

Abstract
The pharmacokinetics (PK) and biodistribution of nanoparticles (NPs) are controlled by a complex array of interrelated, physicochemical and biological factors of NPs. The lipid-bilayer core structure of the Lipid-Calcium-Phosphate (LCP) NPs allows us to examine the effects of the density of polyethylene glycol (PEG) and the incorporation of various lipids onto the surface on their fate in vivo. Fluorescence quantification estimated that up to 20% (molar percent of outer leaflet lipids) could be grafted on the surface of LCP NPs. Contrary to the common belief that high level of PEGylation could prevent the uptake of NPs by the reticuloendothelial system (RES) organs such as liver and spleen, a significant amount of the injected dose was observed in the liver. Confocal microscopy revealed that LCP NPs were largely localized in hepatocytes not Kupffer cells. It was further demonstrated that the delivery to hepatocytes was dependent on both the concentration of PEG and the surface lipids. LCP NPs could be directed from hepatocytes to Kupffer cells by decreasing PEG concentration on the particle surface. In addition, LCP NPs with 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) exhibited higher accumulation in the hepatocytes than LCP NPs with dioleoylphosphatidylcholine (DOPC). Analysis of the proteins bound to NPs suggested that apolipoprotein E (apoE) might serve as an endogenous targeting ligand for LCP-DOTAP NPs, but not LCP-DOPC NPs. The significant uptake of NPs by the hepatocytes is of great interest to formulation design for oncologic and hepatic drug deliveries.
Keywords: nanoparticles, polyethylene glycol, pharmacokinetics, biodistribution, hepatocytes, protein binding
1. Introduction
Nanoparticle (NP) therapeutics possess desirable features for medical applications including (i) protection of the cargo from enzymatic or hydrolytic degradation, (ii) increased solubility and drug loading capacity, (iii) sustained and controlled release of drugs, and (iv) preferential accumulation at the site of interest through the enhanced permeability and retention (EPR) effect [1]. Over 20 NP therapeutics have been approved by the FDA for clinical use and many more are in late-phase clinical trials [2, 3]. Despite these advantages, injected NPs are generally eliminated rapidly by the reticuloendothelial system (RES) after administration and accumulate in the liver and spleen. To be useful in vivo, NPs must avoid opsonization and subsequent recognition by macrophages. This can be accomplished through the coupling of polyethylene glycol (PEG) to the surfaces of NPs, a process known as PEGylation [4].
The success of PEGylation critically depends on the steric stabilization conferred by PEG chains on the surface of the NPs. Stabilization is achieved through the highly hydrophilic and flexible nature of the PEG chains, which provide repulsive interactions with biological components in vivo. The ways in which grafted PEG forms a well hydrated barrier layer on the surface, sterically hindering protein adsorption, can be described by relatively straightforward theories of polymer physics that originated from Flory [5] and De Gennes [6]. Surface-grafted PEG adopts two different statistical conformations: “mushroom” and “brush,” which are dictated by the relationship between the distance of two grafting sites (D) and the radius of the random coil the polymer forms in solution (Flory radius, RF = aN3/5, where N is the degree of polymerization and a is the persistence length of the monomer). The polymer assumes a randomly oriented, mushroom-like conformation at D > 2RF. When the density of the grafted PEG increases, the polymer chains begin to interact and assume a configuration in which they extend out from the surface. A brush-like conformation appears when D < RF [7]. The two conformations do not represent sharply separated regimes, but undergo smooth transitions through mushroom/brush intermediates as D and RF values become closer [8]. The brush configuration is favored for drug delivery because it ensures that the entire surface of the NP is covered, leaving few gaps where opsonin proteins can freely penetrate and bind [9, 10]. Many studies indicate that PEG chains must have a minimum molecular weight of 2000 to achieve RES-avoidance characteristics, therefore PEG2000 is the most frequently used PEG polymer [9, 11, 12]. Essentially, the grafting density of PEG chains determines the efficiency of PEGylation and thereby the capability of the resultant NPs to repel proteins. To date, there is no direct method available for quantifying the number of PEG molecules bound to the surface of NPs or for determining the density of PEG [13]. Thus, the exact, density-dependent influence of PEG on NP biodistribution remains unknown.
In comparison to bulk biomaterials, NPs have extremely high surface-to-volume ratios. Therefore, control of the surface properties of NPs critically affects their in vivo performance. When NPs come into contact with biological mediums, they are immediately covered by proteins [14, 15]. The absorption of proteins to the surface of NPs confers a new “biological identity,” which is what cells, tissues and organs actually “see” when interacting with the NPs [16]. This new interface between the NPs and the biological tissues determines cellular and tissue responses and biological consequences [17, 18]. Surface characteristics, such as charge, hydrophilicity and curvature, dictate the extent and specificity of protein binding [16, 19]. Specific protein binding is one of the key elements that affect the biodistribution of NPs [20, 21]. Indeed, a detailed knowledge of NP-protein interactions is critical to their rational formulation, design and optimization. The complete plasma proteome is expected to contain as many as 3700 proteins [18], of which only approximately 50 have been found to be associated with the surface of nanoparticles [16, 22]. For example, opsonins, like fibrinogen, Immunoglobulin G (IgG), or complement factors, are believed to facilitate phagocytosis and remove the NPs from the circulation. On the other hand, dysopsonins, like human serum albumin (HSA), generally prolong circulation time in the blood [23]. The mechanism by which PEG decreases protein interactions is non-specific [24].
We have developed a Lipid-Calcium-Phosphate (LCP) NP formulation (∼35 nm in diameter) to effectively deliver siRNA [25, 26], cDNA and small molecule drugs [27, 28] to both solid and metastatic tumors. The unique, bilayer core-structure allows for surface modification with different lipids and various amounts of PEGylation (Figure 1A). The in vivo biodistribution and pharmacokinetic studies of LCP NPs modified with different amounts of PEGylation and surface lipids were conducted in normal and tumor-bearing mice. We investigated how these surface characteristics would influence the in vivo behavior of LCP NPs. We believe these findings will benefit the rational design and application of PEG and other hydrophilic polymers for the development of effective drug carrier systems.
Figure 1.
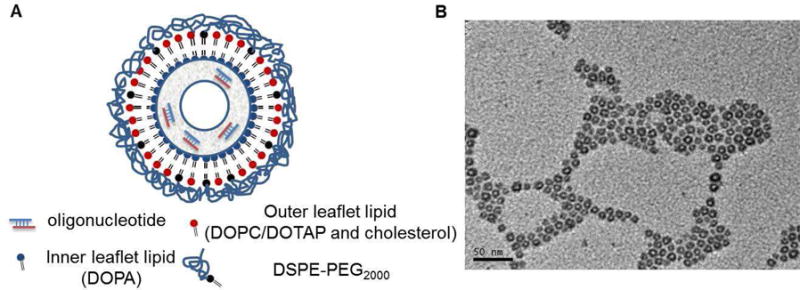
(A) Proposed lipid bilayer-core structure; (B) TEM image of CaP cores.
2. Materials and methods
2.1. Materials
22-mer double-strand oligonucleotides (sense sequence, 5′-CAAGGGACTGGAAGGCTGGG-3′,) labeled with Texas Red (excitation/emission wavelengths of 550/600 nm) were purchased from Sigma, Inc. Oligonucleotides were labeled with 3H through hydrogen exchange with 3H2O at the C8 position of the purine oligonucleotide [29]. The specific radioactivity of the labeled oligonucleotides was 1.03 × 109 cpm/μmol. The radiolabeled compound is stable in biological systems [29]. Both Texas Red and 3H-labeled oligonucleotides were used to mimic siRNA. Dioleoylphosphatidylcholine (DOPC), 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), dioleoylphosphatydic acid (DOPA), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[poly(ethylene glycol)2000] (DSPE-PEG2000), Rhodamine-dioleoyl-phosphatidylethanolamine (Rhodamine-DOPE), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[poly(ethylene glycol)2000-N′-carboxyfluorescein] (DSPE-PEG-CF) were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL).
2.2. Preparation of LCP NPs
LCP NPs were prepared according to the methods described in previous publications [25] with minor modifications. We first prepared two water-in-oil microemulsions: 1) 100 μL of 500 mM CaCl2 and 16 μL of 2 mg/mL Texas Red or 3H-labeled oligonucleotides in 8 mL cyclohexane oil phase (71% cyclohexane with 29% Igepal CO-520 as surfactant), and 2) 100 μL of 100 mM pH 9.0 Na2HPO4 in 8 mL cyclohexane oil phase with 320 μL of 20 mM DOPA added as the inner leaflet lipid. After mixing the two microemulsions for 45 min, 30 mL of absolute ethanol was added and the mixture was centrifuged at 12,500 g for 15 min to precipitate the CaP cores. The precipitated pellets were then washed with ethanol 2–3 times, dispersed in 500 μL of chloroform, and stored in a glass vial for further modification. To create the outer leaflet lipid coating, 200 μL of 20 mM cholesterol, 200 μL of 20 mM DOPC, and 100 μL of 20 mM of DSPE-PEG2000 were mixed with the cores. After the removal of the chloroform, the core was suspended in a small volume of ethanol and then dispersed in water.
2.3. Particle size, Zeta potential and PEGylation analysis
The LCP NPs prepared using the described methods contain excess lipids (e.g. DOPC/DOTAP, cholesterol and DSPE–PEG2000). To determine the accurate concentration of DSPE–PEG2000 on the surface of LCP NPs, sucrose density gradient centrifugation was used to separate LCP NPs from the excess lipids. The discontinuous sucrose gradient was created with 0.9 mL each of 50%, 30%, and 10% sucrose, as well as deionized water layered consecutively from bottom to top in a 4 mL ultracentrifuge tube. The mixture containing LCP NPs and excess lipids was applied between the layers of 10% sucrose and water. The gradients were centrifuged using a Beckman Coulter SW 60Ti rotor at 50,000 rpm for 4 h at 20°C and separated into aliquots removed from top to bottom. The fractions were then diluted with an ethanol and lysis buffer (0.1% Triton-100 and HCl, pH=2.5) for further measurements. The particle size and Zeta potential of the purified LCP NPs were determined by a Malvern ZetaSizer Nano series (Westborough, MA).
To measure the molar ratio of DSPE-PEG2000 in the total, outer leaflet lipid, Rhodamine-DOPE, DSPE-PEG2000-CF and 3H-labeled oligonucleotide were used to label the outer leaflet lipid, PEG2000-DSPE and CaP cores, respectively. The LCP NPs were prepared and purified as described above. The fractions were analyzed with a fluorescence spectrometer and a liquid scintillation counter.
2.4. Pharmacokinetics and biodistribution of 3H-labeled LCP NPs
All work performed on animals was in accordance with and approved by the University of North Carolina Institutional Animal Care and Use Committee. Pharmacokinetic and biodistribution studies of LCP NPs with 20% PEG were performed in normal athymic nude (nu/nu) mice (National Cancer Institute) and mice carrying H460 (ATCC), human lung cancer xenografts. Tumors were allowed to grow to a size of around 0.2 cm3 before injections. Studies of the biodistribution of LCP-DOTAP NPs and LCP-DOPC NPs that had different densities of PEG were performed in CD-1 mice (National Cancer Institute). Animals were intravenously injected with LCP NPs containing 3H-labeled oligonucleotide at a dose of 0.25 mg/kg. At given time intervals, four animals were sacrificed for blood and tissue collection. Radioactivity was measured using liquid scintillation counting.
2.5. Confocal microscopy of frozen tissue sections of liver
Mice were intravenously injected with Texas Red-labeled oligonucleotide encapsulated in different LCP NPs four hours before sacrifice and tissue collection. Tissue blocks were immediately frozen in OCT (Tissue-Tek, Dublin, OH) on dry ice, allowing the generation of ten-μm-thick cryosections. The tissue sections were then mounted on Superfrost Plus slides (Fisher Scientific Co., Houston, TX). After brief rinsing with PBS (to remove any surface embedding medium), as well as fixation with acetone at -20°C, tissue sections were stained with Alexa Fluor 488 phalloidin (Life Technologies) and mounted in a medium containing DAPI (Vector Lab.). Images were captured using an Olympus FV1000 MPE confocal microscope under three channels: DAPI for nuclei, Alexa Fluor 488 for phalloidin, and Texas Red for oligonucleotides.
2.6. Determination of serum protein adsorption by SDS-PAGE and MALDI-TOF-MS
LCP NPs with different surface lipids and PEG densities were prepared and purified as described above. Samples were incubated with 20 or 80% mouse serum for 1.5 h at 37 °C. After the incubation, the samples were centrifuged to pellet the particle-protein complex. The pellets were washed three times with PBS to remove loosely bound proteins and then re-suspended in PBS and a protein loading buffer. Gel electrophoresis was performed at 120V, 400mA for approximately 60 min. The gels were treated with Coomassie blue staining and then destained overnight in 50% methanol, 10% acetic acid [30, 31].
After the separation of proteins by SDS/PAGE, bands were excised from the gel and digested with trypsin. The resulting peptide mixtures were separated and analyzed using MALDI-TOF-MS (ABI 4800 MALDI TOF/TOF). Spectra were analyzed by MASCOT software to identify tryptic peptide sequences matched to the NCBI database (http://www.ncbi.nlm.nih.gov/).
2.7. Biodistribution of LCP NPs in wild-type and apoE-/- mice
ApoE-deficient mice (ApoE-/-, stock #002052) and wild type C57BL/6 mice were obtained from Jackson Laboratories (Bar Harbor, ME). LCP NPs containing 3H-labeled oligonucleotides were administered intravenously via the tail vein at a dose of 0.25 mg/kg. Four hours after the injection, major organs were collected from animals and processed for measurements of radioactivity.
2.8. Statistical analysis
Data were presented as Mean ± (standard deviation) SD. A two-tailed t-test was performed when comparing two groups. Statistical significance was defined by a value of P < 0.05.
3. Results and discussion
3.1. Characterization of LCP NPs
LCP NPs formulation has two distinctive advantages for studying the effect of PEGylation; first, the unique membrane-core structure allows for modification of the surface with various amounts of PEGylation (Figure 1A). Second, LCP NPs can be purified based on the density difference between the particle and the extra excipient, which permits accurate surface characterizations of PEGylation. To determine the accurate concentration of DSPE–PEG2000 on the surface of LCP NPs, sucrose gradient centrifugation was used to separate LCP NPs from the extra lipids (Figure S1). Rhodamine-DOPE, DSPE-PEG2000-CF and 3H-labeled oligonucleotide were used to label the outer leaflet lipid, PEG2000-DSPE and CaP core, respectively. The LCP NPs containing dense CaP cores banded tightly at the interface between the layers of 10 and 30% sucrose, while the unassociated lipids were present as a smear from the top of the gradient to the interface between 10% and 30% sucrose (Figure S1). It was found that the isolated LCP NPs contain around 80% tritium (by liquid scintillation) and calcium (by inductively coupled plasma mass spectrometry, ICP-MS). Rhodamine-DOPE, DSPE-PEG2000-CF and trace amounts of tritium were detected in the fractions of lower sucrose concentrations, suggesting that extra lipids could be separated from the dense NPs using this method. Quantification of DSPE-PEG2000-CF on purified LCP NPs is summarized in Figure 2. The incorporation of PEG-lipid into the outer leaflet of LCP NPs linearly increased with the input amount of the PEG-lipid until 20% of the total lipid. Beyond this level, there was no more increase in the incorporation, suggesting a saturation of the PEG-lipid in the outer leaflet. The bulk lipid composition did not influence the efficiency of PEGylation; both DOPC and DOTAP composition showed the same saturation at 20%. Other characteristics of purified LCP NPs are summarized in Table 1. All NPs were PEGylated at a maximal density of 20 mol % of the total outer leaflet lipid. Both LCP-DOTAP and LCP-DOPC NPs had a hydrodynamic diameter of ∼30 nm. When the particles were formulated with DOPC as the major outer leaflet lipid, the zeta potential was approximately -10 mV. When DOTAP was employed, the surface potential was ∼15 mV. These results were an indication of PEG modification on the NPs, since the zeta potential of non-PEGylated DOPC and DOTAP liposomes were about -20mV and 70 mV, respectively. The presence of PEG on the surface of LCP NPs was also confirmed by X-ray photoelectron spectroscopy (data not shown).
Figure 2.
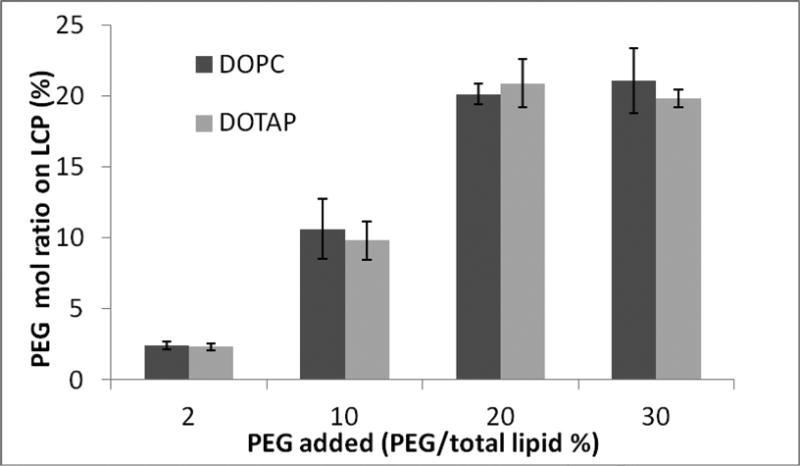
Quantification of DSPE-PEG-CF on purified LCP NPs.
Table 1.
A summary of characteristics of LCP NPs.
| PEG 2000 | |||
|---|---|---|---|
| Size(nm) | Zeta potential(mV) | Maximal PEG Conc. (molar % of outer leaflet lipids) |
|
| DOPC | ∼30 | -10 | ∼ 20 |
| DOTAP | ∼30 | 15 | ∼ 20 |
Our group [32] and others in the early 1990s [33-35] demonstrated that liposomes with PEG incorporated on their surface could achieve RES evasion and prolonged the circulation half-life. This method employs PEG-phospholipid which could be inserted into the lipid membrane by hydrophobic interaction. Due to the amphiphilic nature of the PEG-phospholipid, the degree of surface PEGylation is quite limited, usually less than 6% if the lipid membrane integrity is to be preserved [36]. However, a high density of PEG is necessary to achieve steric shielding of the NPs' surface and create the “stealth” property. By labeling DSPE-PEG2000 with a green fluorescent dye, it was determined that LCP NPs could tolerate as much as 20% PEG-phospholipid. In the LCP formulation, the inner leaflet lipid (DOPA) is known to strongly interact with the Ca ions on the surface of the core. Such strong interaction provides increased stability for the supported bilayer, allowing a high amount of incorporated DSPE-PEG2000, which is a detergent-like surfactant. A supported bilayer has greater stability than the unsupported, conventional liposomal bilayer [37, 38]. This platform provides an opportunity to modify the formulation with a high-density PEG coating and explore the impact of a PEG coating on the in vivo behavior of NP formulation.
3.2. Pharmacokinetics and biodistribution studies in normal and tumor-bearing mice
The aim of this study was to evaluate pharmacokinetics and biodistribution of LCP NPs with 20% PEGylation using the encapsulated 3H-labeled oligonucleotide as a marker. To our best knowledge, the in vivo behavior of NPs with 20% PEG2000 incorporated has never been determined in the previous studies. Blood was collected through parallel sampling. The concentration of NPs in the blood was calculated by assuming that the total blood volume in the mouse is 7% of its body weight. As shown in Figure 3A, no significant differences in the pharmacokinetic profiles were observed between the tumor free and the tumor-bearing mice treated with LCP NPs. NPs in both types of mice showed a rapid distribution phase, in which serum concentrations dropped dramatically within the first 30 min. Following the initial period, concentrations remained relatively steady at least until 4 h after injection. The surface lipid could influence the rate of clearance in the β phase. LCP-DOPC NPs possessed a longer half-life in the β phase than their DOTAP counterparts.
Figure 3.
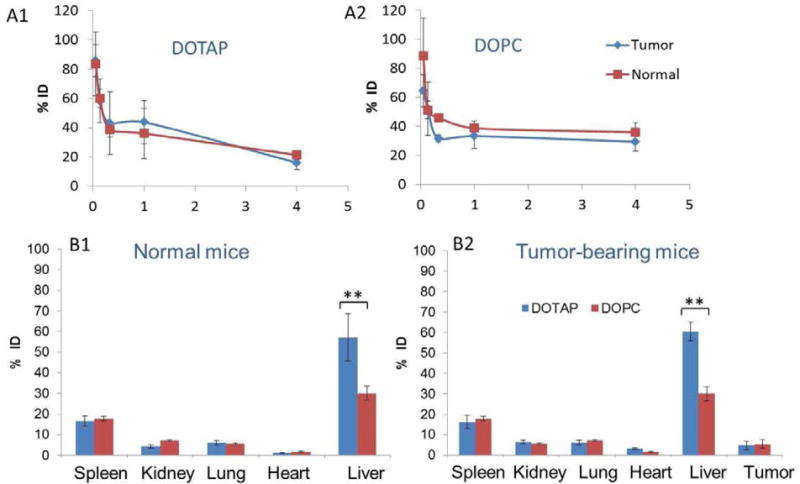
Pharmacokinetics of LCP- DOTAP NPs (A1) and LCP- DOPC NPs (A2) with 20% PEGylation. Biodistribution of LCP-DOPC NPs and LCP-DOTAP NPs with 20% PEGylation in normal (B1) and tumor-bearing mice (B2). Data are shown as % injected dose (ID). Statistical analysis was carried out using a two tailed t-test. ** P < 0.05 for liver accumulation of LCP- DOTAP NPs group compared to LCP- DOPC NPs group in both normal and tumor-bearing mice.
Figure 3B shows the tissue distribution of LCP NPs with 20% PEG 4 h post-injection. The particles were distributed mainly in the liver and spleen. The 3H signal in the liver and spleen reaches a plateau about 1 h after injection (data not shown). About 5% ID was observed in the tumor. This observation is consistent with the fact that these tissues are lined with a discontinuous or “leaky” endothelium that allows for the passive entrapment of foreign particulates [39]. The rapid distribution is not surprising, considering their small size and that their accumulation was mainly caused by their passive entrapment through the discontinuous endothelium of the liver. Extravasation in the liver is plausible because of the presence of fenestrae in the liver sinusoid, which measure 100 nm in diameter in mice [40].
Despite their accumulation in the tumor, biodistribution profiles of the LCP NPs in tumor-bearing mice were not significantly different from those of the normal controls. The surface lipids did not significantly change the global tissue distribution pattern. Interestingly, however, the accumulation level of NPs in the liver was significantly increased by DOTAP compared with DOPC (p < 0.05). It is likely that the faster clearance of LCP-DOTAP NPs in the β phase is due to this enhanced uptake by the liver. Since radioactivity could potentially come from disassembled NPs or metabolized components of the NPs, we previously utilized 111In incorporated into the core of LCP NPs to label the vesicle. The distribution of 111In were very similar to that of 3H [41]. The agreement between the label of the drug (3H-oligonucleotides) and that of the drug carrier (111In) suggested that this biodistribution pattern accurately represented the in vivo behavior of LCP NPs.
We varied the density of PEGylation on the surface of LCP NPs and systematically observed the effects on the tissue and cell-type-specific distribution in the liver. Clearance from the blood and uptake of LCP NPs into the spleen following injection showed a marked PEG density dependence, with % ID in the blood increasing and spleen uptake decreasing as PEG density increased. LCP-DOPC NPs showed a longer circulation time in the blood than LCP-DOTAP NPs, with around 40% ID remaining in the circulation 4h after administration. On the other hand, the amount of the injected dose that was observed in the kidneys, lungs and heart was generally independent of PEG density. Interestingly, the % ID of LCP NPs accumulated in the liver was also independent of PEG density. On the average, approximately 50% ID of LCP-DOTAP NPs that had various amounts of PEGylation accumulated in the liver, while only about 30% ID of non-PEGylated LPC-DOPC NPs accumulated. To obtain direct evidence illustrating whether the LCP NPs that accumulated in the liver entered the hepatocytes or were sequestered by Kupffer cells, we injected mice with LCP NPs containing Texas Red-labeled oligonucleotide and studied the tissue sections with confocal microscopy.
3.3. Cell-type localization of LCP NPs within the liver
Cell-type specific distribution of LCP NPs in the liver and its correlation with surface chemistry of LCP NPs was explored. Confocal microscopy of liver sections taken from mice 4 h after the injection of NPs containing Texas Red-labeled oligonucleotide are shown in Figure 5. Tissue sections were stained with Alexa488-phalloidin (green) to visualize the cytoskeleton of hepatocytes and with DAPI (blue) for all nuclei. Significantly, preferential accumulation of the Texas Red-labeled oligonucleotide in hepatocytes (also called liver parenchymal cells, which contain large nuclei) was observed in mice treated with LCP NPs containing a high density of PEG (Figure 5A and 5B 5-6). Distribution was generally homogenous throughout the different zones and different liver lobules. In general, the replacement of DOTAP with DOPC on the NPs resulted in the significant reduction of hepatocyte uptake (Figure 5A and 5B). Decreasing the amount of PEG on the surface of the NPs markedly reduced hepatocyte uptake. LCP NPs without PEG, or with a lower density of PEG, rarely entered hepatocytes (Figure 5A2-3 and 5B2-4). Instead, they experienced phagocytic uptake by Kupffer cells and reside in the hepatic sinusoids (a region between hepatocytes as indicated by the arrows in Figure 5A2-3 and 5B2-4). These results are evidence that delivery to the hepatocytes is afforded by grafting a dense PEG layer on the surface of LCP NPs. They also suggest that PEG concentration determines cell-type-specific localization at the tissue level. We conclude that LCP NPs with a high (∼20%) PEG density completely evaded RES and accumulated primarily in the hepatocytes.
Figure 5.
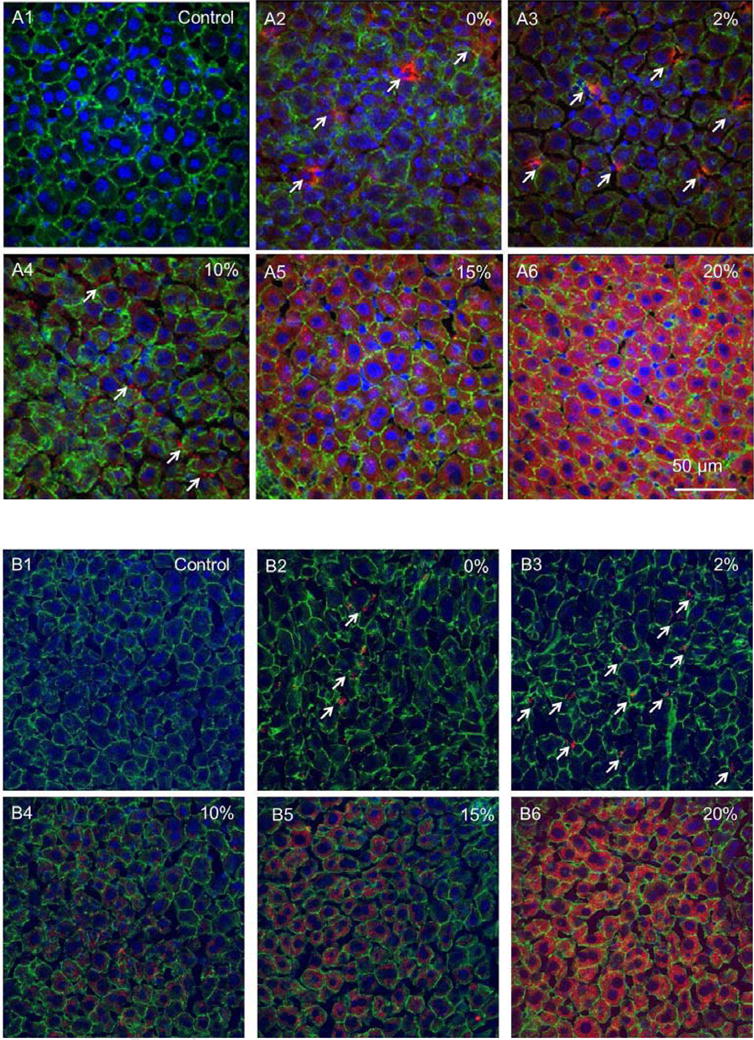
Cell-type specific localization of LCP-DOTAP NPs (A) and LCP-DOPC NPs (B) in the liver. DAPI for nuclei, Alexa 488 for phalloidin, and Texas Red for oligonucleotides. Percentages indicate amount of PEG-DSPE2000 incorporated in the outer leaflet of the wrapping lipid bilayer of LCP NPs. Arrows indicate representative Kupffer cell uptake.
These results and the plausibility of extravasation in the liver suggest that the distribution of small, long-circulating NPs to tissues with discontinuous endothelium could become a competing kinetic process, which are dependent on the properties of the NPs, the vasculature permeability, and blood flow within the tissues. A highly perfused organ with a discontinuous endothelium, such as the liver, can thus become the major distribution site of NPs. The significant uptake by the hepatocytes is of great interest to formulation design in biomedical application due to its importance in many infectious and metabolic disorders. On the other hand, it highlights a potentially important complication in the development of NPs for imaging and therapeutic delivery to extrahepatic tissues, such as the tumor. Recently, increasing evidence indicates that small NPs, in the range of 10-30 nm in diameter, can more effectively penetrate the interstitial matrix and the physiological barriers imposed by tumor vasculature than the larger ones [42, 43]. Avoiding rapid distribution to the liver will be another critical design criterion for future NP systems targeting tumor sites. Furthermore, the ongoing interest in NPs based on cationic lipids for the delivery of siRNA, antisense, or plasmids requires the examination of how these systems interact with the biological environment [44].
3.4. Determination of the protein corona composition
To investigate mechanisms of increased uptake of LCP-DOTAP NPs by hepatocytes, LCP-DOTAP and -DOPC NPs with different amounts of PEGylation were incubated with serum and the resulting protein corona was analyzed for protein identification. NPs were incubated with 20 and 80% mouse serum. The NP-protein complexes were washed three times to remove the proteins with low affinity for the NP surface. The washed complexes were further analyzed. We hypothesize that LCP NPs with 20% PEG could gradually shed their PEG coating, exposing the substrate lipid after administration, due to the sink conditions provided by the serum proteins [45, 46]. Thus, we had prepared LCP NPs with both 20 and 5% PEGylation for the study.
SDS-PAGE gel analysis of serum proteins obtained from LCP NP-protein complexes is shown in Figure 4. The main bands on the gels were albumin, IgG, and the apolipoproteins. For particles made of DOPC with different PEG densities, the qualitative compositions of the patterns of plasma protein adsorption were similar. This observation was consistent with some data that has been previously published indicating that despite the net decrease in the amount of proteins bound with PEGylated NPs, protein profiles of the PEGylated NPs were not significantly different than their uncoated controls [31]. When increasing the PEG content in the NPs to 20 %, a decrease in Complement C3 adsorption was achieved. Since C3 is a major opsonin [47], a decrease in C3 adsorption should bring about reduced uptake of LCP NPs by the RES. This was indeed the case as shown by the data in Figure 5. Specific bands of apolipoproteins Apo E and Apo A-II (Figure 6, band 5 and 7) were observed in LCP-DOTAP NPs with 5% PEGylation. Thus, we hypothesized that NPs based on cationic lipids may recruit ApoE as an endogenous ligand in vivo. Once attached to the surface of hepatocytes, LCP-DOTAP NPs with Apo E can enter the cells via receptor-mediated endocytosis. In contrast, LCP-DOPC NPs could merely transiently associate with the liver and were re-distributed out of this organ without significant internalization. Multiple receptors have been associated with ApoE-mediated uptake, such as low-density lipoprotein receptors (LDLR) and scavenger receptors, which are also expressed on the surface of hepatocytes. The formulations of these systems usually contain less than 5 mol % PEG-lipid in total [48]. However, with 20% PEG on the surface, the substrate lipid still plays a critical role in determining the in vivo fate of NPs. The exact mechanism for this observation needs to be elucidated by further experiments. One possible explanation is that the PEG coating is gradually shed during circulation and, consequently, the presence of surface charge may facilitate specific, protein-membrane interactions. PEG shedding from liposome surface has been previously proposed to control the circulation time of stealth liposomes [45, 49]. This proposed mechanism was summarized in Figure S2.
Figure 4.
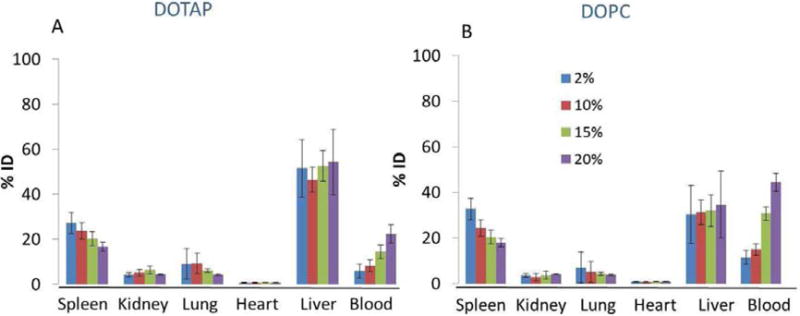
Biodistribution of LCP-DOTAP NPs (A) and LCP-DOPC NPs (B) with different PEG density in normal mice (n=4). Data are plotted as % injected dose (ID).
Figure 6.
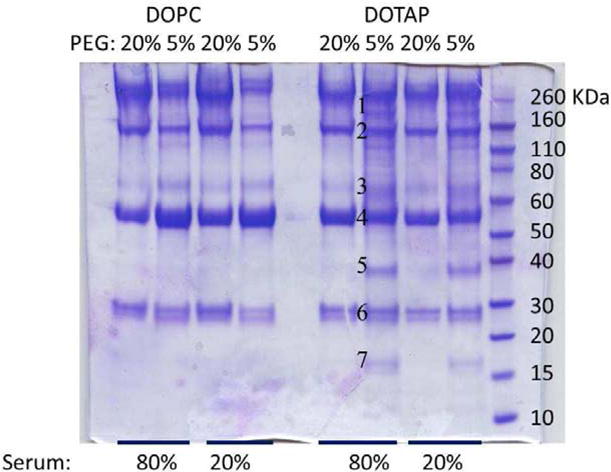
SDS-PAGE gel of serum proteins obtained from LCP NP-protein complexes following incubation at different serum concentrations. The molecular weights of the proteins in the standard ladder are reported on the right for reference. The numbers close to the gel bands for LCP-DOTAP NPs with 5% PEG in 80% serum indicate that those bands were cut out and analyzed with mass spectrometry.
3.5. ApoE dependency of LCP NPs
To determine whether apoE is indeed responsible for the specific delivery of LCP NPs to the hepatocytes, biodistribution studies were performed in wild-type and apoE−/− mice. The enhanced liver uptake of LCP-DOTAP over LCP-DOPC NPs occurred in the wild-type mice (p < 0.05). However, this is not observed in the apoE−/− mice. The biodistribution in apoE−/− mice demonstrated identical levels of hepatic accumulation for both LCP-DOTAP and LCP-DOPC NPs (Figure 7). No significant differences were found in the signals detected in other major organs. These results validated the findings regarding protein adsorption, and suggested the possibility of an Apo E-dependent uptake of LCP-DOTAP NPs by hepatocytes.
Figure 7.
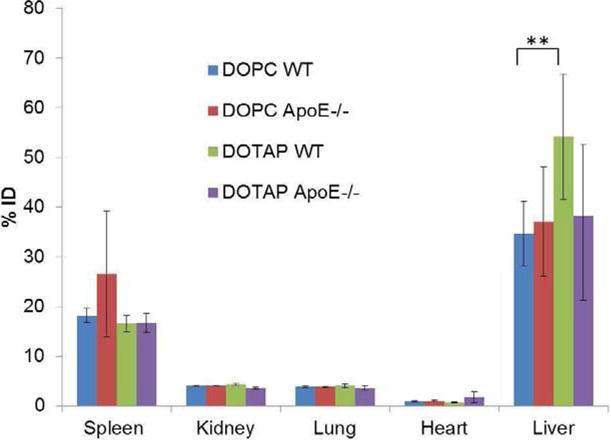
Tissue distribution of LCP NPs in wild-type and apoE−/− mice (n=4). Statistical analysis was carried out using a two-tailed t-test. ** P < 0.05 for liver accumulation of LCP- DOTAP NPs group compared to LCP- DOPC NPs group in wide type mice.
4. Conclusions
In this study, we demonstrated the ability to formulate a membrane-core structured LCP NPs with varied coverage of PEG on the surface. The implication of high density PEGylation of NPs was investigated using this platform. LCP NPs that have their surface modified with different PEG density and lipids exhibit different in vivo behaviors after they were intravenously injected. This study established a relationship between the physicochemical properties of LCP NPs and their pharmacokinetics and biodistribution profile, which may provide important information for a rational formulation development approach. Because of the diversity of engineered NPs and large variation in their surface coatings, interactions between proteins and particulate systems might be a useful tool in investigating their pharmacokinetics, biodistribution, potential therapeutic or toxicological effects.
Supplementary Material
Table 2.
Representative proteins associated with LCP-DOTAP NPs with 5% PEG incubated in 80% serum, as identified by LC MS/MS.
| Band number | Gel band Mw (kDa) | Protein identity |
|---|---|---|
| 1 | 190 | Complement C3 |
| 2 | 150 | IgG Apolipoprotein B |
| 3 | 65 | Serotransferrin Clusterin |
| 4 | 55 | Albumin |
| 5 | 38 | Apolipoprotein E |
| 6 | 28 | Apolipoprotein A-I (HDL) |
| 7 | 16 | Apolipoprotein A-II (HDL) |
Acknowledgments
The authors would like to thank Dr. Yuhua Wang for his help with SDS-PAGE. Confocal microscopy was performed at the UNC Michael Hooker Microscopy Facility. The protein identification was performed by the UNC Proteomics Core facility. The work was supported by NIH grants CA151652, CA149363 and DK100664. Kelly Racette helped to edit the manuscript.
List of Abbreviation
- apoE
apolipoprotein E
- DOPA
Dioleoylphosphatydic acid
- DOPC
Dioleoylphosphatidylcholine
- DOPE
Dioleoyl-phosphatidylethanolamine
- DOTAP
1,2-dioleoyl-3-trimethylammonium-propane
- DSPE-PEG2000
1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[poly(ethylene glycol)2000]
- DSPE-PEG-CF
1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[poly(ethylene glycol)2000-N′-carboxyfluorescein
- EPR
enhanced permeability and retention
- LCP
Lipid/Calcium/Phosphate
- ICP-MS
inductively coupled plasma mass spectrometry
- LDL
low-density lipoprotein
- IgG
Immunoglobulin G
- ID
injected dose
- MALDI
matrix-assisted laser desorption/ionization
- MS
mass spectrometry
- MW
molecular weight
- NPs
nanoparticles
- PAGE
polyacrylamide gel electrophoresis
- PEG
polyethylene glycol
- PK
pharmacokinetics
- RES
reticuloendothelial system
- TEM
transmission electron microscope
- XPS
X-ray photoelectron spectroscopy
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wagner V, Dullaart A, Bock AK, Zweck A. The emerging nanomedicine landscape. Nat Biotechnol. 2006;24:1211–7. doi: 10.1038/nbt1006-1211. [DOI] [PubMed] [Google Scholar]
- 2.Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771–82. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 3.Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7:653–64. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allen C, Dos Santos N, Gallagher R, Chiu GN, Shu Y, Li WM, et al. Controlling the physical behavior and biological performance of liposome formulations through use of surface grafted poly(ethylene glycol) Biosci Rep. 2002;22:225–50. doi: 10.1023/a:1020186505848. [DOI] [PubMed] [Google Scholar]
- 5.Flory PJ. Principles of Polymer Chemistry. Cornell Univ. Press; Ithaca, NY: 1971. [Google Scholar]
- 6.de Gennes P. Conformations of polymers attached to an interface. Macromolecules. 1980;13:1069–75. [Google Scholar]
- 7.Marsh D, Bartucci R, Sportelli L. Lipid membranes with grafted polymers: physicochemical aspects. Biochim Biophys Acta. 2003;1615:33–59. doi: 10.1016/s0005-2736(03)00197-4. [DOI] [PubMed] [Google Scholar]
- 8.Kenworthy AK, Hristova K, Needham D, McIntosh TJ. Range and magnitude of the steric pressure between bilayers containing phospholipids with covalently attached poly(ethylene glycol) Biophys J. 1995;68:1921–36. doi: 10.1016/S0006-3495(95)80369-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Owens DE, 3rd, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307:93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 10.Vonarbourg A, Passirani C, Saulnier P, Benoit JP. Parameters influencing the stealthiness of colloidal drug delivery systems. Biomaterials. 2006;27:4356–73. doi: 10.1016/j.biomaterials.2006.03.039. [DOI] [PubMed] [Google Scholar]
- 11.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5:505–15. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moghimi SM, Szebeni J. Stealth liposomes and long circulating nanoparticles: critical issues in pharmacokinetics, opsonization and protein-binding properties. Prog Lipid Res. 2003;42:463–78. doi: 10.1016/s0163-7827(03)00033-x. [DOI] [PubMed] [Google Scholar]
- 13.Xia X, Yang M, Wang Y, Zheng Y, Li Q, Chen J, et al. Quantifying the coverage density of poly(ethylene glycol) chains on the surface of gold nanostructures. ACS Nano. 2012;6:512–22. doi: 10.1021/nn2038516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Engel MF, Visser AJ, van Mierlo CP. Conformation and orientation of a protein folding intermediate trapped by adsorption. Proc Natl Acad Sci U S A. 2004;101:11316–21. doi: 10.1073/pnas.0401603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cedervall T, Lynch I, Lindman S, Berggard T, Thulin E, Nilsson H, et al. Understanding the nanoparticle-protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc Natl Acad Sci U S A. 2007;104:2050–5. doi: 10.1073/pnas.0608582104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mahmoudi M, Lynch I, Ejtehadi MR, Monopoli MP, Bombelli FB, Laurent S. Protein-nanoparticle interactions: opportunities and challenges. Chem Rev. 2011;111:5610–37. doi: 10.1021/cr100440g. [DOI] [PubMed] [Google Scholar]
- 17.Nel AE, Madler L, Velegol D, Xia T, Hoek EM, Somasundaran P, et al. Understanding biophysicochemical interactions at the nano-bio interface. Nat Mater. 2009;8:543–57. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 18.Cedervall T, Lynch I, Foy M, Berggard T, Donnelly SC, Cagney G, et al. Detailed identification of plasma proteins adsorbed on copolymer nanoparticles. Angew Chem Int Ed Engl. 2007;46:5754–6. doi: 10.1002/anie.200700465. [DOI] [PubMed] [Google Scholar]
- 19.Moghimi SM, Hunter AC, Andresen TL. Factors controlling nanoparticle pharmacokinetics: an integrated analysis and perspective. Annu Rev Pharmacol Toxicol. 2012;52:481–503. doi: 10.1146/annurev-pharmtox-010611-134623. [DOI] [PubMed] [Google Scholar]
- 20.Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev. 2001;53:283–318. [PubMed] [Google Scholar]
- 21.Dobrovolskaia MA, Aggarwal P, Hall JB, McNeil SE. Preclinical studies to understand nanoparticle interaction with the immune system and its potential effects on nanoparticle biodistribution. Mol Pharm. 2008;5:487–95. doi: 10.1021/mp800032f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim HR, Andrieux K, Delomenie C, Chacun H, Appel M, Desmaele D, et al. Analysis of plasma protein adsorption onto PEGylated nanoparticles by complementary methods: 2-DE, CE and Protein Lab-on-chip system. Electrophoresis. 2007;28:2252–61. doi: 10.1002/elps.200600694. [DOI] [PubMed] [Google Scholar]
- 23.Ogawara K, Furumoto K, Nagayama S, Minato K, Higaki K, Kai T, et al. Pre-coating with serum albumin reduces receptor-mediated hepatic disposition of polystyrene nanosphere: implications for rational design of nanoparticles. J Control Release. 2004;100:451–5. doi: 10.1016/j.jconrel.2004.07.028. [DOI] [PubMed] [Google Scholar]
- 24.Cullis PR, Chonn A, Semple SC. Interactions of liposomes and lipid-based carrier systems with blood proteins: Relation to clearance behaviour in vivo. Adv Drug Deliv Rev. 1998;32:3–17. doi: 10.1016/s0169-409x(97)00128-2. [DOI] [PubMed] [Google Scholar]
- 25.Li J, Yang Y, Huang L. Calcium phosphate nanoparticles with an asymmetric lipid bilayer coating for siRNA delivery to the tumor. J Control Release. doi: 10.1016/j.jconrel.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Y, Li J, Liu F, Huang L. Systemic delivery of siRNA via LCP nanoparticle efficiently inhibits lung metastasis. Mol Ther. 2012;20:609–15. doi: 10.1038/mt.2011.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu Y, Haynes MT, Wang Y, Liu F, Huang L. A Highly Efficient Synthetic Vector: Nonhydrodynamic Delivery of DNA to Hepatocyte Nuclei in Vivo. ACS Nano. 2013 doi: 10.1021/nn4012384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y, Kim WY, Huang L. Systemic delivery of gemcitabine triphosphate via LCP nanoparticles for NSCLC and pancreatic cancer therapy. Biomaterials. 2013;34:3447–58. doi: 10.1016/j.biomaterials.2013.01.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Graham MJ, Freier SM, Crooke RM, Ecker DJ, Maslova RN, Lesnik EA. Tritium labeling of antisense oligonucleotides by exchange with tritiated water. Nucleic Acids Res. 1993;21:3737–43. doi: 10.1093/nar/21.16.3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Monopoli MP, Walczyk D, Campbell A, Elia G, Lynch I, Bombelli FB, et al. Physical-chemical aspects of protein corona: relevance to in vitro and in vivo biological impacts of nanoparticles. J Am Chem Soc. 2011;133:2525–34. doi: 10.1021/ja107583h. [DOI] [PubMed] [Google Scholar]
- 31.Gref R, Luck M, Quellec P, Marchand M, Dellacherie E, Harnisch S, et al. ‘Stealth’ corona-core nanoparticles surface modified by polyethylene glycol (PEG): influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf B Biointerfaces. 2000;18:301–13. doi: 10.1016/s0927-7765(99)00156-3. [DOI] [PubMed] [Google Scholar]
- 32.Klibanov AL, Maruyama K, Torchilin VP, Huang L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990;268:235–7. doi: 10.1016/0014-5793(90)81016-h. [DOI] [PubMed] [Google Scholar]
- 33.Woodle MC, Lasic DD. Sterically stabilized liposomes. Biochim Biophys Acta. 1992;1113:171–99. doi: 10.1016/0304-4157(92)90038-c. [DOI] [PubMed] [Google Scholar]
- 34.Blume G, Cevc G. Liposomes for the sustained drug release in vivo. Biochim Biophys Acta. 1990;1029:91–7. doi: 10.1016/0005-2736(90)90440-y. [DOI] [PubMed] [Google Scholar]
- 35.Papahadjopoulos D, Allen TM, Gabizon A, Mayhew E, Matthay K, Huang SK, et al. Sterically stabilized liposomes: improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc Natl Acad Sci U S A. 1991;88:11460–4. doi: 10.1073/pnas.88.24.11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garbuzenko O, Barenholz Y, Priev A. Effect of grafted PEG on liposome size and on compressibility and packing of lipid bilayer. Chem Phys Lipids. 2005;135:117–29. doi: 10.1016/j.chemphyslip.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 37.Swain PS, Andelman D. Supported membranes on chemically structured and rough surfaces. Phys Rev E Stat Nonlin Soft Matter Phys. 2001;63:051911. doi: 10.1103/PhysRevE.63.051911. [DOI] [PubMed] [Google Scholar]
- 38.Lipowsky R. The conformation of membranes. Nature. 1991;349:475–81. doi: 10.1038/349475a0. [DOI] [PubMed] [Google Scholar]
- 39.Senior JH. Fate and behavior of liposomes in vivo: a review of controlling factors. Crit Rev Ther Drug Carrier Syst. 1987;3:123–93. [PubMed] [Google Scholar]
- 40.Braet F, Wisse E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: a review. Comp Hepatol. 2002;1:1. doi: 10.1186/1476-5926-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Y, Tseng Yc, Huang L. Biodistribution Studies of Nanoparticles Using Fluorescence Imaging: A Qualitative or Quantitative Method? Pharmaceutical Research. 2012;29:3273–7. doi: 10.1007/s11095-012-0818-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wong C, Stylianopoulos T, Cui J, Martin J, Chauhan VP, Jiang W, et al. Multistage nanoparticle delivery system for deep penetration into tumor tissue. Proc Natl Acad Sci U S A. 2011;108:2426–31. doi: 10.1073/pnas.1018382108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6:815–23. doi: 10.1038/nnano.2011.166. [DOI] [PubMed] [Google Scholar]
- 44.Huang L, Liu Y. In vivo delivery of RNAi with lipid-based nanoparticles. Annu Rev Biomed Eng. 2011;13:507–30. doi: 10.1146/annurev-bioeng-071910-124709. [DOI] [PubMed] [Google Scholar]
- 45.Wheeler JJ, Palmer L, Ossanlou M, MacLachlan I, Graham RW, Zhang YP, et al. Stabilized plasmid-lipid particles: construction and characterization. Gene Ther. 1999;6:271–81. doi: 10.1038/sj.gt.3300821. [DOI] [PubMed] [Google Scholar]
- 46.Li SD, Huang L. Stealth nanoparticles: high density but sheddable PEG is a key for tumor targeting. J Control Release. 2010;145:178–81. doi: 10.1016/j.jconrel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Devine DV, Wong K, Serrano K, Chonn A, Cullis PR. Liposome-complement interactions in rat serum: implications for liposome survival studies. Biochim Biophys Acta. 1994;1191:43–51. doi: 10.1016/0005-2736(94)90231-3. [DOI] [PubMed] [Google Scholar]
- 48.Akinc A, Querbes W, De S, Qin J, Frank-Kamenetsky M, Jayaprakash KN, et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol Ther. 2010;18:1357–64. doi: 10.1038/mt.2010.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holland JW, Hui C, Cullis PR, Madden TD. Poly(ethylene glycol)--lipid conjugates regulate the calcium-induced fusion of liposomes composed of phosphatidylethanolamine and phosphatidylserine. Biochemistry. 1996;35:2618–24. doi: 10.1021/bi952000v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.