation
Consistent with its chromatin targeting nature, Brd4 was originally identified as a mitotic chromosome associated protein (MCAP) as it was found persistently associated with acetylated chromosomes during mitosis in a number of cell lines [45, 46]. Later studies demonstrated that this association is critical for the rapid expression of early G1 genes upon exiting mitosis [47–50]. Although the mechanism is still unclear, Brd4 has been proposed to play an important role in transmitting the epigenetic memory across cell division by bookmarking the transcriptionally active genes [49, 51, 52]. Besides the bookmarking function, the association of Brd4 with mitotic chromosome is also important for Brd4's function in maintaining chromatin compaction [50, 53] and in facilitating the even segregation of viral DNAs into daughter cells during cell division [54].
Distinct from the other BET proteins, Brd4 contains a unique PID domain at its extreme C-terminus for interacting with P-TEFb [55]. Increasing evidence indicates that for the expression of many signal-inducible genes, the rate-limiting step is not transcription initiation but elongation [56]. After initiation is accomplished, RNAPII is poised at the promoter-proximal regions, and signal-induced activation and recruitment of P-TEFb to these regions convert the stalled RNAPII into productive elongation mode [57, 58].
In cell, P-TEFb is tightly regulated by being sequestrated in the nucleoplasma as an inactive ribonucleoprotein complex that contains the 7SK snRNA, HEXIM1, LARP7, and MePCE [59, 60] (Figure 1). Upon stimulation, P-TEFb is liberated from the inactive 7SK complex by the dephosphorylation of CDK9 T-loop through the concerted actions of PP2B and PP1α (protein phosphatase 2B and 1α) pathways or by the phosphorylation of HEXIM1 through PI3D/Akt or PKC kinase signaling pathways [61–63]. Subsequently, the liberated P-TEFb is recruited by Brd4 onto promoter-proximal region to modulate the processivity of RNAPII [29, 35, 36, 61, 64]. In line with its role in recruiting P-TEFb for stimulation of transcriptional elongation, Brd4 has been shown to be indispensable for cell proliferation [47–49], as well as the integration and transcription of HIV-1 [65–70], inflammatory response [8, 71, 72], cardiac hypertrophy [73, 74], and DNA damage repair [75, 76].
2.2. Brd4, a Novel Drug Target for Cancer Therapy
Accumulating studies have revealed the critical roles of Brd4 in cancer development [77, 78]. The first clue linking Brd4 with cancer was the finding of Brd4-NUT fusion oncogene which was recognized as an important mechanism in NUT midline carcinoma (NMC), an aggressive form of squamous carcinoma [79, 80]. NMC is genetically defined by the chromosomal rearrangements of the gene NUT (C15 or f55). In about 75% of NMCs, most of the NUT gene's coding region on chromosome 15q14 is fused with Brd4 [81–83]. Besides the chromosomal rearrangement-induced NUT midline carcinomas, other studies have also indicated that Brd4 may contribute to cancer development through different mechanisms. For example, deleting the proline-rich region of Brd4 could promote epithelial-to-mesenchymal transition and stem cell-like conversion [84, 85]. The interaction of the first bromodomain (BD1) of Brd4 with acetylated NF-κB/RelA leads to constitutively active NF-κB that enhances cancer cell proliferation [86]. Moreover, a study using shRNA library targeting 243 known chromatin regulators identified Brd4 as a required factor for the maintenance of acute myeloid leukemia (AML). Knockdown of Brd4 exhibited a robust antileukemic activity against AML in vitro and in vivo [87, 88]. Other recent studies with small molecule inhibitors of the BET proteins, such as JQ1 and I-BET 151, revealed the critical role of Brd4 in the development of several hematopoietic and somatic cancers, such as Burkitt's lymphoma, multiple myeloma [88–91], melanoma [92], colon [93], and breast cancer [84]. The mode of action of the BET inhibitors may be, at least in part, due to the inhibition of transcription of the oncogene, C-MYC [89–91]. A recent study shows that treatment with JQ1 results in preferential loss of Brd4 at super-enhancers and consequent transcription elongation defects that preferentially impacted genes with super-enhancers, including MYC [94]. Accordingly, small molecule inhibitors targeting Brd4 have been proven to be a promising drug for cancer therapy [77, 95].
2.3. The Role Switching of Brd4: From Chromatin Targeting to Transcriptional Regulation
The diverse biological roles of Brd4 have been proposed to rely on its functional transition between chromatin targeting and transcriptional regulation [43, 51]. Consistent with this notion, our recent study showed that, besides P-TEFb, the availability of Brd4 is also highly regulated [64]. Surveys of a variety of cell lines show that almost all Brd4 is bound to chromatin in interphase and there is limited chromatin-free Brd4 available in the unstimulated state. Upon stress induction (such as UV or HMBA treatment), Brd4 is released from chromatin and subsequently interacts and recruits signal-activated P-TEFb to enhance transcriptional elongation of inducible genes [64]. Intriguingly, stress treatment also triggers global deacetylation of acetylated-lysine 5 and 8 of nucleosomal histone H4 (H4K5ac/K8ac) [64]. In our recent study, these two acetylated sites are identified as the Brd4 binding sites (unpublished data), suggesting that the signal-controlled dynamic modification of nucleosomal histone is important for Brd4's role transition from chromatin targeting to transcriptional regulation.
Since the release of chromatin-bound Brd4 correlates with the signal-induced deacetylation of Brd4 binding sites H4k5ac/K8ac, the epigenetic regulation of Brd4's role switching apparently could simply be the deacetylation of Brd4's binding sites by histone deacetylases (HDACs). Through in vivo experiments and in vitro biochemical assays, we unexpectedly find that both PP1α and HDACs (HDAC1, 2, and 3 of class I HDAC) signaling pathways are essential for releasing chromatin-bound Brd4. Moreover, this releasing process relies on trans-histone crosstalk between H3S10ph (phosphorylated-serine 10 of histone H3) and H4K5ac/K8ac, which connects PP1α and HDACs to control the functional transition of Brd4 (Figure 2). In the nonstressed state, a H3S10ph-associated factor (referred as “X factor”) prevents HDAC1/2/3 from accessing H4K5ac/K8ac for deacetylation and hence locks up the majority of Brd4 onto chromatin (Figure 2(a)). During stress response, the PP1α pathway dephosphorylates H3S10ph, and the “X factor” departs from the nucleosome. This allows stress-activated HDAC1/2/3 to access and deacetylate H4K5ac/K8ac, thereby releasing chromatin-bound Brd4 for subsequent stimulation of inducible gene expression (Figure 2(b)). In this context, the dephosphorylation of H3S10ph governs Brd4's role switching from chromatin targeting to regulating inducible gene expression.
Figure 2.
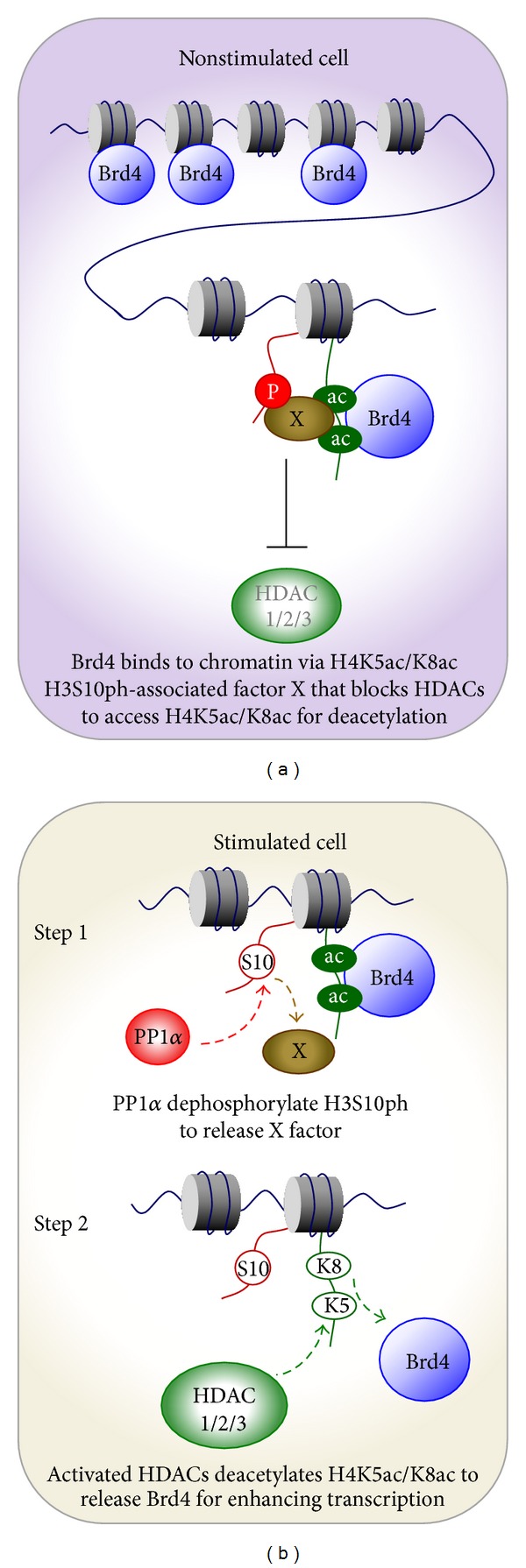
Regulation of the association between Brd4 and chromatins in the nonstimulated (a) and stimulated (b) cells.
For more than two decades, the signal-induced phosphorylation of H3S10 has been regarded as a positive epigenetic mark for transcriptional activation of inducible genes [96–100]. In contrast, we found that the dephosphorylation of H3S10ph is the key to Brd4-mediated expression of inducible genes ([64] and unpublished data). Although apparently contradictory, these events are likely to occur at different regions of the genome. We discover that, despite a significant decrease in global H3S10ph during stress treatments, there is an increased chromatin binding of 14-3-3 (unpublished data), an H3S10ph-associated protein that functions as a scaffold for recruiting chromatin remodeling factors [78, 100]. This result indicates that 14-3-3 and Brd4 are associated with different nucleosomes, and hence 14-3-3 is unlikely to be the “X factor”. Moreover, signal-induced H3S10 phosphorylation usually correlates with the chromatin remodeling of promoter [97], which occurs prior to transcription initiation. However, we observed that stress-induced H3S10ph dephosphorylation enables Brd4 to augment transcription elongation [64], an event subsequent to initiation. Hence, one may envision that for a small fraction of nucleosomes located at the promoter or enhancer regions of inducible genes, the signal-induced H3S10 phosphorylation facilitates chromatin remodeling for subsequent transcription initiation, whereas the global decline in H3S10ph may have distinct function in governing Brd4's release for subsequent P-TEFb recruitment and transcription elongation. As Brd4 represents a novel and promising drug target for cancer therapy, it is crucial to thoroughly elucidate its role in cell cycle progression and in epigenetic and transcriptional regulation.
3. HEXIM1
HEXIM1 was first identified in 1999 and the potential involvement of HEXIM1 in differentiation and cardiac development was suggested [101, 102]. However, the major biological function of HEXIM1 as the inhibitor of P-TEFb was revealed four years later by two research groups led by Olivier Bensaude and Qiang Zhou [29, 30]. Recent evidence demonstrated a role of HEXIM1 in cancers and regulation of the p53 pathway through the P-TEFb-dependent and -independent mechanisms [38, 103].
3.1. HEXIM1 as a Potential Tumor Suppressor
The potential involvement of HEXIM1 in cancers was first reported by Montano and coworkers in 2003. They identified HEXIM1 as a novel binding protein of estrogen receptor α (ERα) in a yeast two-hybrid screen using a cDNA library of MCF7 breast cancer cells. Since estrogens downregulated HEXIM1 expression at both protein and mRNA levels, HEXIM1 was also named estrogen downregulated gene 1 (EDG1) [104]. Estrogens are known to play a critical role in the growth of breast cancers and exert their effects by binding to their specific nuclear receptors. The ligand-bound ERs function as a transcriptional activator and upregulate several genes required for cell proliferation, such as cyclin D1 and C-MYC [105, 106].
ERα has been widely targeted in breast cancer therapy since it is present in more than half of breast tumors [107, 108]. Therefore, a potential role of HEXIM1 in regulating ERα in breast cancers was suggested. Breast cancer cells exhibited lower HEXIM1 expression when compared to normal breast epithelial tissue, and overexpression of HEXIM1 inhibited growth of both normal and breast cancer cells [104]. The molecular mechanism of cell growth inhibition by HEXIM1 was later elucidated. HEXIM1 interacted with ERα through its C-terminal region and inhibited the activity of ligand-bound ERα [109]. In addition, interaction between cyclin T1 and ERα was identified, suggesting the requirement of P-TEFb in regulating ERα activity during transcriptional activation [109]. Based on these findings, it was proposed that ERα might compete with HEXIM1 for binding to cyclin T1. Thus, the transcriptional activity of ERα depends on its association with P-TEFb (i.e., through the association with cyclin T1) and such a process is modulated by HEXIM1 [109].
Tamoxifen (Nolvadex), a selective ER modulator, has been used for more than 30 years to treat patients with advanced stage breast cancers [110]. However, tamoxifen resistance is a major problem in the treatment of ER-positive breast cancer patients. A recent study demonstrated that treatment with tamoxifen induced HEXIM1 recruitment to the promoters of ER target genes [111]. Since HEXIM1 was known to compete with cyclin T1 for binding to ERα, as expected, decreases in the abundance of cyclin T1 and serine 2-phosphorylated RNAPII on the coding regions of ER target genes were detected after tamoxifen treatment [111]. Furthermore, downregulation of HEXIM1 led to attenuation of the inhibition caused by tamoxifen on estrogen-induced gene expression and cell proliferation. Immunohistochemical studies using the samples of breast cancer patients showed that lower expression of HEXIM1 was associated with tumor recurrence in tamoxifen-receiving patients, suggesting that HEXIM1 is a critical determinant of tamoxifen resistance [111].
HEXIM1 is also involved in tumor progression. Angiogenesis is a physiological process governing the formation of new blood vessels from pre-existing vessels and an essential step in the transition of tumors from a benign state to a malignant one. Vascular endothelial growth factor (VEGF) functions as a major contributor to angiogenesis and its expression can be induced by estrogens (via ERα) or by hypoxia (via hypoxia inducible factor-1 alpha (HIF-1α)) [112]. HEXIM1 was found to regulate estrogen-induced VEGF transcription by inhibiting the recruitment of ERα to the VEGF promoter. Interestingly, HEXIM1 regulated this process in a P-TEFb-independent fashion [113]. Under hypoxic conditions, overexpression of HEXIM1 inhibited estrogen-induced expression of hypoxia-inducible factor-1 alpha (HIF-1α) protein and blocked the recruitment of HIF-1α to the promoter region of the VEGF gene [113]. HEXIM1 was later found to directly interact with HIF-1α and increase ubiquitination of HIF-1α, resulting in downregulation of HIF-1α protein expression [114].
Metastasis is the spreading of cancer cells from one organ or tissue to another. The development of metastases and angiogenesis are intrinsically connected [115]. The involvement of HEXIM1 in angiogenesis suggests a potential role of HEXIM1 in metastasis. A lower expression level of HEXIM1 was detected in metastatic breast cancers when compared with matched primary breast tumors [116]. Overexpression of HEXIM1, either by transgene expression or HMBA treatment, significantly inhibited metastasis and angiogenesis [116]. In addition, knockdown of HEXIM1 stimulated the invasion of MCF7 cells [116]. Taken together, these results suggest the tumor suppressor function of HEXIM1.
3.2. Regulation of HEXIM1 by NPM and MDM2
Results obtained from mass spectrometry in search for novel HEXIM1 binding proteins have led to the discovery of a functional interaction between HEXIM1 and the p53 pathway. Nucleophosmin (NPM; encoded by the NPM1 gene), a nucleolar protein and a key regulator of p53, was identified as a HEXIM1 binding partner [117]. NPM enhances p53 activity directly by binding to and stabilizing p53 or indirectly by stimulating the induction of p53 through binding to other p53 regulators, such as HDM2 and ARF [118–121]. Mutation of the NPM 1 gene is the most frequent mutation in acute myeloid leukemia (AML). About 35% of AML patients carrying NPMc+, the cytoplasmic-mislocated mutant form of NPM, indicate the pathological significance of this gene [122]. A distinct gene expression profile in NPMc+ AML cells was reported in an early study [123]; however, the connection between NPM mutation and transcriptional regulation remained to be elucidated.
We found that overexpression of NPM resulted in proteasome-dependent degradation of HEXIM1 and activation of P-TEFb [117]. This result demonstrated the functional significance of the HEXIM1-NPM interaction. The interaction between HEXIM1 and NPMc+ was also detected by immunoprecipitation (IP) [117]. In addition, using a green fluorescent protein (GFP) tagged NPMc+ fusion protein, immunofluorescence studies demonstrated that GFP-NPMc+ sequestered a portion of HEXIM1 in the cytoplasm [117]. Such mislocalization of HEXIM1 in the cytoplasm would have a significant impact on the equilibrium between active and inactive P-TEFb complexes in the nuclei by increasing the amounts of active P-TEFb complexes. In agreement with this observation, the cytoplasmic localization of HEXIM1 was only detected in an AML cell line carrying the NPMc+ mutation (i.e., AML3 cell line) [117]. Furthermore, comparing the AML cell line with wild-type NPM, lower HEXIM1 protein level was detected in AML3 cells [124]. As expected, an increase in P-TEFb-mediated transcription was detected in this NPMc+ AML cell line [117]. Collectively, these findings demonstrate a role for the NPMc+ mutation in transcriptional regulation and suggest the potential involvement of HEXIM1/P-TEFb in the tumorigenesis of AML.
MDM2 (or HDM2, the human homolog) is best-known as the negative regulator of p53. HDM2 ubiquitinates p53 and leads to proteasome-mediated degradation of p53 [125, 126]. Six lysine residues, Lys-370, -372, -373, -381, -382, and -386, located within the C-terminal negative (NEG) domain of p53 are the target residues of HDM2 ubiquitination [127] (Figure 3). Recently, HEXIM1 was identified as a novel substrate of HDM2. Unexpectedly, the ubiquitination of HEXIM1 by HDM2 does not lead to proteasome-dependent degradation but still affects HEXIM1's function [128]. Compared to wild-type HEXIM1, ubiquitinated HEXIM1 exhibits a stronger inhibitory effect on P-TEFb activity, suggesting a potential role for HDM2 on regulation of P-TEFb [128]. Six lysine residues located in the middle region of HEXIM1 were identified as the major sites of HDM2 ubiquitination [128]. Sequence alignment of the ubiquitination sites between p53 (amino acids 370–386) and HEXIM1 (amino acids 150-161) exhibits similar distribution of the lysine residues (Figure 3), raising the possibility that HEXIM1 may compete with p53 in binding to HDM2 and regulate the stability of p53.
Figure 3.

Alignment of the HDM2 ubiquitination sites between p53 (amino acids 370–386) and HEXIM1 (amino acids 150–161). The HDM2-ubiquitinated lysine residues are indicated in black boxes.
3.3. HEXIM1 as a Positive Regulator of p53
Associations between HEXIM1 and two p53 regulators, NPM and HDM2, suggest the possible involvement of HEXIM1 in the p53 pathway. Indeed, the interaction between endogenous HEXIM1 and p53 proteins was detected by IP. The direct binding between HEXIM1 and p53 was demonstrated by an in vitro GST pull-down assay using the purified recombinant proteins [129]. Overexpression of HEXIM1 upregulated the expression levels of p53 and p53 target genes by blocking p53 ubiquitination mediated by HDM2 [129]. This was further supported by domain study, showing that HEXIM1 is bound to p53 through the NEG region of p53 (Figure 4). As stated earlier, the NEG domain contains six lysine residues ubiquitinated by HDM2 [127] (Figure 4). This helps to explain that through the interaction with the NEG region, HEXIM1 prevents p53 ubiquitination by HDM2 and enhances the stability of p53. In addition, it was noted that p53 interacted with the 7SK snRNA-free HEXIM1, indicating that HEXIM1 might positively regulate p53 in a P-TEFb-independent manner [129].
Figure 4.

HEXIM1 stabilizes p53 by blocking the HDM2-mediated ubiquitination of p53. Human p53 protein can be divided into five domains: transactivation (TA), proline rich (PR), DNA binding (DB), oligomerization (OLI), and negative (NEG) domains. The HDM2 ubiquitination sites are located with the NEG domain. HEXIM1 interacts with the NEG domain of p53 and prevents the binding between p53 and HDM2.
Importantly, HEXIM1 is an essential factor that regulates the induction of p53. UV radiation or treatments with the anticancer agents, such as doxorubicin, etoposide, flavopiridol, roscovitine, and nutlin-3, are known to induce and activate p53. In all conditions examined, elevated protein levels of p53 were found to associate with the increased p53-HEXIM1 interaction [129]. In contrast, knockdown of HEXIM1 completely blocked p53 induction and released the cell cycle arrest caused by p53 [129]. These findings reveal a novel role of HEXIM1 in the activation of p53 induced by anticancer agents and may lead to potential development of new anticancer strategies. As the requirement of p53 for angiogenesis and metastasis is wellestablished, it is possible that HEXIM1 inhibits tumor progression through activation of p53.
4. Conclusion
Involvement of Brd4 and HEXIM1 in tumorigenesis through the P-TEFb-dependent and -independent mechanisms opens a new and exciting venue for cancer research. Development of small molecules or other strategies to block the chromatin-binding of Brd4 or to induce the expression of HEXIM1 may provide novel therapeutic options against cancer.
Acknowledgments
This work was supported by National Natural Science Foundation of China (NSFC, 30930046, 31270809 and 812111455 to Ruichuan Chen), the National Basic Research Program of China (973 Programs, 2013CB917802 to Ruichuan Chen), and the Agency for Science, Technology and Research (A*STAR), Singapore (to Sheng-Hao Chao).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Marshall NF, Price DH. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. The Journal of Biological Chemistry. 1995;270(21):12335–12338. doi: 10.1074/jbc.270.21.12335. [DOI] [PubMed] [Google Scholar]
- 2.Henriques T, Gilchrist DA, Nechaev S, et al. Stable pausing by RNA polymerase II provides an opportunity to target and integrate regulatory signals. Molecular Cell. 2013;52(4):517–528. doi: 10.1016/j.molcel.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wada T, Takagi T, Yamaguchi Y, Watanabe D, Handa H. Evidence that P-TEFb alleviates the negative effect of DSIF on RNA polymerase II-dependent transcription in vitro. The EMBO Journal. 1998;17(24):7395–7403. doi: 10.1093/emboj/17.24.7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yamaguchi Y, Takagi T, Wada T, et al. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell. 1999;97(1):41–51. doi: 10.1016/s0092-8674(00)80713-8. [DOI] [PubMed] [Google Scholar]
- 5.Cheng B, Li T, Rahl PB, et al. Functional association of gdown1 with RNA polymerase II poised on human genes. Molecular Cell. 2012;45(1):38–50. doi: 10.1016/j.molcel.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fujinaga K, Irwin D, Huang Y, Taube R, Kurosu T, Peterlin BM. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Molecular and Cellular Biology. 2004;24(2):787–795. doi: 10.1128/MCB.24.2.787-795.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ivanov D, Kwak YT, Guo J, Gaynor RB. Domains in the SPT5 protein that modulate its transcriptional regulatory properties. Molecular and Cellular Biology. 2000;20(9):2970–2983. doi: 10.1128/mcb.20.9.2970-2983.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hargreaves DC, Horng T, Medzhitov R. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell. 2009;138(1):129–145. doi: 10.1016/j.cell.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zippo A, Serafini R, Rocchigiani M, Pennacchini S, Krepelova A, Oliviero S. Histone crosstalk between H3S10ph and H4K16ac generates a histone code that mediates transcription elongation. Cell. 2009;138(6):1122–1136. doi: 10.1016/j.cell.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 10.Chao S-H, Fujinaga K, Marion JE, et al. Flavopiridol inhibits P-TEFb and blocks HIV-1 replication. The Journal of Biological Chemistry. 2000;275(37):28345–28348. doi: 10.1074/jbc.C000446200. [DOI] [PubMed] [Google Scholar]
- 11.Chao S-H, Price DH. Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. The Journal of Biological Chemistry. 2001;276(34):31793–31799. doi: 10.1074/jbc.M102306200. [DOI] [PubMed] [Google Scholar]
- 12.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130(1):77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeitlinger J, Stark A, Kellis M, et al. RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nature Genetics. 2007;39(12):1512–1516. doi: 10.1038/ng.2007.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muse GW, Gilchrist DA, Nechaev S, et al. RNA polymerase is poised for activation across the genome. Nature Genetics. 2007;39(12):1507–1511. doi: 10.1038/ng.2007.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwak H, Fuda NJ, Core LJ, Lis JT. Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science. 2013;339(6122):950–953. doi: 10.1126/science.1229386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Core LJ, Waterfall JJ, Gilchrist DA, et al. Defining the status of RNA polymerase at promoters. Cell Reports. 2012;2(4):1025–1035. doi: 10.1016/j.celrep.2012.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laspia MF, Rice AP, Mathews MB. HIV-1 Tat protein increases transcriptional initiation and stabilizes elongation. Cell. 1989;59(2):283–292. doi: 10.1016/0092-8674(89)90290-0. [DOI] [PubMed] [Google Scholar]
- 18.Wimmer J, Fujinaga K, Taube R, et al. Interactions between Tat and TAR and human immunodeficiency virus replication are facilitated by human cyclin T1 but not cyclins T2a or T2b. Virology. 1999;255(1):182–189. doi: 10.1006/viro.1998.9589. [DOI] [PubMed] [Google Scholar]
- 19.Garber ME, Wei P, KewalRamani VN, et al. The interaction between HIV-1 Tat and human cyclin T1 requires zinc and a critical cysteine residue that is not conserved in the murine CycT1 protein. Genes and Development. 1998;12(22):3512–3527. doi: 10.1101/gad.12.22.3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen D, Fong Y, Zhou Q. Specific interaction of Tat with the human but not rodent P-TEFb complex mediates the species-specific Tat activation of HIV-1 transcription. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(6):2728–2733. doi: 10.1073/pnas.96.6.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peng J, Marshall NF, Price DH. Identification of a cyclin subunit required for the function of Drosophila P-TEFb. The Journal of Biological Chemistry. 1998;273(22):13855–13860. doi: 10.1074/jbc.273.22.13855. [DOI] [PubMed] [Google Scholar]
- 22.Wei P, Garber ME, Fang S-M, Fischer WH, Jones KA. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell. 1998;92(4):451–462. doi: 10.1016/s0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- 23.Shim EY, Walker AK, Shi Y, Blackwell TK. CDK-9/cyclin T (P-TEFb) is required in two postinitiation pathways for transcription in the C. elegans embryo. Genes and Development. 2002;16(16):2135–2146. doi: 10.1101/gad.999002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ni Z, Schwartz BE, Werner J, Suarez J-R, Lis JT. Coordination of transcription, RNA processing, and surveillance by P-TEFb kinase on heat shock genes. Molecular Cell. 2004;13(1):55–65. doi: 10.1016/s1097-2765(03)00526-4. [DOI] [PubMed] [Google Scholar]
- 25.Bartkowiak B, Liu P, Phatnani HP, et al. CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes and Development. 2010;24(20):2303–2316. doi: 10.1101/gad.1968210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shore SM, Byers SA, Maury W, Price DH. Identification of a novel isoform of Cdk9. Gene. 2003;307(1-2):175–182. doi: 10.1016/s0378-1119(03)00466-9. [DOI] [PubMed] [Google Scholar]
- 27.Peng J, Zhu Y, Milton JT, Price DH. Identification of multiple cyclin subunits of human P-TEFb. Genes and Development. 1998;12(5):755–762. doi: 10.1101/gad.12.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fu T-J, Peng J, Lee G, Price DH, Flores O. Cyclin K functions as a CDK9 regulatory subunit and participates in RNA polymerase II transcription. The Journal of Biological Chemistry. 1999;274(49):34527–34530. doi: 10.1074/jbc.274.49.34527. [DOI] [PubMed] [Google Scholar]
- 29.Yik JHN, Chen R, Nishimura R, Jennings JL, Link AJ, Zhou Q. Inhibition of P-TEFb (CDK9/cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Molecular Cell. 2003;12(4):971–982. doi: 10.1016/s1097-2765(03)00388-5. [DOI] [PubMed] [Google Scholar]
- 30.Michels AA, Nguyen VT, Fraldi A, et al. MAQ1 and 7SK RNA interact with CDK9/cyclin T complexes in a transcription-dependent manner. Molecular and Cellular Biology. 2003;23(14):4859–4869. doi: 10.1128/MCB.23.14.4859-4869.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jeronimo C, Forget D, Bouchard A, et al. Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme. Molecular Cell. 2007;27(2):262–274. doi: 10.1016/j.molcel.2007.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krueger BJ, Jeronimo C, Roy BB, et al. LARP7 is a stable component of the 7SK snRNP while P-TEFb, HEXIM1 and hnRNP A1 are reversibly associated. Nucleic Acids Research. 2008;36(7):2219–2229. doi: 10.1093/nar/gkn061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Michels AA, Fraldi A, Li Q, et al. Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. The EMBO Journal. 2004;23(13):2608–2619. doi: 10.1038/sj.emboj.7600275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barboric M, Kohoutek J, Price JP, Blazek D, Price DH, Peterlin BM. Interplay between 7SK snRNA and oppositely charged regions in HEXIM1 direct the inhibition of P-TEFb. The EMBO Journal. 2005;24(24):4291–4303. doi: 10.1038/sj.emboj.7600883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang Z, Yik JHN, Chen R, et al. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Molecular Cell. 2005;19(4):535–545. doi: 10.1016/j.molcel.2005.06.029. [DOI] [PubMed] [Google Scholar]
- 36.Jang MK, Mochizuki K, Zhou M, Jeong H-S, Brady JN, Ozato K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Molecular Cell. 2005;19(4):523–534. doi: 10.1016/j.molcel.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 37.Zhou Q, Yik JHN. The Yin and Yang of P-TEFb regulation: implications for human immunodeficiency virus gene expression and global control of cell growth and differentiation. Microbiology and Molecular Biology Reviews. 2006;70(3):646–659. doi: 10.1128/MMBR.00011-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dey A, Chao S-H, Lane DP. HEXIM1 and the control of transcription elongation: from cancer and inflammation to AIDS and cardiac hypertrophy. Cell Cycle. 2007;6(15):1856–1863. doi: 10.4161/cc.6.15.4556. [DOI] [PubMed] [Google Scholar]
- 39.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 40.Sanchez R, Zhou M-M. The role of human bromodomains in chromatin biology and gene transcription. Current Opinion in Drug Discovery and Development. 2009;12(5):659–665. [PMC free article] [PubMed] [Google Scholar]
- 41.Filippakopoulos P, Picaud S, Mangos M, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149(1):214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muller S, Filippakopoulos P, Knapp S. Bromodomains as therapeutic targets. Expert Reviews in Molecular Medicine. 2011;13:p. e29. doi: 10.1017/S1462399411001992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chiang CM. Brd4 engagement from chromatin targeting to transcriptional regulation: selective contact with acetylated histone H3 and H4. F1000 Biology Reports. 2009;1:p. 98. doi: 10.3410/B1-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rahman S, Sowa ME, Ottinger M, et al. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Molecular and Cellular Biology. 2011;31(13):2641–2652. doi: 10.1128/MCB.01341-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dey A, Chitsaz F, Abbasi A, Misteli T, Ozato K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(15):8758–8763. doi: 10.1073/pnas.1433065100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dey A, Ellenberg J, Farina A, et al. A bromodomain protein, MCAP, associates with mitotic chromosomes and affects G2-to-M transition. Molecular and Cellular Biology. 2000;20(17):6537–6549. doi: 10.1128/mcb.20.17.6537-6549.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mochizuki K, Nishiyama A, Jang MK, et al. The bromodomain protein Brd4 stimulates g1 gene transcription and promotes progression to S phase. The Journal of Biological Chemistry. 2008;283(14):9040–9048. doi: 10.1074/jbc.M707603200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang Z, He N, Zhou Q. Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G 1 gene expression and cell cycle progression. Molecular and Cellular Biology. 2008;28(3):967–976. doi: 10.1128/MCB.01020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dey A, Nishiyama A, Karpova T, McNally J, Ozato K. Brd4 marks select genes on mitotic chromatin and directs postmitotic transcription. Molecular Biology of the Cell. 2009;20(23):4899–4909. doi: 10.1091/mbc.E09-05-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao R, Nakamura T, Fu Y, Lazar Z, Spector DL. Gene bookmarking accelerates the kinetics of post-mitotic transcriptional re-activation. Nature Cell Biology. 2011;13(11):1295–1304. doi: 10.1038/ncb2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Devaiah BN, Singer DS. Two faces of BRD4: mitotic bookmark and transcriptional lynchpin. Transcription. 2012;4(1):13–17. doi: 10.4161/trns.22542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Voigt P, Reinberg D. BRD4 jump-starts transcription after mitotic silencing. Genome Biology. 2011;12(11, article 133) doi: 10.1186/gb-2011-12-11-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang R, Li Q, Helfer CM, Jiao J, You J. Bromodomain protein Brd4 associated with acetylated chromatin is important for maintenance of higher-order chromatin structure. The Journal of Biological Chemistry. 2012;287(14):10738–10752. doi: 10.1074/jbc.M111.323493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.You J, Croyle JL, Nishimura A, Ozato K, Howley PM. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell. 2004;117(3):349–360. doi: 10.1016/s0092-8674(04)00402-7. [DOI] [PubMed] [Google Scholar]
- 55.Bisgrove DA, Mahmoudi T, Henklein P, Verdin E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(34):13690–13695. doi: 10.1073/pnas.0705053104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nechaev S, Adelman K. Pol II waiting in the starting gates: regulating the transition from transcription initiation into productive elongation. Biochimica et Biophysica Acta. 2011;1809(1):34–45. doi: 10.1016/j.bbagrm.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou Q, Li T, Price DH. RNA polymerase II elongation control. Annual Review of Biochemistry. 2012;81:119–143. doi: 10.1146/annurev-biochem-052610-095910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guo J, Price DH. RNA polymerase II transcription elongation control. Chemical Reviews. 2013;113(11):8583–8603. doi: 10.1021/cr400105n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Diribarne G, Bensaude O. 7SK RNA, a non-coding RNA regulating P-TEFb, a general transcription factor. RNA Biology. 2009;6(2):122–128. doi: 10.4161/rna.6.2.8115. [DOI] [PubMed] [Google Scholar]
- 60.Xue Y, Yang Z, Chen R, Zhou Q. A capping-independent function of MePCE in stabilizing 7SK snRNA and facilitating the assembly of 7SK snRNP. Nucleic Acids Research. 2009;38(2):360–369. doi: 10.1093/nar/gkp977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen R, Liu M, Li H, et al. PP2B and PP1α cooperatively disrupt 7SK snRNP to release P-TEFb for transcription in response to Ca2+ signaling. Genes and Development. 2008;22(10):1356–1368. doi: 10.1101/gad.1636008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Contreras X, Barboric M, Lenasi T, Peterlin BM. HMBA releases P-TEFb from HEXIM1 and 7SK snRNA via PI3K/Akt and activates HIV transcription. PLoS Pathogens. 2007;3(10):1459–1469. doi: 10.1371/journal.ppat.0030146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fujinaga K, Barboric M, Li Q, Luo Z, Price DH, Peterlin BM. PKC phosphorylates HEXIM1 and regulates P-TEFb activity. Nucleic Acids Research. 2012;40(18):9160–9170. doi: 10.1093/nar/gks682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ai N, Hu X, Ding F, et al. Signal-induced Brd4 release from chromatin is essential for its role transition from chromatin targeting to transcriptional regulation. Nucleic Acids Research. 2011;39(22):9592–9604. doi: 10.1093/nar/gkr698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gupta SS, Maetzig T, Maertens GN, et al. Bromo and ET domain (BET) chromatin regulators serve as co-factors for murine leukemia virus integration. Journal of Virology. 2013 doi: 10.1128/JVI.01942-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sharma A, Larue RC, Plumb MR, et al. BET proteins promote efficient murine leukemia virus integration at transcription start sites. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(29):12036–12041. doi: 10.1073/pnas.1307157110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Margolis DM, Hazuda DJ. Combined approaches for HIV cure. Current Opinion in HIV and AIDS. 2013;8(3):230–235. doi: 10.1097/COH.0b013e32835ef089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Karn J. A new BET on the control of HIV latency. Cell Cycle. 2013;12(4):545–546. doi: 10.4161/cc.23679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou M, Huang K, Jung K-J, et al. Bromodomain protein Brd4 regulates human immunodeficiency virus transcription through phosphorylation of CDK9 at threonine 29. Journal of Virology. 2009;83(2):1036–1044. doi: 10.1128/JVI.01316-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen R, Liu M, Zhang K, Zhou Q. Isolation and functional characterization of P-TEFb-associated factors that control general and HIV-1 transcriptional elongation. Methods. 2011;53(1):85–90. doi: 10.1016/j.ymeth.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang G, Liu R, Zhong Y, et al. Down-regulation of NF-kappaB transcriptional activity in HIV-associated kidney disease by BRD4 inhibition. The Journal of Biological Chemistry. 2012;287(34):28840–28851. doi: 10.1074/jbc.M112.359505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang B, Yang X-D, Zhou M-M, Ozato K, Chen L-F. Brd4 coactivates transcriptional activation of NF-κB via specific binding to acetylated RelA. Molecular and Cellular Biology. 2009;29(5):1375–1387. doi: 10.1128/MCB.01365-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Anand P, Brown JD, Lin CY, et al. BET bromodomains mediate transcriptional pause release in heart failure. Cell. 2013;154(3):569–582. doi: 10.1016/j.cell.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Spiltoir JI, Stratton MS, Cavasin MA, et al. BET acetyl-lysine binding proteins control pathological cardiac hypertrophy. Journal of Molecular and Cellular Cardiology. 2013;63:175–179. doi: 10.1016/j.yjmcc.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Floyd SR, Pacold ME, Huang Q, et al. The bromodomain protein Brd4 insulates chromatin from DNA damage signalling. Nature. 2013;498(7453):246–250. doi: 10.1038/nature12147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Choi S, Bakkenist CJ. Brd4 shields chromatin from ATM kinase signaling storms. Science Signaling. 2013;6(293):p. pe30. doi: 10.1126/scisignal.2004622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Barbieri I, Cannizzaro E, Dawson MA. Bromodomains as therapeutic targets in cancer. Briefings in Functional Genomics. 2013;12(3):219–230. doi: 10.1093/bfgp/elt007. [DOI] [PubMed] [Google Scholar]
- 78.Belkina AC, Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nature Reviews Cancer. 2012;12(7):465–477. doi: 10.1038/nrc3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.French CA, Miyoshi I, Kubonishi I, Grier HE, Perez-Atayde AR, Fletcher JA. BRD4-NUT fusion oncogene: a novel mechanism in aggressive carcinoma. Cancer Research. 2003;63(2):304–307. [PubMed] [Google Scholar]
- 80.French CA, Miyoshi I, Aster JC, et al. BRD4 bromodomain gene rearrangement in aggressive carcinoma with translocation t(15;19) American Journal of Pathology. 2001;159(6):1987–1992. doi: 10.1016/S0002-9440(10)63049-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Polsani A, Braithwaite KA, Alazraki AL, Abramowsky C, Shehata BM. NUT midline carcinoma: an imaging case series and review of literature. Pediatric Radiology. 2012;42(2):205–210. doi: 10.1007/s00247-011-2272-3. [DOI] [PubMed] [Google Scholar]
- 82.French CA. Pathogenesis of NUT midline carcinoma. Annual Review of Pathology. 2012;7:247–265. doi: 10.1146/annurev-pathol-011811-132438. [DOI] [PubMed] [Google Scholar]
- 83.French CA. NUT midline carcinoma. Cancer Genetics and Cytogenetics. 2010;203(1):16–20. doi: 10.1016/j.cancergencyto.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Alsarraj J, Hunter KW. Bromodomain-Containing protein 4: a dynamic regulator of breast cancer metastasis through modulation of the extracellular matrix. International Journal of Breast Cancer. 2012;2012:7 pages. doi: 10.1155/2012/670632.670632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Alsarraj J, Walker RC, Webster JD, et al. Deletion of the proline-rich region of the murine metastasis susceptibility gene Brd4 promotes epithelial-to-mesenchymal transition- and stem cell-like conversion. Cancer Research. 2011;71(8):3121–3131. doi: 10.1158/0008-5472.CAN-10-4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zou Z, Huang B, Wu X, et al. Brd4 maintains constitutively active NF-kappaB in cancer cells by binding to acetylated RelA. Oncogene. 2013 doi: 10.1038/onc.2013.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Blobel GA, Kalota A, Sanchez PV, Carroll M. Short hairpin RNA screen reveals bromodomain proteins as novel targets in acute myeloid leukemia. Cancer Cell. 2011;20(3):287–288. doi: 10.1016/j.ccr.2011.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zuber J, Shi J, Wang E, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478(7370):524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dawson MA, Prinjha RK, Dittmann A, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478(7370):529–533. doi: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Delmore JE, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mertz JA, Conery AR, Bryant BM, et al. Targeting MYC dependence in cancer by inhibiting BET bromodomains. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(40):16669–16674. doi: 10.1073/pnas.1108190108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Segura MF, Fontanals-Cirera B, Gaziel-Sovran A, et al. BRD4 sustains proliferation and represents a new target for epigenetic therapy in melanoma. Cancer Research. 2013;73(20):6264–6276. doi: 10.1158/0008-5472.CAN-13-0122-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rodriguez RM, Huidobro C, Urdinguio RG, et al. Aberrant epigenetic regulation of bromodomain Brd4 in human colon cancer. Journal of Molecular Medicine. 2012;90(5):587–595. doi: 10.1007/s00109-011-0837-0. [DOI] [PubMed] [Google Scholar]
- 94.Loven J, Hoke HA, Lin CY, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–334. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Khochbin S. When are the BET factors the most sensitive to bromodomain inhibitors? Transcription. 2013;4(2):54–57. doi: 10.4161/trns.23191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Banerjee T, Chakravarti D. A peek into the complex realm of histone phosphorylation. Molecular and Cellular Biology. 2011;31(24):4858–4873. doi: 10.1128/MCB.05631-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nowak SJ, Corces VG. Phosphorylation of histone H3: a balancing act between chromosome condensation and transcriptional activation. Trends in Genetics. 2004;20(4):214–220. doi: 10.1016/j.tig.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 98.Rossetto D, Avvakumov N, Cote J. Histone phosphorylation: a chromatin modification involved in diverse nuclear events. Epigenetics. 2012;7(10):1098–1108. doi: 10.4161/epi.21975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sawicka A, Seiser C. Histone H3 phosphorylation—a versatile chromatin modification for different occasions. Biochimie. 2012;94(11):2193–2201. doi: 10.1016/j.biochi.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mahadevan LC, Willis AC, Barratt MJ. Rapid histone H3 phosphorylation in response to growth factors, phorbol esters, okadaic acid, and protein synthesis inhibitors. Cell. 1991;65(5):775–783. doi: 10.1016/0092-8674(91)90385-c. [DOI] [PubMed] [Google Scholar]
- 101.Kusuhara M, Nagasaki K, Kimura K, et al. Cloning of hexamethylene-bis-acetamide-inducible transcript, HEXIM1, in human vascular smooth muscle cells. Biomedical Research. 1999;20(5):273–279. [Google Scholar]
- 102.Ghatpande S, Goswami S, Mathew S, et al. Identification of a novel cardiac lineage-associated protein(cCLP-1): a candidate regulator of cardiogenesis. Developmental Biology. 1999;208(1):210–221. doi: 10.1006/dbio.1998.9180. [DOI] [PubMed] [Google Scholar]
- 103.Lew QJ, Chu KL, Chia YL, Cheong N, Chao SH. HEXIM1, a New Player in the p53 Pathway. Cancers. 2013;5:838–856. doi: 10.3390/cancers5030838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wittmann BM, Wang N, Montano MM. Identification of a novel inhibitor of breast cell growth that is down-regulated by estrogens and decreased in breast tumors. Cancer Research. 2003;63(16):5151–5158. [PubMed] [Google Scholar]
- 105.Castro-Rivera E, Samudio I, Safe S. Estrogen regulation of cyclin D1 gene expression in ZR-75 breast cancer cells involves multiple enhancer elements. The Journal of Biological Chemistry. 2001;276(33):30853–30861. doi: 10.1074/jbc.M103339200. [DOI] [PubMed] [Google Scholar]
- 106.Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103(6):843–852. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 107.Ali S, Coombes RC. Estrogen receptor alpha in human breast cancer: occurrence and significance. Journal of Mammary Gland Biology and Neoplasia. 2000;5(3):271–281. doi: 10.1023/a:1009594727358. [DOI] [PubMed] [Google Scholar]
- 108.Katzenellenbogen BS, Frasor J. Therapeutic Targeting in the Estrogen Receptor Hormonal Pathway. Seminars in Oncology. 2004;31(1, supplement 3):28–38. doi: 10.1053/j.seminoncol.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 109.Wittmann BM, Fujinaga K, Deng H, Ogba N, Montano MM. The breast cell growth inhibitor, estrogen down regulated gene 1, modulates a novel functional interaction between estrogen receptor alpha and transcriptional elongation factor cyclin T1. Oncogene. 2005;24(36):5576–5588. doi: 10.1038/sj.onc.1208728. [DOI] [PubMed] [Google Scholar]
- 110.Lewis-Wambi JS, Jordan VC. Treatment of postmenopausal breast cancer with selective estrogen receptor modulators (SERMs) Breast Disease. 2005;24(1):93–105. doi: 10.3233/bd-2006-24108. [DOI] [PubMed] [Google Scholar]
- 111.Ketchart W, Ogba N, Kresak A, Albert JM, Pink JJ, Montano MM. HEXIM1 is a critical determinant of the response to tamoxifen. Oncogene. 2011;30(33):3563–3569. doi: 10.1038/onc.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ferrara N, Gerber H-P, LeCouter J. The biology of VEGF and its receptors. Nature Medicine. 2003;9(6):669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 113.Ogba N, Doughman YQ, Chaplin LJ, et al. HEXIM1 modulates vascular endothelial growth factor expression and function in breast epithelial cells and mammary gland. Oncogene. 2010;29(25):3639–3649. doi: 10.1038/onc.2010.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Yeh IJ, Ogba N, Bensigner H, Welford SM, Montano MM. HEXIM1 downregulates Hypoxia Inducible Factor-1 alpha protein stability. Biochemical Journal. 2013;456:195–204. doi: 10.1042/BJ20130592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Folkman J. Role of angiogenesis in tumor growth and metastasis. Seminars in Oncology. 2002;29(6, supplement 16):15–18. doi: 10.1053/sonc.2002.37263. [DOI] [PubMed] [Google Scholar]
- 116.Ketchart W, Smith KM, Krupka T, et al. Inhibition of metastasis by HEXIM1 through effects on cell invasion and angiogenesis. Oncogene. 2013;32(33):3829–3839. doi: 10.1038/onc.2012.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gurumurthy M, Tan CH, Ng R, et al. Nucleophosmin interacts with HEXIM1 and regulates RNA polymerase II transcription. Journal of Molecular Biology. 2008;378(2):302–317. doi: 10.1016/j.jmb.2008.02.055. [DOI] [PubMed] [Google Scholar]
- 118.Colombo E, Marine J-C, Danovi D, Falini B, Pelicci PG. Nucleophosmin regulates the stability and transcriptional activity of p53. Nature Cell Biology. 2002;4(7):529–533. doi: 10.1038/ncb814. [DOI] [PubMed] [Google Scholar]
- 119.Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D. Nucleolar Arf sequesters Mdm2 and activates p53. Nature Cell Biology. 1999;1(1):20–26. doi: 10.1038/8991. [DOI] [PubMed] [Google Scholar]
- 120.Kamijo T, Zindy F, Roussel MF, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19(ARF) Cell. 1997;91(5):649–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- 121.Kurki S, Peltonen K, Latonen L, et al. Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell. 2004;5(5):465–475. doi: 10.1016/s1535-6108(04)00110-2. [DOI] [PubMed] [Google Scholar]
- 122.Falini B, Mecucci C, Tiacci E, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. The New England Journal of Medicine. 2005;352(3):254–266. doi: 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]
- 123.Alcalay M, Tiacci E, Bergomas R, et al. Acute myeloid leukemia bearing cytoplasmic nucleophosmin (NPMc+ AML) shows a distinct gene expression profile characterized by up-regulation of genes involved in stem-cell maintenance. Blood. 2005;106(3):899–902. doi: 10.1182/blood-2005-02-0560. [DOI] [PubMed] [Google Scholar]
- 124.Lew QJ, Tan CH, Gurumurthy M, et al. NPMc+ AML cell line shows differential protein expression and lower sensitivity to DNA-damaging and p53-inducing anti-cancer compounds. Cell Cycle. 2011;10(12):1978–1987. doi: 10.4161/cc.10.12.15859. [DOI] [PubMed] [Google Scholar]
- 125.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88(3):323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 126.Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Letters. 1997;420(1):25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- 127.Rodriguez MS, Desterro JMP, Lain S, Lane DP, Hay RT. Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Molecular and Cellular Biology. 2000;20(22):8458–8467. doi: 10.1128/mcb.20.22.8458-8467.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Lau J, Lew QJ, Diribarne G, et al. Ubiquitination of HEXIM1 by HDM2. Cell Cycle. 2009;8(14):2247–2254. doi: 10.4161/cc.8.14.9015. [DOI] [PubMed] [Google Scholar]
- 129.Lew QJ, Chia YL, Chu KL, et al. Identification of HEXIM1 as a positive regulator of p53. The Journal of Biological Chemistry. 2012;287(43):36443–36454. doi: 10.1074/jbc.M112.374157. [DOI] [PMC free article] [PubMed] [Google Scholar]