

The Stemona alkaloids represent a class of more than eighty structurally diverse alkaloids isolated from plants belonging to the Stemonaceae family.[1] Structurally these alkaloids are characterized by the conserved pyrrolo[1,2-a]azepine core usually linked to a terminal lactone. These plants have been used in folk medicine in East Asia for thousands of years to treat the symptoms of bronchitis, pertussis, and tuberculosis and have been used as anti-parasitics in humans and animals.
Recently, Lie et al. disclosed the structures of two new Stemona alkaloids from the root extracts of Stemona aphylla: stemaphylline (1) and stemaphylline N-oxide (2) (Figure 1), featuring the central pyrrolo[1,2-a]azepine core linked to a 3,5-disubstituted γ-lactone moiety.[2] Stemaphylline was found to have low acetylcholinesterase (AChE) inhibitory activity, pronounced insecticidal activity, weak antimicrobial activity against Escherichia coli, Staphylococcus aureus, Pseudomonas auruginosa and weak antifunal activity against Candida albicans.
Figure 1.
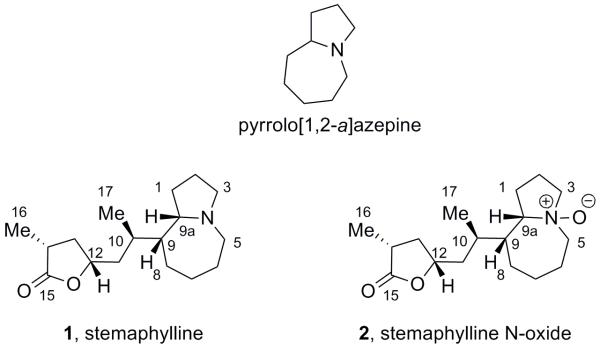
Pyrrolo[1,2-a]azepine core and stemaphylline (1) and stemaphylline N-oxide (2).
Due to the unique properties of azabicyclic containing natural products, our lab has developed general methods for their asymmetric synthesis and applied these methods towards the total synthesis of azabicycle containing natural products (Scheme 1).[3] This method utilizes nucleophilic addition of a Grignard reagent containing a protected aldehyde to an Ellman chiral aldimine (3),[4] acid mediated deprotection and acetal cleavage, and subsequent reductive amination to form the pyrrolidine ring (4).[4b] Ring closing metathesis (RCM) furnishes the azabicyclic ring system (5).
Scheme 1.
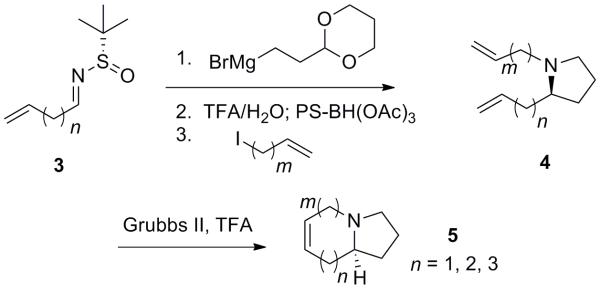
Approach for the enantioselective synthesis of azabicyclic ring systems.[3]
Based on this approach we planned an enantioselective total synthesis of stemaphylline which would allow for further evaluation of its biological activity and the preparation of unnatural analogs. In addition to construction of the azepine ring through ring closing metathesis, our retrosynthetic analysis included concomitant cyclization of the γ-lactone from tetraene 7 to form 6, which upon olefin reduction would afford stemaphylline (1). Chiral tetraene 7 would be formed as outlined in Scheme 2 from chiral N-sulfinyl imine 8. Intermediate 8 represented an attractive point of entry into the preparation of unnatural analogs, as the nitrogen stereocenter can be inverted by preparation of the opposite chiral Ellman N-sulfinyl imine. Preparation of both diastereomers of aldimine 8 would also provide an opportunity to study the ability of the chiral Ellman auxiliary to control the asymmetric Grignard addition to a substrate possessing a significant Felkin-Ahn substrate bias. The C10 and C9 stereogenic centers in intermediate 8 could be installed by oxazolidine-mediated asymmetric conjugate addition followed by asymmetric α-allylation of α,β-unsaturated ester 9. A Nagao aldol reaction would be used to install the allylic alcohol stereogenic center at C12.[5]
Scheme 2.

Retrosynthetic analysis of stemaphylline (1). TBDPS = tert-butyl-diphenylsilyl.
Our synthesis began with the precedented Nagao aldol reaction of thiazolidine thione 10 with acrolein (90% yield and 5:1 dr,[5] and was followed by silyl protection of the allylic alcohol to afford 11. Removal of the chiral auxiliary with LiBH4, Swern oxidation, and Horner-Wadsworth Emmons olefination with triethyl phosphonoacetate using the conditions of Masamune and Roush[6] led to the rapid construction of α,β-unsaturated ester 12 in 82% yield over 3 steps. Ester hydrolysis and coupling of the resultant carboxylic acid with Evans auxiliary 13 led to oxazolidinone 9. After some experimentation, conjugate addition of monoorganocuprate species Li[MeCuI] in the presence of TMSI, as developed by Bergdahl,[7] was found to provide better stereocontrol than the addition of higher order cuprates, and successfully installed the C10 methyl stereocenter in 90% yield and 17:1 dr. Subsequent enolate formation with LiHMDS in the presence of HMPA, and allylation with allyl iodide afforded intermediate 14 in 87% yield and 15:1 dr (Scheme 3).
Scheme 3.

Synthesis of aldimines 8 and 18. (a) TiCl4, DIEA, CH2Cl2, −45 °C; then acrolein, −78 °C, 90%, dr 5:1; (b) TBDPSCl, imidazole, CH2Cl2, 0 °C to rt, 92%; (c) LiBH4, THF/MeOH, 0 °C, 93%; (d) (COCl)2, DMSO, Et3N, CH2Cl2, −78 °C to rt, 98%; (e) DBU, LiCl, triethyl phosphonate, MeCN, rt, 90%; (f) LiOH, THF/H2O (3:1), 55 °C, 89%; (g) Et3N, PivCl, THF, −78 °C to 0 °C; then 13, nBuLi, THF, 78 °C to 0 °C, 95%; (h) CuI-DMS, MeLi, TMSI, THF, −78 °C, 90%, dr 17:1; (i) LiHMDS, HMPA, THF, −78 °C; then allyl iodide, −45 °C, 87%, dr 15:1; (j) H2O2, LiOH, THF/H2O (3:1), 0 °C to rt, 96%; (k) LAH, THF/Et2O (4:1), 0 °C, 84%; (l) TPAP, NMO, 4 Å M.S., CH2Cl2, 85%; (m) (S)- or (R)-tert-butanesulfinamide, Ti(OEt)4, THF, 40 °C, 81%. TBDPS = tert-butyl-diphenylsilyl; DBU = 1,8-Diazabicyclo[5.4.0]undec-7-ene; Piv = pivaloyl; DMS = dimethyl sulfide; LiHMDS = Lithium bis(trimethylsilyl)amide; HMPA = hexamethylphosphoramide; LAH = lithium aluminum hydride; TPAP = Tetrapropylammonium perruthenate; NMO = N-methylmorpholine oxide.
Removal of the oxazolidine chiral auxiliary and reduction to the primary alcohol in the presence of LiBH4 or LAH proved to be problematic. Poor yields were obtained (~30%) and degradation of starting material was observed, presumably due to the steric congestion adjacent to the oxazolidinone. A similar observation was made by Wee and co-workers with a sterically encumbered substrate, and circumvented through adoption of a 2-step protocol involving saponification to the carboxylic acid in the presence of LiOH/H2O2 and LAH reduction to the desired alcohol.[8] Employing this procedure proved effective for substrate 14, affording the desired primary alcohol in 81% yield over 2 steps. Serendipitously, in our efforts to modify oxazolidine 14 we isolated ring opened intermediate 15 which could be crystallized to yield X-ray suitable crystals. This crystal structure confirmed the correct configuration of the C9, C10, and C12 stereocenters.
Ley oxidation and condensation with (S)- and (R)-tert-butanesulfinamides provided access to chiral N-sulfinyl imines 8 and 18 (Scheme 4). With these substrates in hand we next studied the effect of Felkin-Ahn substrate control in the Ellman asymmetric Grignard addition. Initial efforts revealed the large degree of Felkin control for this substrate, as formation of (2-(1,3-dioxan-2-yl)ethyl)magnesium bromide (19) in THF and addition to N-sulfinyl imine 8 yielded poor selectivity (dr = 1:1, Scheme 3). In contrast, addition to N-sulfinyl imine 18, proceeded with much improved selectivity (dr = 10:1). These observations are consistent with the known sense of stereoinduction observed when employing (2-(1,3-dioxan-2-yl)ethyl)magnesium bromide 193b,4b and the Felkin control dictated by the C9 stereocenter in aldimines 8 and 18. In an attempt to improve the selectivity with aldimine 8 to obtain increased quantities of desired stereoisomer 20a, we further investigated the asymmetric Grignard reaction (Table 1). Hypothesizing that the chelating ability of the solvent could play a role in stereocontrol, we attempted to form Grignard reagent 19 in less-coordinating Et2O,[4b] but were unsuccessful. Minimizing the amount of THF present by increasing the concentration of the Grignard reagent to 3.0 M and performing the reaction in CH2Cl2 led to a slight increase in selectivity (1:1.5); however, in favour of the undesired stereoisomer. Switching to formation of Grignard 19 in 2-MeTHF further increased selectivity for the undesired stereoisomer 20b (entry 4), and increased dilution of aldimine 8 in CH2Cl2 exacerbated this effect (entry 5). These results suggest that non-coordinating solvents may deteriorate or reverse the selectivity of the Ellman chiral auxiliary in substrates such as 8.
Scheme 4.

Asymmetric Grignard addition to aldimines 8 and 18. (a) 19 (1.0 M soln in THF), THF, −45 °C to rt, 86%. TBDPS = tert-butyl-diphenylsilyl.
Table 1.
Conditions for Grignard addition of 19 to aldimine 8.
| Entry | Aldimine 8 Conc.. |
RMgX Solvent | RMgX Cone. | % Yield | dr.. (20a: 20b) |
|---|---|---|---|---|---|
| 1 | 02 M | THF | 1.0 M | 90 | 1:1 |
| 2 | 0 2 M | Et20 | 10 M | no rxn | - |
| 3 | 0 2 M | THF | 3 0 M | 61 | 1:1.5 |
| 4 | 0 2 M | 2-MeTHF | 3 0 M | 83 | 1:2.7 |
| 5 | 0 .1 M | 2-MeTHF | 3 0 M | 86 | 1:4 |
*ln all cases, to a solution of aldimine 8 in CH2Cl2 at −45 °C was added Grignard 19 followed by warming to rt and stirring for 18h.
Unable to improve the selectivity for desired Grignard addition product 20a we moved forward with our effort toward the total synthesis of stemaphylline as shown in Scheme 5. Silyl deprotection with TBAF, followed by ester formation with methacryclic anhydride yielded intermediate 22. Nitrogen deprotection and concomitant acetal cleavage with TFA/H2O, followed by reductive amination in the presence of PS-BH(OAc)3,[3a, 4b] and pyrrolidine allylation with allyl iodide yielded tetraene 7 in 47% yield for the 3-step sequence. The corresponding stereoisomer 23 bearing the opposite amine stereocenter was also synthesized in analogous fashion with similar yield.
Scheme 5.

Synthesis of pyrrolidines 7 and 23. (a) TBAF, THF, 0 °C to rt, 97%; (b) methacrylic anhydride, Et3N, DMAP, CH2Cl2rt, 82%; (c) (i) TFA/H2O, (95:5) (ii) PS-BH(OAc)3, DCE, rt, (iii) allyl iodide, K2CO3, DMF, rt, 47% over 3 steps. DMAP = 4-Dimethylaminopyridine; PS = polymer supported.
With tetraene pyrrolidines 7 and 23 in hand we began exploring conditions for the tandem ring closing metathesis to construct the azepine ring and γ-lactone. Having larger quantities of tetraene 23 from our studies on the asymmetric Grignard addition we chose to first explore the tandem ring closing metathesis with this substrate. Ring closing metathesis of amine bearing substrates are known to be difficult and require the use of acid additives to prevent coordination of the amine to the ruthenium which can result in catalyst poisoning. Previously we have successfuly employed Grubbs II catalyst in the presence of TFA and heating to close azabicylic ring systems.[3c]
Unfortunately, these conditions were unsuccessful. While consumption of starting material and formation of a product corresponding to the mass of a single ring closure was observed by LC/MS, no progression toward the desired bis-ring closure product was observed and significant decomposition of starting material occured.
Grela and coworkers have reported Ru catalyst 24 with improved reactivity and mild reaction conditions for challenging ring closing metathesis reactions including a protected nitrogen-containing azepane ring as well as a trisubstituted α,β-unsaturated δ lactone.[9] Based on these reports we hoped to selectively form the azepine ring system by reaction at room temperature, and then to close the γ-lactone under elevated temperatures. Excitingly, this proved to be successful, as reaction with Grela catalyst 24 in the presence of TFA (1 equiv) stirring first at room temperature and then at 90 °C afforded the desired bis-ring closure product in 26% yield (Scheme 6). With this result we next studied the effect of the acid on the closure of the azepine ring, examining acids with a range of pKa values (see Table 2). Without an acid additive the reaction did not progress, and CSA posessing a pKa value in the middle of the acids surveyed was found to provide the greatest degree of conversion as measured by LC/MS (entry 5). Doubling the amount of CSA to 2 equiv was found to greatly improve the efficiency of the reaction, resulting in complete consumption of tetraene 23 within 6 h, and affording 65% isolated yield of pyrroloazepine 25.
Scheme 6.

Tandem ring closing metathesis of 23. (a) TFA (1 equiv), toluene; then 24 (15 mol %), rt. (b) 24 (25 mol %), toluene, 90 °C, 26% yield over 2 steps. (c) CSA (2 equiv), toluene; then 24 (10 mol %), rt, 65%. (d) 24 (3 × 10 mol%), 90 °C, 52% yield over 2 steps. TFA = trifluoroacetic acid; CSA = camphorsulfonic acid; Mes= mesityl.
Table 2.
Acid screen for tandem ring closing reaction.
| Entry | Acid (pKa) | Equiv. | Ratio 25:23 |
|---|---|---|---|
| 1 | - | - | no conv. |
| 2 | TFA (−0 25) | 1 | 1:4 5 |
| 3 | PTSA (−2.8) | 1 | 1:2.2 |
| 4 | AcOH (4 76) | 1 | >1:20 |
| 5 | CSA (12) | 1 | 1:1.3 |
| 6 | CSA (12) | 2 | >20:1 |
*To a solution of tetraene 23 in toluene was added an acid.. After stirring for 10 minutes Grela catalyst (10%) was added and stirring was continued at rt for 7 h..
With optimized conditions for closure of the azepine ring, we applied these conditions (Table 2, entry 6) to the tandem ring closure of tetraene 23, heating the reaction to 90 °C after complete conversion to 25. While conversion to bis-ring closure product was observed by LC/MS (~20% conversion), the use of additional catalyst (2 × 10 mol %) and extended heating was required for full conversion of 25 to 26. A small selection of metathesis catalys were screened with these optimized conditions (Hoveyda Grubbs II, Zhan 1B, Grubbs II, Schrock catalyst). Grela catalyst and Hoveyda Grubbs II were found to be optimal and exhibit comparable activity. Thus, reaction of tetraene 23 in the presence of CSA (2 equiv) and Grela catalyst 24 (3 × 10 mol %) afforded 26 in 52% isolated yield (Scheme 6).
As shown in Scheme 7, intermediate 26 was carried forward to unnatural analog 9a-epi-stemaphylline 27 through olefin reduction employing Pearlman’s catalyst,[10] which also set the final C14 stereocenter in 95% yield and 7:1 dr. Reaction of 27 in the presence of O3 led to the corresponding N-oxide 28 (53% yield).[11]
Scheme 7.
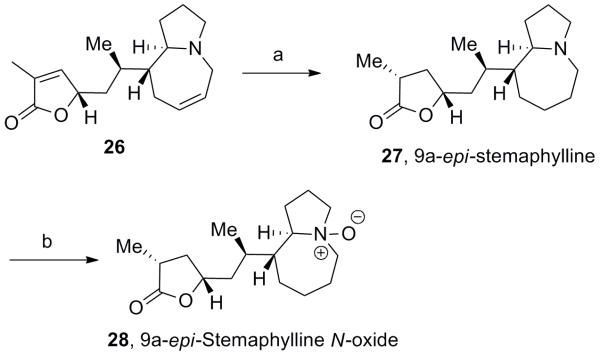
Synthesis of 9a-epi-stemaphylline and 9a-epi-stemaphylline N-oxide. a) Pd(OH)2/C, H2, THF, rt, 95%. b) O3, CH2Cl2, −78 °C, 54%.
With successful conditions for the tandem ring closing metathesis reaction we turned our attention toward the synthesis of stemaphylline, subjecting pyrrolidine tetraene 7 to the optimized tandem ring closing metathesis reaction conditions with the Grela catalyst (Scheme 8). Suprisingly, the ring closure proceeded more slowly and upon isolation an inseparable mixture of products was obtained. In consideration that olefin migration could be leading to epimerization, we tested the addition of 1,4-benzoquinone reported to suppress undesirable olefin migration,[12] however a mixture of bis-ring closure products was still observed, wherein the C12 carbon of the lactone was epimerized.
Scheme 8.
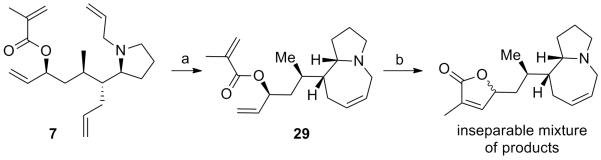
Tandem ring closing metathesis of 7. (a) CSA (2 equiv), 1,4-benzoquinone (50 mol %) toluene; then Hoveyda Grubbs II (3 × 10 mol %), rt, 48%. (b) Hoveyda Grubbs II (2 × 10 mol%), 90 °C. CSA = camphorsulfonic acid.
To elucidate when the suspected epimerization was occuring the reaction was stopped after closure of the azepine ring in the presence of Hoveyda Grubbs II, CSA, and 1,4-benzoquinone at room temperature. Isolation of mono-ring closure product 29 demonstrated that a single product was formed and that closure of the γ-lactone was the problematic step (Scheme 8). Unfortunately, subjection of 29 to a variety of acid additives and RCM catalysts was uneffective in suppressing the formation of the mixture of products.
In an attempt to gain insight into the differences in reactivity between tetraene 7 and 23 and azepeines 29 and 25 the four compounds were sketched and minimized using the MMFF force field to an energy gradient of <0.01 while preserving the stereochemistry. These calculations were performed using MOE (v2012.10; Chemical Computing Group; www.chemcomp.com). The resulting global minima 3-dimensional structures revealed that tetraene 23 possesses an intramolecular hydrogen bond which could pre-organize the substrate for ring closing metathesis. In contrast tetraene 7 possesses a more disordered conformation with the olefins splayed apart providing a possible rationale for the lower reactivty of tetraene 7 in the RCM reaction. Examination of azepines 25 and 29 revealed a striking difference in the orientation of the α,β-unsaturated ester and its proximity to the olefin RCM partner. While the two olefins in intermediate 25 are in close proximity, the reacting olefins in 29 are pointed away from one another. This disfavorable oreintation of the γ-lactone in 29 then allows for competing reaction pathways or epimerization of the C12 stereocenter during the course of the reaction (Figure 3). These data suggest a conformational bias that supports the observed experimental outcomes; however, there are many freely rotatable bonds and solvation is not accounted for, thus, this ground state analysis is laden with caveats, yet provides a partial explanation for the differential substrate reactivity.
Figure 3.
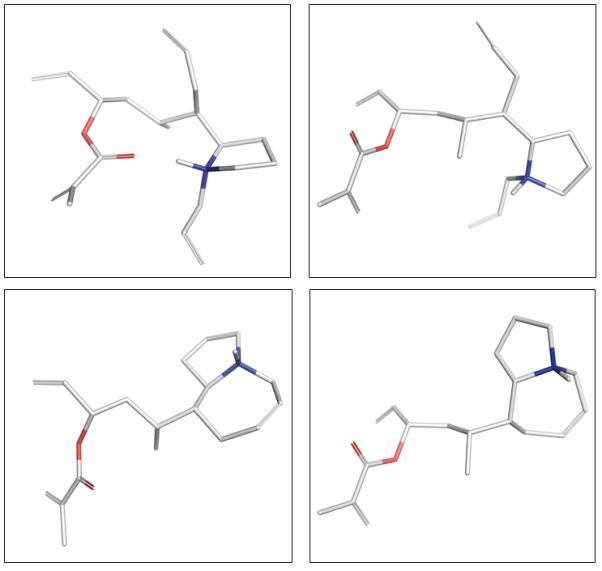
Energy minimized 3-dimensional structures of 23 (top left), 7 (top right), 25 (bottom left), and 29 (bottom right).
From this analysis we began investigation of methods to expidite the closure of the γ-lactone in order to prevent the suspected epimerization. Recently Hoye and coworkers have developed a relay ring closing metathesis (RRCM) strategy[13] to accelerate reactivity for difficult substrates and as a means to preload the ruthenium metathesis catalyst at a desired olefin to improve selectivity. Incorporating dienes 30[14] and 32[15] into intermediate 20a (Scheme 9) we arrived at RRCM substrate 33 in order to determine if directing the formation of the ruthenium alkylidene would prove beneficial. Reaction at room temperature revealed significant amounts of truncation products, presumably intermediates 7 and 29. However, vigorous reflux and argon sparge led to rapid formation of the desired ring closing metathesis product 6, and excitingly with only minor epimerization (10:1). Reduction with Pearlman’s catalyst[10] afforded stemaphylline in 62% yield and with 4:1 selectivity at the C12 stereocenter. Spectral and rotation data of the synthetic material was generally in agreement with that reported for the natural product 1, though complicated by the presence of 4:1 ratio of inseperable diastereomeric products.[2] Further confirmation was achieved by conversion to stemaphylline N-oxide 2. Attempts to form the N-oxide in the presence of O3 was ineffective; however formation with mCPBA in CH2Cl2 at 0 °C afforded stemaphylline N-oxide 2 in 74% yield, isolated as a single isomer by reverse phase chromatorgraphy. The synthetic 2 was in complete agreement with the spectral and rotation data reported, thus completing the first total synthesis of 3 and, by inference, 2[2].
Scheme 9.

Synthesis of RRCM substrate 33 and synthesis of stemaphylline 1 and stemaphylline N-oxide 2. (a) TBAF, THF, 0 °C to rt, 97%; (b) 30, MNBA, Et3N, DMAP, CH2Cl2, reflux, 98%; (c) (i) TFA/H2O, (95:5) (ii) PS-BH(OAc)3, DCE, rt, (iii) 32, K2CO3, DMF, rt, 40% over 3 steps. (d) CSA (2 equiv), toluene, rt; then Hoveyda Grubbs II (20 mol %) reflux, argon sparge, 37%. (e) Pd(OH)2/C, H2, THF, rt, 62%. f) mCPBA, CH2Cl2, 0 °C, 74%. MNBA = 2-methyl-6-nitrobenzoic acid; DMAP = 4-dimethyl-aminopyridine; CSA = camphorsulfonic acid.
In conclusion, we have completed the first total synthesis of both stemaphylline 1 (19 steps) and stemaphylline N-oxide 2 (20 steps), via a tandem bis-RCM strategy, as well as unnatural 9a-epi-stemaphylline 27 and 9a-epi-stemaphylline N-oxide 28. The linear tetraene substrate leading to 27 and 28 was suggested by modelling to be pre-organized for the bis-tandem RCM, and smoothly afforded the desired unnatural products; however, the tetraene corresponding to natural 1 and 2 was disorganized, leading to lower reactivity and ruthenium alklyidene-mediated epimerization. Thus, adopting a relay ring closing metathesis (RRCM) strategy enabled access to 1 and 2. Biological evaluation of both natural and unnatural analogs is underway and will be reported in due course.
Supplementary Material
Figure 2.
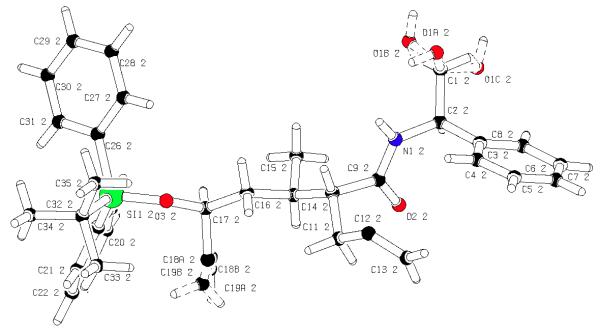
Crystal structure of 15 and confirmation of C9, C10, and C12 stereocenters.
Acknowledgements
The authors acknowledge the Vanderbilt Department of Pharmacology, VUMC and the NIH for funding our research, and in particular the MLPCN (U54MH084659) for the Vanderbilt Specialized Chemistry Center. The financial support from the Warren Foundation in the form of the William K. Warren, Jr. Endowed Chair is gratefully acknoweldged.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
Experimental Section
Please see the Supporting Information Section for full experimental details
References
- [1].a) Pilli RA, Rosso GB, da C. F. de Oliveira M. Nat. Prod. Rep. 2010;27:1908–1937. doi: 10.1039/c005018k. [DOI] [PubMed] [Google Scholar]; b) Pyne SG, Ung AT, Jatisatienr A, Mungkornasawakul P. Mj. Int. J. Sci. Tech. 2007;01(2):157–165. [Google Scholar]; c) Greger H. Planta Med. 2006;72:99–113. doi: 10.1055/s-2005-916258. [DOI] [PubMed] [Google Scholar]
- [2].Mungkornasawakul P, Chaiyong S, Sastrauji T, Jatisatienr A, Jatisatienr C, Pyne SG, Ung AT, Korth J, Lie W. J. Nat. Prod. 2009;72:848–851. doi: 10.1021/np900030y. [DOI] [PubMed] [Google Scholar]
- [3].a) Fadeyi OO, Senter TJ, Hahn KN, Lindsley CW. Chem. Eur. J. 2012;18:5826–5831. doi: 10.1002/chem.201200629. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Senter TJ, Fadeyi OO, Lindsley CW. Org. Lett. 2012;14:1869–1871. doi: 10.1021/ol300466a. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Senter TJ, Schulte ML, Konkol LC, Wadzinski TE, Lindsley CW. Tetrahedron Lett. 2013;54:1645–1648. doi: 10.1016/j.tetlet.2013.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Hahn KN, Fadeyi OO, Cho HP, Lindsley CW. Tet. Lett. 2012;53:3577–3580. doi: 10.1016/j.tetlet.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].a) Cogan DA, Liu G, Ellman JA. Tetrahedron. 1999;55:8883–8904. [Google Scholar]; b) Brinner KM, Ellman JA. Org. Biomol. Chem. 2005;3:2109–2113. doi: 10.1039/b502080h. [DOI] [PubMed] [Google Scholar]; c) Robak MT, Herbage MA, Ellman JA. Chem. Rev. 2010;110:3600–3740. doi: 10.1021/cr900382t. [DOI] [PubMed] [Google Scholar]
- [5].a) Nagao Y, Hagiwara Y, Kumagai T, Ochic M. J. Org. Chem. 1986;51:2391–2393. [Google Scholar]; b) Bock M, Dehn R, Kirschning A. Angew. Chem. Int. Ed. 2008;47:9134–9137. doi: 10.1002/anie.200803271. [DOI] [PubMed] [Google Scholar]
- [6].Blanchette MA, Choy W, Davis JT, Essenfeld AP, Masamune S, Roush WR. Tetrahedron Lett. 1984;25:2183–2186. [Google Scholar]
- [7].Dambacher J, Anness R, Pollock P, Berhdahl M. Tetrahedron. 2004;60:2097–2110. [Google Scholar]
- [8].Wee AG, Yu Q. J. Org. Chem. 2001;66:8935–8943. doi: 10.1021/jo010751z. [DOI] [PubMed] [Google Scholar]
- [9].Grela K, Harutyunyan S, Michrowska A. Angew. Chem. Int. Ed. 2002;41:4038–4040. doi: 10.1002/1521-3773(20021104)41:21<4038::AID-ANIE4038>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- [10].a) Herdeis C, Lütsch K. Tetrahedron: Asymm. 1993;4:121–131. [Google Scholar]; b) Bates RH, Shotwell JB, Roush WR. Org. Lett. 2008;10:4343–4346. doi: 10.1021/ol801852j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ye Y, Velten RF. Tetrahedron Lett. 2003;44:7171–7173. [Google Scholar]
- [12].Hong SH, Sanders DP, Lee CW, Grubbs RH, R. H. J. Am. Chem. Soc. 2005;127:17160–17161. doi: 10.1021/ja052939w. [DOI] [PubMed] [Google Scholar]
- [13].Hoye TR, Jeffrey CS, Tennakoon MA, Wang J, Zhao H. J. Am. Chem. Soc. 2004;126:10210–10211. doi: 10.1021/ja046385t. [DOI] [PubMed] [Google Scholar]; b) Wallace DJ. Angew. Chem. Int. Ed. 2005;44:1912–1915. doi: 10.1002/anie.200462753. [DOI] [PubMed] [Google Scholar]
- [14].Fujioka K, Yokoe H, Yoshida M, Shishido K. Org. Lett. 2012;14:244–247. doi: 10.1021/ol203021c. [DOI] [PubMed] [Google Scholar]
- [15].Meek SJ, Malcolmson SJ, Li B, Schrock RR, Hoveyda AH. J. Am. Chem. Soc. 2009;131:16407–16409. doi: 10.1021/ja907805f. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.