

Abstract
LigAB from Sphingomonas paucimobilis SYK-6 is the only structurally characterized dioxygenase of the largely uncharacterized superfamily of Type II extradiol dioxygenases (EDO). This enzyme catalyzes the oxidative ring-opening of protocatechuate (3,4-dihydroxybenzoic acid or PCA) in a pathway allowing the degradation of lignin derived aromatic compounds (LDACs). LigAB has also been shown to utilize two other LDACs from the same metabolic pathway as substrates, gallate, and 3-O-methyl gallate; however, kcat/KM had not been reported for any of these compounds. In order to assess the catalytic efficiency and get insights into the observed promiscuity of this enzyme, steady-state kinetic analyses were performed for LigAB with these and a library of related compounds. The dioxygenation of PCA by LigAB was highly efficient, with a kcat of 51 s−1 and a kcat/KM of 4.26 × 106 M−1s−1. LigAB demonstrated the ability to use a variety of catecholic molecules as substrates beyond the previously identified gallate and 3-O-methyl gallate, including 3,4-dihydroxybenzamide, homoprotocatechuate, catechol, and 3,4-dihydroxybenzonitrile. Interestingly, 3,4-dihydroxybenzamide (DHBAm) behaves in a manner similar to that of the preferred benzoic acid substrates, with a kcat/Km value only ~4-fold lower than that for gallate and ~10-fold higher than that for 3-O-methyl gallate. All of these most active substrates demonstrate mechanistic inactivation of LigAB. Additionally, DHBAm exhibits potent product inhibition that leads to an inactive enzyme, being more highly deactivating at lower substrate concentration, a phenomena that, to our knowledge, has not been reported for another dioxygenase substrate/product pair. These results provide valuable catalytic insight into the reactions catalyzed by LigAB and make it the first Type II EDO that is fully characterized both structurally and kinetically.
The desire to discover and develop renewable alternatives to fossil fuels for the production of energy and as chemical feedstocks has been ever increasing in the past decade, leading to research into the natural ability of microbes to process a wide range of carbon sources. Much attention has been focused on accessing the carbon locked in cellulose, the most abundant natural polymer, while lignin, the second most abundant natural polymer,1 has not been as extensively investigated. Small aromatic molecules produced by the lignin depolymerization process, lignin derived aromatic compounds (LDACs), have been shown to be accessible for use in central metabolism in some bacteria,2–4 thereby unleashing their potential as a possible carbon source for the fermentative production of a wide variety of chemicals. The enzymes capable of oxidative cleavage of the aromatic rings play a key role in the catabolism of these LDACs. Aromatic ring cleaving dioxygenases are generally divided into three classes: intradiol (Class I), extradiol (Class II), and those capable of cleaving aromatics without a diol (proposed Class III).5,6 Extradiol dioxygenases (EDOs) are further divided into types based on superfamily classification: vicinal oxygen chelate (VOC, Type I), cupin (Type III), and a currently undefined superfamily (Type II) hereafter referred to as the protocatechuate dioxygenase (PCAD) superfamily.6,7 EDOs from the VOC and cupin superfamilies have been well characterized by both enzymological and structural methods.8–12
The existence of Type II EDOs, as postulated by protein sequence homology,13,14 has been known for the better part of two decades, and yet, only a handful of these enzymes have been studied beyond sequence alignments, or gene and pathway identification. Ten Type II EDOs have been isolated and characterized in varying degrees either functionally or kinetically (2,3-dihydroxyphenylpropionate 1,2-dioxygenase from E. coli (MhpB)14 and Alcaligenes eutrophus (MpcI),14,15 protocatechuate 4,5-dioxygenases from Sphingomonas paucimobilis SYK-6 (LigAB), Comamonas testosteroni Pt-L5,16 and Comamonas testosteroni T-2 (PmdAB),17 protocatechuate 2,3-dioxygenase (2,3-PCD) from Paenibacillus sp. (formerly Bacillus macerans),18,19 2-aminophenol 1,6-dioxygenase (1,6-ApDO) from Pseudomonas pseudoalcaligenes,20,21 2′-aminobiphenyl-2,3-diol-1,2-dioxygenase (CarBab) from Pseudomonas stutzeri,22,23 the polycyclic arene diol dioxygenase (PhnC) from Burkholderia sp.,24 and gallate 2,3-dioxygenase (GDO)25 from Pseudomonas putida KT2440). Further, only five other enzymes in this superfamily have been functionally identified by molecular biology techniques (homoprotocatechuate 2,3-dioxygenase (HpcB) from E. coli,26 an acid catechol dioxygenase (HppB) from Rhodococcus globerulus PWD1,27 an extradiol dioxygenase EdoD from Rhodococcus sp.,28 and protocatechuate 4,5-dioxygenases from Pseudomonas straminea (originally ochraceae NGJ1) (ProOab)29 and Anthrobacter keyseri 12B (PcmA)30). Additionally, the protocatechuate 4,5-dioxygenase LigAB from Sphingomonas paucimobilis SYK-6 is the only structurally characterized EDO from this largely undefined superfamily.31 Until now, full kinetic characterization and mechanistic investigation of Type II EDOs has only been performed for MhpB by Bugg,32,33 and 2,3-PCD by Lipscomb,18 though 2,3-PCD was only identified as a Type II dioxygenase a decade later by Masai.19 Kinetic analysis beyond specific activity is largely lacking for LigAB and other Type II EDOs.
LigAB was identified as a member of the bacterial LDAC catabolic pathway of S. paucimobilis SYK-6,4,34–37 and it catalyzes the extradiol aromatic ring cleavage of protocatechuate by the insertion of O2 across the C4–C5 bond of the aromatic ring to form 4-carboxy-2-hydroxymuconate-6-semialdehyde, CHMS (Scheme 1). LigAB has been shown to have some promiscuous activity with PCA substrate analogues gallate (GA) and 3-O-methyl gallate (3OMG), two compounds also found in the LDAC catabolic pathway of S. paucimobilis SYK-6. This activity has been demonstrated through knockout studies and in vitro assays of specific activity;38–41 however, the breadth of substrate specificity is previously unknown.
Scheme 1.
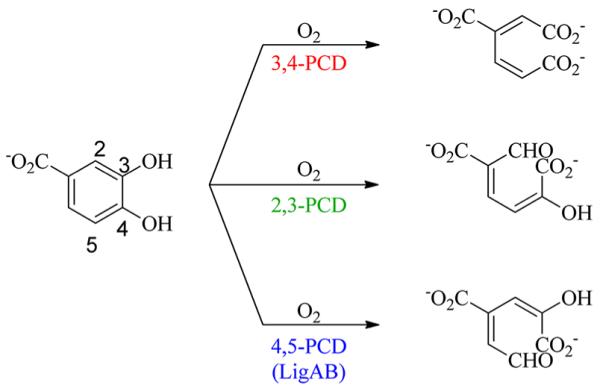
Ring Cleavage Reactions Catalyzed by Known PCA Dioxygenases
In this work, we report the kinetic parameters for anaerobically purified LigAB with both of its physiological substrates, PCA and O2, as well as an investigation of other substrates containing the catechol scaffold, including GA and 3OMG, with full kinetic characterization of the best substrates. From these analyses, we aim to establish the structural and oxidation potential requirements of substrates for LigAB. Additionally, steady-state kinetic studies are used to establish the pH and metal dependence of LigAB. Furthermore, we demonstrate that anaerobic purification of this enzyme is necessary for optimal stability and kinetic efficiency.
MATERIALS AND METHODS
Commercially available reagents and solvents were purchased from Aldrich or Alfa Aesar and used without purification unless otherwise noted. 3-O-Methyl gallate was purchased from ChromaDex. Several substrates were synthesized as previously described: 3,4-dihydroxybenzamide,42 1,2-dihydroxy-4-aminobenzene hydrobromide,43 and methyl-3,4-dihydroxybenzoate.44 A Varian Mercury 400 MHz NMR was used to collect all 1H NMR spectra. Absorption spectra were collected with a Varian Cary 100 Bio UV–visible spectrophotometer (Palo Alto, CA). Bradford and Ferene-S assays were conducted in 96-well plate format and absorption values recorded on a Molecular Devices SpectraMax M5 plate reader (Sunnyvale, CA). MAX Efficiency DH5α and One Shot BL21 Star chemically competent E. coli cells were purchased from Life Technologies (Carlsbad, CA). All enzymes used for DNA manipulations were purchased from New England Biolabs (Ipswitch, MA). Centrifugation and ultracentrifugation were performed on a DuPont Instruments (Wilmington, DE) Sorvall RC-5B centrifuge and Beckman (Brea, CA) L7-80 Ultracentrifuge with a type 60-Ti rotor, respectively. All cells were lysed using a SIMO-Aminco Industry Inc. (Rochester, NY) French press.
Protein Expression
Unable to acquire genomic DNA for Sphingomonas paucimobilis SYK-6 or the plasmids containing the genes for LigAB, we retrieved the DNA sequence encoding the LigA-LigB gene cluster from the NCBI data bank. A construct for gene synthesis was prepared with insertion of an upstream NdeI endonuclease site and additional stop codons followed by a BamHI endonuclease site downstream of the LigA and LigB gene cluster. The modified sequence was synthesized by DNA2.0 (Menlo Park, CA). The synthetic gene sequence was subsequently removed from the provided pJ241 vector by endonuclease cutting with NdeI and BamHI, ligated into pET-15b (EMD Millipore, Billerica, MA) in the NdeI and BamHI cloning sites, and transformed by heat shock separately into DH5α and BL21 competent E. coli cells.
His-tagged protein (α subunit N-terminally tagged and β subunit untagged) was expressed from a pET15b plasmid by induction of BL21 competent cells with a final concentration of 1 mM isopropyl β-d-1-thiogalactopyranose (IPTG) at OD600 = 0.6 under aerobic conditions at 37 °C. After 20 h, the cell cultures were centrifuged at 5,000 rpm. If not used immediately, cell pellets were stored at −80 °C.
Aerobic Purification
Pelleted cells were resuspended in 50 mM Tris (pH 7.0) containing 1:9 glycerol/H2O with one tablet of cOmplete-EDTA free protease inhibitor cocktail (Roche, Basel, Switzerland). Resuspended cells were lysed by 3 passes through a French-press at 13,000 psi. The cell lysate was subsequently centrifuged at 25,000 rpm for 45 min in an ultracentrifuge.
The supernatant was applied to a gravity column loaded with HisPur Ni-NTA resin (10 mL, Thermo Scientific) previously charged with Ni2+ and equilibrated with 5 column volumes (CV) of bind buffer (50 mM HEPES, 300 mM NaCl, and 10 mM imidazole at pH 7.5) followed by 5 CV of wash buffer (50 mM HEPES, 300 mM NaCl, and 20 mM imidazole at pH 7.5) to remove unbound protein. The tagged enzyme was eluted by a step gradient of a buffer containing 50 mM HEPES (pH 8.0) and 300 mM NaCl with, sequentially, 62.5 mM (2.5 CV), 125 mM (2.5 CV), and 250 mM (5 CV) imidazole. Fractions (10 mL) were collected for all wash and elution steps. Aliquots (30 μL) of each fraction were analyzed for purity by SDS–PAGE.
Fractions containing the copurified LigAB α and β subunits were pooled, concentrated using an Amicon concentrator stirred cell with a regenerated cellulose 10 kDa molecular weight cutoff ultrafiltration membrane (EMD Millipore), and buffer exchanged into 50 mM Tris (pH 7.5), 0.5 mM DTT, and 0.5 mM Fe(NH4)2(SO4)2·6H2O. Exchanged protein was filtered with a 0.45 μm syringe filter and subsequently flash frozen in liquid nitrogen and stored at −80 °C. From 6 L of cells, 70 mg of protein was typically obtained and concentrated to ~7 mg/mL.
Anaerobic Purification
Anaerobic purification of LigAB was performed as described above with modification to maintain an oxygen free environment. After cell lysis, the lysate was transferred to centrifuge tubes, and the head space was flushed with N2 gas prior to centrifugation at 25,000 rpm for 45 min in an ultracentrifuge. The supernatant was decanted to a serum vial, flushed with N2 gas, sealed, and transferred into a Vacuum Atmospheres Company (Hawthorne, CA) HE-493/MO-5 glovebox under N2 atmosphere, operated at <5 ppm O2.
All buffers, water, and column resins to be used in the glovebox were prepared as described and thoroughly degassed, to remove O2, by three cycles of degassing under vacuum followed by N2 gas bubbling. Degassed buffers were allowed to equilibrate in the glovebox atmosphere overnight prior to use.
The supernatant was applied to a gravity column loaded with HisPur Ni-NTA resin inside the glovebox, and protein was eluted from the column as described above. Fractions (10 mL) were collected for all wash and elution steps. Aliquots (30 μL) of each fraction were removed from the glovebox and analyzed for purity by SDS–PAGE. Fractions containing the copurified LigAB α and β subunits were pooled, concentrated using an Amicon concentrator stirred cell with a regenerated cellulose 10 kDa molecular weight cutoff ultrafiltration membrane (EMD Millipore), and buffer exchanged into 50 mM Tris at pH 8.
Phosphate buffered saline (PBS) was added to the protein solution to a concentration of 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4·2H2O, and 2 mM KH2PO4 at pH 7.4 followed by bovine derived thrombin (MP Bio) to a concentration of 1 U/mg purified protein to remove the His-tag. His-tag cleaved protein was buffer exchanged into 50 mM Tris (pH 8) to remove excess salt prior to application to a Source-Q ion exchange column (10 mL, GE Healthcare). LigAB was applied to the column with 50 mM Tris (pH 8), washed with 50 mM Tris (pH 8) and 100 mM NaCl, and eluted with 50 mM Tris (pH 8) and 350 mM NaCl. Fractions (5 mL) were collected for both wash and elution steps. Aliquots (30 μL) of each fraction were removed from the glovebox and analyzed for purity by SDS–PAGE. Those fractions containing LigAB α and β subunits were pooled, concentrated, and buffer exchanged into 50 mM Tris (pH 7.5), 0.5 mM DTT, and 0.5 mM Fe(NH4)2(SO4)2·6H2O. Exchanged protein was filtered with a 0.45 μm syringe filter and removed from the glovebox in an airtight syringe with the needle sealed by a rubber stopper and subsequently flash frozen in liquid nitrogen and stored at −80 °C. From 6 L of cells, 70 mg of protein was typically obtained and concentrated to ~7 mg/mL.
Steady-State Kinetics Assays
The rate of the enzymatic reaction was determined by measuring O2 consumption using an O2-sensitive Clark-type electrode with computer integration via an Oxygraph electrode control unit (Hansatech, King’s Lynn, Norfolk, England). Prior to each assay, the electrode was standardized with air-saturated water and water depleted of O2 by the addition of sodium hydrosulfite as described by the manufacturer. Stock solutions and buffers were prepared fresh daily. Prior to each experiment, 100 μL of LigAB (60 μM) was thawed and buffer exchanged into degassed 50 mM Tris (pH 7.5) containing 1:9 t-butanol/H2O using a 3 mL Sephadex G-25 desalting gel (GE Healthcare) column in a glovebag flushed with high purity N2 for 1 h. Exchanged enzyme (3–7 μM) was kept under inert atmosphere (N2) in a sealed vial on ice for the duration of each experiment.
The effect of pH on the rate of LigAB O2 consumption was investigated in the pH range of 6–10 at 23 °C. The buffer solutions (50 mM) used to span this pH range were phosphate (pH 6 to 8), Tris (pH 7.5 to 9), BICINE (pH 8 to 9), and CAPSO (pH 9 to 10). The initial rate assays were performed in a final volume of 1 mL of air-saturated 50 mM buffer of desired pH and 250 μM PCA. The reaction was initiated by the addition of 1–2 μL LigAB (3–7 nM final concentration) after the equilibration of all other components for at least 1 min to obtain a constant background O2 consumption rate of ±0.5 μM/min. Reaction velocities were calculated from the slope of the first 30 s of data after LigAB addition and corrected for background O2 consumption using 30 s of data immediately prior to LigAB addition. The precise enzyme concentration for each experiment was determined by the Bradford assay (Bio-Rad) after completion. The pKa values of effected active site residues were determined by a fit of the reaction velocities as a function of pH to eq 1 (1 acidic pKa and 1 basic pKa) and eq 2 (2 indistinguishable acidic pKa’s and 1 basic pKa).
| (1) |
| (2) |
Steady-state kinetic parameters for LigAB with respect to the organic substrate were determined by measuring the rate of O2 consumption in the presence of varying concentrations of organic substrate (1 μM to 5000 μM). An aqueous stock solution (25 mM) of the desired organic substrate (PCA, GA, or 3OMG) was prepared immediately prior to use in a 10 mL volumetric flask. Stock solutions of 3OMG (25 mM) were prepared in 1:9 DMSO/H2O. The initial rate assays were performed, as described above, in air-saturated 50 mM Tris (pH 7.5) and initiated by the addition of 1–2 μL of LigAB (3–7 nM). Steady-state kinetic parameters with respect to O2 were measured in 50 mM Tris (pH 7.5), 1 mM PCA, and 40–450 μM O2 and initiated by the addition of LigAB (3–7 nM). The buffer was equilibrated prior to each reaction with a fixed mixture of O2 and N2 gas using a Cole-Parmer gas proportioner (Vernon Hills, IL), and the reaction chamber was maintained under an atmosphere of the same O2/N2 mixture. The precise enzyme concentration for each experiment was determined by the Bradford assay (Bio-Rad) after completion. Kinetic parameters were determined by a least-squares fitting of the Michaelis–Menten equation to the data using KaleidaGraph (Synergy).
Substrate Induced Enzyme Inactivation
The partition ratios and rates of enzyme inactivation in the presence of PCA, GA, 3OMG, and DHBAm were calculated using methods previously described for mechanism based inactivation of dioxygenases.5,45 The apparent rate of inactivation in air-saturated buffer during catalytic turnover was determined from the curvature of O2 consumption progress curves of reactions performed at varying substrate concentrations using anaerobically purified enzyme. The rate constant of inactivation at a given substrate concentration (js) was determined by fitting individual progress curves to eq 3 using KaleidaGraph.5,45 In this equation, Pt is the concentration of the product formed at time t, P∞ is the concentration of the product formed at reaction completion, and Pi is the concentration of the product at the start of the reaction.
| (3) |
The apparent rate of inactivation was subsequently determined by a fit of eq 4 to the values of js as a function of substrate concentration, for substrates showing a substrate concentration dependent inactivation. For substrates not showing a concentration dependence of the rate of inactivation, was calculated as the average of the js values over the range of substrate concentrations.
| (4) |
The partition ratio was then calculated using the determined from the steady-state kinetics assays and the value determined above using eq 5.
| (5) |
Metal Dependence
The influence of different divalent metals on PCA dioxygenase activity was tested via a method adapted from Diaz, et al.25 Anaerobically purified enzyme was buffer exchanged into degassed 100 mM Tris (pH 7.5) containing 1:9 t-butanol/H2O and subsequently incubated under N2 in a sealed vial with 2 mM 2,2′-dipyridyl (2,2′-bipyridine, bipy) at 4 °C for 1 and 24 h. Apo-enzyme reactivation was attempted by the incubation of the apoenzyme solution in a 2 mM solution of the desired divalent metal salt (FeSO4, CuSO4, CoSO4, MnSO4·H2O, MgSO4·7H2O, NiSO4, or ZnSO4) under anaerobic conditions for 1 h and/or 24 h at 4 °C. Activities of untreated enzyme (no bipy added after 1 and 24 h), apoenzyme samples, and samples incubated with metal salts were determined by O2 consumption assays of 250 μM PCA in 50 mM Tris (pH 7.5). The concentration of enzyme in the buffer exchanged stock was determined by the Bradford assay, and the concentration of enzyme in bipy and metal salt treated samples was calculated by calculating the dilution of the buffer exchanged stock.
Product Determination
LigAB reaction products were identified by two different methods. The reaction product of the PCA starting material was generated under O2 saturated conditions in 1 mL of 50 mM phosphate buffer (pH 7.5), 2 mM PCA, and incubation with ~10 μg (50 μL of a 4.5 μM stock) of anaerobically purified LigAB added portion-wise over 1 h. The reaction was monitored by 1H NMR for the disappearance of the PCA aromatic chemical shifts, with 3-(trimethylsilyl)-propionic-2,2,3,3-d4 acid sodium salt (DSS, Aldrich) as a reference. Additionally, the product, CHMS, was converted to 2,4-lutidinic acid as previously described by the addition of 200 μL of concentrated NH4OH.46–48 After stirring at room temperature for 2 h, the 1H NMR spectrum was recorded. All aqueous 1H NMR spectra were recorded in 1:9 D2O/H2O with water suppression. The UV–vis spectra of the CHMS reaction product were recorded under reaction (pH 7.5), acidic (pH 1), and alkaline (pH 14) conditions by the addition of 4 M HCl or 4 M NaOH, respectively. The UV–vis spectrum of the product formed by the reaction of CHMS with ammonium hydroxide was also recorded.
Substrate Promiscuity Assessment
A wide range of potential substrates were screened using methods analogous to those previously described, using anaerobically purified LigAB and 5 mM compound. Reactions were performed in 1 mL of air-saturated 50 mM Tris (pH 7.5) with 5 mM substrate at 25 °C and monitored by oxygen consumption. LigAB was added to initiate the reaction as previously described. In addition to stock solutions of PCA and gallate (25 mM in H2O), and 3OMG (25 mM in 1:9 DMSO/H2O), stock solutions of the organic substrates used for alternative substrate determination were prepared as 25 mM solutions in H2O (3,4-dihydroxybenzaldehyde, 2,3,4-trihydroxybenzoic acid, 2,3-dihydroxybenzoic acid, homoprotocatechuate, 4-hydroxybenzoic acid, 3,4-dihydroxybenzamide, 1,2-dihydroxy-4-aminobenzene, 4-nitrocatechol, catechol, 4-methyl catechol, 3-methoxycatechol, 2-aminophenol, and 3,4-dihydroxybenzonitrile), 1:9 DMSO/H2O (benzoic acid, vanillic acid, syringic acid, 2-iodobenzoic acid, vanillin, 3,4-dimethoxy benzaldehyde, syringaldehyde, 3-ethoxy-4-hydroxy benzaldehyde, methyl-3,4-dihydroxybenzoate, 2-methoxy-4-methylphenol, 3-methoxyphenol, and veratrol), or as 12.5 mM solutions in 1:4 DMSO/H2O (caffeic acid and p-coumaric acid). In an effort to see activity from potentially poor substrates, varying concentrations of LigAB (3–350 nM) were added to assays of the organic substrates. Assays with an addition of anaerobic enzyme greater than 2 μL exhibited O2 dilution effects upon addition. The dilution effect was corrected by subtraction of the progress curve for the addition of an equivalent volume of degassed 50 mM Tris and 1:9 t-butanol/H2O buffer containing no enzyme. Kinetic parameters were determined for substrates identified to have significant reactivity. Substrates found to have low levels of O2 consumption activity are reported as a percentage of the activity of LigAB with 5 mM PCA.
The oxidation potentials of a series of PCA analogues where the C1 substituent was varied were calculated using Gaussian 09 (Wallingford, CT).49–51 Initial structures were minimized by a molecular mechanics procedure, starting from several different initial geometries to ensure the location of the global minimum, followed by complete geometry optimization at the density functional B3LYP/6-31+g(d)-PCM level. Because solvation is especially important with charged species, solvation energies of all species were computed by the commonly used polarized continuum method of Tomasi.51 The computed free energies (in kcal/mol) of each parent anion and the corresponding neutral species formed by the removal of an electron from the parent were computed. The difference (ΔGcomp) between the two energies is a measure of the ease of electron removal from the anion. ΔGcomp was then converted to the absolute oxidation potential (independent of reference electrode) of the substance through the relationship (ΔGcomp = −τ(ΔGcomp), in which Faraday (τ, the proportionality constant between electrochemical and chemical free energies) = 23.06 kcal/volt. Absolute potentials have been shown to be highly linearly correlated with experimental oxidation potentials in other systems.52
Inhibition Assessment
All molecules that exhibited no oxygen consumption upon incubation with LigAB were examined for their ability to bind to and inhibit the dioxygenation of PCA by LigAB. Stock solutions of all molecules were prepared as previously described. Reactions were performed in 1 mL of air-saturated 50 mM Tris (pH 7.5) with 500 μM PCA and 1 mM potential inhibitor at 25 °C, and monitored by oxygen consumption; LigAB was added to initiate the reaction (3–7 nM). Inhibition was determined by comparison of the rates of inhibitor containing assays to the rates obtained from assays performed directly before and after where only 500 μM PCA (and no potential inhibitor) was present.
pH Effect on Iron Binding
Thawed anaerobically purified enzyme was buffer exchanged using Sephadex G-25 desalting gel as described above into degassed 50 mM Tris (pH 7.5) and 1:9 DMSO/H2O to remove excess iron from storage. A 30 μL aliquot of each enzyme sample was removed for use in a Bradford assay to determine the enzyme concentration. Desalted enzyme was subsequently buffer exchanged under ambient conditions into a 50 mM buffer of pH 6 (phosphate), 7.5 (phosphate), and 10 (CAPSO) using Amicon Ultra 0.5 mL centrifugal filters with a 10 kDa molecular weight cutoff (Millipore). After buffer exchanging, the protein samples were diluted to their original volume with water, and a Ferene-S assay adapted for the 96-well plate format was performed, as described by Capyk, et al.,53 to determine the iron content of all samples. The iron concentration was divided by the concentration of enzyme determined from the Bradford assay to give the relative iron–enzyme ratios.
RESULTS AND DISCUSSION
LigAB has been the subject of several studies since it was first reported as a protocatechuate 4,5-dioxygenase in 1990 by Noda, et al.54 The crystal structure, the first for this superfamily, and reaction kinetics (specific activity) of LigAB have been previously reported and are often cited by those investigating dioxygenase enzymes believed to be members of the PCAD superfamily. However, in these previous studies, LigAB was expressed and purified aerobically.37–39 Our attempts to mimic these purification conditions did indeed lead to the production of active enzyme; however, experiments to confirm the kinetic parameters of the enzyme revealed inconsistent initial rates at a given organic substrate concentration as well as loss of activity (~20% per hour) after thawing from storage (Figure 1). Incubation of aerobically purified enzyme with reductants, such as dithiothreitol or ascorbate, failed to recover or enhance the activity. The adoption of the anaerobic protein purification techniques described here and by others55–57 has produced pure copurified LigAB with greater than 10-fold higher activity over that of the aerobically purified enzyme and lower loss of activity over time after thawing from storage. Additionally, no loss of activity has been observed in the anaerobically purified enzyme stored at −80 °C over a period of 6 months.
Figure 1.

(A) Steady-state kinetics of LigAB with PCA: anaerobically purified (red circles) and aerobically purified (blue squares). (B) Steady-state kinetics of aerobically purified LigAB (blue squares) and the time dependent inactivation of aerobically purified enzyme after thawing (green triangles) at pH 7.5, 25 °C.
pH Effects on Rate
Despite exhaustive searching, a previous determination of the pH rate maximum of LigAB was not found. The pH rate maximum of a related enzyme, gallate 3,4-dioxygenase (DesB), from the same LDAC degradation pathway was determined, by oxygen consumption, to be 8.5 in the presence of gallate.39 Kinetic parameters (specific activity) were previously determined for LigAB as well as DesB and DesZ (3-O-methylgallic acid 3,4-dioxygenase) using the pH optimum conditions for DesB. Here, LigAB was found to have a pH rate maximum of 7.5 in phosphate buffer for the consumption of O2 in the presence of PCA (Figure 2A) when fit to eq 2. This pH rate maximum is consistent with pH values found for other Type II EDOs, including PCA and homoprotocatechuate (HPCA) dioxygenases.8,14,17,18,25 The pKa values determined from the fit of eq 2 suggest that there are two residues with a pKa of 6.3 and a third residue with a pKa of 9.7. The slope of the acidic arm, 1.8, from the plot of log(kcat) as a function of pH and fit to eq 2 (Figure 2B) also supports the presence of two residues with acidic pKa values, and the slope of the basic arm, 0.6, supports the presence of one residue with a basic pKa.
Figure 2.

pH rate profiles of LigAB with PCA at 25 °C, using buffers (orange triangle) Tris, (blue circle) phosphate, (red square) BICINE, (green diamond) CAPSO. (A) kcat vs pH with fitting to eq 1 (solid line) and fitting to eq 2 (dashed line). (B) log(kcat) vs pH fit to log(eq 1) and log(eq 2), solid and dashed lines, respectively.
In addition to the pH rate effect, changes in buffering agents also influenced the reaction rate, as can be seen in the pH region of 7.5 to 9 (Figure 2A). The effect was most evident at pH 7.5 where the initial rate of reactions performed in phosphate buffer displayed a small rate enhancement (20%) over the initial rate of reactions performed in Tris buffer. The small size and the diol like structure of Tris may contribute to the ability of Tris to possibly bind and occupy the LigAB active site, thus causing a mild inhibitory effect. Despite the rate enhancement in phosphate buffer, assays of LigAB activity were performed in Tris buffer to remain consistent with previous studies, and the pH was adjusted to 7.5 corresponding to the pH rate maximum found here.
LigAB contains two active site histidine residues, His127β and His195β, which based upon homology to mechanistically characterized Type I and Type III EDOs are believed to aid in substrate alignment and play a role as a catalytic base and catalytic acid, respectively (Figure 3). His127β is likely one of the two active site residues that becomes protonated and leads to decreased activity at lower pH. The second residue to be protonated may be one of the iron chelating residues, most likely Glu242β, although His12β and His61β are also candidates, leading to a loss of iron from the active site and subsequently a decrease in activity (see below for a discussion of metal dissociation experiments). The loss of activity at high pH corresponds to a single deprotonation event. Although His195β has been suggested to play a role in substrate alignment in the active site, it is possible that this residue plays an additional role as a catalytic acid, based upon homology to the mechanisms of other dioxygenases.32,58 Additional explanations for the decrease in activity at high pH corresponding to the relatively high pKa of 9.7 could include oxidation of the active site iron or deprotonation of pKa depressed Ser269β. Deprotonation of Ser269β would create an unfavorable charge–charge interaction with the carboxylic acid of PCA, preventing proper alignment in the active site. Further mechanistic analysis, including mutagenesis of these residues, will be performed to test mechanistic assignments.
Figure 3.
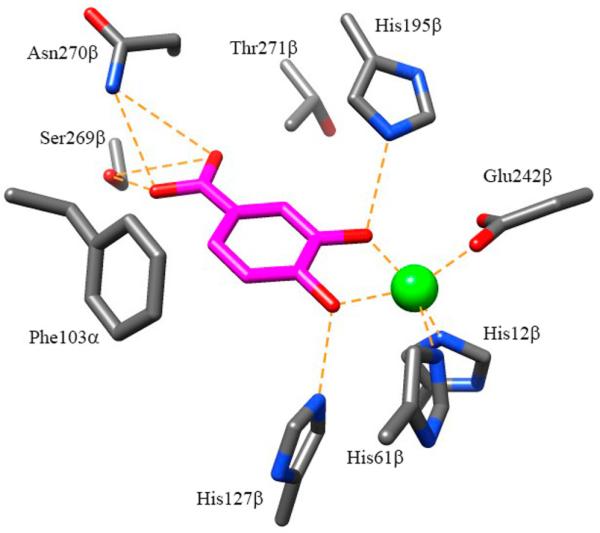
Active site of LigAB (1B4U.pdb) with PCA bound (magenta).
Metal Dependence
In metaloenzymes, the relative promiscuity of active site ligands to bind a variety of different metal ions of the same oxidation state makes it necessary to carefully determine which metal(s) are able to promote catalysis. In a variety of enzyme superfamilies, like the amidohydrolase superfamily, the number of metal ions necessary for catalysis and also the identity of the metal ion(s) can be an important diagnostic for membership in an enzyme subtype, which can be informative when trying to determine the reaction catalyzed.59,60 Since nonheme iron extradiol dioxygenases have been observed to be catalytically active with a variety of metal ions, most commonly Fe2+, but also with other divalent metals such as Mn2+, Co2+, Cu2+, Zn2+, or Ni2+, we sought to determine the metal ion dependency of LigAB.20,25,57,61–63 Since many nonheme iron dioxygenases are expressed and purified aerobically, and subsequently dosed with an iron(II) salt (or other divalent metal ions) to reconstitute the enzyme activity, LigAB was grown and purified in the absence of any exogenous metal. Despite the absence of any metals in the growth media or lysis buffer, the enzyme copurified with metal under both aerobic and anaerobic conditions, in a catalytically active state.
In order to obtain apo enzyme, LigAB was incubated alternately with EDTA and bipy to remove the liganded metal. Incubation for 1 h with either bipy or EDTA, as described by Diaz,25 was insufficient to completely eliminate LigAB activity suggesting that LigAB has a more tightly bound and/or less accessible iron center than other dioxygenases (LigAB maintained 65% activity upon incubation with EDTA and 50% activity upon incubation with bipy, after 1 h of incubation at pH 7.5). In the case of LigAB, complete loss of activity was only achieved by incubation of the enzyme with 2 mM bipy for 24 h. Incubation of LigAB with EDTA (1 or 2 mM) for the same time frame yielded the enzyme, which maintained 35% activity. Recovery of activity for the apo enzyme (generated with 24 h incubation with bipy) was only observed with a 1 h incubation of Fe2+ under anaerobic conditions; however, only 5% of the native activity was recovered. Longer incubation times with Fe2+ had no positive effect on the recovery of activity. Additionally, no recovery of activity was observed with any of the other metal ions. We speculate that based upon our observation that LigAB activity is lost over time and that the long incubation times needed to strip the enzyme of metal leads to a protein solution that is partially dead, which would explain the only partial recovery (5%) of enzyme activity. Interestingly, LigAB stripped of 50% activity (generated by incubation with 1 mM EDTA for 2 h) and reconstituted with 2 mM Cu2+ for 1 h lost activity further to 8% of that of the native enzyme. Incubation of a partially active enzyme with either Co2+ or Mn2+ salts did not have this added effect of activity loss. This result suggests that, while not able to form an active complex, Cu2+ may be able to out-compete Fe2+ for the metal binding ligands in the active site. Inhibition by the addition of non-native metal ions (Co2+, Mn2+, Ni2+, or Cu2+) has been observed in previous studies of other 4,5-PCDs.48,64
Iron Dissociation
To investigate the potential contribution of Fe2+ dissociation to the decreases in rate observed in the pH rate profile, the iron content of enzyme exchanged and incubated at pH 6.5, 7.5, and 10 was analyzed (Table 3). At these pH values, the iron to enzyme ratios were found to be 0:1, 1:5, and 1:1, respectively. (Note: the enzyme assayed directly after the desalting column was found to have an iron to enzyme ratio of 4:1, suggesting that some free or adventitiously bound iron is also present, leading to the routine use of the Bradford assay to determine the enzyme concentration.) The results of the pH incubation and buffer exchange suggest that at low pH, a decrease in catalytic rate, in addition to the rate effect from a protonated catalytic base residue, may be caused by one or more iron binding residues being protonated leading to an increased rate of dissociation of Fe2+ from the active site. This is consistent with the slope of 1.8 observed in the acidic arm of the plot of pH vs log(kcat). The slope greater than 1 suggests additional mechanisms of inactivation beyond the protonation of a catalytic base.
Table 3.
pH Dependent Binding of Fe by LigAB
| concentration (μM) |
ratio |
||
|---|---|---|---|
| condition | LigAB | Fe | Fe/LigAB |
| desalted | 3.4 ± 0.6 | 14.92 ± 0.5 | 4.4 |
| pH 6 | 3.5 ± 0.3 | 0 | 0 |
| pH 7.5 | 5.5 ± 0.5 | 1.0 ± 0.2 | 0.2 |
| pH 10 | 3.9 ± 0.2 | 4.1 ± 0.3 | 1.04 |
The 1:1 ratio of iron to enzyme found at pH 10 suggests that the iron binding residues remain deprotonated and able to bind Fe at higher pH. At this high pH, oxidation of Fe2+ to Fe3+ occurs much more rapidly than at a pH of 6 or even 7.5,65 and so, the Fe remaining bound to LigAB at this pH is likely in the Fe3+ state. The reduced kcat at high pH observed in the pH rate profile may then have a contribution from iron oxidation in addition to the rate reduction caused by a deprotonated catalytic acid residue. The slope corresponding to the basic arm of the pH rate profile is less than 1, and so, the contributions of either mechanism of deactivation are unclear. Additionally, the 1:1 Fe/enzyme ratio at pH 10, in comparison to the ratio of 1:4 observed at pH 7.5, suggests that the inactive Fe3+ state is more tightly bound to LigAB than Fe2+. While the nonstoichiometric occupancy of Fe at pH 7.5 may seem surprising, the buffer exchanging process likely reduces the concentration of Fe2+ in the solution below LigAB’s Kd for Fe2+ allowing the ion to dissociate more freely. At pH 6, no residual iron was observed after buffer exchanging, suggesting that iron, when not bound to a complex larger than 10 kDa, is capable of being reduced to undetectable levels by this method. Thus, at pH 7.5 the remaining iron is likely enzyme bound, and the reduced iron content is due to a Kd for Fe2+ that is higher than the residual iron concentration after complete buffer exchange. To the best of our knowledge, the pH related dissociation of Fe from a dioxygenase of any type has not previously been measured; however, Takemori et al. previously analyzed the pH dependence of holoenzyme reconstitution for the catechol 2,3-dioxygenase from Pseudomonas putida (XylE) and observed that enzyme reactivation occurred most quickly at pH 6.0 ± 0.2, as compared with the exchange rates observed at pH 6.6.66 We speculate that these differences in enzyme coordination behavior could be related to membership in different dioxygenase superfamilies, among other possible reasons. Further, Kd values for Fe2+ at constant pH are rare, and values for Fe3+ have not been found in literature searches. The mononuclear nonheme dioxygenases cysteine dioxygenase (CDO) from rat and β-diketone cleaving enzyme (Dke1) from Acinetobacter johnsonii were found to have Kd values for Fe2+ of 5.2 μM and 5.8 μM, respectively.67,68 While these values may not be directly applicable to LigAB since CDO and Dke1 are cupin dioxygenases with a 3-His iron binding motif in contrast with the 2-His-1-Glu motif found in LigAB, they provide a value for reference that is not commonly measured for dioxygenase enzymes. While an iron Kd value for LigAB is not elucidated through this experiment, thawed enzyme buffer exchanged via the desalting gel column, has an [Fe2+]Tot = 14.9 ± 0.5 μM (higher than the Kd values determined for CDO and Dke1) suggesting that LigAB should be fully occupied assuming a similar Kd.
Product Identification
The product of the dioxygenation of PCA by LigAB, 4-carboxy-2-hydroxymuconate-6-semialdehyde (CHMS), was identified by UV–vis spectroscopy and 1H NMR, aided by a secondary conversion of CHMS to 2,4-lutidinic acid. Since CHMS is involved in an extensive equilibrium of structural isomeric forms, its isolation is difficult. The UV–vis spectra of CHMS under multiple pH conditions have been previously reported, and the spectra recorded here (Figure 4A) are consistent with previous data showing a λmax of 298 nm and a second λmax of 410 nm at pH 7.5.17,47,48,69,70 Additionally, the spectra of PCA at pH 7.5 is reported here. The spectral changes observed upon reducing the pH to 1 or increasing the pH to 14 are also consistent with previous observations of CHMS (Figure 4B). The conversion of CHMS to 2,4-lutidinic acid also provides a diagnostic UV–vis spectral change that is consistent with the conversion to this product. In addition to the UV–vis spectral changes, the reaction was monitored by 1H NMR spectroscopy (Figure S1, Supporting Information). The disappearance of PCA can be clearly observed on the NMR time scale; however, peaks corresponding to product formation were not observed in the 1H NMR spectrum of the reaction at completion due to the equilibrium of multiple product isomers. Addition of NH4OH to convert CHMS to 2,4-lutidinic acid results in the appearance of three aromatic proton signals consistent with a spectrum of 2,4-lutidinic acid from the Sigma-Aldrich spectral library.71
Figure 4.
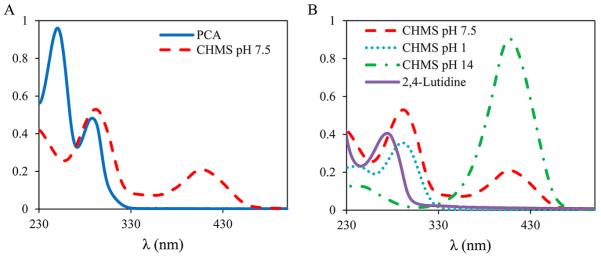
UV–vis spectra of LigAB catalysis with PCA. (A) Spectra of PCA (solid line) and CHMS at reaction completion (dashed line) at pH 7.5. (B) pH dependent UV–vis spectral changes of CHMS (pH 7.5, dashed line; pH 2, dotted line; pH 14, dashed-dotted line) and the spectrum of 2,4-lutidinic acid (solid) formed upon the addition of NH4OH.
Steady-State Kinetics
Although Masai and co-workers previously established the relative rates of reaction for LigAB with PCA, GA, and 3OMG, these experiments were conducted under less than optimal conditions with aerobically purified enzyme and a pH optimum determined for a different, yet related, dioxygenase.39 Also, the kinetic parameters of kcat and kcat/Km were not determined. Results of the steady-state kinetics for LigAB with PCA, gallate, and 3OMG obtained in this study are shown in Figure 5. The steady-state kinetic parameters are listed in Table 1. The values of for these substrates are consistent with those previously determined; however, here we have found that 3OMG binds much less tightly than previously observed: 2319 ± 316 μM compared to 937 ± 18 μM, respectively. The values of determined here for these substrates demonstrate a similar relative ratio of activities to the previously determined specific activity with PCA having the highest turnover, followed by GA, and then 3OMG. The C5-substituent effect on reactivity and substrate specificity is quite apparent from this series, likely due to the steric clash with Phe103α (Figure 3); however, not all steric and electronic contributions to substrate specificity are evident from these data. Significantly, as described above, the LigAB purified anaerobically in this study is greater than 10-fold more active than the enzyme expressed from the same source but purified aerobically. While the previously reported results for LigAB are significant and inspired our work with this enzyme, anaerobic purification techniques allow for a more accurate characterization of LigAB.
Figure 5.
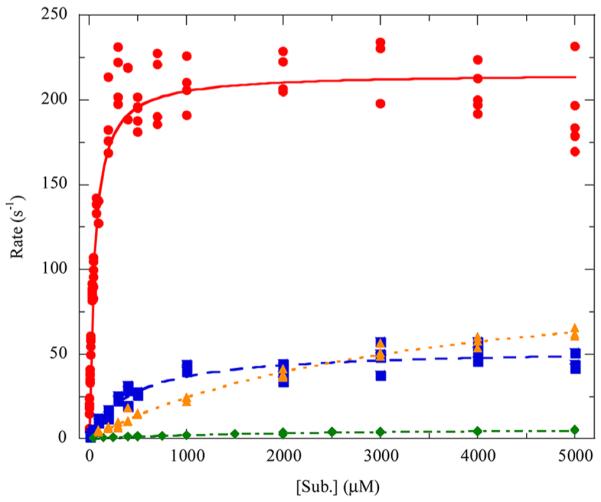
Steady-state kinetics of LigAB with PCA and substrate analogues: (red circle) PCA, (blue square) gallate, (green diamond) 3OMG, (orange triangle) DHBAm. The dependence of the initial rates on the organic substrate concentration was determined in air-saturated buffer (250–275 μM O2, pH 7.5, 25 °C), and the curves are fit to the Michaelis–Menten equation.
Table 1.
LigAB Steady-State Kinetic Parameters Measured with PCA and Analogous Substrates in Air-Saturated Buffer
| substrate | (μM) | (s−1) | (M−1 s−1) | partition ratioa | (s−1) |
|---|---|---|---|---|---|
| PCA | 51 ± 4 | 216 ± 3 | 4.26 × 106 | ~18000 | 0.012c |
| gallate | 441 ± 55 | 53 ± 2 | 1.21 × 105 | ~2500 | 0.021b |
| 3OMG | 2319 ± 316 | 7.3 ± 0.5 | 3.13 × 103 | ~5600 | 0.0013c |
| DHBAm | 3263 ± 327 | 104 ± 5 | 3.20 × 104 | ~3200 | 0.033d |
Estimated from the and (eq 5).
Calculated from a fit of eq 4 to js values determined from the fitting of progress curves (eq 3) at different concentrations of gallate.
Concentration dependent inactivation was not observed for PCA or 3OMG. was calculated as an average of the js values determined from the fitting of eq 3 to progress curves at different substrate concentrations.
The steady-state kinetic parameters determined for LigAB with the native substrate, PCA, as compared to other dioxygenases with their native substrates are listed in Table S1, Supporting Information. Dioxygenases of all types (I-VOC, II-PCAD, and III-cupin) are quite proficient enzymes, with kcat/Km values ranging from 105 – 108 M−1s−1,5,8,11,12,16,18,33,55,63,72–77 and LigAB (4.3 × 106 M−1s−1) fits well within this trend. Additionally, the kinetic parameters of LigAB coincide well with those determined for 2,3-PCD, MhpB, and 4,5-PCD (C. testosteroni Pt-L5), the only other Type II dioxygenases with a complete set of steady-state kinetic parameters determined.16,18,33 The KmO2 for LigAB (162 ± 16 μM), determined from Figure 6, was also found to be in the range of those determined for other extradiol dioxygenases (Table S1, Supporting Information) and is nearly identical to that determined for the Type II dioxygenase 2,3-PCD (142 ± 14 μM).
Figure 6.
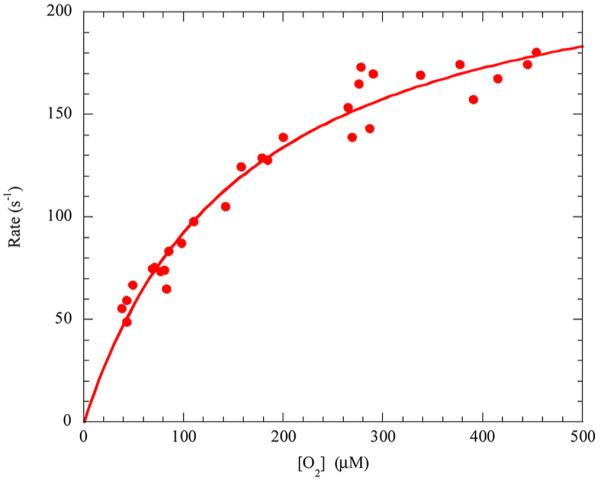
Steady-state kinetics of LigAB with varying O2 concentrations. Dependence of the initial rates on the O2 concentration was measured at high substrate concentration (1000 μM PCA, pH 7.5, 25 °C), and the curve is a fit of the Michaelis–Menten equation to the data.
Substrate Specificity and Inhibition
Intradiol and extradiol dioxygenases have long been known to exist for the preferential cleavage of a wide variety of substrates. Substrate classes include catechols, protocatechuates, gentisates, 2-aminophenols, quinones, aromatic amino acids, and even halogenated aromatics.7 Dioxygenase specificities range from the highly specific to the quite promiscuous. LigAB has been shown here and in previous studies to be a promiscuous enzyme capable of efficient catalytic turnover of several substrates in the LDAC catabolic pathway of S. paucimobilis (PCA, GA, and 3OMG). The promiscuity of LigAB with GA and 3OMG contrasts with another Type II dioxygenase, gallate dioxygenase (GDO) from Pseudomonas putida KT2440 (39% sequence identity to LigB). GDO was found to have no oxygen consumption activity with PCA and is believed to be a gallate specific dioxygenase.25 Additionally, several PCA 4,5-dioxygenases (4,5-PCD) have been identified for which substrate specificity, substrate uptake by live cells, and the inhibition effect of other substrates have been studied; however, reaction kinetics remain largely unstudied for these 4,5-PCDs17,29,46,48,61,64,78–80 with the exception of the 4,5-PCD from Comamonas testosteroni Pt-L5 (formerly Pseudomonas testosteroni Pt-L5) (Table S1, Supporting Information).16 Despite the evidence of promiscuity in LigAB, the range of accepted substrates was never explored beyond the PCA analogues identified from within its pathway. Here, we have tested multiple classes of substrates analogous to the basic catechol scaffold with substitutions primarily at C1, C5, and functional modification of the catecholic hydroxyl groups (Table 2 and Figure S2, Supporting Information). In addition to the three known substrates, LigAB was demonstrated to utilize four new substrates: 3,4-dihydroxybenzamide (DHBAm), homoprotocatechuate (HPCA), catechol, and 3,4-dihydroxybenzonitrile. DHBAm was the most active of the newly identified substrates, having steady-state kinetic parameters similar to those found for the previously identified substrates (Table 1). LigAB was found to have the following trend for specificity with highly active substrates: PCA > GA > DHBAm > 3OMG. LigAB was found to turn over DHBAm more quickly than GA but with a larger than that of 3OMG. Although DHBAm is a good substrate, O2 consumption kinetic traces displayed very fast deactivation of LigAB, and in most cases, only ~10 s of the data was used to obtain a rate after the initiation of the reaction by the addition of LigAB. The rate of inactivation (js) during the turnover of DHBAm shows substrate concentration dependence; however, DHBAm is more inactivating at lower substrate concentrations than at high substrate concentrations and so cannot be fit using eq 4. Assays, performed in the presence of the DHBAm product formed in situ, show enhanced inactivation over assays performed with no product initially present. These observations are consistent with the turnover of DHBAm leading to a product that competes with substrate to bind LigAB and resulting in an inactivated enzyme by a dead-end product inhibition mechanism.
Table 2.
Library of Alternative Substrates: Observed Activity, Inhibition, and Calculated Oxidation Potentials for Analogues of PCA
| substrate | activity observed | % activitya | % inhibitionb | Eabsolute oxidation (eV) |
|---|---|---|---|---|
| (HPCA) Homprotocatechuate | Y | 0.2 | 5.40 | |
| 4-methylcatechol | N | 28 | 5.71 | |
| PCA | Y | 100 | 5.82 | |
| catechol | Y | 0.17 | 5.88 | |
| 3,4-dihydroxybenzamide | Y | 33 | 6.11 | |
| 3,4-dihydroxybenzaldehyde | N | 6.19 | ||
| 3,4-dihydroxybenzonitrile | Y | 0.04 | 6.30 | |
| 4-nitrocatechol | N | 31 | 6.46 | |
| gallate | Y | 24 | ||
| 3-O-methylgallate | Y | 3 | ||
| 2,3,4-trihydroxybenzoic acid | N | 95 | ||
| 2,3-dihydroxybenzoic acid | N | 36 | ||
| 3-methoxycatechol | N | 26 |
Measured with 5 mM substrate in air-saturated 50 mM Tris buffer at 25 °C, and pH 7.5. Calculated as a percentage of the activity of LigAB with 5 mM PCA.
Measured with 500 μM PCA and 1 mM inhibitor in air-saturated 50 mM Tris buffer at 25 °C and pH 7.5. Calculated as a percentage of the activity of LigAB with 500 μM PCA with no inhibitor present.
HPCA, 3,4-dihydroxybenzonitrile, and catechol were poorer substrates, displaying only a small fraction of the activity (less than 1%) as seen for LigAB with PCA (Table 2). Interestingly, a number of molecules were identified that were not substrates but showed competitive inhibition of the LigAB reaction with PCA, including 4-nitrocatechol and 4-methylcatechol (Table 2). All of these molecules are analogues of PCA, where the C1 substituent (using the PCA numbering scheme) is varied. To explain these observations, we initially performed calculations to determine if the electronic influence of the C1 substituent was correlated with the catalytic efficiency, using Gaussian 09 (Table 2). From the data, it is clear that a range of organic substrate oxidation potentials (5.40–6.30 eV) is tolerated for accepted substrates; however, the incorporation of O2 is most facile when the oxidation potential of the organic substrate is 5.82–6.11 eV, as for PCA and 3,4-dihydroxybenamide. 4-Methyl catechol and catechol, with oxidation potentials of 5.71 and 5.88 eV, respectively, are close to or within the facile range, yet oxygen consumption is not seen or is very low. Some molecules on the fringe of the oxidation potential range such as HPCA (5.40 eV) and 3,4-dihydroxybenzonitrile (6.30 eV) are tolerated as substrates by LigAB, but the activity is severely diminished. 4-Nitrocatechol with the highest oxidation potential of the C1 PCA analogues is not a substrate but is rather an inhibitor, suggesting that compounds that have a higher potential than 6.30 eV are not electronically accessible by LigAB.
Since a clear correlation between catalytic efficiency and oxidation potential for the library of C1 analogues of PCA could not be established for all of the compounds in this series, we then evaluated the likely binding interactions of the various molecules with the LigAB active site residues. We noted that the two worst substrates pyrocatechol and 3,4-dihydroxybenzonitrile as well as one of the inhibitors 4-methylcatechol would all be unable to have favorable hydrogen bonding interactions with the NH2 group of Asn270β, which is predicted to hydrogen bond to the carboxylate anion of PCA (Figure 3). Sterics could also explain the relatively low catalytic efficiency of LigAB with HPCA (a C1 substrate analogue which moves the carboxylate further into the pocket by the addition of a methylene) and the absence of catalysis or inhibition with methyl-3,4-dihydroxybenzoate and caffeic acid, which are likely just too big to fit into the binding site, perhaps due to steric conflict with Thr271β (Figure 3). Conversely, the inhibitory nature of 4-nitrocatechol, which is isosteric with PCA and could accept hydrogen bonds from the NH2 group of Asn270β, is likely dominated by electronic effects. In other systems, it has been observed that substrate/inhibitor scope is generally smaller than that observed for LigAB. In the 4,5-PCD from C. testosteroni, sulfonylcatechol (a C1 sulfonyl substitution of the PCA carboxylic acid) was the only known substrate analogue with a C1 modification to be turned over.16 Additionally, catechol (C1-H) and 4-methylcatechol (C1-Me) were observed to be inhibitors of the 4,5-PCD of Pseudomonas sp.61 but nonsubstrates of the 4,5-PCD of C. testosteroni.48,64
Interestingly, 3,4-dihydroxybenzaldehyde was neither a substrate nor an inhibitor of LigAB despite the structural similarity to PCA and its seemingly acceptable oxidation potential. The inability to turnover 3,4-dihydroxybenzaldehyde has been observed in 4,5-PCDs from Pseudomonas sp.61 and C. testosteroni;64 however, in these instances 3,4-dihydroxybenzaldehyde does act as an inhibitor. Examination of other aldehyde containing compounds including vanillin revealed that none of these compounds were substrates or inhibitors for LigAB despite their structural similarity to viable LigAB substrates. Knowing that vanillin is the substrate of the enzyme two ahead of LigAB in the S. paucimobilis LDAC catabolic pathway, we hypothesize that perhaps LigAB has evolved to preclude the binding of aldehydes in the active site.
Taken together, the results of the library suggest that there are both structural and electronic requirements which are necessary for organic substrate activity with LigAB. The first requirement is an electron withdrawing group in the C1 position such that the oxidation potential of the substrate is 5.40–6.30 eV (~5.8 eV being the sweet spot). Second, a 3,4 or a 4,5 diol is required. Functional modification of hydroxyls in these positions, such that hydroxyls ortho to one another are not present, eliminates activity. For example, 3OMG remains a viable substrate because the 4,5-diol motif is conserved, yet vanillate and syringate, which have a methylether rather than a hydroxyl ortho to the C4-hydroxyl, displayed no activity. Mutagenesis of various active site residues will be pursued to test for possible gain of function for the various substrates that are believed to be electronically allowed but sterically disfavored.
Mechanism Based Inactivation in Steady-State Kinetics
As has been previously reported for other dioxygenase enzymes,5,7,21,45,81 LigAB is susceptible to inactivation during catalytic turnover under air-saturated conditions. While inactivation of a Type II dioxygenase has not previously been reported, the rates of inactivation for LigAB (Table 1) are similar to those reported for the well characterized Type I dioxygenases 2,3-dihydroxybiphenyl 1,2-dioxygenase (DHBD)45 and 2,6-dichlorohydroquinone 1,2-dioxygenase (PcpA).5 Inactivation was seen with each of the four most preferred substrates (PCA, GA, 3OMG, and DHBAm); however, only gallate showed substrate concentration dependent inactivation rates that could be fit to eq 4, resulting in a of 0.021 s−1. Inactivation during the turnover of PCA (0.012 s−1) and 3OMG (0.0013 s−1) was independent of substrate concentration within the experimental concentration range. While there has been some debate over how mechanism based inactivation occurs, the strongest evidence suggests that the active site Fe(II) becomes irreversibly oxidized to Fe(III) due to the release of superoxide.45 Superoxide release can occur before or after organic substrate binding, though it tends to occur more slowly in the free enzyme due to a lower affinity (higher KmO2) for oxygen in the absence of substrate as was observed for DHBD.45,55 Additionally, while inactivation tends to be more pronounced in the presence of poor substrates, inactivation has also been observed in the presence of the preferred substrate(s).45 While the data presented here does not strictly follow this trend (Table 1), the rate of inactivation for the three most preferred substrates (PCA, gallate, and DHBAm) is consistent with this observation with PCA being the least inactivating and DHBAm being the most inactivating of these substrates. 3OMG, despite being the poorest of the preferred substrates, displayed the least inactivation. The increased sterics of 3OMG likely prevent this substrate from properly binding; however, once bound in a geometry suitable for catalysis, 3OMG may be more active toward C–O bond formation with superoxide thus preventing premature release and slowing the oxidation of the LigAB active site iron.
CONCLUSIONS
Steady-state kinetic studies have demonstrated that LigAB is highly specific for catecholic substrates with an electron withdrawing/hydrogen bond accepting substituent on C1. While LigAB was previously known to have promiscuous activity, this promiscuity has now been expanded beyond molecules of the LDAC degradation pathway of S. paucimobilis SYK-6. Analysis of the structure and oxidation potentials of the accepted substrates suggests that LigAB is highly specific for diol containing substrates with an oxidation potential between 5.40 and 6.30 eV. Additionally, steric and hydrogen bonding interactions by active site residues (such as Phe103α, Thr271β, or Ser269β) likely impart some of LigAB’s substrate specificity, modulating the catalysis of accepted substrates and preventing some substrate-like molecules that meet these parameters from being acceptable substrates. Mutagenesis of LigAB will likely reveal the relative importance of these residues in controlling substrate specificity. LigAB, like other dioxygenases, is subject to mechanistic inactivation, although not all substrates show a concentration dependence on the rate of inactivation. In the case of the newly discovered substrate DHBAm, dead-end product inhibition dominates the mechanisms of inactivation. While inhibition of LigAB is not directly applicable to our research goals, a number of competitive inhibitors were also identified in this study. These results could provide insight toward identifying lead compounds in a system where dioxygenase inhibition is desired. Furthermore, the pH rate studies suggest this enzyme would have either two catalytic bases or that Fe(II) coordination is highly pH sensitive. Further mechanistic analyses, including mutagenesis, might shed light on these results and allow comparisons to mechanisms proposed for dioxygenases from other enzyme superfamilies. In total, these results make LigAB the first Type II dioxygenase to be fully characterized both structurally and kinetically and solidify its position as the defining member of the Type II dioxygenase superfamily.
Supplementary Material
ACKNOWLEDGMENTS
We thank Professor Lindsay D. Eltis and Jenna K. Capyk from the University of British Columbia for their advice and training in regard to anaerobic protein purification methods and the use of an O2-sensitive electrode. We also thank Professor Albert J. Fry from Wesleyan University for determining the oxidation potentials of the PCA substrate analogues.
Funding This work was supported in part by grant DE-SC0005267 from the Department of Energy Office of Biological and Environmental Research (DOE BER) and the N.I.H. Doctoral Studies in Molecular Biophysics Training Grant 2T32GM008271-24. We also thank the Wesleyan University Chemistry Department for financial support.
Footnotes
Supporting Information Steady-state kinetic parameters for Type I, II, and III extradiol dioxygenases, NMR characterization of CHMS, and UV–vis spectra of the PCA LigAB reaction. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes The authors declare no competing financial interest.
REFERENCES
- (1).Gosselink RJA, de Jong E, Guran B, Abächerli A. Co-ordination network for lignin - standardisation, production and applications adapted to market requirements (EUROLIGNIN) Ind. Crops Prod. 2004;20:121–129. [Google Scholar]
- (2).Vicuña R. Bacterial degradation of lignin. Enzyme Microb. Technol. 1988;10:646–655. [Google Scholar]
- (3).Bugg TDH, Ahmad M, Hardiman EM, Singh R. The emerging role for bacteria in lignin degradation and bio-product formation. Curr. Opin. Biotechnol. 2010;22:1–7. doi: 10.1016/j.copbio.2010.10.009. [DOI] [PubMed] [Google Scholar]
- (4).Katayama Y, Nishikawa S, Murayama A, Yamasaki M, Morohoshi N, Haraguchi T. The metabolism of biphenyl structures in lignin by the soil bacterium (Pseudomonas paucimobilis SYK-6) FEBS Lett. 1988;233:129–133. [Google Scholar]
- (5).Machonkin TE, Doerner AE. Substrate specificity of Sphingobium chlorophenolicum 2,6-dichlorohydroquinone 1,2-dioxygenase. Biochemistry. 2011;50:8899–8913. doi: 10.1021/bi200855m. [DOI] [PubMed] [Google Scholar]
- (6).Vaillancourt FH, Bolin JT, Eltis LD. The ins and outs of ring-cleaving dioxygenases. Crit. Rev. Biochem. Mol. Biol. 2006;41:241–267. doi: 10.1080/10409230600817422. [DOI] [PubMed] [Google Scholar]
- (7).Vaillancourt FH, Bolin JT, Eltis LD. Ring-Cleavage Dioxygenases. Pseudomonas. 2004;3:359–395. [Google Scholar]
- (8).Siani L, Viggiani A, Notomista E, Pezzella A, Di Donato A. The role of residue Thr249 in modulating the catalytic efficiency and substrate specificity of catechol-2,3-dioxygenase from Pseudomonas stutzeri OX1. FEBS J. 2006;273:2963–2976. doi: 10.1111/j.1742-4658.2006.05307.x. [DOI] [PubMed] [Google Scholar]
- (9).Kovaleva EG, Lipscomb JD. Crystal structures of Fe2+ dioxygenase superoxo, alkylperoxo, and bound product intermediates. Science. 2007;316:453–457. doi: 10.1126/science.1134697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Mbughuni MM, Chakrabarti M, Hayden JA, Meier KK, Dalluge JJ, Hendrich MP, Muenck E, Lipscomb JD. Oxy intermediates of homoprotocatechuate 2,3-dioxygenase: Facile electron transfer between substrates. Biochemistry. 2011;50:10262–10274. doi: 10.1021/bi201436n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Schaab MR, Barney BM, Francisco WA. Kinetic and spectroscopic studies on the quercetin 2,3-dioxygenase from Bacillus subtilis. Biochemistry. 2006;45:1009–1016. doi: 10.1021/bi051571c. [DOI] [PubMed] [Google Scholar]
- (12).Veldhuizen EJA, Vaillancourt FH, Whiting CJ, Hsiao MMY, Gingras G, Xiao YF, Tanguay RM, Boukouvalas J, Eltis LD. Steady-state kinetics and inhibition of anaerobically purified human homogentisate 1,2-dioxygenase. Biochem. J. 2005;386:305–314. doi: 10.1042/BJ20041370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Eltis LD, Bolin JT. Evolutionary relationships among extradiol dioxygenases. J. Bacteriol. 1996;178:5930–5937. doi: 10.1128/jb.178.20.5930-5937.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Spence EL, Kawamukai M, Sanvoisin J, Braven H, Bugg TDH. Catechol dioxygenases from Escherichia coli (MhpB) and Alcaligenes eutrophus (MpcI): Sequence analysis and biochemical properties of a third family of extradiol dioxygenases. J. Bacteriol. 1996;178:5249–5256. doi: 10.1128/jb.178.17.5249-5256.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kabisch M, Fortnagel P. Nucleotide sequence of metapyrocatechase I (catechol 2,3-oxygenase I) gene mpcI from Alcaligenes eutrophus JMP222. Nucleic Acids Res. 1990;18:3405–3405. doi: 10.1093/nar/18.11.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Arciero DM, Orville AM, Lipscomb JD. Protocatechuate 4,5-dioxygenase from Pseudomonas testosteroni. Methods Enzymol. 1990;188:89–95. doi: 10.1016/0076-6879(90)88017-5. [DOI] [PubMed] [Google Scholar]
- (17).Mampel J, Providenti MA, Cook AM. Protocatechuate 4,5-dioxygenase from Comamonas testosteroni T-2: biochemical and molecular properties of a new subgroup within class III of extradiol dioxygenases. Arch. Microbiol. 2005;183:130–139. doi: 10.1007/s00203-004-0755-4. [DOI] [PubMed] [Google Scholar]
- (18).Wolgel SA, Dege JE, Perkins-Olson PE, Juarez-Garcia CH, Crawford RL, Munck E, Lipscomb JD. Purification and characterization of protocatechuate 2,3-dioxygenase from Bacillus macerans: A new extradiol catecholic dioxygenase. J. Bacteriol. 1993;175:4414–4426. doi: 10.1128/jb.175.14.4414-4426.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kasai D, Fujinami T, Abe T, Mase K, Katayama Y, Fukuda M, Masai E. Uncovering the protocatechuate 2,3-Cleavage pathway genes. J. Bacteriol. 2009;191:6758–6768. doi: 10.1128/JB.00840-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lendenmann U, Spain JC. 2-Aminophenol 1,6-dioxygenase: A novel aromatic ring cleavage enzyme purified from Pseudomonas pseudoalcaligenes JS45. J. Bacteriol. 1996;178:6227–6232. doi: 10.1128/jb.178.21.6227-6232.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Davis JK, He ZQ, Somerville CC, Spain JC. Genetic and biochemical comparison of 2-aminophenol 1,6-dioxygenase of Pseudomonas pseudoalcaligenes JS45 to meta-cleavage dioxygenases: divergent evolution of 2-aminophenol meta-cleavage pathway. Arch. Microbiol. 1999;172:330–339. doi: 10.1007/s002030050787. [DOI] [PubMed] [Google Scholar]
- (22).Sato S, Ouchiyama N, Kimura T, Nojiri H, Yamane H, Omori T. Cloning of genes involved in carbazole degradation of Pseudomonas sp. strain CA10: Nucleotide sequences of genes and characterization of meta-cleavage enzymes and hydrolase. J. Bacteriol. 1997;179:4841–4849. doi: 10.1128/jb.179.15.4841-4849.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Larentis AL, Almeida RV, Rossle SC, Cardoso AM, Almeida WI, Bisch PM, Alves TLM, Martins OB. Expression and homology modelling of 2 ′-aminobiphenyl-2,3-diol-1,2-dioxygenase from Pseudomonas stutzeri carbazole degradation pathway. Cell Biochem. Biophys. 2006;44:530–538. doi: 10.1385/CBB:44:3:530. [DOI] [PubMed] [Google Scholar]
- (24).Laurie AD, Lloyd-Jones G. The phn genes of Burkholderia sp. strain RP007 constitute a divergent gene cluster for polycyclic aromatic hydrocarbon catabolism. J. Bacteriol. 1999;181:531–540. doi: 10.1128/jb.181.2.531-540.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Nogales J, Canales A, Jimenez-Barbero J, Garcia JL, Diaz E. Molecular characterization of the gallate dioxygenase from Pseudomonas putida KT2440 - The prototype of a new subgroup of extradiol dioxygenases. J. Biol. Chem. 2005;280:35382–35390. doi: 10.1074/jbc.M502585200. [DOI] [PubMed] [Google Scholar]
- (26).Roper DI, Cooper RA. Subcloning and nucleotide sequence of the 3,4-dihydroxyphenylacetate (homoprotocatechuate) 2,3-dioxygenase gene from Escherichia coli C. FEBS Lett. 1990;275:53–57. doi: 10.1016/0014-5793(90)81437-s. [DOI] [PubMed] [Google Scholar]
- (27).Barnes MR, Duetz WA, Williams PA. A 3-(3-hydroxyphenyl)propionic acid catabolic pathway in Rhodococcus globerulus PWD1: Cloning and characterization of the hpp operon. J. Bacteriol. 1997;179:6145–6153. doi: 10.1128/jb.179.19.6145-6153.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kulakov LA, Delcroix VA, Larkin MJ, Ksenzenko VN, Kulakova AN. Cloning of new Rhodococcus extradiol dioxygenase genes and study of their distribution in different Rhodococcus strains. Microbiology (Reading, U.K.) 1998;144:955–963. doi: 10.1099/00221287-144-4-955. [DOI] [PubMed] [Google Scholar]
- (29).Maruyama K, Shibayama T, Ichikawa A, Sakou Y, Yamada S, Sugisaki H. Cloning and characterization of the genes encoding enzymes for the protocatechuate meta-degradation pathway of Pseudomonas ochraceae NGJ1. Biosci., Biotechnol., Biochem. 2004;68:1434–1441. doi: 10.1271/bbb.68.1434. [DOI] [PubMed] [Google Scholar]
- (30).Eaton RW. Plasmid-encoded phthalate catabolic pathway in Arthrobacter keyseri 12B. J. Bacteriol. 2001;183:3689–3703. doi: 10.1128/JB.183.12.3689-3703.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Sugimoto K, Senda T, Aoshima H, Masai E, Fukuda M, Mitsui Y. Crystal structure of an aromatic ring opening dioxygenase LigAB, a protocatechuate 4,5-dioxygenase, under aerobic conditions. Structure (London) 1999;7:953–965. doi: 10.1016/s0969-2126(99)80122-1. [DOI] [PubMed] [Google Scholar]
- (32).Mendel S, Arndt A, Bugg TDH. Acid-base catalysis in the extradiol catechol dioxygenase reaction mechanism: Site-directed mutagenesis of His-115 and His-179 in Escherichia coli 2,3-dihydroxyphenylpropionate 1,2-dioxygenase (MhpB) Biochemistry. 2004;43:13390–13396. doi: 10.1021/bi048518t. [DOI] [PubMed] [Google Scholar]
- (33).Schlosrich J, Eley KL, Crowley PJ, Bugg TDH. Directed evolution of non-heme-iron-dependent extradiol catechol dioxygenase: Identification of mutants with intradiol oxidative cleavage activity. ChemBioChem. 2006;7:1899–1908. doi: 10.1002/cbic.200600296. [DOI] [PubMed] [Google Scholar]
- (34).Katayama Y, Nishikawa S, Nakamura M, Yano K, Yamasaki M, Morohoshi N, Haraguchi T. Cloning and expression of Pseudomonas paucimobilis SYK-6 genes involved in the degradation of vanillate and protocatechuate in P. putida. Mokuzai Gakkaishi. 1987;33:77–79. [Google Scholar]
- (35).Masai E, Katayama Y, Nishikawa S, Yamasaki M, Morohoshi N, Haraguchi T. Detection and localization of a new enzyme catalyzing the β-aryl ether cleavage in the soil bacterium (Pseudomonas paucimobilis SYK-6) FEBS Lett. 1989;249:348–352. doi: 10.1016/0014-5793(89)80656-8. [DOI] [PubMed] [Google Scholar]
- (36).Masai E, Katayama Y, Nishikawa S, Fukuda M. Characterization of Sphingomonas paucimobilis SYK-6 genes involved in degredation of lignin-related compounds. J. Ind. Microbiol. Biotechnol. 1999;23:364–373. doi: 10.1038/sj.jim.2900747. [DOI] [PubMed] [Google Scholar]
- (37).Sugimoto K, Aoshima H, Senda T, Masai E, Fukuda M, Mitsui Y. Purification and crystallization of a protocatechuate 4,5-dioxygenase LigAB from Sphingomonas paucimobilis SYK-6. Protein Pept. Lett. 1999;6:55–58. [Google Scholar]
- (38).Kasai D, Masai E, Miyauchi K, Katayama Y, Fukuda M. Characterization of the 3-O-methylgallate dioxygenase gene and evidence of multiple 3-O-methylgallate catabolic pathways in Sphingomonas paucimobilis SYK-6. J. Bacteriol. 2004;186:4951–4959. doi: 10.1128/JB.186.15.4951-4959.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kasai D, Masai E, Miyauchi K, Katayama Y, Fukuda M. Characterization of the gallate dioxygenase gene: Three distinct ring cleavage dioxygenases are involved in syringate degradation by Sphingomonas paucimobilis SYK-6. J. Bacteriol. 2005;187:5067–5074. doi: 10.1128/JB.187.15.5067-5074.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kasai D, Masai E, Katayama Y, Fukuda M. Degredation of 3-O-methylgallate in Sphingomonas paucimobilis SYK-6 by pathways involving protocatechuate 4,5-dioxygenase. FEMS Microbiol. Lett. 2007;274:323–328. doi: 10.1111/j.1574-6968.2007.00855.x. [DOI] [PubMed] [Google Scholar]
- (41).Masai E, Katayama Y, Fukuda M. Genetic and biochemical investigations on bacterial catabolic pathways for lignin-derived aromatic compounds. Biosci., Biotechnol., Biochem. 2007;71:1–15. doi: 10.1271/bbb.60437. [DOI] [PubMed] [Google Scholar]
- (42).Saito S, Kawabata J. Effects of electron-withdrawing substituents on DPPH radical scavenging reactions of protocatechuic acid and its analogues in alcoholic solvents. Tetrahedron. 2005;61:8101–8108. [Google Scholar]
- (43).Rubino MT, Maggi D, Laghezza A, Loiodice F, Tortorella P. Identification of novel matrix metalloproteinase inhibitors by screening of phenol fragments library. Arch. Pharm. (Weinheim) 2011;344:557–563. doi: 10.1002/ardp.201000350. [DOI] [PubMed] [Google Scholar]
- (44).Pandey RK, Jarvis GG, Low PS. Efficient synthesis of the siderophore petrobactin via antimony triethoxide mediated coupling. Tetrahedron Lett. 2012;53:1627–1629. [Google Scholar]
- (45).Vaillancourt FH, Labbe G, Drouin NM, Fortin PD, Eltis LD. The mechanism-based inactivation of 2,3-dihydroxybiphenyl 1,2-dioxygenase by catecholic substrates. J. Biol. Chem. 2002;277:2019–2027. doi: 10.1074/jbc.M106890200. [DOI] [PubMed] [Google Scholar]
- (46).Cain RB. New aromatic ring-splitting enzyme, protocatechuic acid 4,5-oxygenase. Nature. 1962;193:842–844. doi: 10.1038/193842a0. [DOI] [PubMed] [Google Scholar]
- (47).Maruyama K, Ariga N, Tsuda M, Deguchi K. Purification and properties of α-hydroxy-γ-carboxymuconic-ε-semialdehyde dehydrogenase. J. Biochem. 1978;83:1125–1134. doi: 10.1093/oxfordjournals.jbchem.a132002. [DOI] [PubMed] [Google Scholar]
- (48).Dagley S, Geary PJ, Wood JM. The metabolism of protocatechuate by Pseudomonas testosteroni. Biochem. J. 1968;109:559–568. doi: 10.1042/bj1090559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr., Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09. Gaussian, Inc.; Wallingford, CT: 2009. [Google Scholar]
- (50).Tomasi J, Mennucci B, Cammi R. Quantum mechanical continuum solvation models. Chem. Rev. 2005;105:2999–3093. doi: 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- (51).Tomasi J, Persico M. Molecular interactions in solution - An overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994;94:2027–2094. [Google Scholar]
- (52).Davis AP, Fry AJ. Experimental and computed absolute redox potentials of polycyclic aromatic hydrocarbons are highly linearly correlated over a wide range of structures and potentials. J. Phys. Chem. A. 2010;114:12299–12304. doi: 10.1021/jp106088n. [DOI] [PubMed] [Google Scholar]
- (53).Capyk JK, D’Angelo I, Strynadka N, Eltis LD. Characterization of 3-ketosteroid 9α-hydroxylase, a Rieske-oxygenase in the cholesterol degradation pathway of Mycobacterium tuberculosis. J. Biol. Chem. 2009;284:9937–9946. doi: 10.1074/jbc.M900719200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Noda Y, Nishikawa S, Shiozuka KI, Kadokura H, Nakajima H, Yoda K, Katayama Y, Morohoshi N, Haraguchi T, Yamasaki M. Molecular-cloning of the protocatechuate 4,5-dioxygenase genes of Pseudomonas paucimobilis. J. Bacteriol. 1990;172:2704–2709. doi: 10.1128/jb.172.5.2704-2709.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Vaillancourt FH, Han S, Fortin PD, Bolin JT, Eltis LD. Molecular basis for the stabilization and inhibition of 2,3-dihydroxybiphenyl 1,2-dioxygenase by t-butanol. J. Biol. Chem. 1998;273:34887–34895. doi: 10.1074/jbc.273.52.34887. [DOI] [PubMed] [Google Scholar]
- (56).Horsman GP, Jirasek A, Vaillancourt FH, Barbosa CJ, Jarzecki AA, Xu CL, Mekmouche Y, Spiro TG, Lipscomb JD, Blades MW, Turner RFB, Eltis LD. Spectroscopic studies of the anaerobic enzyme - Substrate complex of catechol 1,2-dioxygenase. J. Am. Chem. Soc. 2005;127:16882–16891. doi: 10.1021/ja053800o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Emerson JP, Kovaleva EG, Farquhar ER, Lipscomb JD, Que L. Swapping metals in Fe- and Mn-dependent dioxygenases: Evidence for oxygen activation without a change in metal redox state. Proc. Natl. Acad. Sci. U.S.A. 2008;105:7347–7352. doi: 10.1073/pnas.0711179105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Groce SL, Lipscomb JD. Conversion of extradiol aromatic ring-cleaving homoprotocatechuate 2,3-dioxygenase into an intradiol cleaving enzyme. J. Am. Chem. Soc. 2003;125:11780–11781. doi: 10.1021/ja0368103. [DOI] [PubMed] [Google Scholar]
- (59).Seibert CM, Raushel FM. Structural and catalytic diversity within the amidohydrolase superfamily. Biochemistry. 2005;44:6383–6391. doi: 10.1021/bi047326v. [DOI] [PubMed] [Google Scholar]
- (60).Cummings JA, Fedorov AA, Xu C, Brown S, Fedorov E, Babbitt PC, Almo SC, Raushel FM. Annotating enzymes of uncertain function: the deacylation of d-amino acids by members of the amidohydrolase superfamily. Biochemistry. 2009;48:6469–6481. doi: 10.1021/bi900661b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Ono K, Nozaki M, Hayaishi O. Purification and some properties of protocatechuate 4,5-dioxygenase. Biochim. Biophys. Acta. 1970;220:224–238. doi: 10.1016/0005-2744(70)90008-2. [DOI] [PubMed] [Google Scholar]
- (62).Whiting AK, Boldt YR, Hendrich MP, Wackett LP, Que L. Manganese(II)-dependent extradiol-cleaving catechol dioxygenase from Arthrobacter globiformis CM-2. Biochemistry. 1996;35:160–170. doi: 10.1021/bi951979h. [DOI] [PubMed] [Google Scholar]
- (63).Fielding AJ, Kovaleva EG, Farquhar ER, Lipscomb JD, Que L. A hyperactive cobalt-substituted extradiol-cleaving catechol dioxygenase. J. Biol. Inorg. Chem. 2011;16:341–355. doi: 10.1007/s00775-010-0732-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Zabinski R, Wood JM, Champion PM, Munck E. Kinetic and Mossbauer studies on mechanism of protocatechuic acid 4,5-oxygenase. Biochemistry. 1972;11:3212. doi: 10.1021/bi00767a012. [DOI] [PubMed] [Google Scholar]
- (65).Morgan B, Lahav O. The effect of pH on the kinetics of spontaneous Fe(II) oxidation by O-2 in aqueous solution - basic principles and a simple heuristic description. Chemosphere. 2007;68:2080–2084. doi: 10.1016/j.chemosphere.2007.02.015. [DOI] [PubMed] [Google Scholar]
- (66).Takemori S, Komiyama T, Katagiri M. Apo- and reconsitututed holoenzymes of metapyrocatechase from Pseudomonas putida. Eur. J. Biochem. 1971;23:178–184. doi: 10.1111/j.1432-1033.1971.tb01606.x. [DOI] [PubMed] [Google Scholar]
- (67).Tchesnokov EP, Wilbanks SM, Jameson GNL. A strongly bound high-spin iron(II) coordinates cysteine and homocysteine in cysteine dioxygenase. Biochemistry. 2012;51:257–264. doi: 10.1021/bi201597w. [DOI] [PubMed] [Google Scholar]
- (68).Leitgeb S, Straganz GD, Nidetzky B. Biochemical characterization and mutational analysis of the mono-nuclear non-haem Fe2+ site in Dke1, a cupin-type dioxygenase from Acinetobacter johnsonii. Biochem. J. 2009;418:403–411. doi: 10.1042/BJ20081161. [DOI] [PubMed] [Google Scholar]
- (69).Enya M, Aoyagi K, Hishikawa Y, Yoshimura A, Mitsukura K, Maruyama K. Molecular and catalytic properties of 2,4′-dihydroxyacetophenone dioxygenase from Burkholderia sp AZ11. Biosci., Biotechnol., Biochem. 2012;76:567–574. doi: 10.1271/bbb.110867. [DOI] [PubMed] [Google Scholar]
- (70).Dagley S, Evans WC, Ribbons DW. New pathways in the oxidative metabolism of aromatic compounds by micro-organisms. Nature. 1960;188:560–566. doi: 10.1038/188560a0. [DOI] [PubMed] [Google Scholar]
- (71). [accessed Jan 25, 2013];Sigma-Aldrich, 2,4-Pyridine Dicarboxylic Acid Monohydrate. http://www.sigmaaldrich.com/spectra/fnmr/FNMR011311.PDF.
- (72).Ishida T, Tanaka H, Horiike K. Quantitative structure-activity relationship for the cleavage of C3/C4-substituted catechols by a prototypal extradiol catechol dioxygenase with broad substrate specificity. J. Biochem. 2004;135:721–730. doi: 10.1093/jb/mvh089. [DOI] [PubMed] [Google Scholar]
- (73).Ishida T, Senda T, Tanaka H, Yamamoto A, Horiike K. Single-turnover kinetics of 2,3-dihydroxybiphenyl 1,2-dioxygenase reacting with 3-formylcatechol. Biochem. Biophys. Res. Commun. 2005;338:223–229. doi: 10.1016/j.bbrc.2005.08.218. [DOI] [PubMed] [Google Scholar]
- (74).Harpel MR, Lipscomb JD. Gentisate 1,2-dioxygenase from Pseudomonas - Purification, characterization, and comparison of the enzymes from Pseudomonas testosteroni and Pseudomonas acidovorans. J. Biol. Chem. 1990;265:6301–6311. [PubMed] [Google Scholar]
- (75).Feng YM, Khoo HE, Poh CL. Purification and characterization of gentisate 1,2-dioxygenases from Pseudomonas alcaligenes NCIB 9867 and Pseudomonas putida NCIB 9869. Appl. Environ. Microbiol. 1999;65:946–950. doi: 10.1128/aem.65.3.946-950.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Dai Y, Pochapsky TC, Abeles RH. Mechanistic studies of two dioxygenases in the methionine salvage pathway of Klebsiella pneumoniae. Biochemistry. 2001;40:6379–6387. doi: 10.1021/bi010110y. [DOI] [PubMed] [Google Scholar]
- (77).Sauter M, Lorbiecke R, Bo OY, Pochapsky TC, Rzewuski G. The immediate-early ethylene response gene OsARD1 encodes an acireductone dioxygenase involved in recycling of the ethylene precursor S-adenosylmethionine. Plant J. 2005;44:718–729. doi: 10.1111/j.1365-313X.2005.02564.x. [DOI] [PubMed] [Google Scholar]
- (78).Shimoni E, Baasov T, Ravid U, Shoham Y. The trans-anethole degradation pathway in an Arthrobacter sp. J. Biol. Chem. 2002;277:11866–11872. doi: 10.1074/jbc.M109593200. [DOI] [PubMed] [Google Scholar]
- (79).Yun S-H, Yun C-Y, Kim SI. Characterization of protocatechuate 4,5-dioxygenase induced from p-hydroxybenzoate cultured Pseudomonas sp. K82. J. Microbiol. 2004;42:152–155. [PubMed] [Google Scholar]
- (80).Yoon Y-H, Park S-H, Leem S-H, Kim SI. Cloning of p-hydroxybenzoate degradation genes and the over-expression of protocatechuate 4,5-dioxygenase from Pseudomonas sp. K82. J. Microbiol. Biotechnol. 2006;16:1995–1999. [Google Scholar]
- (81).Klecka GM, Gibson DT. Inhibition of catechol 2,3-dioxygenase from Pseudomonas putida by 3-chlorocatechol. Appl. Environ. Microbiol. 1981;41:1159–1165. doi: 10.1128/aem.41.5.1159-1165.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.