

Abstract
G protein-coupled receptor desensitization and trafficking are important regulators of opioid receptor signaling that can dictate overall drug responsiveness in vivo. Furthermore, different μ-opioid receptor (μOR) ligands can lead to varying degrees of receptor regulation, presumably because of distinct structural conformations conferred by agonist binding. For example, morphine binding produces a μOR with low affinity for β-arrestin proteins and limited receptor internalization, whereas enkephalin analogs promote robust trafficking of both β-arrestins and the receptors. Here, we evaluate μOR trafficking in response to activation by a novel μ-selective agonist derived from the naturally occurring plant product, salvinorin A. It is interesting that this compound, termed herkinorin, does not promote the recruitment of β-arrestin-2 to the μOR and does not lead to receptor internalization. Moreover, whereas G protein-coupled receptor kinase overexpression can promote morphine-induced β-arrestin interactions and μOR internalization, such manipulations do not promote herkinorin-induced trafficking. Studies in mice have shown that β-arrestin-2 plays an important role in the development of morphine-induced tolerance, constipation, and respiratory depression. Therefore, drugs that can activate the receptor without recruiting the arrestins may be a promising step in the development of opiate analgesics that distinguish between agonist activity and receptor regulation and may ultimately lead to therapeutics designed to provide pain relief without the adverse side effects normally associated with the opiate narcotics.
G protein-coupled receptor (GPCR) internalization as a means of receptor regulation has been associated with conditions as wide-ranging as opioid analgesia to opioid addiction (Alvarez et al., 2001; Bohn et al., 2004b; Connor et al., 2004; Gainetdinov et al., 2004; Raehal and Bohn, 2005). Agonist-induced, GPCR kinase (GRK)-mediated phosphorylation, subsequent β-arrestin protein binding, the assembly of clathrin coated vesicles, and vesicular internalization describe a general paradigm for GPCR internalization (Pierce and Lefkowitz, 2001; Shenoy and Lefkowitz, 2003). The μ-opioid receptor (μOR), however, has been shown to be differentially regulated depending on agonist occupancy. For example, whereas both morphine and etorphine are agonists at the μOR and can promote receptor desensitization and analgesic tolerance, morphine seems to be much less effective in promoting receptor phosphorylation, β-arrestin recruitment, and μOR internalization than etorphine (Whistler and von Zastrow, 1998; Zhang et al., 1998; Bohn et al., 2004a). Each of these limitations can be overcome by overexpression of GRK2 in cell culture, suggesting that the agonist occupancy promotes different receptor conformations that can determine overall receptor regulation in a GRK-dependent manner (Zhang et al., 1998; Bohn et al., 2004a).
The β-arrestin proteins, namely β-arrestin-1 (βarr1) and β-arrestin-2 (βarr2), play an important role in GPCR trafficking (Pierce and Lefkowitz 2001). Although the morphine-bound μOR seems to be a poor substrate for βarr2 binding, a combination of both animal and cellular studies suggests that βarr2 is prominently involved in μOR regulation. Mice that lack βarr2 display enhanced and prolonged morphine analgesia with limited development of tolerance (Bohn et al., 1999, 2000b, 2002; Przewlocka et al., 2002). It is somewhat of a surprise that these animals are not as susceptible to the morphine-induced side effects, including constipation and respiratory suppression (Raehal et al., 2005). Therefore, the μOR-βarr2 interactions may represent a point at which agonist-directed signaling events diverge (Raehal and Bohn, 2005; Bruns et al., 2006).
Opioid agonists that do not induce receptor-βarr2 interactions or the subsequent receptor internalization may be promising candidates for dissociating the therapeutic effects from the unwanted side effects that usually confound opiate therapy. Salvinorin A, a diterpene isolated from Salvia divinorum, has been classified as a potent opioid receptor agonist (Ortega and Manchand, 1992; Roth et al., 2002) that produces less receptor trafficking (Wang et al., 2005). Salvinorin A is selective for κ-opioid receptors (κOR) and acts as a potent and efficacious agonist in G protein coupling and adenylyl cyclase assays; however, it promotes 40-fold less efficient κOR internalization than what is seen by the typical κ agonists including U50,488H (Roth et al., 2002; Chavkin et al., 2004; Wang et al., 2005). The chemical structure of the compound is unique in that it lacks a basic nitrogen atom, a property that is conserved among all other known opioid receptor agonists and believed to play a critical role in ligand binding. Together, these findings suggest that the κOR conformation induced by salvinorin A binding is conducive to G protein-mediated signal transduction but resistant to internalization-mediated regulation.
Chemical derivatives of salvinorin A were described recently, and here we report on one such derivative that has greater affinity for the μOR over the κOR (μ/κ = 0.13 from Harding et al., 2005). This unique compound, termed, herkinorin [(2S,4aR,6aR,7R,9S,10aS,10bR)-9-(benzoyloxy)-2-(3-furanyl)dodecahydro-6a,10b-dimethyl-4,10-dioxo-2H-naphtho-[2,1-c]pyran-7-carboxylic acid methyl ester; Harding et al., 2005] activates the μOR, leads to very little receptor phosphorylation, does not recruit βarr2, and does not lead to receptor internalization, even in the presence of overexpressed GRK2. Therefore, compounds based on this structure may provide insight into the mechanisms of ligand-directed μOR regulation and may represent a step in the development of opiate analgesics that promote pain relief with limited adverse effects.
Materials and Methods
Drugs
[d-Ala2,N-MePhe4,Gly5-ol]enkephalin (DAMGO; Tocris, Ellisville, MO), morphine sulfate, and naloxone (Sigma, St. Louis, MO) were prepared as 10 mM stocks in phosphate-buffered saline. Herkinorin (referred to as “analog 13” in Harding et al., 2005) was prepared in DMSO as a 10 mM stock. Dilutions were made directly into minimal essential media (without phenol red for confocal imaging). Herkinorin is subject to degradation; therefore, drugs were produced in small batches, protected from light, and not stored in DMSO or extensively as a powder.
Phospho-ERK1/2 Immunoblot Assay
HEK-293 cells stably expressing a hemagglutinin (HA; N terminus)-tagged mouse μOR (~2 pmol/mg of membrane protein) were assessed for agonist-induced ERK1/2 phosphorylation. Cells were serum-starved at 37°C under 5% CO2 for 30 min before drug treatment. Cells were stimulated for the times indicated, and cell lysates were prepared on ice in lysing buffer [20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 2 mM EDTA, 0.1% SDS, 1% NP-40, 0.25% deoxycholate, 1 mM sodium orthovanadate, 1 mM PMSF, 1 mM NaF, with Complete Mini, EDTA-free protease inhibitor cocktail tablet (Roche Diagnostics, Indianapolis, IN)]. Protein levels were determined with the use of the detergent-compatible protein assay system (Bio-Rad Laboratories, Hercules, CA), and 20 μg of protein per lane was resolved by 1-D gel electrophoresis on 10% Bis-Tris gels (Bio-Rad Laboratories or Invitrogen, Carlsbad, CA). Proteins were transferred to polyvinylidene fluoride (PVDF) membranes (Immobilon P; Millipore, Billerica, MA) and immunoblotted for phosphorylated ERK1/2 (p-ERK E-4; Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Blots were stripped and blotted for total ERK1/2 levels (p44/42 MAP kinase antibody; Cell Signaling Technology, Danvers, MA), which were used to normalize the overall phosphorylation of ERK1/2 between samples (Bohn et al., 2000b). Chemiluminescence was detected and quantified using the Kodak 2000R imaging system (Eastman Kodak Company, Rochester, NY) and Prism software (GraphPad Software, Inc., San Diego, CA). Mouse embryonic fibroblasts expressing (MEF-WT) and lacking endogenous β-arrestins 1 and 2 (MEF βarr1&2-KO) were kindly provided by Dr. Robert Lefkowitz (Kohout et al., 2001; Bohn et al., 2004a). The μOR expression in MEFs was obtained by using retroviral expression vectors. ERK activation studies were performed in parallel to trafficking studies to assure activity of the agonists.
Phosphorylated μOR Immunoprecipitation
HEK-293 cells stably expressing HA-tagged μOR were serum-starved for 15 min and then treated for 10 min with saline, DAMGO (1 μM), morphine (10 μM), or herkinorin (10 μM). A mock transfection treated with DAMGO (1 μM) was included as a control. Cells were washed on ice and then collected in 250 μl of lysing buffer (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 2 mM EDTA, 0.1% SDS, 1% NP-40, 0.25% deoxycholate, 1 mM sodium orthovanadate, 1 mM PMSF, 1 mM NaF, and protease inhibitor cocktail; Roche Diagnostics). Cell lysates were collected and solubilized at 4°C for 1 h. The insoluble fraction was removed with a 20,000g spin at 4°C for 30 min. Protein content was measured using Bio-Rad Laboratories detergent-compatible protein assay, and samples were diluted to equal concentrations. Equal amounts of protein (700–1000 μg) or buffer only (for “no protein” control) were incubated with 70 μl of a 1:1 suspension of monoclonal anti-HA-agarose beads (Sigma, St. Louis, MO) overnight at 4°C with rotation. The immunoprecipitated complex was collected and washed according to the manufacturer’s instructions. Proteins were eluted from anti-HA-agarose in 30 μl of 4× XT Sample buffer (Bio-Rad Laboratories) (62.5 mM Tris-HCl, pH 6.8, 25% glycerol, 2% SDS, and 0.01% bromphenol blue) with 5% β-mercaptoethanol at 95°C for 4 min. Samples were resolved on 10% Bis-Tris XT Precast Gels (Bio-Rad Laboratories) and transferred to PVDF membranes (Immobilon-P; Millipore). Membranes were incubated with a phospho-μOR antibody (1:500) that recognizes phosphorylated serine 375 of the mouse μOR (p-μOR Ser375; Cell Signaling). Chemiluminescence was visualized using a Kodak 2000R image station. Membranes were stripped and blotted with a primary antibody against the C terminus of the μOR (1:1000; Sigma, St. Louis, MO) to determine total levels of receptor per lane. Densitometry was assessed using the Kodak imaging software, and p-μOR levels were normalized to the total receptor per lane and to the degree of stimulation compared with saline-treated controls in each blot. Data were analyzed using Prism software (GraphPad Software).
Cross-Linking and Coimmunoprecipitation
HEK-293 cells stably expressing the μOR tagged at the N terminus with HA were used, and the procedure is based on those described by Shenoy et al., (2006) and Gesty-Palmer et al., (2006). Cells were washed with phosphate-buffered saline (PBS) + 10 mM HEPES and then incubated in PBS-HEPES buffer for 20 min at 37°C. Cells were then treated with vehicle (0.1% DMSO), 1 μM DAMGO, or 10 μM herkinorin for 5 min. The cross-linking reagent, dithiobis(succinimidylpropionate) (DSP; Pierce Chemical, Rockford, IL) was prepared in DMSO and administered drop-wise to the plates (2 mM final DSP concentration at <10% DMSO); plates were then kept at room temperature for 20 min with constant agitation. The cross-linking reaction was stopped by the addition of 1 M Tris-HCl, pH 7.4, to give a final concentration of 50 mM. Cells generally came off of the plates with the agitation, so they were collected and centrifuged at 2000 rpm and then washed four times in Tris-buffered saline. Cells were resuspended in lysing buffer [50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% NP-40, 1 mM sodium orthovanadate, 1 mM PMSF, 1 mM NaF, and protease inhibitor pellet (1/10 ml; Roche)] and solubilized overnight at 4°C with rotation. Lysates were cleared by 12,000 rpm centrifugation and then immunoprecipitated with anti-HA conjugated agarose beads (Sigma) for 2 h at 4°C with rotation. Proteins were eluted in SDS sample buffer (Bio-Rad Laboratories) with 5% 2-mercaptoethanol and 100 mM dithiothreitol by boiling for 10 min at 100°C. Proteins were resolved by SDS-PAGE under denaturing conditions and transferred to PVDF membranes. The A1CT antibody was kindly provided by Dr. Robert Lefkowitz and was used to detect β-arrestins 1 and 2 (Gesty-Palmer et al., 2006; Shenoy et al., 2006). Enhanced chemiluminescence was used to detect bands as described above. Controls included reprobing the blots for equal pull-down of μOR, lysates of HEK-293 cells transfected with HA-μOR for μOR immunoblotting, and MEF-WT or MEF βarr1&2-KO for β-arrestin immunoblotting (Kohout et al., 2001).
Cellular Trafficking
HEK-293 cells were transiently transfected with combinations of the following cDNA as indicated in the figure legends: hemagglutinin (HA-N terminus)-tagged mouse μOR (10 μg of cDNA); β-arrestin2 tagged with green fluorescent protein (βarr2-GFP) (2 μg of cDNA); mouse μOR tagged at the C terminus with yellow fluorescent protein (μOR-YFP); and GRK2 (5 μg of cDNA). In some cases, cells stably expressing the μOR were used, and no differences were observed compared with transiently transfected cells. Cell media were changed 10 to 20 min before the addition of drug to serum-free and phenol red-free MEM. Cells were visualized using a confocal microscope with green-helium neon and argon lasers (Olympus, Tokyo, Japan). Multiple cells were recorded per dish after more than four separate transfection experiments; representative cells are shown. Transfection by electroporation and confocal imaging were performed as described previously (Bohn et al., 2004a).
Immunofluorescence of Cell Surface Receptors
Stably transfected HA-N terminus-tagged μOR-expressing HEK-293 cells were plated on a 96-well optical plate (collagen coated) and treated with either DAMGO or herkinorin for the times indicated. Cells were washed two times with MEM, fixed with 4% paraformaldehyde at room temperature for 20 min, and blocked in MEM plus 5% goat serum for 1 h. Cells were then incubated with an anti-HA antibody (monoclonal clone 12CA5; Roche Diagnostics) at 1:500 dilution in blocking buffer overnight at 4°C. Cells were washed three times in blocking buffer and incubated for 1 h with the Alexa Fluor 488 goat anti-mouse secondary antibody (Molecular Probes/Invitrogen, Carlsbad, CA) at room temperature. After three washes in phosphate-buffered saline, immunofluorescence was assessed using a Fusion Plate Reader (PerkinElmer Life and Analytical Sciences, Boston, MA). Data are normalized to the controls in which no agonists were added. Nonspecific secondary antibody interactions were subtracted from each point. Images were taken after the experiment using a 10× objective to ensure that cells had remained uniformly attached; images were also taken of these cells. If cells came off the plate, the measurements were deleted for that well. Cells were also treated in parallel to this experiment in small confocal dishes. After 60 min of treatment, cells were processed in the same manner as above, and immunofluorescent images were taken using a 40× objective via confocal microscopy.
Biotinylated Receptor Internalization
HEK-293 cells stably expressing the μOR tagged at the N terminus with HA were used for cell surface biotinylation experiments based on methods described previously (Cao et al., 1998). Cells were washed once with ice-cold PBS, and then sulfo-NHS-SS-biotin (Pierce, 600 μg/ml in PBS) was added to each dish, and cells were incubated for 30 min at 4°C. Cells generally came off of the plates and were therefore collected, combined, and spun down at 2000 rpm for all subsequent washes. Cells were washed in Tris-buffered saline (50 mM Tris-HCl, pH 7.4, and 150 mM NaCl) to quench unreacted biotin. Cells were divided into identical aliquots to receive the following treatments. For drug treatment, cells were resuspended in MEM containing either vehicle (0.1% DMSO), 1 μM DAMGO, or 10 μM herkinorin and incubated for 1 h in a 37°C water bath. After incubation, surface biotinylation was stripped away using glutathione stripping buffer (50 mM glutathione, 75 mM NaCl, 75 mM NaOH, and 10% fetal bovine serum, in H2O) for 15-min incubation on ice. Glutathione is quenched with 15-min incubation on ice in iodoacetamide buffer (50 mM iodoacetamide, and 1% bovine serum albumin in PBS, pH 7.4). Cells were resuspended in Triton X-100 extraction buffer [10 mM Tris, pH 7.5, 120 mM NaCl, 25 mM KCl, 1 mM PMSF, 1 protease inhibitor pellet/10 ml (Roche), 1 mg/ml iodoacetamide, and 0.5% Triton X-100] and solubilized on ice for 2 h. Lysates were cleared by 12,000 rpm spin and then added to avidin-conjugated agarose beads (Pierce) for immunoprecipitation overnight at 4°C with rotation. Immunoprecipitates were centrifuged at 12,000 rpm and washed three times with Triton X-100 extraction buffer and once with PBS plus 0.1% SDS. Immunoprecipitates were washed, and proteins were eluted and resolved as described in the coimmunoprecipitation experiments. Immunoblots were performed using the μOR C-terminal directed antibody (Neuromics, Edina, MN), donkey anti-rabbit secondary antibody, and enhanced chemiluminescence detection as described above. Several controls were performed, and they are as follows: 1) “100%” refers to total surface receptors and represents cells lysed directly after biotinylation and washes; 2) “strip” refers to cells that have been biotinylated and then stripped, and this serves as a control for the effectiveness of the glutathione stripping buffer; 3) “mock” is the same as 100% but for cells that are stably mock-transfected with empty vector (no μOR); and 4) “no protein” refers to immunoprecipitation in the absence of cellular lysate.
Statistics
Statistical analyses were performed using Prism software (GraphPad Software), and the specific tests used are presented in the figure legends.
Results
The chemical synthesis of herkinorin from salvinorin A has been described previously; the chemical structures of herkinorin and salvinorin A are presented in Fig. 1 for reference (Harding et al., 2005). Receptor binding assays revealed that herkinorin has a greater affinity for μOR over κOR and very little affinity for the δOR; G protein-coupling assays determined that herkinorin preferentially activates the μOR (Harding et al., 2005). To assess herkinorin agonist activity in parallel with the current studies, we used the phosphorylation of the downstream MAP kinases (ERK1/ERK2) as an indicator of receptor activation. In Fig. 2, we show that like other μOR agonists, such as the enkephalin analog, DAMGO, herkinorin dose-dependently activates the MAP kinases ERK1/2 in a μOR antagonist-reversible manner as shown in Fig. 2C (Belcheva et al., 1998). Similar results were also obtained using Chinese hamster ovary cells expressing the human μOR (data not shown). A comparison of ERK phosphorylation between DAMGO and herkinorin reveals comparable potencies, although there is a tendency for herkinorin to continue to activate ERK past the efficacy observed with DAMGO (Fig. 2). To test whether β-arrestins are required to mediate μOR signaling to ERK, we examined ERK1/2 phosphorylation in MEFs that lack both β-arrestin1 and β-arrestin2 and found that ERK1/2 could be activated in the absence of β-arrestins (Fig. 2D) (Kohout et al., 2001; Bohn et al., 2004b).
Fig. 1.
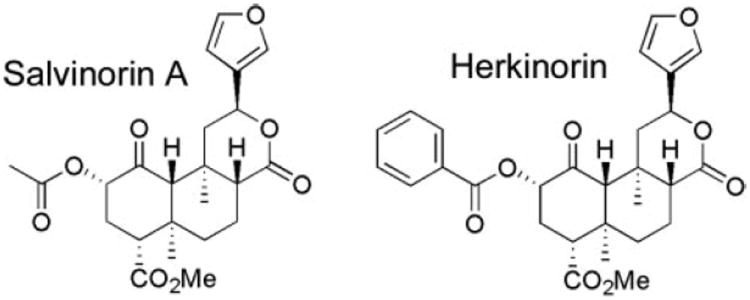
Chemical structures of salvinorin A and herkinorin.
Fig. 2.
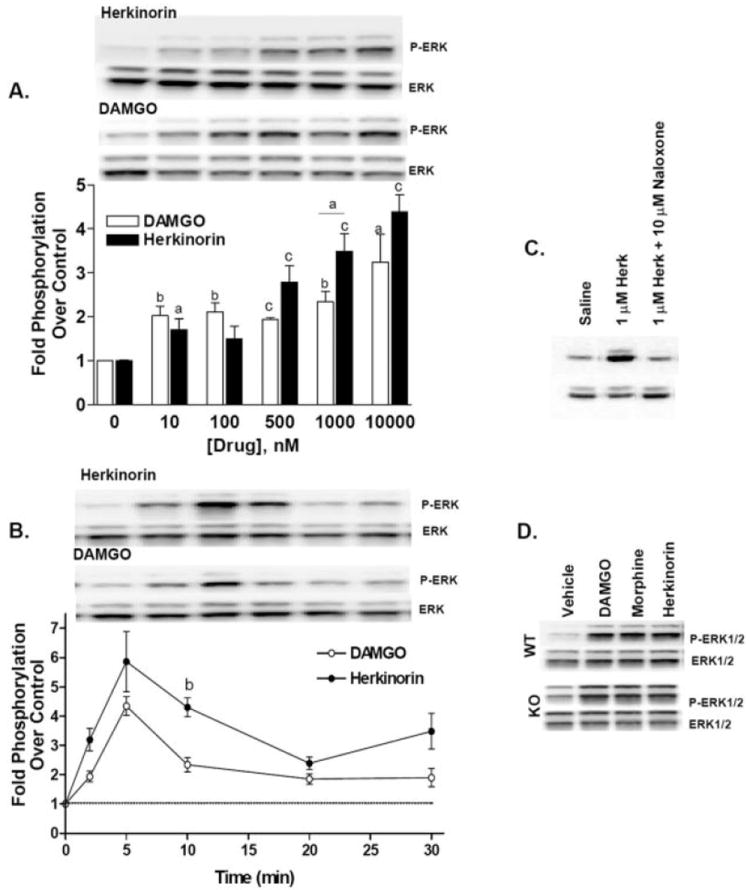
Herkinorin promotes ERK1/2 phosphorylation. Herkinorin and DAMGO induce ERK1/2 phosphorylation in HEK-293 cells stably expressing a hemagglutinin (HA-N terminus)-tagged mouse μOR. A, dose-response of herkinorin and DAMGO activation of ERK. Stimulation was for 10 min at the concentrations indicated. The dose-response comparison reveals a difference between DAMGO and herkinorin as assessed by two-way ANOVA: p < 0.05 (n = 2–6, n = 2 for 100 nM). Statistical legend: a, p < 0.05; b, p < 0.01; c, p < 0.0001 versus vehicle control, or between groups indicated by a bar, Student’s t test. B, time course of herkinorin- and DAMGO-induced phosphorylation of ERK. Cells were treated with 10 μM herkinorin or 1 μM DAMGO at the times indicated. Phopsho-ERK1/2 bands were analyzed by densitometry and normalized to total ERK levels (bottom bands) and densitometry is presented as fold stimulation (mean ± S.E.M.) over saline-treated controls. The time course comparison reveals a difference between DAMGO and herkinorin as assessed by two-way ANOVA, p < 0.05 (n = 3–5) and a significant difference between the compounds at the 10-min point. b, p < 0.01 Student’s t test, (n = 3–5). C, pretreatment with naloxone (10 μM) during the 30-min serum starvation blocked 1 μM herkinorin-induced ERK activation. D, activation of μOR in the absence of β-arrestins (βarr1&2-KO MEFs) still leads to ERK1/2 phosphorylation. Immunoblots were performed as described above using lysates from MEFs.
Because agonist activation of many GPCRs leads to GRK-mediated receptor phosphorylation, β-arrestin recruitment, and internalization, we compared herkinorin with other opiate agonists for their ability to regulate the μOR in HEK-293 cells, a model system routinely used to study opioid receptor regulation and trafficking. Although both morphine and DAMGO activate the μOR, morphine is much less effective in promoting receptor phosphorylation (Fig. 3), which is believed to underlie the decreased receptor trafficking that results from morphine binding (Whistler and von Zastrow, 1998; Zhang et al., 1998; Bailey et al., 2003; Bohn et al., 2004a; Schulz et al., 2004; Koch et al., 2005). Herkinorin resembles morphine in that it does not promote robust phosphorylation of the μOR at serine 375 (Fig. 3).
Fig. 3.
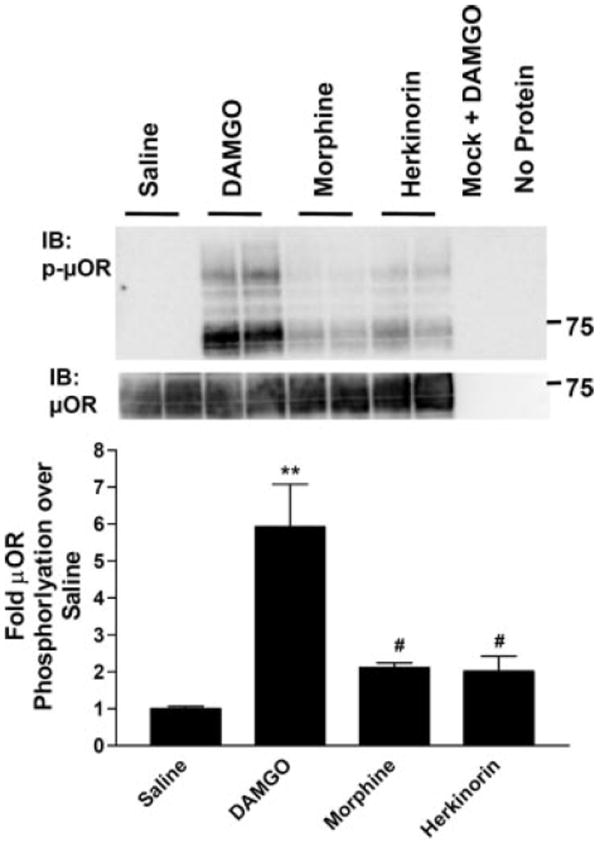
μ-Opioid receptor phosphorylation at serine 375 after agonist treatment. HEK-293 cells stably expressing μOR were treated with saline, 1 μM DAMGO, 10 μM morphine, or 10 μM herkinorin for 10 min. The receptor was immunoprecipitated from cell lysates using an anti-HA antibody-agarose bead complex. Representative Western blots using antibodies that recognize the μOR phosphorylated at serine 375 (top) or the total μOR (C-terminal antibody) from the same blot (bottom) are shown. Densitometric analyses of three independent experiments performed in duplicate or triplicate were normalized to total receptor per lane and expressed as fold stimulation over saline control for each blot. Data are presented as the mean ± S.E.M. **, p < 0.001; #, p < 0.01 versus DAMGO one-way ANOVA analysis of variance with Bonferroni’s multiple comparison test (n = 4–6).
Agonist-induced βarr2 translocation in response to morphine, DAMGO, or herkinorin was assessed by confocal imaging of live cells. HEK-293 cells expressing both the μOR and βarr2-GFP were stimulated with agonist for the times indicated. Images were collected from the same plate of cells before and after drug treatment. Shown here are representative images at the indicated time points. The translocation of βarr2-GFP to the receptor is robust with DAMGO compared with morphine (Fig. 4A) (Zhang et al., 1998; Bohn et al., 2004a). Herkinorin resembles morphine in that it does not seem to induce βarr2-GFP translocation to the μOR (Fig. 4A). Cells were also observed at earlier (real-time video every 30 s for the first 10 min) and later time points (15, 20, 30, 60, and 120 min), but no translocation was observed with morphine or herkinorin under either condition (data not shown). To assess whether herkinorin could recruit endogenous β-arrestins to the receptors, we stimulated the μOR-expressing cells with drug, stabilized the β-arrestin receptor complex using a cross-linking reagent, immunoprecipitated the HA-tagged μOR, and blotted for β-arrestins using the A1CT β-arrestin antibody (Gesty-Palmer et al., 2006; Shenoy et al., 2006). Although DAMGO promoted μOR-β-arrestin interactions, vehicle and herkinorin did not (Fig. 4B).
Fig. 4.
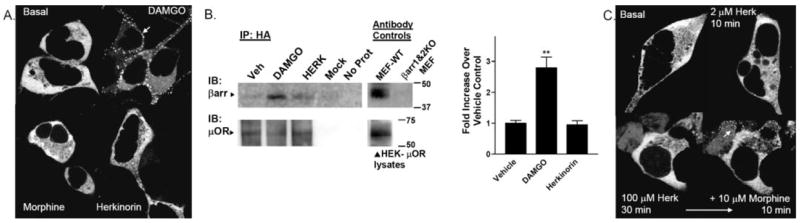
Agonist-induced β-arrestin interactions with μOR in HEK-293 cells. HEK-293 cells transiently transfected with μOR and βarr2-GFP were imaged in real time after agonist treatment at room temperature. The cytosolic distribution of βarr2-GFP is shown in the untreated cells in the top left. A, βarr2-GFP translocation to μOR in HEK-293 cells. DAMGO (1 μM) treatment leads to βarr2-GFP translocation within 5 min (white arrow, punctate accumulation at membrane) whereas morphine (10 μM, 10 min) does not. Herkinorin (2 μM, 10 min) does not induce βarr2-GFP translocation. B, coimmunoprecipitation of β-arrestins and μOR after drug treatment. Cells were treated with vehicle (0.1% DMSO), 1 μM DAMGO, or 10 μM herkinorin for 5 min. Cells were then cross-linked using a cell-permeable cross-linking reagent (DSP). Cell lysates were immunoprecipitated by anti-HA-conjugated agarose beads, and proteins were resolved by SDS-PAGE under denaturing conditions. Blots (left), immunoblotting was performed using the β-arrestin antibody (A1CT); blots were stripped and then reprobed with the μOR antibody (Neuromics). Representative blots are shown. Mock refers to HEK cells transfected with empty vector. “No protein” contained no cell lysate in the immunoprecipitation. Antibody controls were performed on lysates run on the same gel as those shown for the immunoprecipitation. Densitometry (right), densitometry was measured from a total of three to four samples of each treatment prepared on 2 separate days. Shown are the means ± S.E.M. **, p < 0.01 versus WT or herkinorin (Herk), Student’s t test. C, βarr2-GFP translocation to μOR in HEK-293 cells overexpressing GRK2. Herkinorin does not promote βarr2-GFP translocation at 2 μM after 10 min or at 100 μM after 30 min. The same cells were treated with morphine (10 μM, 10 min) and βarr2-GFP translocates demonstrating that these cells do overexpress GRK2 because morphine does not induce visible translocation otherwise.
Morphine can induce βarr2-GFP translocation when GRK2 is overexpressed in HEK-293 cells (Zhang et al., 1998; Bohn et al., 2004a). Although this cellular manipulation is sufficient to promote morphine-induced translocation, the overexpression of GRK2 does not promote herkinorin-induced βarr2-GFP translocation to the μOR (Fig. 4C). To be certain that the cells imaged had been transfected with GRK2, morphine was applied after herkinorin treatment, and translocation was observed. Morphine does not normally induce βarr2-GFP translocation in the absence of GRK2 overexpression (Fig. 4A); therefore, a positive response to morphine was taken as evidence that the cell was overexpressing GRK2. Conversely, a combination of morphine and herkinorin was not sufficient to induce translocation in cells that did not overexpress GRK2 (data not shown).
Because β-arrestin-GPCR interactions precede receptor vesicular internalization, it is not surprising that DAMGO leads to robust receptor internalization, whereas morphine does not (Fig. 5A) (Whistler and von Zastrow, 1998; Zhang et al., 1998; Bailey et al., 2003; Bohn et al., 2004a; Schulz et al., 2004; Koch et al., 2005). HEK-293 cells expressing YFP-tagged μOR were treated with each of the indicated opioids to assess receptor trafficking. Herkinorin, like morphine, does not promote μOR-YFP internalization in the HEK-293 cells (Fig. 5A). The lack of herkinorin-induced receptor internalization is further demonstrated by immunolabeling the N terminus of receptors in intact, nonpermeabilized HEK-293 cells after drug treatment over 2 h. Although DAMGO-treated cells show a loss of ~50% of cell surface receptors, the herkinorin-treated cells maintain the same level of cell surface receptor expression over the 2-h time period (Fig. 5, B and C). A cell surface protein biotinylation approach was also used to assess receptor internalization after drug treatment (Fig. 5D) (Cao et al., 1998). DAMGO induced a significant increase in internalized receptors, whereas vehicle and herkinorin did not.
Fig. 5.
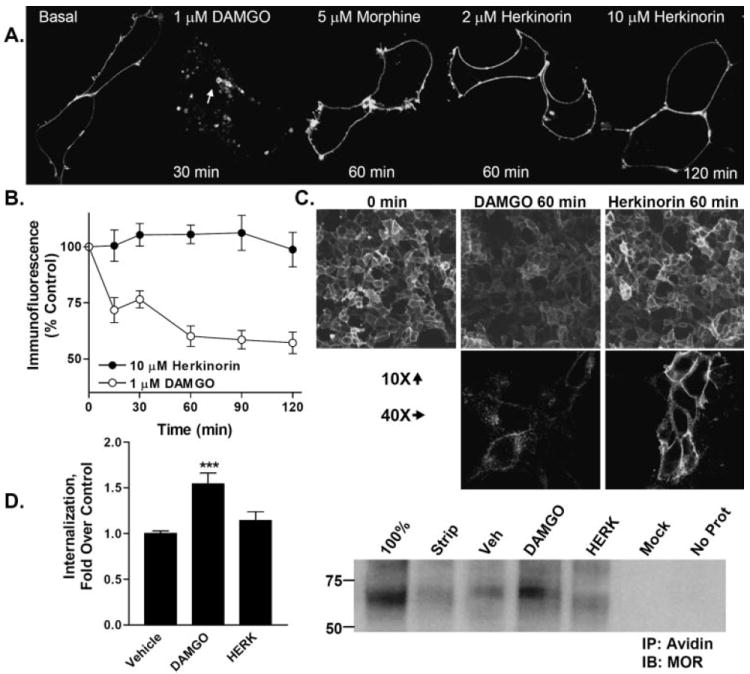
Agonist-induced internalization of μOR-YFP in HEK-293 cells. A, HEK-293 cells were transiently transfected with μOR (2 μg of cDNA) tagged at the C terminus with YFP. Cells were treated with the agonists indicated. Internalization can be seen after DAMGO treatment as indicated by the appearance of donut-like intracellular vesicles (white arrow) and the disappearance of membrane receptor localization as seen in the basal panel. Herkinorin and morphine do not induce receptor internalization. B, cell surface expression of receptor as determined by remaining N-terminal HA-immunoreactivity in nonpermeabilized cells after drug treatment. DAMGO, but not herkinorin, leads to a loss of cell surface expression after 60 to 120 min of drug treatment. Two-way ANOVA analysis reveals that the curves differ (p < 0.001) and that the DAMGO-treated cells display fewer surface receptors at each time point as determined by Bonferroni post hoc analysis (p < 0.001 at each time point after 0). C, immunocytochemistry of HA-MOR expression on cell surface after 60 min of agonist treatment. Top, 10× objective, images are taken directly from the 96-well plates assayed in 5B. Bottom, 40× objective images in parallel dishes. D, internalization of receptors as determined by cell surface biotinylation assay. Right blot, cell surface proteins were biotinylated and treated with indicated drug or vehicle (vehicle, 0.1% DMSO; 1 μM DAMGO or 10 μM herkinorin) for 1 h. Remaining surface proteins were then subjected to glutathione cleavage of biotin. Cells were then lysed, and remaining biotinylated proteins were immunoprecipitated with avidin-conjugated agarose beads. Proteins were resolved via SDS-PAGE under reducing conditions. Immunoblotting was performed with the C-terminal specific μOR antibody (Neuromics); 100% represents cells that were not stripped with glutathione; Strip represents remaining surface-biotinylated μOR after glutathione treatment and without drug treatment. Mock represents DAMGO treated HEK-293 cells that were transfected with empty vector. No Prot represents no cell lysate present in the immunoprecipitation. Densitometry (left), densitometry was measured from a total of five to seven samples of each treatment prepared on 3 separate days. ***, p < 0.001 versus vehicle; DAMGO induced significantly more internalization than herkinorin, p < 0.05. Herkinorin did not differ from vehicle, p > 0.1; Student’s t test.
The overexpression of GRK2 can promote morphine-induced μOR internalization as it does βarr2-GFP translocation (Fig. 4C), suggesting that the agonist occupancy promotes a conformation that differs between DAMGO and morphine at the level of GRK-mediated phosphorylation of the agonist-bound receptor (Zhang et al., 1998; Bohn et al., 2004a). However, overexpression of GRK2 is insufficient to promote herkinorin-induced μOR internalization (Fig. 6A), further demonstrating the differences between morphine and herkinorin with respect to agonist-induced μOR trafficking.
Fig. 6.
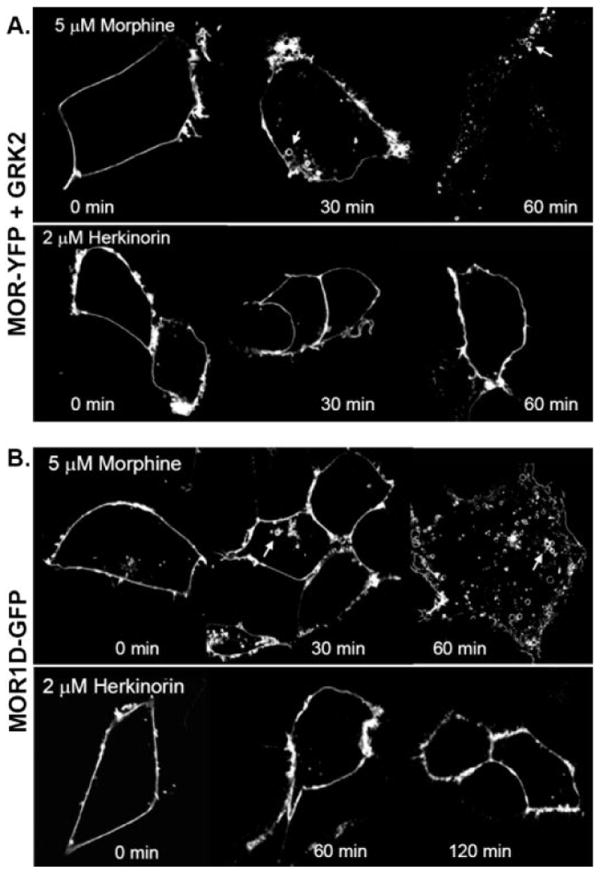
Receptor internalization under conditions in which morphine will internalize μOR. A, agonist-induced μOR-YFP internalization in cells overexpressing GRK2. Experiments were performed as described in Fig. 5A with the addition of cotransfecting 5 μg of GRK2 cDNA. Morphine induces a redistribution of membrane-associated μOR-YFP to intracellular vesicle (white arrows); herkinorin does not. B, agonist induced μOR1-d-GFP internalization. μOR1-d-GFP (2 μg of cDNA) was transiently transfected into HEK-293 cells. Morphine treatment internalizes the receptor as indicated by the appearance of intracellular vesicles (white arrows), whereas herkinorin does not. Experiments were performed on at least three separate transfections; representative cells are shown.
GPCR phosphorylation and internalization can be altered by changing the serine and threonine numbers in the C-terminal tail (Oakley et al., 2001). A naturally occurring splice variant of the mouse μOR, μOR1-D, differs from the μOR only in the C-terminal sequence and has been shown to recruit βarr2-GFP and internalize after morphine treatment (L. M. Bohn, unpublished observations; Pan et al., 1999; Koch et al., 2001). The μOR1-D internalizes with morphine treatment, whereas herkinorin fails to internalize this receptor variant (Fig. 6B). Taken together, our findings demonstrate that the herkinorin-bound μOR does not internalize or recruit βarr2-GFP in HEK-293 cells and, unlike morphine, this cannot be overcome by overexpressing GRK2 or by substituting the μOR1-D splice variant.
Discussion
Herkinorin is a μOR agonist that does not promote βarr2-GFP translocation or μOR internalization. It differs from morphine, which can promote βarr2-GFP translocation and receptor internalization when GRK2 is overexpressed in the μOR-expressing HEK-293 cells. Herkinorin could be classified as a biased agonist of the μOR because it induces signaling through one pathway (MAP kinase), whereas it does not promote an interaction with another pathway (β-arrestin translocation and receptor internalization). Agonists that can activate the receptor may be therapeutically useful considering that suppression of the β-arrestin2-μOR interaction attenuates morphine antinociceptive tolerance and morphine-induced side effects in genetically modified mouse models (Bohn et al., 2000b; Przewlocka et al., 2002; Raehal et al., 2005).
Nearly a decade ago, Burford et al. (1998) first described how morphine and DAMGO could be considered as “biased agonists” because both ligands could efficiently activate G proteins yet only DAMGO induced robust μOR internalization in HEK-293 cells. Biased agonism based on receptor signaling versus arrestin recruitment has also been described. Several recent reports from the Lefkowitz group have demonstrated that agonists can mediate distinct G protein- and arrestin-dependent downstream signaling (Gesty-Palmer et al., 2006; Shenoy et al., 2006). An earlier study by Kohout et al. (2004) describes a similar effect for CCL7 receptor activation by the two agonists CCL19 and CCL21, wherein they show that although both agonists activate G protein mediated signaling, only CCL19 places the receptor into a conformation that can be phosphorylated and desensitized by β-arrestins. Phosphorylation at the C terminus was found to be important for the CCL19-mediated desensitization, and it was likely that both GRKs and protein kinase C (PKC) could play a role in the phosphorylation. The μOR is also phosphorylated by both GRKs and PKCs. Extensive work by the Chavkin group has indicated several potential residues by which the μOR can be phosphorylated by GRKs and by PKC, and they have further shown that the phosphorylation profiles are tightly linked to agonist-specific regulation (Celver et al., 2001, 2004; Lowe et al., 2002). We have not evaluated the role of other protein kinases on regulation of the herkinorin-bound μOR, yet it is likely that some form of receptor regulation will occur. Furthermore, our studies have examined phosphorylation of only one residue implicated in desensitization, and it is likely that other phosphorylation sites in the receptor can affect receptor regulation. Further studies of this compound and its derivatives are ongoing.
To test for agonist activity of the compound in parallel with the trafficking studies, we used a MAP kinase assay and found that herkinorin could lead to phosphorylation of ERK1/2 in a dose-dependent, antagonist-reversible manner (Fig. 2). A recent report by Wang et al. (2006) reports that β-arrestins can serve as a molecular switch in the activation of ERK via SRC mediated by α2 adrenergic receptor activation. In the absence of β-arrestins, they found that ERK phosphorylation persisted over time and that the phospho-ERK was predominantly localized to the nucleus at the later time points. Herkinorin treatment activates ERK in a manner that seems to be more efficacious than DAMGO, which may be due to a more persistent signal. Whether the altered ERK activation profile is due to a mechanism similar to those described for the α2 adrenergic receptors in the absence of β-arrestins remains to be determined. We have found that the μOR activates ERK in the absence of β-arrestins using mouse embryonic fibroblasts that lack endogenous β-arrestin1 and β-arrestin2, and these cells will be useful for further elucidating the contribution of the β-arrestins to the μOR activation of MAP kinase pathways. Moreover, the physiological significance of such molecular events downstream of μOR remain to be determined.
The implications of how opiates that can activate the receptor without recruiting β-arrestins will act in vivo remain to be seen. In studies using mice lacking β-arrestin2, morphine produced enhanced and prolonged antinociception with very little morphine tolerance (Bohn et al., 1999, 2000b, 2002). Therefore, opioid agonists that do not promote receptor-β-arrestin interactions may mimic the effect of blocking the β-arrestins and result in analgesics that do not promote tolerance. Moreover, agonists that activate the μOR without the recruitment of β-arrestins may have additional benefits besides preventing antinociceptive tolerance. Morphine-induced side effects, including constipation and respiratory suppression, have been shown to be greatly diminished in mice lacking βarr2 (Raehal et al., 2005). The mechanisms that determine how one μOR-mediated physiological response can be enhanced while another is suppressed due to the ablation of βarr2 is not known; however, these observations speak to the diverse roles that β-arrestins may play in receptor signaling and regulation (Shenoy and Lefkowitz, 2003). If βarr2-opioid receptor interaction is required for opioid-induced constipation and respiratory suppression, then compounds that can activate the μOR and not recruit the β-arrestins may be promising opioid analgesics with much fewer side effects (Bruns et al., 2006).
The question remains as to whether the β-arrestin translocation and internalization profiles observed in the cellular system are preserved at the biological sites of action in vivo. This is an important consideration because morphine has been shown to promote efficient internalization profiles in certain neuronal populations while promoting no internalization in other neurons (Keith et al., 1996; Haberstock-Debic et al., 2003, 2005). Here we have used the HEK-293 cellular system to assess the herkinorin-induced μOR trafficking profiles because this model has been the most extensively used to study opioid receptor trafficking and should allow for the best overall comparison of herkinorin with other opioids. In the end, receptor trafficking induced by herkinorin and other opiates should be compared in neurons that mediate opiate-induced antinociceptive effects.
Salvinorin A is biologically active in vivo because it produces hallucinations by its actions at the κOR, yet these effects are very short-lasting (Roth et al., 2002). Systemic injections of herkinorin into mice and rats do not produce analgesic responses; however, peripheral injections do produce localized analgesia that can be blocked by the opioid antagonist naloxone (T.E. Prisinzano and K. Tidgewell, unpublished observations). This may be because herkinorin, like salvinorin A, is susceptible to degradation by esterase, or by other metabolic routes, in vivo and may not be readily available in the central nervous system. Moreover, poor solubility of the compound makes it difficult to administer high concentrations systemically. Further modifications of the herkinorin template are being generated with hopes of identifying a compound with greater stability and solubility than herkinorin.
The structures of herkinorin and salvinorin A and its related compounds are unique among opioid receptor ligands in that they lack a basic nitrogen atom. This unique chemical backbone might provide the framework for the generation of new opioid agonists that take advantage of the differences in opioid receptor regulation induced by alterations in ligand-receptor interactions. Future agonists built on this novel chemical structure may shed light on the molecular determinants of opioid receptor signaling and may lead to the fine-tuning of opioid analgesics by limiting adverse narcotic effects.
Acknowledgments
We thank Dr. M. G. Caron for the βarr2-GFP cDNA construct and Drs. G. Pasternak and Y. X. Pan for the μOR1-D cDNA construct. We are also grateful to Dr. Robert Lefkowitz for providing the MEF-WT and MEF-βarr1&2-KO cells and the A1CT antibody. We thank Lori Hudson for excellent technical support.
This work was supported by National Institute on Drug Abuse grants DA14600, DA18860 (to L.M.B), and DA18151 (to T.E.P).
ABBREVIATIONS
- GPCR
G protein-coupled receptor
- GRK
G protein-coupled receptor kinase
- βarr
β-arrestin
- μOR
μ-opioid receptor
- YFP
yellow fluorescent protein
- GFP
green fluorescent protein
- DAMGO
[d-Ala2,N-MePhe4,Gly-ol5]enkephalin
- herkinorin
(2S,4aR,6aR,7R,9S,10aS,10bR)-9-(benzoyloxy)-2-(3-furanyl)dodecahydro-6a,10b-dimethyl-4,10-dioxo-2H-naphtho-[2,1-c]pyran-7-carboxylic acid methyl ester
- DMSO
dimethyl sulfoxide
- HEK
human embryonic kidney
- PMSF
phenylmethylsulfonyl fluoride
- PVDF
polyvinylidene difluoride
- MAP
mitogen-activated protein
- NP-40
Nonidet P-40
- PBS
phosphate-buffered saline
- PAGE
polyacrylamide gel electrophoresis
- MEM
minimal essential medium
- PKC
protein kinase C
- ANOVA
analysis of variance
- WT
wild type
- HA
hemagglutinin
- ERK1/2
extracellular signal-regulated kinases 1 and 2
- MEF-WT
mouse embryonic fibroblasts expressing endogenous β-arrestins 1 and 2
- MEF βarr1&2-KO
mouse embryonic fibroblasts lacking endogenous β-arrestins 1 and 2
- DSP
dithiobis(succinimidylpropionate)
- U50,488H
(trans)-3,4-dichloro-N-methyl-N-[2-(1-pyrrolidinyl)-cyclohexyl]benzeneacetamide
References
- Alvarez V, Arttamangkul S, Williams JT. A RAVE about opioid withdrawal. Neuron. 2001;32:761–763. doi: 10.1016/s0896-6273(01)00530-x. [DOI] [PubMed] [Google Scholar]
- Bailey CP, Couch D, Johnson E, Griffiths K, Kelly E, Henderson G. Mu-opioid receptor desensitization in mature rat neurons: lack of interaction between DAMGO and morphine. J Neurosci. 2003;23:10515–10520. doi: 10.1523/JNEUROSCI.23-33-10515.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belcheva MM, Vogel Z, Ignatova E, Avidor-Reiss T, Zippel R, Levy R, Young EC, Barg J, Coscia CJ. Opioid modulation of extracellular signal-regulated protein kinase activity is ras-dependent and involves Gbetagamma subunits. J Neurochem. 1998;70:635–645. doi: 10.1046/j.1471-4159.1998.70020635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Belcheva MM, Coscia CJ. Mitogenic signaling via endogenous kappa-opioid receptors in C6 glioma cells: evidence for the involvement of protein kinase C and the mitogen-activated protein kinase signaling cascade. J Neurochem. 2000a;74:564–573. doi: 10.1046/j.1471-4159.2000.740564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Dykstra LA, Lefkowitz RJ, Caron MG, Barak LS. Relative opioid efficacy is determined by the complements of the G protein-coupled receptor desensitization machinery. Mol Pharmacol. 2004a;66:106–112. doi: 10.1124/mol.66.1.106. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Caron MG. G protein-coupled receptor kinase/beta-arrestin systems and drugs of abuse: psychostimulant and opiate studies in knockout mice. Neuromol Med. 2004b;5:41–50. doi: 10.1385/NMM:5:1:041. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature (Lond) 2000b;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Caron MG. Differential mechanisms of morphine antinociceptive tolerance revealed in (beta)arrestin-2 knock-out mice. J Neurosci. 2002;22:10494–10500. doi: 10.1523/JNEUROSCI.22-23-10494.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science (Wash DC) 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- Bruns IR, Chhum S, Dinh AT, Doerr H, Dunn NR, Ly YT, Mitman CL, Rickards HD, Sol C, Wan EW, et al. A potential novel strategy to separate therapeutic- and side-effects that are mediated via the same receptor: beta-arrestin2/G-protein coupling antagonists. J Clin Pharm Ther. 2006;31:119–128. doi: 10.1111/j.1365-2710.2006.00714.x. [DOI] [PubMed] [Google Scholar]
- Burford NT, Tolbert LM, Sadee W. Specific G protein activation and mu-opioid receptor internalization caused by morphine, DAMGO and endomorphin I. Eur J Pharmacol. 1998;342:123–126. doi: 10.1016/s0014-2999(97)01556-2. [DOI] [PubMed] [Google Scholar]
- Cao TT, Mays RW, von Zastrow M. Regulated endocytosis of G-protein-coupled receptors by a biochemically and functionally distinct subpopulation of clathrin-coated pits. J Biol Chem. 1998;273:24592–24602. doi: 10.1074/jbc.273.38.24592. [DOI] [PubMed] [Google Scholar]
- Celver J, Xu M, Jin W, Lowe J, Chavkin C. Distinct domains of the mu-opioid receptor control uncoupling and internalization. Mol Pharmacol. 2004;65:528–537. doi: 10.1124/mol.65.3.528. [DOI] [PubMed] [Google Scholar]
- Celver JP, Lowe J, Kovoor A, Gurevich VV, Chavkin C. Threonine 180 is required for G-protein-coupled receptor kinase 3- and beta-arrestin 2-mediated desensitization of the μ-opioid receptor in Xenopus oocytes. J Biol Chem. 2001;276:4894–4900. doi: 10.1074/jbc.M007437200. [DOI] [PubMed] [Google Scholar]
- Chavkin C, Sud S, Jin W, Stewart J, Zjawiony JK, Siebert DJ, Toth BA, Hufeisen SJ, Roth BL. Salvinorin A, an active component of the hallucinogenic sage salvia divinorum is a highly efficacious κ-opioid receptor agonist: structural and functional considerations. J Pharmacol Exp Ther. 2004;308:1197–1203. doi: 10.1124/jpet.103.059394. [DOI] [PubMed] [Google Scholar]
- Connor M, Osborne PB, Christie MJ. Mu-opioid receptor desensitization: is morphine different? Br J Pharmacol. 2004;143:685–696. doi: 10.1038/sj.bjp.0705938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainetdinov RR, Premont RT, Bohn LM, Lefkowitz RJ, Caron MG. Desensitization of G protein-coupled receptors and neuronal functions. Annu Rev Neurosci. 2004;27:107–144. doi: 10.1146/annurev.neuro.27.070203.144206. [DOI] [PubMed] [Google Scholar]
- Gesty-Palmer D, Chen M, Reiter E, Ahn S, Nelson CD, Wang S, Eckhardt AE, Cowan CL, Spurney RF, Luttrell LM, et al. Distinct β-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J Biol Chem. 2006;281:10856–10864. doi: 10.1074/jbc.M513380200. [DOI] [PubMed] [Google Scholar]
- Haberstock-Debic H, Kim KA, Yu YJ, von Zastrow M. Morphine promotes rapid, arrestin-dependent endocytosis of mu-opioid receptors in striatal neurons. J Neurosci. 2005;25:7847–7857. doi: 10.1523/JNEUROSCI.5045-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberstock-Debic H, Wein M, Barrot M, Colago EE, Rahman Z, Neve RL, Pickel VM, Nestler EJ, von Zastrow M, Svingos AL. Morphine acutely regulates opioid receptor trafficking selectively in dendrites of nucleus accumbens neurons. J Neurosci. 2003;23:4324–4332. doi: 10.1523/JNEUROSCI.23-10-04324.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding WW, Tidgewell K, Byrd N, Cobb H, Dersch CM, Butelman ER, Rothman RB, Prisinzano TE. Neoclerodane diterpenes as a novel scaffold for mu opioid receptor ligands. J Med Chem. 2005;48:4765–4771. doi: 10.1021/jm048963m. [DOI] [PubMed] [Google Scholar]
- Keith DE, Murray SR, Zaki PA, Chu PC, Lissin DV, Kang L, Evans CJ, von Zastrow M. Morphine activates opioid receptors without causing their rapid internalization. J Biol Chem. 1996;271:19021–19024. doi: 10.1074/jbc.271.32.19021. [DOI] [PubMed] [Google Scholar]
- Koch T, Schulz S, Pfeiffer M, Klutzny M, Schroder H, Kahl E, Hollt V. C-terminal splice variants of the mouse mu-opioid receptor differ in morphine-induced internalization and receptor resensitization. J Biol Chem. 2001;276:31408–31414. doi: 10.1074/jbc.M100305200. [DOI] [PubMed] [Google Scholar]
- Koch T, Widera A, Bartzsch K, Schulz S, Brandenburg LO, Wundrack N, Beyer A, Grecksch G, Hollt V. Receptor endocytosis counteracts the development of opioid tolerance. Mol Pharmacol. 2005;67:280–287. doi: 10.1124/mol.104.004994. [DOI] [PubMed] [Google Scholar]
- Kohout TA, Lin FS, Perry SJ, Conner DA, Lefkowitz RJ. β-Arrestin 1 and 2 differentially regulate heptahelical receptor signaling and trafficking. Proc Natl Acad Sci USA. 2001;98:1601–1606. doi: 10.1073/pnas.041608198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohout TA, Nicholas SL, Perry SJ, Reinhart G, Junger S, Struthers RS. Differential desensitization, receptor phosphorylation, beta-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7. J Biol Chem. 2004;279:23214–23222. doi: 10.1074/jbc.M402125200. [DOI] [PubMed] [Google Scholar]
- Lowe JD, Celver JP, Gurevich VV, Chavkin C. μ-Opioid receptors desensitize less rapidly than δ-opioid receptors due to less efficient activation of arrestin. J Biol Chem. 2002;277:15729–15735. doi: 10.1074/jbc.M200612200. [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-β-arrestin complexes after receptor endocytosis. J Biol Chem. 2001;276:19452–19460. doi: 10.1074/jbc.M101450200. [DOI] [PubMed] [Google Scholar]
- Ortega A, Blount JF, Manchand PS. Salvinorin, a new trans-neoclerodane diterpene from Salvia-divinorum (Labiatae) J Chem Soc Perkin Trans I. 1982;(10):2505–2508. [Google Scholar]
- Pan YX, Xu J, Bolan E, Abbadie C, Chang A, Zuckerman A, Rossi G, Pasternak GW. Identification and characterization of three new alternatively spliced μ-opioid receptor isoforms. Mol Pharmacol. 1999;56:396–403. doi: 10.1124/mol.56.2.396. [DOI] [PubMed] [Google Scholar]
- Pierce KL, Lefkowitz RJ. Classical and new roles of beta-arrestins in the regulation of G-protein-coupled receptors. Nat Rev Neurosci. 2001;2:727–733. doi: 10.1038/35094577. [DOI] [PubMed] [Google Scholar]
- Przewlocka B, Sieja A, Starowicz K, Maj M, Bilecki W, Przewlocki R. Knockdown of spinal opioid receptors by antisense targeting beta-arrestin reduces morphine tolerance and allodynia in rat. Neurosci Lett. 2002;325:107–110. doi: 10.1016/s0304-3940(02)00246-x. [DOI] [PubMed] [Google Scholar]
- Raehal KM, Bohn LM. Mu opioid receptor regulation and opiate responsiveness. AAPS J. 2005;7:E587–E591. doi: 10.1208/aapsj070360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raehal KM, Walker JK, Bohn LM. Morphine side effects in β-arrestin 2 knockout mice. J Pharmacol Exp Ther. 2005;314:1195–1201. doi: 10.1124/jpet.105.087254. [DOI] [PubMed] [Google Scholar]
- Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Salvinorin A: a potent naturally occurring nonnitrogenous κ opioid selective agonist. Proc Natl Acad Sci USA. 2002;99:11934–11939. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz S, Mayer D, Pfeiffer M, Stumm R, Koch T, Hollt V. Morphine induces terminal micro-opioid receptor desensitization by sustained phosphorylation of serine-375. EMBO (Eur Mol Biol Organ) J. 2004;23:3282–3289. doi: 10.1038/sj.emboj.7600334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, Lefkowitz RJ. β-Arrestin-dependent, G protein-independent ERK1/2 activation by the β2 adrenergic receptor. J Biol Chem. 2006;281:1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- Shenoy SK, Lefkowitz RJ. Multifaceted roles of beta-arrestins in the regulation of seven-membrane-spanning receptor trafficking and signalling. Biochem J. 2003;375:503–515. doi: 10.1042/BJ20031076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Lu R, Zhao J, Limbird LE. Arrestin serves as a molecular switch, linking endogenous α2-adrenergic receptor to SRC-dependent, but not SRC-independent, ERK activation. J Biol Chem. 2006;281:25948–25955. doi: 10.1074/jbc.M605415200. [DOI] [PubMed] [Google Scholar]
- Wang Y, Tang K, Inan S, Siebert D, Holzgrabe U, Lee DY, Huang P, Li JG, Cowan A, Liu-Chen LY. Comparison of pharmacological activities of three distinct κ ligands (Salvinorin A, TRK-820 and 3FLB) on κ opioid receptors in vitro and their antipruritic and antinociceptive activities in vivo. J Pharmacol Exp Ther. 2005;312:220–230. doi: 10.1124/jpet.104.073668. [DOI] [PubMed] [Google Scholar]
- Whistler JL, von Zastrow M. Morphine-activated opioid receptors elude desensitization by β-arrestin. Proc Natl Acad Sci USA. 1998;95:9914–9919. doi: 10.1073/pnas.95.17.9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ferguson SS, Barak LS, Bodduluri SR, Laporte SA, Law PY, Caron MG. Role for G protein-coupled receptor kinase in agonist-specific regulation of mu-opioid receptor responsiveness. Proc Natl Acad Sci USA. 1998;95:7157–7162. doi: 10.1073/pnas.95.12.7157. [DOI] [PMC free article] [PubMed] [Google Scholar]