

Abstract
Free radicals originate from both exogenous environmental sources and as by-products of the respiratory chain and cellular oxygen metabolism. Sustained accumulation of free radicals, beyond a physiological level, induces oxidative stress that is harmful for the cellular homeodynamics as it promotes the oxidative damage and stochastic modification of all cellular biomolecules including proteins. In relation to proteome stability and maintenance, the increased concentration of oxidants disrupts the functionality of cellular protein machines resulting eventually in proteotoxic stress and the deregulation of the proteostasis (homeostasis of the proteome) network (PN). PN curates the proteome in the various cellular compartments and the extracellular milieu by modulating protein synthesis and protein machines assembly, protein recycling and stress responses, as well as refolding or degradation of damaged proteins. Molecular chaperones are key players of the PN since they facilitate folding of nascent polypeptides, as well as holding, folding, and/or degradation of unfolded, misfolded, or non-native proteins. Therefore, the expression and the activity of the molecular chaperones are tightly regulated at both the transcriptional and post-translational level at organismal states of increased oxidative and, consequently, proteotoxic stress, including ageing and various age-related diseases (e.g. degenerative diseases and cancer). In the current review we present a synopsis of the various classes of intra- and extracellular chaperones, the effects of oxidants on cellular homeodynamics and diseases and the redox regulation of chaperones.
Abbreviations: α(2)M, α(2)-Macroglobulin; AGEs, Advanced Glycation End Products; ALS, Autophagy Lysosome System; AP-1, Activator Protein-1; CLU, apolipoprotein J/Clusterin; EPMs, Enzymatic Protein Modifications; ER, Endoplasmic Reticulum; ERAD, ER-Associated protein Degradation; GRP78, Glucose Regulated Protein of 78 kDa; GPx7, Glutathione Peroxidase 7; Hb, Haemoglobin; HSF1, Heat Shock transcription Factor-1; HSP, Heat Shock Protein; Keap1, Kelch-like ECH-associated protein 1; NADH, Nicotinamide Adenine Dinucleotide; NEPMs, Non-Enzymatic Protein Modifications; NOS, Nitric Oxide Synthase; NOx, NAD(P)H Oxidase; Nrf2, NF-E2-related factor 2; PDI, Protein Disulfide Isomerase; PDR, Proteome Damage Responses; PN, Proteostasis Network; RNS, Reactive Nitrogen Species; ROS, Reactive Oxygen Species; UPR, Unfolded Protein Response; UPS, Ubiquitin Proteasome System
Keywords: Chaperones, Diseases, Free radicals, Oxidative stress, Proteome, Redox signalling
Graphical abstract
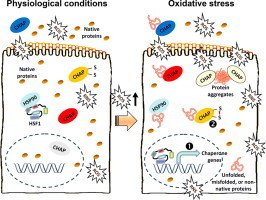
Differential regulation of chaperones activity, under physiological conditions or during oxidative stress-mediated proteome instability.
Highlights
-
•
Free radicals originate from various sources and at physiological concentrations are essential for the modulation of cell signalling pathways.
-
•
Abnormally high levels of free radicals induce oxidative stress and damage all cellular biomolecules, including proteins.
-
•
Molecular chaperones facilitate folding of nascent polypeptides, as well as holding, folding, and/or degradation of damaged proteins.
-
•
The expression and the activity of chaperones during oxidative stress are regulated at both the transcriptional and post-translational level.
1. Introduction
Cellular respiratory chain is the most important redox reaction where reactive oxygen species are produced. During the spatially and temporally separated series of redox reactions and electrons which are transferred from one molecule donor to an acceptor molecule of the electron transport chain in mitochondria, a small percentage of electrons do not complete the cycle and instead leak directly to oxygen [1]. Excessive amounts of free radicals and radical-derived reactive species may also arise from the activity of NAD(P)H oxidases (NOx) and/or xanthine oxidase, as well as from nitric oxide synthase (NOS), P450 metabolism and peroxisomes. Typical products of redox signalling are oxygen free radicals known as reactive oxygen species (ROS) and reactive nitrogen species (RNS). Free radicals and their derivatives are highly reactive and (among others) are comprised of superoxide anion (), hydroxyl radical (•OH) and the nitric oxide (NO•) radical [1–3]. Notably, free radicals may also arise from exogenous sources including various types of radiations (e.g. UV light, X-rays or gamma rays), atmospheric pollutants and metal-catalyzed reactions [1–3].
Free radicals were originally believed to be harmful, but it has been realized that in physiological concentrations they serve as redox messengers, which are essential in the regulation of intracellular signalling and significant cellular processes including metabolism, antioxidant defenses and responses to pathogens [1,4]. Nevertheless, sustained increased levels of oxygen free radicals beyond a physiological threshold that exceed cellular antioxidant capacity are toxic and introduce oxidative stress that affects all cellular biomolecules. Antioxidant pathways are then activated to buffer the excess amount of free radicals and re-establish normal cellular homeodynamics [5–7].
Proteome is modified post-translationally by either numerous highly regulated enzymatic protein modifications (EPMs) (e.g. phosphorylation, acetylation, ubiquitination, methylation, etc.) or by non-enzymatic protein modifications (NEPMs), which are mostly stochastic and increase with ageing or in age-related diseases (Fig. 1). NEPMs include (among others) protein oxidation and/or the formation of Advanced Glycation End Products (AGEs) that are formed mostly by reducing sugars and reactive aldehydes via the Maillard reaction and the non-enzymatic glycation of free amino groups [7–9]. EPMs alter the targeted proteins, which however remain fully functional, while NEPMs may induce protein unfolding or misfolding resulting in increased proteome instability. Proteins and protein machines are particularly vulnerable to oxidative stress as they are among the major target group of ROS. Reportedly, in a cell under oxidative stress proteins consist ~69% of the oxidized molecules; the remaining 31% accounts for lipids and DNA [10]. Free radicals-derived protein modification can result in either gain- or loss-of-function due to the protein misfolding or unfolding. Specifically, oxidized proteins may lose their secondary or tertiary structure and thus their stability, activity and/or function are compromised [7,9,11]. Given the fact that proteins and their macromolecular assemblies (that form protein machines) are virtually the entities that perform all major cellular functions it is not surprising that cells invest significant energetic effort to retain a fully functional proteome.
Fig. 1.
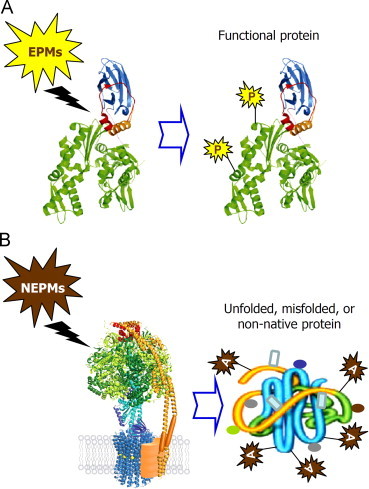
Proteome is modified post-translationally by either highly regulated enzymatic protein modifications (EPMs) [e.g. phosphorylation (P), acetylation, ubiquitination, etc.] or by non-enzymatic protein modifications (NEPMs), which are mostly stochastic and increase with ageing or in age-related diseases. NEPMs include (among others) protein oxidation and the formation of Advanced Glycation End Products [AGEs; (A)] that are formed by reducing sugars and reactive aldehydes via the non-enzymatic glycation of free amino groups (the Maillard reaction). EPMs modify targeted proteins, which however remain fully functional, while NEPMs may induce protein unfolding or misfolding resulting in increased proteome instability.
Proteome stability and, most importantly functionality, is achieved by the action of a cellular modular, yet integrated system which ensures general proteome quality control and it is called the proteostasis network (PN) [7,12]. The PN curates the basal functionality and homeodynamics of the proteome and responds to conditions of proteotoxic stress by addressing the triage decision of fold, hold, or degrade. Specifically, PN is constituted of several complex protein machines that under conditions of proteotoxic stress aim to, firstly identify, and then, either rescue (if feasible) or degrade unfolded, misfolded or non-native polypeptides. Additional integrated modules of the PN can be considered the protein synthesis module and the regulatory pathways of the cellular stress (e.g. heat or oxidative) responses which mobilize the proteome caretakers; mitotic cells can also dilute proteome damage by mitosis (Fig. 2).
Fig. 2.
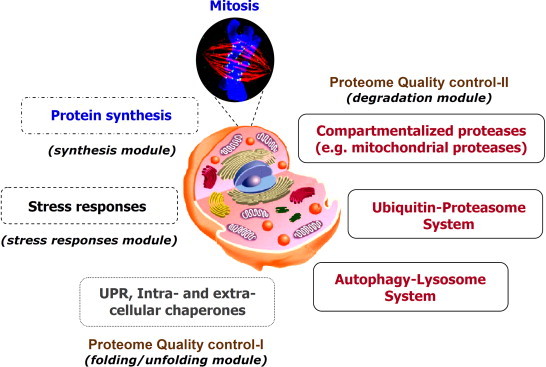
Main components of the proteostasis network (PN) and of the proteome damage responses (PDR). In the case of proteome damage cells launch a massive response by activating the PN, which functions by addressing the triage decision fold, hold or degrade. PN is composed of several modules including the protein synthesis machinery, the UPR of the ER and the armada of the intra- and extra-cellular chaperones, and finally, a number of proteases, which (among others) detoxify cells from non-repairable proteins; mitotic cells can also dilute proteome damage by mitosis. Part of PDR can be also considered the stress responses module, which regulates cellular responses to oxidative and electrophilic stress (e.g. the Nrf2/Keap1 signalling pathway). Molecular chaperones function in all PN modules and they are key players of the PN and PDR since apart from facilitating folding of nascent polypeptides they are also involved in holding, folding, and/or degradation of unfolded, misfolded and/or non-native proteins during stressful conditions (e.g. redox imbalance).
Central to the PN functionality is the armada of the chaperone machines which function in a coordinated order to prevent protein aggregation and either refold unfolded proteins or drive them for degradation through the main proteolytic systems, namely the ubiquitin proteasome (UPS) or the autophagy lysosome (ALS) systems [7,12]. Therefore, both the expression and the activity of the molecular chaperones are tightly regulated at both the transcriptional and post-translational level under conditions of increased oxidative and, consequently, proteotoxic stress. In the current review we present a synopsis of the various classes of chaperones, their functional implication in proteome stability, as well as the effects of oxidants on cellular homeodynamics, diseases and on the regulation of chaperones.
2. Main classes of molecular chaperones
Molecular chaperones [also referred as heat shock proteins (HSPs)] are families of multidomain proteins that are involved in protein folding, unfolding and remodelling. In particular, they assist newly synthesized proteins to reach their native fold; mediate either assembly of complexes or unfolding and disassembly while protecting them from various types of stress (e.g. heat and oxidative stress) and protein aggregation [13,14]. To achieve their goal(s) chaperones function in different ways by co-operating with many different proteins, binding to different substrates and performing changes in the structure of proteins to achieve correct folding. Due to their “multitasking” on a wide range of substrates and the interactions or different conformations that the chaperone domains undergo in order to achieve correct protein folding, they are often referred to as molecular machines. Chaperone machines are in most cases composed of highly coordinated moving parts (involving displacements and rotations) which depend on energy input (i.e. cycles of ATP binding and hydrolysis) for their function and, although they rather have a broad spectrum of substrates, their specificity is enough to ensure proteome stability [14,15].
At the systemic level (i.e. the organism) chaperones curate the proteome both intracellularly as well as at the extracellular milieu (i.e. the biological fluids). Given the immense importance of proteome stability, it is not surprising that intracellular chaperones are found in virtually all subcellular compartments including the nucleus, the cytosol, the mitochondria and the endoplasmic reticulum (ER), while during protein synthesis chaperones keep proteins from misfolding as they are still being translated [16]. The diverse family of the prominent HSPs is divided into the small heat shock proteins (sHSPs) that modulate proteome stability in an ATP-independent fashion (referred also as “holdases”) and to those that depend on energy consumption (i.e. ATP) for their functionality (e.g. members of the Hsp60, Hsp70, Hsp90 family; often termed as “foldases”) [13,14].
In those organismal states (e.g. ageing or diseases) where the chaperone network becomes deregulated, the accumulating non-native, misfolded or unfolded proteins can form (among others) fibrils, amyloids or large amorphous aggregates [15]. These aggregates are responsible for many severe diseases like Alzheimer׳s, Parkinson׳s, Huntington׳s and prion disease since their accumulation becomes toxic and further disrupts the PN introducing global proteome instability and downstream proteome damage responses (PDR) [15]. In other diseases characterized by increased oxidative and proteotoxic stress (e.g. cancer) high expression levels of the chaperones contribute to tumor cells detoxification and proteome stability thus increasing tumor cells resistance to therapeutic approaches [7]. Considering that most of the aforementioned pathological states (including ageing) are characterized by increased oxidative stress which may then promote proteotoxic stress, it is essential to explore the mechanisms of redox regulation of chaperones in order to provide new insights on how these diseases evolve and how to treat them effectively.
2.1. Small heat shock proteins
sHSPs are ATP-independent members of the heat shock protein family that form high molecular weight oligomers [17,18]. These chaperones provide high-affinity binding platforms for unfolded proteins and prevent protein aggregation specifically during stress conditions. There are several sHSPs encoded by the human genome and even though they are a poorly conserved family all sHSPs have a similar range of low molecular weight (~10–40 kDa) and share a number of structural and functional characteristics including a conserved α-crystallin domain that is critical for sHSPs dimerization and function; the capacity to form large oligomers, the potent induction under conditions of stress and a dynamic quaternary structure [19,20]. sHSPs are involved in preventing aggregation of partially misfolded proteins, while they also associate and modulate the assembly of most major cytoskeletal components [21–23]. Upon heat shock, αB-crystallin translocates from the nuclear speckles to the nucleoplasm, thereby contributing in the stability of the nuclear proteome [24]. sHSPs are tightly regulated by EPMs (e.g. phosphorylation) that enable them to respond upon stress and to perform their chaperone activities [25,26]. Furthermore, HSP27 and αB-crystallin target damaged or mutated proteins to UPS for degradation [27,28], while along with other sHSPs they interact with many cytoskeletal parts to inhibit aggregation of misfolded proteins [29]. Moreover, HSP22 stimulates autophagy-mediated degradation of protein aggregates in an eIF2α-dependent manner [30]. sHSPs are overexpressed upon many different types of stress as they are key components of the PN and PDR. Specifically, they have been found overexpressed in many different diseases including various types of cancer where they contribute in reducing proteotoxic stress [7]. Because sHSPs do not depend on ATP for their function they are not typical druggable targets and thus current efforts aim to inhibit their function and provide therapeutic tools via the development of antisense oligonucleotides that suppress their expression; this approach has been studied in prostate and bladder cancer with encouraging, so far, results [31,32].
2.2. Heat shock proteins 60, 70 and 90
The HSP60 family of chaperones (or chaperonins) are oligomeric proteins categorized in two subfamilies; group I that is found in bacteria, mitochondria and chloroplasts, and group II, which is found in the eukaryotic cytosol and in archaea [33]. Group I includes the bacterial chaperonin 60 (GroEL) and co-chaperonin 10 (GroES), as well as, the chloroplast and mitochondrion specific HSP60 proteins and their co-chaperonins [34]. Chaperonins are well-established protein machines that ensure correct protein folding by providing an isolation chamber. GroEL, which is extensively studied, forms a barrel like structure, comprised of two tightly close rings that surround open cavities and constitute a line of continuous hydrophobic surfaces. This open structure that resembles a “lid” is where non-native polypeptides with exposed hydrophobic surfaces are captured. Upon ATP-dependent binding, GroES (a single heptamer ring) which is recruited by a GroEL ring, closes the “lid” thereby transforming the open ring into an isolated chamber with a hydrophilic band [35]. GroEL and GroES act on the substrate and their coordinated ATP-dependent action results in unfolding of the enclosed misfolded protein [36] while the power of this action also forces the protein away from the hydrophobic sites and at the same time it retranslocates it inside the GroES-capped hydrophilic chamber for folding [37]. In this isolated site the unfolded/misfolded protein cannot misfold further due to the lack of hydrophobic sites or other proteins for aggregation, and can either remain in this state or fold properly according to its amino acid sequence. After a step of slow ATP hydrolysis, the “lid” is re-opened and if the protein is correctly folded it will be released or it will otherwise be recaptured by another chaperonin ring [37]. Group II chaperonins include the eukaryotic cytosolic chaperonin-containing TCP1 (CCT; also known as TriC) and the archaeal thermosome. CCT is consisted of rings each one having eight subunits, which are encoded by eight related but distinct genes [38]; CCT is known to bind to cytoskeletal proteins among other substrates [39].
HSP70s is an abundant heat shock protein family that is found in all cellular compartments. HSP70 chaperones have a diverse array of cellular functions but their major role is to ensure correct folding of newly synthesized proteins and to perform the refolding of proteins that are misfolded and/or aggregated. This broad spectrum of cellular functions is achieved through the amplification and diversification of HSP70 genes and also by specialized co-chaperones (e.g. HSP40) that support HSP70s function [40]. The HSP70 proteins are consisted of two domains, namely an amino-terminal ATPase domain and a carboxy-terminal substrate-binding domain which is comprised of a helical lid segment and a β-sandwich subdomain; the latter recognizes proteins׳ hydrophobic amino acids [41,42]; notably, the exposure of this patch of hydrophobic amino acids correlate with a proteins׳ tendency to aggregate [43]. When ATP is bound, the HSP70 helical lid is open and only when ATP hydrolysis takes place the lid can close [44]. ATP hydrolysis is fastened by HSP40, which also interacts with unfolded peptides and oxidized proteins and mediates HSP70 recruitment to protein substrates. Upon release of the substrate, peptides that need time for folding will rebind to HSP70 until they reach their correct folded state and therefore will be prevented from aggregation [43]. Moreover, HSP70 cooperates with HSP100 ATPases in disaggregating large aggregates; HSP100 disassembly machines are unfoldases and “disaggregatases”, which deliver substrates to compartmentalized proteases (e.g. the proteasome) or disassemble aggregates containing misfolded proteins [45,46]. Proteins that cannot reach their correct folded state after many cycles of binding to HSP70 might be translocated to the specialized chaperonin cage-like structure for correct folding. HSP70 also functions in the nucleus where it is imported upon thermal stress to facilitate nuclear proteome stability [47]. A direct connection between HSP70 and the proteolytic systems involves CHIP (Carboxyl terminus of HSP70-Interacting Protein) that targets denatured proteins bound to HSP70 (or to HSP90) to proteasome for degradation [48].
The HSP90 class of ATP-dependent chaperones are ubiquitously expressed and account for ~1–2% of total cellular proteins in non-stressed normal cells. These chaperones function downstream to HSP70 and display a wide range of biological roles and many “client” proteins. HSP90 exists as a homodimer and functions in the regulation of signalling and cell cycle by binding client proteins and enabling their interaction with other proteins or their ligands. Each monomer protein contains an N-terminal ATPase domain that is also involved in the interaction with co-chaperones, a central domain that modulates binding of client proteins and ATP hydrolysis and a C-terminal high affinity dimerization domain [49]. Among the proteins that are substrates of the HSP90 system are hormone receptors, kinases and oncogenic proteins [49]. In general, HSP90s are more specialized than other chaperones and are essential for survival in eukaryotic cells as they also are capable of binding non-native polypeptides (at the late stages of their folding) and preventing their aggregation [14]. Specifically, HSP90 interacts with co-chaperones and co-regulators, many of which use tetratricopeptide repeat (TPR) domains to dock onto HSP90, and can bind to non-native proteins in order to prevent their aggregation [50]. The TPR-motif containing protein, HSC70-HSP90-organizing protein (HOP, also known as STI1) is one of these co-regulators that provides a direct link between HSP70 and HSP90, allowing substrate transfer [50–52]; STI1 also translocates to the nucleus upon DNA damage implying a potential role for this protein in genotoxic stress and DNA damage responses [53].
2.3. Endoplasmic reticulum and extracellular chaperones
The intracellular proteins of the secretory pathway and those of the extracellular millieu are synthesized and processed at the extracellular-like environment of the ER. Here, unfolded or misfolded non-repairable proteins will be targeted for ER-associated protein degradation (ERAD) via ER-attached proteasomes [54]. Misfolded proteins in the ER are recognized in mammals by the ER-chaperone, glucose regulated protein of 78 kDa (GRP78/BiP/HSPA5), which binds to hydrophobic residues in the unfolded part of proteins [55]. GRP78 (the ER homologue of HSP70) is consisted of an ATPase domain, a peptide-binding domain and a KDEL ER-retention signal [56]. Under physiological conditions GRP78 is kept at an inert state by binding to three ER proteins, namely the activating transcription factor-6 (ATF6), the inositol-requiring transmembrane kinase/endoribonuclease 1 (IRE1) and the double-stranded RNA (PKR)-activated protein kinase-like eukaryotic initiation factor 2 kinase (PERK) [7,56]. Upon ER proteotoxic stress, GRP78 dissociates from its binding partners, which are then free to trigger the Unfolded Protein Response (UPR) by regulating specific gene responses aiming to restore ER proteome stability. The three sensors of ER proteotoxic stress facilitate contradictory responses since they either promote cell survival by decreasing the misfolded protein and/or oxidative load, or, if UPR fails, they promote the activation of apoptotic pathways that eventually result in cell death [57]. Other molecular chaperones of the ER are ERp72, GRP94, GRP170/ORP150, protein disulfide isomerase (PDI), cyclophilin B, CaBP1 (calcium binding protein) and SDF2- L1 [58].
The extracellular space (that is a continuous with the lumen of ER and Golgi) faces various challenges in retaining proteostasis as it virtually lacks ATP [59]; the abundance of intracellular chaperones (e.g. HSP70) is normally extremely low [60] and it is more oxidized than the nucleo-cytosolic compartments. Moreover, it is readily affected by environmental stressors and it is also subject to shear stress due to the continuous pumping of plasma around the body [61]. A number of secreted glycoproteins, namely haptoglobin [62], α(2)-Macroglobulin [α(2)M] [63] and apolipoprotein J/Clusterin (CLU) [64] have been recently proposed to act as extracellular chaperones, due to their demonstrated ATP-independent chaperone properties. Moreover, it was shown that the concentration of the extracellular heat shock protein 72 (eHSP72) increases during exercise-heat stress [65]. It has been proposed that these chaperones contribute to proteome stability in the biological fluids by capturing and leading unfolded or misfolded proteins for lysosomal proteolysis via endocytosis [66]. In relation to proteostasis regulation, haptoglobin was shown to act as a potential extracellular chaperone for β2-microglobulin in acidic conditions during inflammation [67], while it also contributes to hemoglobin (Hb) structural stabilization and possibly exerts an anti-oxidative function during peroxidative stress [68]. α(2)M also reacts with proteases and following its activation it inhibits amorphous and fibrillar protein aggregation [69]. CLU is a heterodimeric secreted glycoprotein which is ubiquitously expressed in human tissues and seems to exert its sHSPs-like chaperone function both extracellularly as well as at intracellular compartments [70,71]. It is thus not surprising that CLU has been functionally involved in many physiological processes including ageing, as well as several age-related diseases such as neurodegeneration, metabolic diseases and cancer [72]. Although unrelated in their etiology and clinical manifestation, all of these diseases represent states of increased oxidative stress, which in turn, promotes increased genomic instability, amorphous aggregation of target proteins and high rates of cellular death. Based on this realization and the combined presence of many potential regulatory elements in the CLU gene promoter, including a Heat Shock transcription Factor-1 (HSF1) and an Activator Protein-1 (AP-1) element, we recently proposed that the CLU gene is an extremely sensitive cellular biosensor of even minute alterations in the cellular oxidative load [72,73].
3. Redox biology and chaperones
Free radicals may have opposing effects in organisms׳ homeostasis as in physiological levels they are essential for cell signalling whereas at abnormally high levels they affect a wide variety of cellular processes.
3.1. Sources of oxidants and cellular responses
The most important redox reaction in cells is the respiratory chain that takes place in the mitochondrion, where electron transport chain and oxidative phosphorylation are coupled to synthesize ATP. Most of ROS are generated as undesirable side products during the process where nicotinamide adenine dinucleotide (NADH) or succinate delivers electrons to the respiratory chain and the four complexes transfer electrons to finally reduce O2 by forming water [1,2,74]. Excessive amounts of ROS may also arise from inflammatory processes [75], excessive stimulation of NAD(P)H oxidases [76], P450 metabolism and activity of peroxisomes [1,2,74]. Also, a number of exogenous sources facilitate the production of free radicals including UV light, gamma rays, X-rays, atmospheric pollutants, smoke and metal catalyzed reactions [1,2,74]. As mentioned, at physiological cellular levels ROS act as secondary messengers or signalling molecules and are essential in intracellular signalling. Specifically, ROS have been shown to regulate a wide variety of signalling pathways including anti-inflammatory responses and adaptation to hypoxia [77,78], autophagy [79], immune cell function [80], cellular differentiation [81], integrins [82], as well as oncogenes signalling [83]. Thus, a very delicate balance between free radicals production and removal or neutralization occurs that confers cellular homeostasis.
An increase of intracellular or extracellular concentration of ROS results in deregulation of redox-sensitive signalling pathways and a series of primary and secondary responses begins in order to re-establish the former balance. Primary cellular defensive mechanisms include enzymes like the superoxide dismutases, SOD1 (Cu–Zn SOD) and SOD2 (MnSOD) that convert superoxide to H2O2 and catalase or glutathione peroxidase that convert H2O2 to H2O; additional non-enzymatic antioxidant defences are vitamins, carotenoids, thiol antioxidants and natural flavonoids [3,5,7]. A main secondary antioxidant and cellular detoxification program is also executed by the NF-E2-related factor 2 (Nrf2)-Kelch-like ECH-associated protein 1 (Keap1) signalling pathway. Under normal conditions, Nrf2 is retained in the cytoplasm by the actin-binding protein Keap1; a substrate adaptor protein for the Cullin3-containing E3-ligase complex, which targets Nrf2 for ubiquitination and degradation by the proteasome [84,85]. Keap1 is redox sensitive as its 27 cysteine residues act as sensitive sensors of oxidative stress since they can be modified by different oxidants and electrophiles [86]. Oxidative stress abrogates the Keap1-mediated degradation of Nrf2 which in turn accumulates in the nucleus where it heterodimerizes with a small musculoaponeurotic fibrosarcoma (Maf) protein on antioxidant response elements (AREs) to stimulate the expression of a wide arrays of phase II and antioxidant enzymes including NAD(P)H quinone oxidoreductase 1 (Nqo1), heme oxygenase 1 (Hmox1), glutamane-cysteine ligase (GCL) and glutathione S transferases (GSTs) [84,85,87,88]. Moreover, Nrf2 contributes to cellular proteostasis by regulating the expression of molecular chaperones [89], as well as of additional players of proteome stability and maintenance, namely the proteasome subunits [90–92].
3.2. Impact of oxidative stress to cellular proteostasis and chaperones activity
In those cases where the amount of oxygen free radicals exceeds the antioxidant capacity of the cell, due to reduced levels of antioxidants or malfunctioning antioxidant pathways, imbalance between ROS generation and detoxification occurs and risky oxidative stress arise. Accumulating free radicals can then be harmful in cells by introducing stochastic damage to most cellular biomolecules including lipids, nucleic acids and proteins [3,7,93].
Protein oxidation results in reversible or irreversible protein modifications affecting their secondary or tertiary structure and thus their activity, solubility and stability [7,9,94]. Proteome oxidation and instability has been associated with ageing and the progression of several age-related diseases, including cardiovascular disorders, neurodegeneration, and cancer [7,95,96]. ROS can modify proteins by either affecting protein amino acids, especially cysteine residues, or by modifying non-protein components such as lipids, which can then react with amino- and thiol-groups of proteins [96–98]. Free radicals can directly modify histidine residues to generate unstable amino acids while cysteine and methionine residues can be converted to disulfides and methionine sulfoxide residues, respectively. A frequent oxidative modification of proteins is irreversible carbonylation which can occur by either direct oxidation where oxidants act and leave a functional carbonyl group on amino acid side chains or in the protein backbone, or, indirectly, by protein conjugation with oxidation products of polyunsaturated fatty acids and carbohydrates [98]. Under conditions of reduced antioxidant capacity superoxide anion can be readily converted to hydroxyl radicals via Fenton chemistry. These molecules are responsible for the production of lipid aldehydes, such as 4-hydroxy trans-2,3-nonenal (4-HNE) and 4-oxo trans-2,3-nonenal (4-ONE), which can react with various amino acids enabling protein carbonylation [9,98–100]. Heavily carbonylated proteins tend to form aggregates that are resistant to degradation and accumulate as unfolded or damaged proteins [101]. These protein aggregates promote global proteome instability as they (among others) disrupt the functionality of proteolytic pathways [102,103]. Large insoluble aggregates, amyloids and partially denatured proteins activate the HSP70 system to disassemble the protein skein. HSP70 proteins bind their substrates and protein aggregates with low affinity when they are in the ATP-bound state, whereas the ADP-bound state has high affinity for its substrates (see also above); HSP70 function is assisted by co-chaperones like HSP40 or HSP110 [104,105]. Interestingly, HSP70 was recently reported to also mediate dissociation and reassociation of the 26S proteasome during adaptation to oxidative stress [106]; these studies further highlight its crucial role in proteome stability and PDR.
As molecular chaperones activity is central to PN functionality and proteome stability it is not surprising that they are tightly regulated at both the transcriptional and post-translational level under conditions of increased oxidative and, consequently, proteotoxic stress. Specifically, the expression of chaperone genes is mainly regulated by a small group of transcription factors named heat-shock factors (HSFs). Among the four HSFs that have been identified in mammals, HSF1 is the dominant highly conserved transcription factor [107]. HSF1 binds to a consensus heat shock element (HSE) within the promoter regions of HSP genes resulting in the activation of HSPs׳ gene expression and the control of cellular responses to oxidative and proteotoxic stress [108]. As in the case of Nrf2, the activation of HSF1 during stress is a multi-step process. Specifically, under normal conditions HSF1 molecules remain in an inert state as inactive monomers by being bound to various chaperones/co-chaperones including HSP90, HSP70, HSP40 and others [109] (Fig. 3). During elevated stress the chaperones within the repressive HSF1-containing multi-chaperone complexes bind the unfolded proteins and thus the liberated monomeric HSF1s undergo phosphorylation, trimerization and nuclear localization with increased transcriptional activity [109]. HSF1 is subject to additional enzymatic post-translational modifications including sumoylation [110] as well as acetylation [111], while at the post-translational level of redox regulation, HSF1 can also sense oxidative stress via two cysteine residues within the HSF1׳s DNA-binding domain that are engaged in redox-sensitive disulfide bonds [112].
Fig. 3.
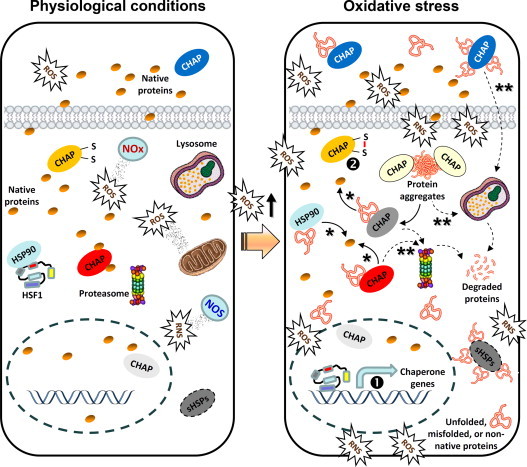
Free radicals (e.g. ROS or RNS) are derived from exogenous (e.g. environmental) as well as intracellular (e.g. mitochondria, NOS, NOx) sources and at physiological concentrations play a significant role as regulatory mediators in signalling pathways. Under these conditions of minimal proteotoxic stress, chaperones (CHAP) are expressed at basal levels and HSF1 is at an inert state by binding to chaperones (e.g. HSP90). A sustained increase in ROS beyond a physiological threshold (redox imbalance) results in increased oxidative and proteotoxic stress. Consequently, HSF1 liberates from HSP90 (which now preferentially binds stressed proteins) and translocates to the nucleus to enhance the expression levels of chaperone genes (transcriptional regulation; ❶). Also, chaperones can be activated at a post-translational level by cycling between a low- and high-affinity substrate binding state, depending on the redox state of their cysteines (❷). Chaperone bound extra- or intra-cellular stressed polypeptides can be either refolded into their native form (⁎) or targeted to proteases for degradation (⁎⁎); similarly, accumulating protein aggregates can be disaggregated to unfolded intermediates and either refolded or targeted for degradation.
Cysteine residues are the most redox-sensitive side chains of amino acids functioning as redox switches in proteins since they are the first amino acids that are oxidized under conditions of increased oxidative stress; thus they are actively involved in post-translational regulatory processes of redox imbalance responses [113]. Reversible thiol oxidation reactions that reportedly play a role in redox regulation of proteins include the formation of intra- or intermolecular disulfide bonds (R–S–S–R), stable sulfenic acid intermediates (R-SOH), mixed disulfides with the small tripeptide glutathione (R–S–S–G) and the over-oxidation of cysteine thiols to sulfinic acid (R–SO2H) [114]. Thus, proteins with redox-sensitive amino acid side chains (such as Keap1 or HSF1) are used as sensors of cellular oxidation status.
In the case of chaperones, the abundant ER molecular chaperone PDI is a critical player in oxidative folding of ER and extracellular proteins. PDI is a redox sensitive chaperone that acts not only as a sensor but also as a protein involved in the processing of oxidized proteins and in preventing misfolding and/or aggregation of proteins. PDI binds to oxidized proteins through their exposed hydrophobic or aromatic residues and its folding activity is stimulated by disulfide bonds created at proper sites [115]. Although it was controversial whether the chaperone activity of PDI is redox-regulated it was shown recently that both the chaperone activity and the overall conformation of human PDI are redox-regulated via conformational changes that release the compact conformation of the molecule and expose the shielded hydrophobic areas to facilitate its high chaperone activity [116]. In support, exposure of human cells to cigarette smoke affected the formation of disulfide bonds through excessive posttranslational oxidation of PDI resulting in increased amounts of complexes between PDI and its client proteins [117]. Moreover, H2O2 oxidizes Cys57 of Glutathione Peroxidase 7 (GPx7) to sulfenic acid, which can be resolved by Cys86 to form an intramolecular disulfide bond. Both the disulfide form and sulfenic acid form of GPx7 can oxidize PDI for catalyzing oxidative folding [118]. Another member of the GPx family, namely NPGPx transmits oxidative stress signals by forming the disulfide bond between its Cys57 and Cys86 residues. This oxidized form of NPGPx binds to GRP78 and forms covalent bonding intermediates between Cys86 of NPGPx and Cys41/Cys420 of GRP78; subsequently, the formation of the disulfide bond between Cys41 and Cys420 of GRP78 enhances its chaperone activity [119].
Additional paradigms of post-translational redox-mediated modifications that guarantee an immediate response to oxidative stress conditions refer to the Hsp33 chaperone in bacteria as well as to typical 2-Cys peroxiredoxins in eukaryotes [120]. In the case of Hsp33, oxidative stress-mediated disulfide bond formation triggers a partial unfolding of the chaperone, which leads to the exposure of a high-affinity binding site for unfolded proteins and thus the protein becomes a highly efficient holdase that binds tightly to unfolding proteins [121]. This rapid conformational change inhibits bacterial functional amyloid assembly [122] and it protects bacteria cells from hypochlorite (bleach) treatment-mediated proteotoxic stress [123,124]. Regarding the highly abundant eukaryotic typical 2-Cys peroxiredoxins (which are found in various subcellular compartments) the oxidative stress-mediated formation of sulfinic acid turns the peroxidase into a molecular chaperone [125]. During this process a catalytically active cysteine attacks the O–O bond of the ROOH substrate, thereby forming the ROH product and a sulfenic acid (CP-SOH) on the catalytically active cysteine [126]. The mechanism of active cysteine regeneration (which is different among the individual members of the peroxiredoxin family) involves the attack of CP-SOH by a free thiol and the release of water [127]. Additional studies in mammalian peroxiredoxins showed that over-oxidation induces the formation of high molecular weight oligomers which function as potent chaperones and prevent protein aggregation [128,129]; these findings indicate that typical 2-Cys peroxiredoxins might act as the eukaryotic counterpart of bacterial Hsp33 by using a unique “sulfinic acid switch” to convert from a peroxidase under non-stress conditions to a molecular chaperone under severe oxidative stress conditions [128].
These observations highlight the dual role of chaperones in PN stability as both proteome caretakers, as well as sensors of disrupted cellular redox homeodynamics.
4. Conclusive remarks
A large body of evidence supports the direct implication of abnormally high levels of free radicals in the progression and pathogenesis of several unrelated diseases. Central to the progression of these diseases is the free radicals-mediated induction of proteome instability that is accompanied by deregulation of the PN modules. This realization is in line with reports showing constant activation of the Nrf2 antioxidant program and high expression levels of chaperones in advanced tumors as well as in degenerative diseases [7]. In fact this process refers to global systemic alterations that eventually enable abnormal (e.g. cancer) cells to exploit PN as a mean to suppress proteotoxic stress and to shape the ultimate landscape for the progression of the disease. The comprehension of this process offers novel therapeutic opportunities, as it seems that in some cases diseased cells have exhausted their capacity to survive under conditions of increased oxidative and proteotoxic stress. In support, it was recently showed that a novel compound (piperlongumine) increased ROS in both cancer and normal cells but it had selective in vivo anti-tumor effects with no apparent toxicity in normal mice [130]. Moreover, a plethora of HSP90 inhibitors are currently investigated in clinical or preclinical trials as anti-tumor drugs [131–134]; 17-AAG, an HSP90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration [135], while an antisense oligonucleotide that targets CLU gene expression is currently in advanced clinical trials for several types of cancer [136]. Furthermore, a GRP78/BiP-targeting cytotoxin treatment along with photodynamic therapy eliminates lung cancer cell lines [137] and this chaperone is also under investigation for the treatment of neurodegenerative diseases [138,139]. Additionally, the enhancement of HSF1 activity or chaperones expression by genetic techniques or pharmacological manipulation has been shown to restore proteostasis in several models of degenerative diseases or to delay ageing [140–142]. In conclusion proteome stability is a key determinant of cellular homeostasis in the healthy organism and thus the expansion of our current knowledge on PN regulation and PDR will also provoke innovative new strategies for the treatment of human diseases.
Acknowledgements
This work was supported by the Empirikion Foundation and the FP7-EU project INsPiRE/REGPOT-CT-2011-284460.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Droge W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 2.Valko M., Rhodes C.J., Moncol J., Izakovic M., Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 3.Sesti F., Tsitsilonis O.E., Kotsinas A., Trougakos I.P. Oxidative stress-mediated biomolecular damage and inflammation in tumorigenesis. In Vivo. 2012;26:395–402. [PubMed] [Google Scholar]
- 4.Cai Z., Yan L.J. Protein oxidative modifications: beneficial roles in disease and health. J. Biochem. Pharmacol. Res. 2013;1:15–26. [PMC free article] [PubMed] [Google Scholar]
- 5.Martindale J.L., Holbrook N.J. Cellular response to oxidative stress: signaling for suicide and survival. J. Cell Physiol. 2002;192:1–15. doi: 10.1002/jcp.10119. [DOI] [PubMed] [Google Scholar]
- 6.Klaunig J.E., Kamendulis L.M. The role of oxidative stress in carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 2004;44:239–267. doi: 10.1146/annurev.pharmtox.44.101802.121851. [DOI] [PubMed] [Google Scholar]
- 7.Trougakos I.P., Sesti F., Tsakiri E., Gorgoulis V.G. Non-enzymatic post-translational protein modifications and proteostasis network deregulation in carcinogenesis. J. Proteomics. 2013;92:274–298. doi: 10.1016/j.jprot.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 8.Lushchak V.I. Free radical oxidation of proteins and its relationship with functional state of organisms. Biochemistry (Mosc) 2007;72:809–827. doi: 10.1134/s0006297907080020. [DOI] [PubMed] [Google Scholar]
- 9.Nedic O., Rattan S.I., Grune T., Trougakos I.P. Molecular effects of advanced glycation end products on cell signalling pathways, ageing and pathophysiology. Free Radic. Res. 2013;47(Suppl. 1):28–38. doi: 10.3109/10715762.2013.806798. [DOI] [PubMed] [Google Scholar]
- 10.Corcoran A., Cotter T.G. Redox regulation of protein kinases. FEBS J. 2013;280:1944–1965. doi: 10.1111/febs.12224. [DOI] [PubMed] [Google Scholar]
- 11.Perez V.I., Buffenstein R., Masamsetti V., Leonard S., Salmon A.B., Mele J., Andziak B., Yang T., Edrey Y., Friguet B., Ward W., Richardson A., Chaudhuri A. Protein stability and resistance to oxidative stress are determinants of longevity in the longest-living rodent, the naked mole-rat. Proc. Natl. Acad. Sci. USA. 2009;106:3059–3064. doi: 10.1073/pnas.0809620106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roth D.M., Balch W.E. Modeling general proteostasis: proteome balance in health and disease. Curr. Opin. Cell Biol. 2011;23:126–134. doi: 10.1016/j.ceb.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bukau B., Weissman J., Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 14.Saibil H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Cancer. 2013;13:630–642. doi: 10.1038/nrm3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doyle S.M., Genest O., Wickner S. Protein rescue from aggregates by powerful molecular chaperone machines. Nat. Rev. Mol. Cell Biol. 2013;14:617–629. doi: 10.1038/nrm3660. [DOI] [PubMed] [Google Scholar]
- 16.Pechmann S., Willmund F., Frydman J. The ribosome as a hub for protein quality control. Mol. Cell. 2013;49:411–421. doi: 10.1016/j.molcel.2013.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benesch J.L., Ayoub M., Robinson C.V., Aquilina J.A. Small heat shock protein activity is regulated by variable oligomeric substructure. J. Biol. Chem. 2008;283:28513–28517. doi: 10.1074/jbc.M804729200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stromer T., Ehrnsperger M., Gaestel M., Buchner J. Analysis of the interaction of small heat shock proteins with unfolding proteins. J. Biol. Chem. 2003;278:18015–18021. doi: 10.1074/jbc.M301640200. [DOI] [PubMed] [Google Scholar]
- 19.Haslbeck M., Franzmann T., Weinfurtner D., Buchner J. Some like it hot: the structure and function of small heat-shock proteins. Nat. Struct. Mol. Biol. 2005;12:842–846. doi: 10.1038/nsmb993. [DOI] [PubMed] [Google Scholar]
- 20.Garrido C., Paul C., Seigneuric R., Kampinga H.H. The small heat shock proteins family: the long forgotten chaperones. Int. J. Biochem. Cell. Biol. 2012;44:1588–1592. doi: 10.1016/j.biocel.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 21.Somara S., Gilmont R.R., Varadarajan S., Bitar K.N. Phosphorylated HSP20 modulates the association of thin-filament binding proteins: caldesmon with tropomyosin in colonic smooth muscle. Am. J. Physiol. Gastrointest. Liver Physiol. 2010;299:G1164–G1176. doi: 10.1152/ajpgi.00479.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Almeida-Souza L., Asselbergh B., d׳Ydewalle C., Moonens K., Goethals S., de Winter v., Azmi A., Irobi J., Timmermans J.P., Gevaert K., Remaut H., Van Den Bosch L., Timmerman V., Janssens S. Small heat-shock protein HSPB1 mutants stabilize microtubules in Charcot-Marie-Tooth neuropathy. J. Neurosci. 2011;31:15320–15328. doi: 10.1523/JNEUROSCI.3266-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kayser J., Haslbeck M., Dempfle L., Krause M., Grashoff C., Buchner J., Herrmann H., Bausch A.R. The small heat shock protein Hsp27 affects assembly dynamics and structure of keratin intermediate filament networks. Biophys. J. 2013;105:1778–1785. doi: 10.1016/j.bpj.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.den Engelsman J., van de Schootbrugge C., Yong J., Pruijn G.J., Boelens W.C. Pseudophosphorylated alphaB-Crystallin Is a nuclear chaperone imported into the nucleus with help of the SMN complex. PLoS One. 2013;8:e73489. doi: 10.1371/journal.pone.0073489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clements R.T., Sodha N.R., Feng J., Mieno S., Boodhwani M., Ramlawi B., Bianchi C., Sellke F.W. Phosphorylation and translocation of heat shock protein 27 and alphaB-crystallin in human myocardium after cardioplegia and cardiopulmonary bypass. J. Thorac. Cardiovasc. Surg. 2007;134:1461–1470. doi: 10.1016/j.jtcvs.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 26.Song I.S., Kang S.S., Kim E.S., Park H.M., Choi C.Y., Tchah H., Kim J.Y. Heat shock protein 27 phosphorylation is involved in epithelial cell apoptosis as well as epithelial migration during corneal epithelial wound healing. Exp. Eye Res. 2013;118C:36–41. doi: 10.1016/j.exer.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 27.Li M.L., Defren J., Brewer G. Hsp27 and F-box protein β-TrCP promote degradation of mRNA decay factor AUF1. Mol. Cell Biol. 2013;33:2315–2326. doi: 10.1128/MCB.00931-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahner A., Gong X., Schmidt B.Z., Peters K.W., Rabeh W.M., Thibodeau P.H., Lukacs G.L., Frizzell R.A. Small heat shock proteins target mutant cystic fibrosis transmembrane conductance regulator for degradation via a small ubiquitin-like modifier-dependent pathway. Mol. Biol. Cell. 2013;24:74–84. doi: 10.1091/mbc.E12-09-0678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pivovarova A.V., Chebotareva N.A., Chernik I.S., Gusev N.B., Levitsky D.I. Small heat shock protein Hsp27 prevents heat-induced aggregation of F-actin by forming soluble complexes with denatured actin. FEBS J. 2007;274:5937–5948. doi: 10.1111/j.1742-4658.2007.06117.x. [DOI] [PubMed] [Google Scholar]
- 30.Carra S., Brunsting J.F., Lambert H., Landry J., Kampinga H.H. HspB8 participates in protein quality control by a non-chaperone-like mechanism that requires eIF2{alpha} phosphorylation. J. Biol. Chem. 2009;284:5523–5532. doi: 10.1074/jbc.M807440200. [DOI] [PubMed] [Google Scholar]
- 31.Hadaschik B.A., Jackson J., Fazli L., Zoubeidi A., Burt H.M., Gleave M.E., So A.I. Intravesically administered antisense oligonucleotides targeting heat-shock protein-27 inhibit the growth of non-muscle-invasive bladder cancer. BJU Int. 2008;102:610–616. doi: 10.1111/j.1464-410X.2008.07669.x. [DOI] [PubMed] [Google Scholar]
- 32.Hotte S.J., Yu E.Y., Hirte H.W., Higano C.S., Gleave M., Chi K.N. OGX-427, a 2'methoxyethyl antisense oligonucleotide (ASO), against Hsp27: results of a first-in-human trial. J. Clin. Oncol. 2009;27 [Google Scholar]
- 33.Ranford J.C., Coates A.R., Henderson B. Chaperonins are cell-signalling proteins: the unfolding biology of molecular chaperones. Expert Rev. Mol. Med. 2000;2:1–17. doi: 10.1017/S1462399400002015. [DOI] [PubMed] [Google Scholar]
- 34.Horwich A.L., Fenton W.A., Chapman E., Farr G.W. Two families of chaperonin: physiology and mechanism. Annu. Rev. Cell Dev. Biol. 2007;23:115–145. doi: 10.1146/annurev.cellbio.23.090506.123555. [DOI] [PubMed] [Google Scholar]
- 35.Xu Z., Horwich A.L., Sigler P.B. The crystal structure of the asymmetric GroEL-GroES-(ADP)7 chaperonin complex. Nature. 1997;388:741–750. doi: 10.1038/41944. [DOI] [PubMed] [Google Scholar]
- 36.Lin Z., Madan D., Rye H.S. GroEL stimulates protein folding through forced unfolding. Nat. Struct. Mol. Biol. 2008;15:303–311. doi: 10.1038/nsmb.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Motojima F., Chaudhry C., Fenton W.A., Farr G.W., Horwich A.L. Substrate polypeptide presents a load on the apical domains of the chaperonin GroEL. Proc. Natl. Acad. Sci. USA. 2004;101:15005–15012. doi: 10.1073/pnas.0406132101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Leitner A., Joachimiak L.A., Bracher A., Monkemeyer L., Walzthoeni T., Chen B., Pechmann S., Holmes S., Cong Y., Ma B., Ludtke S., Chiu W., Hartl F.U., Aebersold R., Frydman J. The molecular architecture of the eukaryotic chaperonin TRiC/CCT. Structure. 2012;20:814–825. doi: 10.1016/j.str.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muñoz I.G., Yébenes H., Zhou M., Mesa P., Serna M., Park A.Y., Bragado-Nilsson E., Beloso A., de Cárcer G., Malumbres M., Robinson C.V., Valpuesta J.M., Montoya G. Crystal structure of the open conformation of the mammalian chaperonin CCT in complex with tubulin. Nat. Struct. Mol. Biol. 2011;18:14–19. doi: 10.1038/nsmb.1971. [DOI] [PubMed] [Google Scholar]
- 40.Mayer M.P., Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol. Life Sci. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rudiger S., Buchberger A., Bukau B. Interaction of Hsp70 chaperones with substrates. Nat. Struct. Biol. 1997;4:342–349. doi: 10.1038/nsb0597-342. [DOI] [PubMed] [Google Scholar]
- 42.Hartl F.U., Bracher A., Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- 43.Rousseau F., Serrano L., Schymkowitz J.W.H. How evolutionary pressure against protein aggregation shaped chaperone specificity. J. Mol. Biol. 2006;355:1037–1047. doi: 10.1016/j.jmb.2005.11.035. [DOI] [PubMed] [Google Scholar]
- 44.Kampinga H.H., Craig E.A. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 2010;11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seyffer F., Kummer E., Oguchi Y., Winkler J., Kumar M., Zahn R., Sourjik V., Bukau B., Mogk. A. Hsp70 proteins bind Hsp100 regulatory M domains to activate AAA+ disaggregase at aggregate surfaces. Nat. Struct. Mol. Biol. 2012;19:1347–1355. doi: 10.1038/nsmb.2442. [DOI] [PubMed] [Google Scholar]
- 46.Winkler J., Tyedmers J., Bukau B., Mogk. A. Hsp70 targets Hsp100 chaperones to substrates for protein disaggregation and prion fragmentation. J. Cell Biol. 2012;198:387–404. doi: 10.1083/jcb.201201074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Imamoto N., Kose S. Heat-shock stress activates a novel nuclear import pathway mediated by Hikeshi. Nucleus. 2012;3:422–428. doi: 10.4161/nucl.21713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marques C., Guo W., Pereira P., Taylor A., Patterson C., Evans P.C., Shang F. The triage of damaged proteins: degradation by the ubiquitin-proteasome pathway or repair by molecular chaperones. FASEB J. 2006;20:741–743. doi: 10.1096/fj.05-5080fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Whitesell L., Lindquist S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 50.Brychzy A., Rein T., Winklhofer K.F., Hartl F.U., Young J.C., Obermann W.M. Cofactor Tpr2 combines two TPR domains and a J domain to regulate the Hsp70/Hsp90 chaperone system. EMBO J. 2003;22:3613–3623. doi: 10.1093/emboj/cdg362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wegele H., Wandinger S.K., Schmid A.B., Reinstein J., Buchner J. Substrate transfer from the chaperone Hsp70 to Hsp90. J. Mol. Biol. 2006;356:802–811. doi: 10.1016/j.jmb.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 52.Trepel J., Mollapour M., Giaccone G., Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer. 2010;10:537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Soares I.N., Caetano F.A., Pinder J., Roz Rodrigues B., Beraldo F.H., Ostapchenko V.G., Durette C., Schenatto Pereira G., Lopes M.H., Queiroz-Hazarbassanov N., Cunha I.W., Sanematsu P.I., Suzuki S., Bleggi-Torres L.F., Schild-Poulter C., Thibault P., Dellaire G., Martins V.R., Prado V.F., Prado M.A. Regulation of stress-inducible phosphoprotein 1 nuclear retention by PIAS1. Mol. Cell Proteomics. 2013;12:3252–3270. doi: 10.1074/mcp.M113.031005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vashist S., Ng D.T. Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J. Cell Biol. 2004;165:41–52. doi: 10.1083/jcb.200309132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hendershot L., Wei J., Gaut J., Melnick J., Aviel S., Argon Y. Inhibition of immunoglobulin folding and secretion by dominant negative BiP ATPase mutants. Proc. Natl. Acad Sci. USA. 1996;93:5269–5274. doi: 10.1073/pnas.93.11.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gorbatyuk M.S., Gorbatyuk O.S. The molecular chaperone GRP78/BiP as a therapeutic target for neurodegenerative disorders: a mini review. J. Genet. Syndr. Gene Ther. 2013;4:123. doi: 10.4172/2157-7412.1000128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Minamino T., Komuro I., Kitakaze M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circ. Res. 2010;107:1071–1082. doi: 10.1161/CIRCRESAHA.110.227819. [DOI] [PubMed] [Google Scholar]
- 58.Ni M., Lee A.S. ER chaperones in mammalian development and human diseases. FEBS Lett. 2007;581:3641–3651. doi: 10.1016/j.febslet.2007.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Farias M., 3rd, Gorman M.W., Savage M.V., Feigl E.O. Plasma ATP during exercise: possible role in regulation of coronary blood flow. Am. J. Physiol. Heart Circ. Physiol. 2005;288:H1586–H1590. doi: 10.1152/ajpheart.00983.2004. [DOI] [PubMed] [Google Scholar]
- 60.Martin-Ventura J.L., Leclercq A., Blanco-Colio L.M., Egido J., Rossignol P., Meilhac O., Michel J.B. Low plasma levels of HSP70 in patients with carotid atherosclerosis are associated with increased levels of proteolytic markers of neutrophil activation. Atherosclerosis. 2007;194:334–341. doi: 10.1016/j.atherosclerosis.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 61.Bekard I.B., Asimakis P., Bertolini J., Dunstan D.E. The effects of shear flow on protein structure and function. Biopolymers. 2011;95:733–745. doi: 10.1002/bip.21646. [DOI] [PubMed] [Google Scholar]
- 62.Yerbury J.J., Rybchyn M.S., Easterbrook-Smith S.B., Henriques C., Wilson M.R. The acute phase protein haptoglobin is a mammalian extracellular chaperone with an action similar to clusterin. Biochemistry. 2005;44:10914–10925. doi: 10.1021/bi050764x. [DOI] [PubMed] [Google Scholar]
- 63.French K., Yerbury J.J., Wilson M.R. Protease activation of alpha2-macroglobulin modulates a chaperone-like action with broad specificity. Biochemistry. 2008;47:1176–1185. doi: 10.1021/bi701976f. [DOI] [PubMed] [Google Scholar]
- 64.Humphreys D.T., Carver J.A., Easterbrook-Smith S.B., Wilson M.R. Clusterin has chaperone-like activity similar to that of small heat shock proteins. J. Biol. Chem. 1999;274:6875–6881. doi: 10.1074/jbc.274.11.6875. [DOI] [PubMed] [Google Scholar]
- 65.O.R. Gibson, A. Dennis, T. Parfitt, L. Taylor, P.W. Watt, N.S. Maxwell, Extracellular Hsp72 concentration relates to a minimum endogenous criteria during acute exercise-heat exposure, Cell Stress Chaperones (2013). [DOI] [PMC free article] [PubMed]
- 66.Dabbs R.A., Wyatt A.R., Yerbury J.J., Ecroyd H., Wilson M.R. Extracellular chaperones. Top Curr. Chem. 2013;328:241–268. doi: 10.1007/128_2011_262. [DOI] [PubMed] [Google Scholar]
- 67.Sultan A., Bakthisaran R., Rao C.M., Tangirala R. The extracellular chaperone haptoglobin prevents serum fatty acids-promoted amyloid fibril formation of beta2-microglobulin, resistance to lysosomal degradation and cytotoxicity. J. Biol. Chem. 2013;288:32326–32342. doi: 10.1074/jbc.M113.498337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schaer C.A., Deuel J.W., Bittermann A.G., Rubio I.G., Schoedon G., Spahn D.R., Wepf R.A., Vallelian F., Schaer D.J. Mechanisms of haptoglobin protection against hemoglobin peroxidation triggered endothelial damage. Cell Death Differ. 2013;20:1569–1579. doi: 10.1038/cdd.2013.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wyatt A.R., Constantinescu P., Ecroyd H., Dobson C.M., Wilson M.R., Kumita J.R., Yerbury J.J. Protease-activated alpha-2-macroglobulin can inhibit amyloid formation via two distinct mechanisms. FEBS Lett. 2013;587:398–403. doi: 10.1016/j.febslet.2013.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Trougakos I.P., Lourda M., Antonelou M.H., Kletsas D., Gorgoulis V.G., Papassideri I.S., Zou Y., Margaritis L.H., Boothman D.A., Gonos E.S. Intracellular clusterin inhibits mitochondrial apoptosis by suppressing p53-activating stress signals and stabilizing the cytosolic Ku70-Bax protein complex. Clin Cancer Res. 2009;15:48–59. doi: 10.1158/1078-0432.CCR-08-1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Trougakos I.P., Djeu J.Y., Gonos E.S., Boothman D.A. Advances and challenges in basic and translational research on clusterin. Cancer Res. 2009;69:403–406. doi: 10.1158/0008-5472.CAN-08-2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Trougakos I.P. The molecular chaperone apolipoprotein J/clusterin as a sensor of oxidative stress: implications in therapeutic approaches — a mini-review. Gerontology. 2013;59:514–523. doi: 10.1159/000351207. [DOI] [PubMed] [Google Scholar]
- 73.Trougakos I.P., Gonos E.S. Chapter 9: Oxidative stress in malignant progression: the role of Clusterin, a sensitive cellular biosensor of free radicals. Adv. Cancer Res. 2009;104:171–210. doi: 10.1016/S0065-230X(09)04009-3. [DOI] [PubMed] [Google Scholar]
- 74.Bartosz G. Reactive oxygen species: destroyers or messengers? Biochem. Pharmacol. 2009;77:1303–1315. doi: 10.1016/j.bcp.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 75.Khansari N., Shakiba Y., Mahmoudi M. Chronic inflammation and oxidative stress as a major cause of age-related diseases and cancer. Recent Pat. Inflamm. Allergy Drug Discov. 2009;3:73–80. doi: 10.2174/187221309787158371. [DOI] [PubMed] [Google Scholar]
- 76.Jiang F., Zhang Y., Dusting G.J. NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev. 2011;63:218–242. doi: 10.1124/pr.110.002980. [DOI] [PubMed] [Google Scholar]
- 77.Ji L.L., Zhang Y. Antioxidant and anti-inflammatory effects of exercise: role of redox signalling. Free Radic. Res. 2014;48:3–11. doi: 10.3109/10715762.2013.844341. [DOI] [PubMed] [Google Scholar]
- 78.Ward C.W., Prosser B.L., Lederer W.J. Mechanical stretch induced activation of ROS/RNS signaling in striated muscle. Antioxid. Redox Signal. 2014 doi: 10.1089/ars.2013.5517. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dodson M., Darley-Usmar V., Zhang J. Cellular metabolic and autophagic pathways: traffic control by redox signalling. Free Radic. Biol. Med. 2013;63:207–221. doi: 10.1016/j.freeradbiomed.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nathan C., Cunningham-Bussel A. Beyond oxidative stress: an immunologist׳s guide to reactive oxygen species. Nat. Rev. Immunol. 2013;13:349–361. doi: 10.1038/nri3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wen J.W., Hwang J.T., Kelly G.M. Reactive oxygen species and Wnt signalling crosstalk patterns mouse extraembryonic endoderm. Cell Signal. 2012;24:2337–2348. doi: 10.1016/j.cellsig.2012.07.024. [DOI] [PubMed] [Google Scholar]
- 82.Rosc-Schlüter B.I., Häuselmann S.P., Lorenz V., Mochizuki M., Facciotti F., Pfister O., Kuster G.M. NOX2-derived reactive oxygen species are crucial for CD29-induced pro-survival signalling in cardiomyocytes. Cardiovasc. Res. 2012;93:454–462. doi: 10.1093/cvr/cvr348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Leikam C., Hufnagel A., Schartl M., Meierjohann S. Oncogene activation in melanocytes links reactive oxygen to multinucleated phenotype and senescence. Oncogene. 2008;27:7070–7082. doi: 10.1038/onc.2008.323. [DOI] [PubMed] [Google Scholar]
- 84.Sykiotis G.P., Bohmann D. Stress-activated cap׳n’collar transcription factors in aging and human disease. Sci. Signal. 2010;(3):re3. doi: 10.1126/scisignal.3112re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kansanen E., Kuosmanen S.M., Leinonen H., Levonen A.L. The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1:45–49. doi: 10.1016/j.redox.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Holland R., Fishbein J.C. Chemistry of the cysteine sensors in Kelch-like ECH-associated protein 1. Antioxid. Redox Signal. 2010;13:1749–1761. doi: 10.1089/ars.2010.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bryan H.K., Olayanju A., Goldring C.E., Park B.K. The Nrf2 cell defence pathway: Keap1-dependent and -independent mechanisms of regulation. Biochem. Pharmacol. 2013;85:705–717. doi: 10.1016/j.bcp.2012.11.016. [DOI] [PubMed] [Google Scholar]
- 88.Numazawa S., Yoshida T. Nrf2-dependent gene expressions: a molecular toxicological aspect. J. Toxicol. Sci. 2004;29:81–89. doi: 10.2131/jts.29.81. [DOI] [PubMed] [Google Scholar]
- 89.Hensen S.M., Heldens L., van Enckevort C.M., van Genesen S.T., Pruijn G.J., Lubsen N.H. Activation of the antioxidant response in methionine deprived human cells results in an HSF1-independent increase in HSPA1A mRNA levels. Biochimie. 2013;95:1245–1251. doi: 10.1016/j.biochi.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 90.Grimberg K.B., Beskow A., Lundin D., Davis M.M., Young P. Basic leucine zipper protein Cnc-C is a substrate and transcriptional regulator of the Drosophila 26S proteasome. Mol. Cell Biol. 2011;31:897–909. doi: 10.1128/MCB.00799-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tsakiri E.N., Iliaki K.K., Hohn A., Grimm S., Papassideri I.S., Grune T., Trougakos I.P. Diet-derived advanced glycation end products or lipofuscin disrupts proteostasis and reduces life span in Drosophila melanogaster. Free Radic. Biol. Med. 2013;65C:1155–1163. doi: 10.1016/j.freeradbiomed.2013.08.186. [DOI] [PubMed] [Google Scholar]
- 92.Tsakiri E.N., Sykiotis G.P., Papassideri I.S., Terpos E., Dimopoulos M.A., Gorgoulis V.G., Bohmann D., Trougakos I.P. Proteasome dysfunction in Drosophila signals to an Nrf2-dependent regulatory circuit aiming to restore proteostasis and prevent premature aging. Aging Cell. 2013;12:802–813. doi: 10.1111/acel.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dizdaroglu M., Jaruga P. Mechanisms of free radical-induced damage to DNA. Free Radic. Res. 2012;46:382–419. doi: 10.3109/10715762.2011.653969. [DOI] [PubMed] [Google Scholar]
- 94.Mirzaei H., Regnier F. Creation of allotypic active sites during oxidative stress. J. Proteome. Res. 2006;5:2159–2168. doi: 10.1021/pr060021d. [DOI] [PubMed] [Google Scholar]
- 95.Stadtman E.R. Protein oxidation and aging. Free Radic. Res. 2006;40:1250–1258. doi: 10.1080/10715760600918142. [DOI] [PubMed] [Google Scholar]
- 96.Grune T., Shringarpure R., Sitte N., Davies K. Age-related changes in protein oxidation and proteolysis in mammalian cells. J. Gerontol. A Biol. Sci. Med. Sci. 2001;56:B459–B467. doi: 10.1093/gerona/56.11.b459. [DOI] [PubMed] [Google Scholar]
- 97.Gardner H.W. Oxygen radical chemistry of polyunsaturated fatty acids. Free Radic. Biol. Med. 1989;7:65–86. doi: 10.1016/0891-5849(89)90102-0. [DOI] [PubMed] [Google Scholar]
- 98.Moller I.M., Rogowska-Wrzesinska A., Rao R.S. Protein carbonylation and metal-catalyzed protein oxidation in a cellular perspective. J. Proteomics. 2011;74:2228–2242. doi: 10.1016/j.jprot.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 99.Esterbauer H., Schaur R.J., Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 100.Grune T., Davies K.J. The proteasomal system and HNE-modified proteins. Mol. Aspects Med. 2003;24:195–204. doi: 10.1016/s0098-2997(03)00014-1. [DOI] [PubMed] [Google Scholar]
- 101.Dalle-Donne I., Aldini G., Carini M., Colombo R., Rossi R., Milzani A. Protein carbonylation, cellular dysfunction, and disease progression. J. Cell Mol. Med. 2006;10:389–406. doi: 10.1111/j.1582-4934.2006.tb00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Höhn A., Jung T., Grimm S., Catalgol B., Weber D., Grune T. Lipofuscin inhibits the proteasome by binding to surface motifs. Free Radic. Biol. Med. 2011;50:585–591. doi: 10.1016/j.freeradbiomed.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 103.Tsakiri E.N., Iliaki K.K., Höhn A., Grimm S., Papassideri I.S., Grune T., Trougakos I.P. Diet-derived advanced glycation end products or lipofuscin disrupts proteostasis and reduces life span in Drosophila melanogaster. Free Radic. Biol. Med. 2013;65:1155–1163. doi: 10.1016/j.freeradbiomed.2013.08.186. [DOI] [PubMed] [Google Scholar]
- 104.Udan-Johns M., Bengoechea R., Bell S., Shao J., Diamond M.I., True H.L., Weihl C.C., Baloh R.H. Prion-like nuclear aggregation of TDP-43 during heat shock is regulated by HSP40/70 chaperones. Hum. Mol. Genet. 2014;23:157–170. doi: 10.1093/hmg/ddt408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mattoo R.U., Sharma S.K., Priya S., Finka A., Goloubinoff P. Hsp110 is a bona fide chaperone using ATP to unfold stable misfolded polypeptides and reciprocally collaborate with Hsp70 to solubilize protein aggregates. J. Biol. Chem. 2013;288:21399–21411. doi: 10.1074/jbc.M113.479253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Grune T., Catalgol B., Licht A., Ermak G., Pickering A.M., Ngo J.K., Davies K.J. HSP70 mediates dissociation and reassociation of the 26S proteasome during adaptation to oxidative stress. Free Radic. Biol. Med. 2011;51:1355–1364. doi: 10.1016/j.freeradbiomed.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Akerfelt M., Trouillet D., Mezger V., Sistonen L. Heat shock factors at a crossroad between stress and development. Ann. N Y Acad. Sci. 2007;1113:15–27. doi: 10.1196/annals.1391.005. [DOI] [PubMed] [Google Scholar]
- 108.Anckar J., Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu. Rev. Biochem. 2011;80:1089–1115. doi: 10.1146/annurev-biochem-060809-095203. [DOI] [PubMed] [Google Scholar]
- 109.Anckar J., Sistonen L. Heat shock factor 1 as a coordinator of stress and developmental pathways. Adv. Exp. Med. Biol. 2007;594:78–88. doi: 10.1007/978-0-387-39975-1_8. [DOI] [PubMed] [Google Scholar]
- 110.Hietakangas V., Ahlskog J.K., Jakobsson A.M., Hellesuo M., Sahlberg N.M., Holmberg C.I., Mikhailov A., Palvimo J.J., Pirkkala L., Sistonen L. Phosphorylation of serine 303 is a prerequisite for the stress-inducible SUMO modification of heat shock factor 1. Mol. Cell Biol. 2003;23:2953–2968. doi: 10.1128/MCB.23.8.2953-2968.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Westerheide S.D., Anckar J., Stevens S.M., Jr., Sistonen L., Morimoto R.I. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science. 2009;323:1063–1066. doi: 10.1126/science.1165946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ahn S.G., Thiele D.J. Redox regulation of mammalian heat shock factor 1 is essential for Hsp gene activation and protection from stress. Genes Dev. 2003;17:516–528. doi: 10.1101/gad.1044503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Miki H., Funato Y. Regulation of intracellular signalling through cysteine oxidation by reactive oxygen species. J. Biochem. 2012;151:255–261. doi: 10.1093/jb/mvs006. [DOI] [PubMed] [Google Scholar]
- 114.Barford D. The role of cysteine residues as redox-sensitive regulatory switches. Curr. Opin. Struct. Biol. 2004;14:679–686. doi: 10.1016/j.sbi.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 115.Laurindo F.R.M., Pescatore L.A., de Castro Fernandes D. Protein disulfide isomerase in redox cell signaling and homeostasis. Free Radical. Biol. Med. 2012;52:1954–1969. doi: 10.1016/j.freeradbiomed.2012.02.037. [DOI] [PubMed] [Google Scholar]
- 116.Wang C., Yu J., Huo L., Wang L., Feng W., Wang C.C. Human protein-disulfide isomerase is a redox-regulated chaperone activated by oxidation of domain a'. J. Biol. Chem. 2012;287:1139–1149. doi: 10.1074/jbc.M111.303149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kenche H., Baty C.J., Vedagiri K., Shapiro S.D., Blumental-Perry A. Cigarette smoking affects oxidative protein folding in endoplasmic reticulum by modifying protein disulfide isomerise. FASEB J. 2013;27:965–977. doi: 10.1096/fj.12-216234. [DOI] [PubMed] [Google Scholar]
- 118.Wang L., Zhang L., Niu Y., Sitia R., Wang C.C. Glutathione peroxidase 7 utilizes hydrogen peroxide generated by Ero1α to promote oxidative protein folding. Antioxid. Redox Signal. 2013;20:545–556. doi: 10.1089/ars.2013.5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wei P.C., Hsieh Y.H., Su M.I., Jiang X., Hsu P.H., Lo W.T., Weng J.Y., Jeng Y.M., Wang J.M., Chen P.L., Chang Y.C., Lee K.F., Tsai M.D., Shew J.Y., Lee W.H. Loss of the oxidative stress sensor NPGPx compromises GRP78 chaperone activity and induces systemic disease. Mol. Cell. 2012;48:747–759. doi: 10.1016/j.molcel.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kumsta C., Jakob U. Redox-regulated chaperones. Biochemistry. 2009;48:4666–4676. doi: 10.1021/bi9003556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ilbert M., Horst J., Ahrens S., Winter J., Graf P.C., Lilie H., Jakob U. The redox-switch domain of Hsp33 functions as dual stress sensor. Nat. Struct. Mol. Biol. 2007;14:556–563. doi: 10.1038/nsmb1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Evans M.L., Schmidt J.C., Ilbert M., Doyle S.M., Quan S., Bardwell J.C., Jakob U., Wickner S., Chapman M.R. E. coli chaperones DnaK, Hsp33 and Spy inhibit bacterial functional amyloid assembly. Prion. 2011;5:323–334. doi: 10.4161/pri.5.4.18555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Winter J., Ilbert M., Graf P.C., Ozcelik D., Jakob U. Bleach activates a redox-regulated chaperone by oxidative protein unfolding. Cell. 2008;135:691–701. doi: 10.1016/j.cell.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wholey W.Y., Jakob U. Hsp33 confers bleach resistance by protecting elongation factor Tu against oxidative degradation in Vibrio cholera. Mol. Microbiol. 2012;83:981–991. doi: 10.1111/j.1365-2958.2012.07982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rhee S.G., Woo H.A. Multiple functions of peroxiredoxins: peroxidases, sensors and regulators of the intracellular messenger H2O2, and protein chaperones. Antioxid Redox Signal. 2011;15:781–794. doi: 10.1089/ars.2010.3393. [DOI] [PubMed] [Google Scholar]
- 126.Poole L.B. The catalytic mechanism of peroxiredoxins. Subcell Biochem. 2007;44:61–81. doi: 10.1007/978-1-4020-6051-9_4. [DOI] [PubMed] [Google Scholar]
- 127.Knoops B., Loumaye E., Van Der Eecken V. Evolution of the peroxiredoxins. Subcell Biochem. 2007;44:27–40. doi: 10.1007/978-1-4020-6051-9_2. [DOI] [PubMed] [Google Scholar]
- 128.Jang H.H., Lee K.O., Chi Y.H., Jung B.G., Park S.K., Park J.H., Lee J.R., Lee S.S., Moon J.C., Yun J.W., Choi Y.O., Kim W.Y., Kang J.S., Cheong G.W., Yun D.J., Rhee S.G., Cho M.J., Lee S.Y. Two enzymes in one: two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell. 2004;117:625–635. doi: 10.1016/j.cell.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 129.Moon J.C., Hah Y.S., Kim W.Y., Jung B.G., Jang H.H., Lee J.R., Kim S.Y., Lee Y.M., Jeon M.G., Kim C.W., Cho M.J., Lee S.Y. Oxidative stress-dependent structural and functional switching of a human 2-Cys peroxiredoxin isotype II that enhances HeLa cell resistance to H2O2-induced cell death. J. Biol. Chem. 2005;280:28775–28784. doi: 10.1074/jbc.M505362200. [DOI] [PubMed] [Google Scholar]
- 130.Raj L., Ide T., Gurkar A.U., Foley M., Schenone M., Li X., Tolliday N.J., Golub T.R., Carr S.A., Shamji A.F., Stern A.M., Mandinova A., Schreiber S.L., Lee S.W. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011;475:231–234. doi: 10.1038/nature10167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 131.Reddy N., Voorhees P.M., Houk B.E., Brega N., Hinson J.M., Jr., Jillela A. Phase I trial of the HSP90 inhibitor PF-04929113 (SNX5422) in adult patients with recurrent, refractory hematologic malignancies. Clin. Lymphoma Myeloma Leuk. 2013;13:385–391. doi: 10.1016/j.clml.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 132.Goldman J.W., Raju R.N., Gordon G.A., El-Hariry I., Teofilivici F., Vukovic V.M., Bradley R., Karol M.D., Chen Y., Guo W., Inoue T., Rosen L.S. A first in human, safety, pharmacokinetics, and clinical activity phase I study of once weekly administration of the Hsp90 inhibitor ganetespib (STA-9090) in patients with solid malignancies. BMC Cancer. 2013;13:152. doi: 10.1186/1471-2407-13-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Choi H.K., Lee K. Recent updates on the development of ganetespib as a Hsp90 inhibitor. Arch. Pharm. Res. 2012;35:1855–1859. doi: 10.1007/s12272-012-1101-z. [DOI] [PubMed] [Google Scholar]
- 134.Sang J., Acquaviva J., Friedland J.C., Smith D.L., Sequeira M., Zhang C., Jiang Q., Xue L., Lovly C.M., Jimenez J.P., Shaw A.T., Doebele R.C., He S., Bates R.C., Camidge D.R., Morris S.W., El-Hariry I., Proia D.A. Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non-small cell lung cancer. Cancer Discov. 2013;3:430–443. doi: 10.1158/2159-8290.CD-12-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Waza M., Adachi H., Katsuno M., Minamiyama M., Sang C., Tanaka F., Inukai A., Doyu M., Sobue G. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat. Med. 2005;11:1088–1095. doi: 10.1038/nm1298. [DOI] [PubMed] [Google Scholar]
- 136.Chi K.N., Eisenhauer E., Fazli L., Jones E.C., Goldenberg S.L., Powers J., Tu D., Gleave M.E. A phase I pharmacokinetic and pharmacodynamic study of OGX-011, a 2'-methoxyethyl antisense oligonucleotide to clusterin, in patients with localized prostate cancer. J. Natl. Cancer Inst. 2005;97:1287–1296. doi: 10.1093/jnci/dji252. [DOI] [PubMed] [Google Scholar]
- 137.Firczuk M., Gabrysiak M., Barankiewicz J., Domagala A., Nowis D., Kujawa M., Jankowska-Steifer E., Wachowska M., Glodkowska-Mrowka E., Korsak B., Winiarska M., Golab J. GRP78-targeting subtilase cytotoxin sensitizes cancer cells to photodynamic therapy. Cell Death Dis. 2013;4:e741. doi: 10.1038/cddis.2013.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wang M., Ye R., Barron E., Baumeister P., Mao C., Luo S., Fu Y., Luo B., Dubeau L., Hinton D.R., Lee A.S. Essential role of the unfolded protein response regulator GRP78/BiP in protection from neuronal apoptosis. Cell Death Differ. 2010;17:488–498. doi: 10.1038/cdd.2009.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gorbatyuk M.S., Shabashvili A., Chen W., Meyers C., Sullivan L.F., Salganik M., Lin J.H., Lewin A.S., Muzyczka N., Gorbatyuk O.S. Glucose regulated protein 78 diminishes α-synuclein neurotoxicity in a rat model of Parkinson disease. Mol. Ther. 2012;20:1327–1337. doi: 10.1038/mt.2012.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fujimoto M., Takaki E., Hayashi T., Kitaura Y., Tanaka Y., Inouye S., Nakai A. Active HSF1 significantly suppresses polyglutamine aggregate formation in cellular and mouse models. J. Biol. Chem. 2005;280:34908–34916. doi: 10.1074/jbc.M506288200. [DOI] [PubMed] [Google Scholar]
- 141.Fujikake N., Nagai Y., Popiel H.A., Okamoto Y., Yamaguchi M., Toda T. Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J. Biol. Chem. 2008;283:26188–26197. doi: 10.1074/jbc.M710521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Argyropoulou A., Aligiannis N., Trougakos I.P., Skaltsounis A.L. Natural compounds with anti-ageing activity. Nat. Prod. Rep. 2013;30:1412–1437. doi: 10.1039/c3np70031c. [DOI] [PubMed] [Google Scholar]