

The acyl-CoA–binding protein Atg37 is a new component of the pexophagic receptor protein complex that regulates the recruitment of Atg11 by Atg30 in the peroxisomal membrane during pexophagy
Abstract
Autophagy is a membrane trafficking pathway that sequesters proteins and organelles into autophagosomes. The selectivity of this pathway is determined by autophagy receptors, such as the Pichia pastoris autophagy-related protein 30 (Atg30), which controls the selective autophagy of peroxisomes (pexophagy) through the assembly of a receptor protein complex (RPC). However, how the pexophagic RPC is regulated for efficient formation of the phagophore, an isolation membrane that sequesters the peroxisome from the cytosol, is unknown. Here we describe a new, conserved acyl-CoA–binding protein, Atg37, that is an integral peroxisomal membrane protein required specifically for pexophagy at the stage of phagophore formation. Atg30 recruits Atg37 to the pexophagic RPC, where Atg37 regulates the recruitment of the scaffold protein, Atg11. Palmitoyl-CoA competes with Atg30 for Atg37 binding. The human orthologue of Atg37, acyl-CoA–binding domain containing protein 5 (ACBD5), is also peroxisomal and is required specifically for pexophagy. We suggest that Atg37/ACBD5 is a new component and positive regulator of the pexophagic RPC.
Introduction
Autophagy is a major degradative pathway by which cytosol and organelles are delivered to the lytic compartment (the lysosome in mammals or the vacuole in yeasts) for the degradation and recycling of constituent components. Macroautophagy, the most commonly studied mode of autophagy, comprises a series of dynamic membrane-rearrangement reactions that lead to the formation of double-membrane vesicles—autophagosomes—that engulf the cargo destined for lysosomal degradation. Autophagosome formation is mediated by a core set of autophagy-related (Atg) proteins that organize the phagophore assembly site, from which the phagophore, a membrane intermediate in autophagosome formation, expands (Nakatogawa et al., 2009; Yang and Klionsky, 2009). Although autophagy is generally considered to be nonselective, there are many types of selective autophagy, e.g., pexophagy, where specific cargo, peroxisomes in this case, are recognized by the pexophagic receptor protein complex (RPC) and sequestered within a specialized autophagosome, the pexophagosome (Kraft et al., 2009; Mijaljica et al., 2012).
Peroxisomes are ubiquitous organelles that play an essential role in the metabolism of lipids and reactive oxygen species (Wanders et al., 2010; Titorenko and Terlecky, 2011). Peroxisome homeostasis is achieved by balancing peroxisome proliferation and pexophagy (Schrader et al., 2012; Till et al., 2012). Even mild defects in peroxisome proliferation cause pathological conditions commonly referred to as peroxisome biogenesis disorders (PBDs; Ebberink et al., 2010, 2012). Loss of peroxisomes also contributes to the pathogenesis of Alzheimer’s disease (AD), whereas peroxisome proliferation protects from neurodegeneration and reduces various neuropathological markers of AD (Santos et al., 2005; Kou et al., 2011; Inestrosa et al., 2013). Patients with mild forms of PBD or AD with partially dysfunctional or less abundant peroxisomes could potentially benefit from the inhibition of basal pexophagy. However, very little is known about regulation of pexophagy in mammalian cells (Till et al., 2012; Nordgren et al., 2013).
Three pexophagy receptors tag peroxisomes for degradation: Pichia pastoris Atg30, Saccharomyces cerevisiae Atg36, and mammalian NBR1 (Farré et al., 2008, 2013; Motley et al., 2012; Deosaran et al., 2013). Although yeast Atg30 and Atg36 are strictly pexophagy-specific, mammalian NBR1 is also required for aggrephagy, midbophagy, and endocytic trafficking (Kirkin et al., 2009; Mardakheh et al., 2009, 2010; Kuo et al., 2011). Therefore, mammalian proteins controlling pexophagy, but not other types of selective autophagy, are unknown. Here, we describe a novel pexophagy-specific protein, Atg37/acyl-CoA–binding domain containing protein 5 (ACBD5), conserved from yeast to human. Atg37 is a peroxisome membrane–associated acyl-CoA–binding protein, which is specifically required for phagophore formation during pexophagy. Atg37 is recruited to the pexophagic RPC by Atg30 to promote the interaction of Atg30 with Atg11. Palmitoyl-CoA competes with Atg30 for Atg37 binding. Therefore, Atg37 is a new important component and positive regulator of the pexophagic RPC. ACBD5 is the first mammalian protein to be described that is specific to pexophagy.
Results
Atg37 is an integral peroxisomal membrane protein (PMP) that binds Pex3
Pex3 is a PMP that engages peroxisomes in a range of peroxisome-related pathways, including pexophagy (Farré et al., 2008; Motley et al., 2012). To identify new proteins controlling peroxisome homeostasis, we purified a functional Pex3–TAP fusion protein from wild-type (WT) P. pastoris cells grown under peroxisome proliferation conditions (methanol medium). The Pex3-associated proteins were identified by multidimensional protein identification technology (Paoletti et al., 2004). One of the top hits was a 409-aa hypothetical protein, CAY71862, which we named Atg37.
Coimmunoprecipitation experiments confirmed the association of Atg37 with Pex3, but not significantly with other peroxins tested (Fig. 1 A). The higher molecular weight species of Atg37-HA detected by immunoblotting represent phosphorylated forms of the protein, as indicated by their sensitivity to phosphatase treatment, similar to phosphorylated forms of Pex14 (Johnson et al., 2001; Fig. S1 A). In the yeast two-hybrid (Y2H) system, Pex3 interacted with Atg30 (Farré et al., 2008), but not with Atg37. We tried several truncations of Atg37, such as Atg37ΔC (aa 1–278), Atg37-131C (aa 279–409), and Atg37-80C (aa 330–409), but none of them interacted with Pex3 in the Y2H system (Fig. S1 B). However, the in vitro protein binding assays with Glutathione Sepharose beads demonstrated that purified His-Atg37ΔC was pulled down with GST-Pex3ΔN (aa 41–455, corresponding to the cytosolic domain), but not with GST (Fig. 1 B). Therefore, Atg37 directly binds Pex3.
Figure 1.

Atg37 is an integral PMP that binds Pex3. (A) Pex3 coimmunoprecipitates with Atg37. IP, immunoprecipitate. Cells were induced in methanol medium for 4 h. (B) Atg37ΔC binds Pex3ΔN in vitro. I, input; U, unbound; P, pull-down. (C) Atg37 is an integral membrane protein. PNS, postnuclear supernatant; 27S, 27,000 g supernatant; 27P, 27,000 g pellet; S, 200,000 g supernatant; P, 200,000 g pellet. (D) Atg37 localizes to the peroxisomal membranes (in WT) or ER-associated peroxisome remnants labeled with Pex3-mRFP (in Δpex19). DIC, differential interference contrast. Bar, 5 µm. (E) The TMD is essential for peroxisomal localization of Atg37. SKL, Ser-Lys-Leu, a peroxisomal targeting signal. Bar, 5 µm.
Like Pex3 (Wiemer et al., 1996), Atg37-HA was pelleted in the organelle membrane fraction (27P) and was resistant to carbonate extraction (Fig. 1 C). In contrast, the peroxisomal matrix protein, thiolase, was completely solubilized from the organelle pellet by carbonate, as expected. Atg37-GFP completely colocalized with Pex3-mRFP on the peroxisomal membranes in WT or on the ER-associated peroxisome remnants in Δpex19 cells, which are affected at an early stage of peroxisome biogenesis (Snyder et al., 1999; Fig. 1 D). We conclude that Atg37 is a new integral PMP. Consistent with this conclusion, deletion of the predicted transmembrane domain (TMD; aa 290–339) mislocalized Atg37-GFP to the cytosol (Fig. 1 E).
Atg37 is required for pexophagy at the stage of phagophore formation
To gain insight into the function of Atg37, we studied its expression profile. Like many peroxins, Atg37 was repressed in glucose and ethanol media, but induced under peroxisome proliferation conditions in methanol and oleate media (Fig. 2 A). While cells lacking most peroxins, such as Δpex3 cells, fail to grow under peroxisome proliferation conditions, Δatg37 cells grew normally in both methanol and oleate media (Fig. 2, B and C), which suggests that Atg37 is not required for peroxisome biogenesis. We hypothesized that it might be required for pexophagy.
Figure 2.

Atg37 is required for pexophagy at the stage of phagophore formation. (A) Atg37 is expressed strictly under peroxisome proliferation conditions. Glc, glucose medium; Eth, ethanol medium; Mth, methanol medium; Ole, oleate medium; F1β, loading control. (B and C) Atg37 is dispensable for peroxisome biogenesis. Peroxisome biogenesis was assessed by the growth of strains in methanol (B) and oleate (C) media. The data shown are from a single representative experiment out of two repeats. (D and E) Atg37 is required for micropexophagy (D) and macropexophagy (E). Cells were replica plated from methanol to glucose and ethanol plates, respectively. The residual activity of AOX was used to assess pexophagy. (F and G) Atg37 is needed for the delivery of peroxisomes to the vacuole during micropexophagy (F) and macropexophagy (G). Methanol-grown cells were adapted to glucose and ethanol media, respectively. FM 4-64 labeled vacuolar membranes. Arrows point to the WT vacuoles with the peroxisome-associated GFP-SKL. Bars, 5 µm. (H and I) Atg37 is essential for MIPA (H) and pexophagosome (I) formation during micropexophagy and macropexophagy, respectively. Methanol-grown cells were adapted to glucose and ethanol media. Arrows point to MIPA (H) and pexophagosome (I) in the WT cells. Bars, 5 µm.
There are two modes of pexophagy: glucose-induced micropexophagy and ethanol-induced macropexophagy (Tuttle and Dunn, 1995). Both micropexophagy and macropexophagy were severely affected in Δatg37 cells, as deduced from the high residual activity of the peroxisomal enzyme, alcohol oxidase (AOX), after the transfer of cells from methanol to glucose or ethanol plates, respectively (Fig. 2, D and E). At the same time, deletion of another PMP, Pmp47, did not interfere with pexophagy. As in Δatg1 cells (Strømhaug et al., 2001; Mukaiyama et al., 2002; Komduur et al., 2003), peroxisomes were not delivered to the vacuole in Δatg37 cells during either micropexophagy or macropexophagy (Fig. 2, F and G).
Many Atg proteins are involved in distinct steps of phagophore formation. During micropexophagy, the phagophore is a terminal membrane structure called the micropexophagic apparatus (MIPA; Mukaiyama et al., 2004). During macropexophagy, the phagophore expands until it closes into the double-membrane pexophagosome (Mukaiyama et al., 2004). Using GFP-Atg8, which marks both the structures (Mukaiyama et al., 2004), we found that Atg37 is essential for both MIPA and pexophagosome formation, like the peroxisome receptor, Atg30 (Farré et al., 2008; Fig. 2, H and I). These results were independently confirmed with another MIPA marker, GFP-Atg26 (Oku et al., 2003). In contrast to WT, Δatg37 cells failed to form GFP-Atg26–labeled MIPA, similar to the strain devoid of Atg8, which is essential for MIPA formation (Mukaiyama et al., 2004; Fig. S1 C). Collectively, Atg37 is essential for phagophore formation during pexophagy.
Atg37 is dispensable for the cytoplasm-to-vacuole targeting (Cvt) pathway and nonselective autophagy
Despite a strong defect in pexophagy, Δatg37 cells were not affected in the maturation of the precursor of aminopeptidase I (prApe1) under both growth and starvation conditions (Fig. 3 A). At the same time, Δatg11 cells were blocked in the maturation of prApe1 only under growth conditions, Δatg28 cells showed partial maturation of prApe1 under both growth and starvation conditions, and pep4 prb1 cells, deficient in the vacuolar proteases A and B, were completely blocked in the maturation of prApe1 under both growth and starvation conditions, as expected (Farré et al., 2007; Nazarko et al., 2009). These results show that Atg37 is not required for the Cvt pathway and nonselective autophagy.
Figure 3.

Atg37 is dispensable for the Cvt pathway and nonselective autophagy. (A) Atg37 is not required for the maturation of prApe1. Cells were grown in glucose medium (“Growth”) and then adapted to glucose medium without nitrogen for 6 h (“Starvation”). mApe1, mature Ape1; *, nonspecific band. (B) Atg37 is dispensable for cell survival during nitrogen starvation. Aliquots of cultures starved in glucose medium without nitrogen were plated on YPD medium. The number of cells able to form colonies was counted after 2–3 d of incubation. The data shown are from a single representative experiment out of two repeats. (C) Atg37 is not required for GFP-Atg8 processing by autophagy. Glucose-grown cells were adapted to glucose medium without nitrogen (SD-N).
To examine the nonselective autophagy in Δatg37 cells in greater detail, we also studied cell survival and GFP-Atg8 processing under starvation conditions. In contrast to Δatg8 (Farré et al., 2010), the Δatg37 cells retained viability during prolonged nitrogen starvation, like WT cells (Fig. 3 B). Autophagy rates measured in Δatg37 cells by processing of GFP-Atg8 to GFP were also normal (Fig. 3 C). At the same time, Δatg1 cells did not process GFP-Atg8 to GFP, as expected (Nazarko et al., 2009). Therefore, Atg37 is indeed dispensable for autophagy.
Overexpression of Atg37 does not interfere with pexophagy
Because overexpression of Atg30 induces pexophagy even under peroxisome proliferation conditions and slows down the growth of cells (Farré et al., 2008), we tested whether Atg37 behaved similarly. Overexpression of Atg37-GFP from the promoter of the GAPDH gene neither induced pexophagy, judged by the levels of Pex3 and Pex12 (Fig. 4 A), nor retarded the growth of cells (Fig. 4 B) in methanol medium.
Figure 4.

Overexpression of Atg37 does not interfere with pexophagy. (A and B) Overexpression of Atg37 does not induce pexophagy (A) or affect the growth of cells in methanol medium (B). The data shown in B are from a single representative experiment out of two repeats. Ee, endogenous expression; Oe, overexpression. (C) Overexpression of neither N-terminal (GFP-Atg37) nor C-terminal (Atg37-GFP) fusions of Atg37 and GFP affects pexophagy. Methanol-grown cells were adapted to ethanol medium without nitrogen (SE-N). (D) Overexpression of Atg37 does not interfere with micropexophagy and macropexophagy. Cells were replica plated from methanol to glucose and ethanol plates, respectively. The residual activity of AOX was used to assess pexophagy.
We also addressed the effect of Atg37 overexpression under pexophagy conditions. Endogenous expression of either N-terminal (GFP-Atg37) or C-terminal (Atg37-GFP) fusions of Atg37 and GFP complemented the pexophagy defect of the Δatg37 mutant, measured by the restoration of the AOX protein degradation (Fig. 4 C). Comparable complementation was also achieved with corresponding fusions overexpressed from the promoter of the GAPDH gene, despite a dramatic overabundance of the fusion proteins (Fig. 4 C, right). Moreover, both pexophagy modes were equally unaffected by Atg37 overexpression, as overexpression of GFP-Atg37 from the promoter of the GAPDH gene did not interfere with AOX inactivation in WT cells and rescued AOX inactivation in the Δatg37 mutant, after the transfer of cells from methanol to glucose or ethanol plates (Fig. 4 D). Therefore, overexpression of Atg37 does not interfere with pexophagy.
Atg37 binds and requires Atg30 for proper localization
The involvement of Atg37 in pexophagy prompted us to examine its interactions under pexophagy conditions. Atg37 was still associated with Pex3, but not with other peroxins tested, in coimmunoprecipitation experiments (Fig. S2 A). Despite the fact that the Atg37 sequence has three potential Atg8 family–interacting motifs (AIMs; aa 25–28, 119–122, and 198–201), Atg37-HA did not coimmunoprecipitate with mCherry-Atg8 (Fig. S2 B). In the Y2H system, we identified Atg37ΔC self-interaction, which was as strong as the control interaction between Pex19 and Pex34, but did not detect any interaction of Atg37ΔC and Atg8 or truncated Atg8 (Atg8ΔC, aa 1–115), which was used in the Atg30–Atg8 interaction studies (Farré et al., 2013; Fig. S2 C). These results are consistent with the multiple sequence alignment of Atg37 orthologues showing that none of the three AIMs in the P. pastoris protein are conserved (Fig. S3).
The similar localization and function of Atg37 and the pexophagy receptor, Atg30, implied that these proteins might work together. Indeed, Atg37-GFP coimmunoprecipitated with Atg30-HA under pexophagy conditions (Fig. 5 A), and Atg37ΔC interacted with Atg30 in the Y2H system (Fig. 5 B). Because Atg30 and Atg37 interacted and were both PMPs, we tested if they played a redundant role in peroxisome biogenesis. However, normal growth of the Δatg30 Δatg37 double mutant under peroxisome proliferation conditions (Fig. 2, B and C) excluded this possibility. Although Atg30 also binds Pex3 (Farré et al., 2008) and could potentially bridge the Atg37 and Pex3 interaction in vivo, Pex3 still coimmunoprecipitated with Atg37-HA in the Δatg30 mutant under both peroxisome proliferation (Fig. 5 C) and pexophagy (Fig. 5 D) conditions, reaffirming a direct interaction between Atg37 and Pex3 in vivo.
Figure 5.

Atg37 binds and requires Atg30 for proper localization. (A) Atg37 coimmunoprecipitates with Atg30. Methanol-grown cells were adapted to glucose medium for 0.5 h to induce pexophagy. (B) Atg37ΔC (BD-37ΔC) interacts with Atg30 (AD-30) in Y2H. 3-AT, 10 mM 3-amino-1,2,4-triazole; AD, activation domain; BD, DNA binding domain. (C and D) Atg30 does not bridge the interaction between Pex3 and Atg37. Pex3 still coimmunoprecipitated with Atg37 in the absence of Atg30 under both peroxisome proliferation (C) and pexophagy (D) conditions. (E) Atg37 is not required for peroxisomal localization of Atg30. Cells were grown in methanol medium. Bar, 5 µm. (F) Atg30 is required for localization of Atg37 at the peroxisome cluster periphery. Arrows point to the peroxisome cluster with an accumulation of Atg37-GFP in the middle. Bar, 5 µm. (G) Quantitation of data in F presented as the mean + SD (error bars; n = 3; *, P < 0.001 vs. WT). (H) Atg37 accumulates in the middle of the peroxisome cluster in Δatg30 cells. Cells were grown in methanol medium. Z slices of peroxisome clusters are labeled 1–10. Bar, 2 µm.
Next, we examined the localization of Atg30-GFP and Atg37-GFP independently under peroxisome proliferation conditions, and found that the peroxisomal localization of Atg30-GFP was unaffected in the Δatg37 mutant (Fig. 5 E). In contrast, the localization of Atg37 to the peroxisome cluster periphery depended on Atg30: the Atg37-GFP was often mislocalized to the middle of the peroxisome cluster in the Δatg30 mutant (Fig. 5, F and G). Z slices of peroxisome clusters in Δatg30 cells confirmed the accumulation of Atg37-GFP in the middle of the peroxisome cluster (Fig. 5 H). Because WT and Δatg30 mutant cells had comparable levels of Atg37-HA under peroxisome proliferation conditions (Fig. 5 C, Input), the accumulation of Atg37-GFP in the middle of the peroxisome cluster in Δatg30 cells could only be explained by an aberrant redistribution of Atg37-GFP from the peroxisome cluster periphery. Together, these results suggest that Atg30 keeps Atg37 at the periphery of the peroxisome cluster, where phagophore formation is initiated.
Atg30 requires Atg37 for recruitment of Atg11 to the pexophagic RPC
Formation of the phagophore around the peroxisome is a multistep process initiated by the pexophagy receptor, Atg30, via formation of the RPC. To find the role of Atg37 in this process, we first positioned the function of Atg37 relative to Atg30. As mentioned above, overexpression of Atg30 can induce pexophagy in methanol medium, which is disadvantageous for cell growth due to the degradation of peroxisomes required for methanol utilization (Farré et al., 2008). We found that Atg37 is required for this process, as the levels of Pex3, Pex12, and even of Atg30-GFP itself were considerably higher in the Δatg37 mutant than in WT cells, when Atg30-GFP was overexpressed from the promoter of the GAPDH gene (Fig. 6 A). Deletion of the ATG37 gene also improved the growth of the strain overexpressing Atg30-GFP in methanol medium (Fig. 6 B), which suggests that Atg37 acts downstream of Atg30 in pexophagy.
Figure 6.

Atg30 requires Atg37 for recruitment of Atg11 to the pexophagic RPC. (A) Atg37 is required for pexophagy induced by the overexpression of Atg30. (B) Atg37 is necessary for the growth suppression in methanol medium caused by the overexpression of Atg30. The data shown are from a single representative experiment out of two repeats. (C) Atg11 does not coimmunoprecipitate with Atg30 in the absence of Atg37. Methanol-grown cells were adapted to the glucose medium without nitrogen for 0.5 h. The results are presented for two independent colonies of Δatg37. *, nonspecific band. (D) Atg37 is required for the Atg30-dependent localization of Atg11 to the proximity of peroxisomes. Methanol-grown cells were adapted to the glucose medium without nitrogen for 1 h. Arrows point to the accumulation of GFP-Atg11 near the peroxisomes in WT cells and on the vacuolar membrane in the mutant cells. Bar, 5 µm.
To form the RPC, Atg30 must recruit two autophagic scaffold proteins, Atg11 and Atg17, to the periphery of the peroxisome cluster (Farré et al., 2008). We tested if Atg37 was required at this step of the pexophagic RPC formation by coimmunoprecipitation. Under pexophagy conditions, Pex3, FLAG-Atg11, and GFP-Atg17 coimmunoprecipitated with Atg30-TAP in Δypt7 cells (Fig. 6 C), similar to coimmunoprecipitation of these proteins with Atg30-TAP in Δatg26 cells (Farré et al., 2008). Whereas deletion of the ATG37 gene did not affect the ability of Atg30-TAP to pull down Pex3 and GFP-Atg17, the pull-down of FLAG-Atg11 was severely impaired in two independent colonies of the Δypt7 Δatg37 double mutant (Fig. 6 C). Moreover, GFP-Atg11, which normally tags the peroxisomes during pexophagy in WT cells, did not colocalize with the peroxisomes in Δatg37 cells, as in Δatg30 cells (Fig. 6 D). Therefore, we conclude that Atg37 regulates the recruitment of Atg11 to the pexophagic RPC.
Atg37 binds palmitoyl-CoA, which competes with Atg30 for binding to Atg37
Sequence analysis of domains in Atg37 revealed a highly conserved acyl-CoA–binding domain (aa 5–102) at its N terminus. We first determined the topology of Atg37 in the peroxisomal membrane. Protease protection analyses of the organelle fractions containing N- or C-terminal GFP-tagged versions of Atg37 showed that the GFP N terminus was sensitive to proteases, like the integral PMP, Pex12 (Kalish et al., 1996), whereas the C-terminal GFP was resistant to proteases, like the intraperoxisomal peroxin, Pex8 (Zhang et al., 2006; Fig. 7 A). Therefore, the N terminus of Atg37 faces the cytosol, whereas the C terminus faces the peroxisomal matrix.
Figure 7.

Atg37 binds palmitoyl-CoA, which competes with Atg30 for binding to Atg37. (A) The acyl-CoA–binding domain of Atg37 faces the cytosol. The organelle pellet, 27P, was subjected to proteinase K and trypsin treatment. TX100, Triton X-100. Arrow points to the protease-protected C-terminal GFP fragment of Atg37-GFP. (B) Atg37ΔC binds specifically to palmitoyl-CoA. The amounts of [14C]palmitoyl-CoA and [14C]oleoyl-CoA bound by 0.07 µM His-Atg37ΔC are shown as the mean ± SD (error bars; n = 3; *, P < 0.0001 between corresponding concentrations). (C) The ACBS of Atg37ΔC is required to bind palmitoyl-CoA. The amounts of [14C]palmitoyl-CoA (9 µM) bound by 0.07 µM His-Atg37ΔC (WT or acbs mutant) or GST-Atg30 are presented as the mean of the disintegration rates ± SD (error bars; n = 3; *, P < 0.001 vs. WT His-Atg37ΔC). (D and E) The acbs mutant of Atg37ΔC can bind Pex3ΔN (D), but not Atg30 (E). (F) Palmitoyl-CoA inhibits the in vitro binding of Atg30 and Atg37ΔC. The 36 µM and 360 µM palmitoyl-CoA partially and completely inhibits the pull-down of 0.28 µM His-Atg37ΔC by 0.14 µM GST-Atg30, respectively. I, input; U, unbound; P, pull-down.
Because the acyl-CoA–binding activity on the cytosolic side of the peroxisomal membrane could potentially affect phagophore formation, we examined whether Atg37 could bind the acyl-CoA. Using an in vitro binding assay, we demonstrated that His-Atg37ΔC bound palmitoyl-CoA, but not oleoyl-CoA (Fig. 7 B). The acyl-CoA binding of Atg37ΔC depended on the acyl-CoA binding site (ACBS; aa Y40 and K44), as the Y40F K44A mutant of Atg37ΔC (acbs) was severely affected in the palmitoyl-CoA binding (Fig. 7 C). Notably, GST-Atg30 was not able to bind palmitoyl-CoA and served as a negative control. We conclude that Atg37 is a functional acyl-CoA–binding protein.
To uncover a role for acyl-CoA binding, we tested how the mutation of the ACBS affected the Atg37ΔC protein–protein interactions. Using the protein in vitro binding assay, we found that the acbs mutant of His-Atg37ΔC was pulled down with GST-Pex3ΔN, like the WT His-Atg37ΔC protein (Fig. 7 D). However, the pull-down of the acbs mutant of His-Atg37ΔC with GST-Atg30 was severely affected (Fig. 7 E), which suggests that the ACBS might either overlap with, or influence, the Atg30 binding site (30BS). In the former case, palmitoyl-CoA and Atg30 would compete for Atg37ΔC binding. To check whether such a competition really exists, we compared the effects of adding increasing concentrations of palmitoyl-CoA on GST-Atg30 binding to His-Atg37ΔC (Fig. 7 F). At 36 µM, palmitoyl-CoA started to inhibit and, at 360 µM, palmitoyl-CoA completely inhibited this protein–protein interaction. Therefore, palmitoyl-CoA and Atg30 compete for the same binding site on Atg37.
ACBS/30BS is required for Atg37 localization and phagophore formation
As stated earlier, Atg30 keeps Atg37 at the periphery of the peroxisome cluster. Then, the prediction would be that the ACBS/30BS mutant of the Atg37-GFP protein should be mislocalized to the middle of the peroxisome cluster, like the WT Atg37-GFP in the Δatg30 strain (Fig. 5, F and G). Indeed, the acbs mutant of Atg37-GFP was often found redistributed from the peroxisome cluster periphery to the middle (Fig. 8, A and B). Therefore, the ACBS/30BS is required for proper localization of Atg37.
Figure 8.
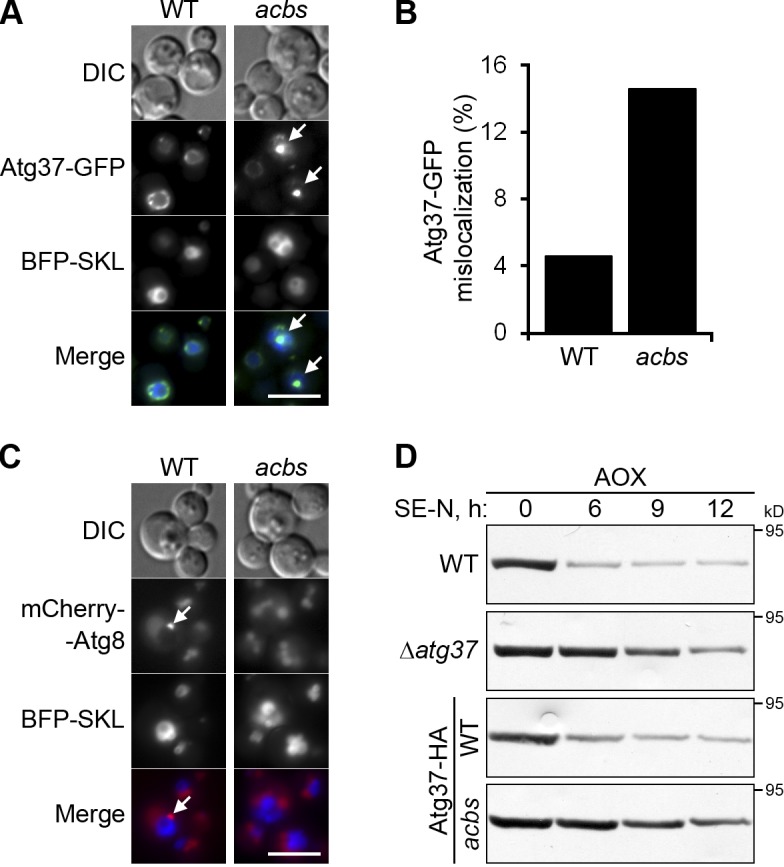
ACBS/30BS is required for Atg37 localization and phagophore formation. (A) The ACBS/30BS is required for proper localization of Atg37. Arrows point to the peroxisome clusters with accumulation of Atg37-GFP in the middle. Bar, 5 µm. (B) Quantitation of data in A. The data shown are from a single representative experiment out of three repeats. (C and D) The ACBS/30BS of Atg37 is also necessary for phagophore formation (C) and pexophagy (D). Methanol-grown cells were adapted to glucose medium (C) or ethanol medium without nitrogen (D; SE-N). Arrows point to the MIPA. Bar, 5 µm.
Because phagophore formation is normally initiated at the periphery of the peroxisome cluster, redistribution of the acbs mutant of Atg37 protein to the middle could potentially affect phagophore formation and pexophagy rates. We found that the Δatg37 strain complemented with the acbs mutant of Atg37-GFP did not form the MIPA, whereas the mCherry-Atg8–labeled MIPAs were readily identifiable in the Δatg37 strain complemented with the WT Atg37-GFP (Fig. 8 C). As a result, the Δatg37 strain complemented with the acbs mutant of Atg37-HA was severely affected in pexophagy, comparable to the Δatg37 strain (Fig. 8 D). We conclude that the ACBS/30BS is essential for phagophore formation during pexophagy.
ATG37/ACBD5 is essential for pexophagy in human cells
The mammalian orthologue of yeast Atg37 is ACBD5 (Fig. S3). Acbd5 was identified in two proteomic studies of rat liver and mouse kidney peroxisomes (Kikuchi et al., 2004; Wiese et al., 2007). To assess the localization of human ACBD5, we expressed the peroxisomally targeted mRFP–Ser-Lys-Leu (SKL) in HeLa cells and immunostained them for ACBD5. The dot-like structures labeled with anti-ACBD5 antibody completely overlapped with the mRFP-SKL–labeled peroxisomes, proving that ACBD5 localizes to peroxisomes (Fig. S4 A). Besides the sequence similarity and peroxisomal localization, P. pastoris Atg37 and human ACBD5 also share the same domain architecture with a predicted N-terminal acyl-CoA–binding domain (aa 8–96 in ACBD5) and C-terminal TMD (aa 458–480 in ACBD5). Based on the evidence presented above, we conclude that ACBD5 is the structural orthologue of PpAtg37 in human.
Next, we addressed the functional role of human ACBD5. To measure pexophagy in human cells, we used the tandem fluorochrome pexophagy assay with mRFP-EGFP-SKL as a reporter. It is based on the same principle as the recently published RG-lysosome pexophagy assay with EGFP-mCherry-TMDPEX26 (Deosaran et al., 2013). In both assays, the “red-green” peroxisomal reporter becomes “red only” after the delivery of the peroxisome to the lysosome, whose pH is acidic and causes quenching of the EGFP fluorescence. In the control treatment of HeLa cells with a nontargeting pool of siRNAs, we could readily detect the cells containing autolysosomes with acid-resistant mRFP fluorescence (Fig. 9, A and B). However, the cells treated with either siATG7 or siACBD5 did not display such “red only” structures, which suggests that, like ATG7, ACBD5 is required for the autophagic delivery of peroxisomes to the lysosomes. We also expressed mRFP-EGFP-SKL in Ntera-2 cells and calculated the ratio of mRFP and EGFP intensities per cell after knockdown of ATG16L1 or ACBD5 (Fig. 9, C and D). This experiment confirmed the appearance of “red only” structures exclusively in the cells treated with control siRNA. The mRFP/EGFP ratio in the cells treated with siACBD5 remained as low as that in the siATG16L1-treated cells, proving that ACBD5 plays an essential role in pexophagy.
Figure 9.

ATG37/ACBD5 is essential for pexophagy in human cells. (A and C) ACBD5 is needed for the delivery of peroxisomes to the lysosomes in HeLa (A) and Ntera-2 (C) cells expressing peroxisomally targeted mRFP-EGFP-SKL. Arrows point to the autolysosomes with acid-resistant mRFP fluorescence. Bars, 10 µm. (B) Quantitation of data in A. The percentage of cells with mRFP-labeled autolysosomes is shown as the mean + SD (error bars; n ≥ 50; *, P < 0.05 vs. control siRNA). (D) Quantitation of data in C. The ratio of mRFP and EGFP intensities per cell reflects the occurrence of pexophagy and is presented as the mean + SD (error bars; n ≥ 10 images; *, P < 0.05 vs. control siRNA).
Because pexophagy decreases the abundance of peroxisomes, it can also be monitored by the levels of peroxisomal marker proteins (e.g., catalase). This approach with catalase was recently used to assess basal pexophagy in HeLa cells (Huang et al., 2011; Deosaran et al., 2013). Efficient knockdown of ATG16L1 or ACBD5 in HeLa cells (Fig. S4 B) significantly increased the total fluorescence intensity of catalase (Fig. S5, A and B), confirming the requirement of ACBD5 for pexophagy. We modified this assay to measure peroxisome abundance in the primary fibroblasts of a human patient with a PBD caused by a missense mutation in the PEX16 gene (Ebberink et al., 2010). To identify transfected cells, the mRFP-SKL plasmid was cotransfected with siATG16L1, siACBD5, or control siRNA. Then, the total fluorescence intensity of peroxisomally targeted mRFP-SKL per cell was used to measure peroxisome abundance. Blocking pexophagy via ATG16L1 or ACBD5 knockdown increased peroxisome abundance in patient fibroblasts (Fig. S5, C and D), which suggests that the lack of peroxisome proliferation in human cells can be compensated by the down-regulation of pexophagy.
ATG37/ACBD5 is dispensable for aggrephagy and nonselective autophagy
To consider ACBD5 for future therapeutic applications, it is important to know how the knockdown of ACBD5 affects other autophagic pathways. We studied the role of ACBD5 in the selective autophagy of protein aggregates (aggrephagy) and nonselective autophagy. For the aggrephagy assay, we expressed the EGFP-tagged mutant huntingtin exon 1 with 72 CAG repeats, EGFP-Htt-Q72, reported to form hyperfluorescent inclusions (Furlong et al., 2000). These bright protein aggregates were easily detected in HeLa cells transfected with siATG7, but not with siACBD5 or control siRNA, 2 d after transfection with EGFP-Htt-Q72 (Fig. 10, A and B). Therefore, ACBD5 is not required for aggrephagy. Consistent with this conclusion, HeLa cells treated with ACBD5 siRNA had the same levels of SQSTM1/p62, the aggrephagy receptor that is degraded together with protein aggregates (Bjørkøy et al., 2005), as the cells treated with control siRNA (Fig. 10 C). In contrast, the knockdown of ATG7 prevented degradation of SQSTM1, as expected.
Figure 10.

ATG37/ACBD5 is dispensable for aggrephagy and nonselective autophagy. (A) ACBD5 is not required for the autophagy of Htt-Q72 aggregates in HeLa cells. Cells expressed EGFP-Htt-Q72. Arrows point to the EGFP-Htt-Q72 aggregates. Bar, 10 µm. (B) Quantitation of data in A. The percentage of cells with EGFP-Htt-Q72 aggregates is shown as the mean + SD (error bars; n ≥ 50; *, P < 0.05 vs. siATG7). (C) ACBD5 does not affect SQSTM1 degradation and LC3 lipidation. Short, short exposure; Long, long exposure; β-Actin, loading control. (D) ACBD5 is dispensable for LC3-positive vesicle formation in HeLa cells expressing fluorescently tagged LC3. Cells were treated with 20 µM chloroquine in EBSS medium for 3 h and permeabilized with 0.025% digitonin to enrich for the vesicle-bound LC3 (indicated by arrows). Bar, 10 µm. (E) Quantitation of data in D. The percentage of cells with the vesicle-bound LC3 is shown as the mean + SD (error bars; n ≥ 75; *, P < 0.05 vs. siATG16L1).
Next, we examined the role of ACBD5 in nonselective autophagy. The lipidation of mammalian orthologue of yeast Atg8, the microtubule-associated protein 1 light chain 3 (MAP1LC3 or LC3), is essential for mammalian autophagy (Tanida et al., 2004). To assess the role of ACBD5 in this process, we monitored the conversion of nonlipidated LC3-I to lipidated LC3-II by immunoblotting. The transfection of HeLa cells with siATG7 increased the amount of LC3-I and decreased the amount of LC3-II, whereas siACBD5 did not affect the lipidation of LC3 (Fig. 10 C, compare short and long exposures). Because the levels of LC3-II correlate with the number of autophagosomes, we also tested the LC3-positive vesicle formation under starvation conditions. To visualize autophagosomes and autolysosomes with fluorescently tagged LC3, Earle’s Balanced Salt Solution (EBSS) medium was supplemented with 20 µM chloroquine to neutralize lysosomal pH. Cells were enriched for LC3-positive vesicles by permeabilization with 0.025% digitonin, which allowed the removal of the nonlipidated, soluble form of LC3 from cells (Kaminskyy et al., 2011). We found that ATG16L1 siRNA severely affected autophagosome formation in HeLa cells, whereas siACBD5 did not interfere with this process (Fig. 10, D and E). Therefore, like yeast Atg37, human ACBD5 is dispensable for autophagosome formation.
Collectively, we conclude that peroxisomal ATG37/ACBD5 is specifically required for pexophagy in human cells.
Discussion
In this study, we describe a new conserved autophagic protein, Atg37/ACBD5, specifically required for pexophagy. Like the pexophagy receptor, Atg30 (Farré et al., 2008), Atg37 is expressed under peroxisome proliferation conditions, localizes to the peroxisomal membrane, and binds Pex3, but is not required for peroxisome biogenesis. Also, similar to Atg30 (Farré et al., 2008), Atg37 is involved in the phagophore formation during pexophagy, but not in the Cvt pathway or nonselective autophagy. Atg30 and Atg37 interact and depend on each other. Atg30 assists in the localization of Atg37 at the peroxisome cluster periphery where phagophore formation is initiated. In turn, Atg37 assists Atg30 in the recruitment of Atg11 to the pexophagic RPC. Therefore, Atg37 is also required for the Atg30 overexpression-induced pexophagy, but overexpression of Atg37 itself neither induces, nor interferes with, pexophagy. Atg37 binds palmitoyl-CoA, which competes with Atg30 for the binding site. Importantly, Atg37/ACBD5 localization and function are conserved from yeast to human.
Atg37 is a peroxisome membrane–associated acyl-CoA–binding protein
Several lines of evidence suggest that Atg37 is an integral PMP: (1) Atg37 binds Pex3 in vitro and coimmunoprecipitates with Pex3 in vivo even in the absence of Atg30 or pexophagy signaling; (2) Atg37 cofractionates with Pex3 in the membrane organelle fraction and, like Pex3 (Wiemer et al., 1996), is resistant to carbonate extraction; and (3) Atg37 colocalizes with Pex3 on the peroxisomal membrane and becomes cytosolic after deletion of its TMD. Topology studies showed that the N-terminal, acyl-CoA–binding domain of Atg37 faces the cytosol, whereas the C terminus of the protein faces the peroxisomal matrix. Atg37 is a functional acyl-CoA–binding protein, as it binds palmitoyl-CoA, and this binding requires the ACBS. The palmitoyl-CoA binding of Atg37 does not interfere with the Pex3 binding. Collectively, Atg37 is a new integral PMP with a cytosolic, acyl-CoA–binding domain.
We also found that human ATG37/ACBD5 colocalizes with mRFP-SKL on peroxisomes, similar to mouse Acbd5 that colocalizes with peroxisomal PMP70 (Wiese et al., 2007). Mouse and rat Acbd5 proteins were originally identified in proteomic studies of kidney and liver peroxisomes, respectively (Kikuchi et al., 2004; Wiese et al., 2007). Mutation in human ACBD5 was initially found in a family with an autosomal-dominant form of inherited thrombocytopenia 2 (Punzo et al., 2010). However, the disease was later shown to be caused by the mutations in the 5′ UTR of the ankyrin repeat domain 26 gene mapped adjacent to ACBD5 (Pippucci et al., 2011). Like yeast Atg37, human ACBD5 has a predicted acyl-CoA–binding domain at the N terminus of the protein with a conserved ACBS. We suggest that Atg37 and ACBD5 belong to a new family of the peroxisome membrane–associated acyl-CoA–binding proteins.
Atg37 is specifically required for phagophore formation during pexophagy
Atg30 (Farré et al., 2008) and Atg37 are the only two autophagic proteins known to localize to the peroxisomal membrane. However, neither of them is required for growth of cells under peroxisome proliferation conditions. They are also not redundant in peroxisome biogenesis, as the Δatg30 Δatg37 double mutant grows, like WT cells, in both methanol and oleate media. Instead, Atg30 (Farré et al., 2008) and Atg37 are required for micropexophagy and macropexophagy, the two pexophagy modes that deliver peroxisomes to the yeast vacuole for degradation and recycling (Tuttle and Dunn, 1995). Micropexophagy and macropexophagy share the de novo formation of a membrane structure that sequesters peroxisomes from the cytosol, the phagophore (Mukaiyama et al., 2004). Like Atg30 (Farré et al., 2008), Atg37 is essential for phagophore formation during both pexophagy modes, as neither MIPA, nor pexophagosomes, are formed in the Δatg37 strain under micropexophagy or macropexophagy conditions, respectively. In contrast, phagophore formation during the Cvt and nonselective autophagy must happen independently of Atg30 (Farré et al., 2008) and Atg37 because these PMPs are not required for those pathways.
The function of Atg37/ACBD5 in pexophagy is conserved from yeast to human. We provide several lines of evidence that human ACBD5 is involved in basal pexophagy: (1) like ATG7 (Iwata et al., 2006), ACBD5 is essential for the delivery of peroxisomes to the lysosomes in HeLa cells; (2) similar to ATG16L1, ACBD5 is essential for the delivery of peroxisomes to the lysosomes in Ntera-2 cells; and (3) like ATG16L1, ACBD5 controls the peroxisome abundance in HeLa cells. Moreover, knockdown of ACBD5 increased the peroxisome abundance in the primary fibroblasts of a human patient with a mild defect of peroxisome proliferation caused by a missense mutation in the PEX16 gene (Ebberink et al., 2010). The same increase of peroxisome abundance in primary fibroblasts could also be achieved with siATG16L1, but in contrast to the core autophagic proteins, such as ATG7 and ATG16L1 (Yang and Klionsky, 2010), ACBD5 was dispensable for other forms of autophagy, including aggrephagy and nonselective autophagy. Therefore, ACBD5 holds promise as a specific target for AD and the mild forms of PBD.
Recently, the lack of ACBD5 was reported to be associated with retinal dystrophy, a major cause of blindness in industrialized countries (Abu-Safieh et al., 2013). Our studies suggest how the lack of ACBD5 in the photoreceptor cells could cause a disease. The absence of ACBD5 abrogates pexophagy. Over time, it would cause the accumulation of damaged and superfluous peroxisomes, which generate the increased amounts of hydrogen peroxide. This hydrogen peroxide would increase the level of oxidative stress, which is known to cause the photoreceptor cell death (Chen et al., 2013). It remains to be tested whether pexophagy, like autophagy and mitophagy (Chen et al., 2013), protects the retina from light-induced degeneration.
Atg37 is a new component and regulator of the pexophagic RPC
Several independent approaches proved a direct interaction between the P. pastoris Atg30 and Atg37 proteins: (1) Atg30 binds Atg37 in vitro, (2) Atg30 and Atg37 interact in the heterologous yeast S. cerevisiae (Y2H system), and (3) Atg30 and Atg37 are coimmunoprecipitated in vivo during pexophagy. However, Atg37 cannot be classified as another pexophagy receptor for several reasons. In contrast to all known pexophagy receptors, Atg30, Atg36, and NBR1 (Farré et al., 2008, 2013; Motley et al., 2012; Deosaran et al., 2013), Atg37 does not interact with the phagophore protein, Atg8. Second, the overexpression of Atg37 does not induce pexophagy under peroxisome proliferation conditions. Finally, the Δatg30 mutant is completely blocked in pexophagy even after prolonged incubation of cells under pexophagy conditions (unpublished data), demonstrating that no other protein is able to substitute for Atg30.
The possibility exists that we are still missing a strictly pexophagy-specific mammalian receptor, the counterpart of yeast Atg30. Indeed, besides its recently established role in pexophagy (Deosaran et al., 2013), NBR1 together with another aggrephagy receptor, SQSTM1, is also required for the selective autophagy of ubiquitinated protein aggregates (Kirkin et al., 2009). Moreover, NBR1 also serves as the midbophagy receptor via direct binding to the midbody protein, CEP55 (Kuo et al., 2011). Additionally, NBR1 can also localize to the late endosomes and participate in the endocytic trafficking (Mardakheh et al., 2009, 2010). Therefore, ACBD5 is currently the only strictly pexophagy-specific protein identified in mammals. Because of its acyl-CoA–binding domain, it is also an excellent drug target for treatment of AD and the mild forms of PBD.
We suggest that Atg37/ACBD5 constitutes a new functional unit of the pexophagic RPC required for the recruitment of the autophagic scaffold protein. Indeed, in the absence of Atg37, Atg30 was unable to recruit Atg11 to the peroxisomes. Our studies also suggest a mechanism for how the pexophagic RPC could be regulated at the level of Atg37. Palmitoyl-CoA and Atg30 compete for the same binding site on Atg37. Several lines of evidence suggest that the ACBS serves as the 30BS: (1) the acbs mutant of Atg37 does not bind Atg30 in vitro, (2) the Atg30 and Atg37 in vitro binding is inhibited by palmitoyl-CoA, and (3) the acbs mutant of Atg37 and Δatg30 cells has the same phenotype in vivo (mislocalization of Atg37, block of phagophore formation, and deficient pexophagy). Therefore, the spatiotemporal increase in the palmitoyl-CoA can potentially displace Atg37 from Atg30 and lead to the disengagement of Atg11. Our study identifies the peroxisomal acyl-CoA–binding protein, Atg37, as a new component and positive regulator of the RPC required for phagophore formation during pexophagy.
Materials and methods
Yeast cells and transformation
We used the P. pastoris strains and plasmids (Table S1). Cells were grown to late exponential phase in YPD (1% wt/vol yeast extract, 2% wt/vol peptone, and 2% wt/vol glucose) medium, diluted 25–50 fold with fresh YPD, and grown to the early/mid-exponential phase. Cells were washed twice with YNB solution (1.7 g/liter yeast nitrogen base without amino acids and ammonium sulfate) and inoculated into peroxisome proliferation (methanol or oleate) medium. To induce pexophagy, methanol-grown cells were washed twice with YNB solution and inoculated into pexophagy (ethanol or glucose) medium. All peroxisome proliferation and pexophagy media were prepared in YNB solution. Histidine (50 mg/liter) and/or arginine (50 mg/liter) were added when needed. All peroxisome proliferation, but not pexophagy, media contained 0.05% wt/vol yeast extract. The concentration of carbon sources were: 0.5% vol/vol methanol, 0.5% vol/vol oleate, 0.5% vol/vol ethanol, or 2% wt/vol glucose. The oleate stock emulsion contained 20% vol/vol oleate and 0.5% vol/vol Tween-80. The concentration of nitrogen sources was 0.25% or 0.5% wt/vol ammonium sulfate. Cells were transformed by electroporation, as described previously (Cregg and Russell, 1998).
Construction of Δatg37 cells
The deletion cassettes containing a 796-bp fragment upstream of the ATG37 ORF, the geneticin or zeocin resistance gene, and a 483-bp fragment downstream of the ATG37 ORF were created to knock out the ATG37 ORF by the gene replacement method. The ATG37 gene was deleted in the WT strain and the Δatg37 mutant was verified by PCR.
Protein purification
Pex3–TAP was purified from the SMY154 strain (Table S1) as follows. Overnight methanol-grown cells were lysed in Dounce buffer (50 mM 2-[N-morpholino]ethanesulfonic acid, pH 6.0, 5 mM EDTA, 1% ethanol, and 1 M sorbitol) containing protease inhibitors (Roche). Differential centrifugation was performed to enrich the organelle fraction (20 min at 4,500 g to eliminate cell debris and nuclei, followed by 30 min at 100,000 g to collect the pellet). The organelle fraction was solubilized in Dounce buffer containing 1% CHAPS and cleansed by 30 min of centrifugation at 100,000 g and overnight dialysis (12–14 kD mol wt cutoff). The Pex3–TAP complex in the dialysate was purified by affinity chromatography using immunoglobulin-G agarose beads (A2909; Sigma-Aldrich). After a wash with IPP150 buffer (10 mM Tris-Cl, pH 8.0, 150 mM NaCl, and 0.1% NP-40), the complex was released from beads by digestion with tobacco etch virus enzyme (10127-017; Invitrogen). The resulting Pex3 complex was further purified using calmodulin beads (214303; Stratagene) in calcium-containing binding buffer (IPP150 buffer plus 10 mM β-mercaptoethanol, 1 mM magnesium acetate, 1 mM imidazole, and 2 mM CaCl2), and finally released by the EGTA-containing elution buffer (binding buffer with 2 mM CaCl2 replaced by 5 mM EGTA).
Atg37ΔC, Pex3ΔN and Atg30 were cloned into the pETDuet-1 (EMD Millipore), pGEX-KG, and pGEX-2TK (GE Healthcare) expression vectors, respectively. His-Atg37ΔC (WT and acbs mutant), GST-Pex3ΔN, GST-Atg30, and GST were purified according to Recombinant Protein Purification Handbook (GE Healthcare). In brief, to purify His-Atg37ΔC (WT and acbs mutant), we used the Escherichia coli BL21 (DE3) cells and Ni-NTA affinity chromatography. Cells were induced with 0.4–0.5 mM IPTG for 3 h and recombinant proteins were purified using the Ni-NTA agarose (30210; QIAGEN). Cells were resuspended in lysis buffer (50 mM Tris-Cl, pH 7.5, and 300 mM NaCl) and disrupted by sonication. The 10,000 g supernatant was allowed to bind to the Ni-NTA agarose. The beads were washed twice with lysis buffer containing 10 mM and 50 mM imidazole, respectively, and bound protein was collected using elution buffer (300 mM imidazole in 50 mM Tris-HCl, pH 7.5). For the purification of the acbs mutant, cells were resuspended in lysis buffer with 6 M urea and bound protein was eluted in elution buffer with 1 M urea.
To purify GST-Pex3ΔN, GST-Atg30, and GST proteins, we used the E. coli Rosetta (DE3) cells and Glutathione affinity chromatography. Cells were induced with 0.3–0.5 mM IPTG for 3–4 h, and recombinant proteins were purified using Glutathione Sepharose 4B media (17-0756-01; GE Healthcare). Cells were resuspended in lysis buffer (50 mM Tris-Cl, pH 7.5, 150 mM NaCl, and 0.05% NP-40) and disrupted by sonication. The 10,000 g supernatant was allowed to bind to the Glutathione Sepharose 4B media. The beads were washed with lysis buffer, and bound protein was collected using elution buffer (10 mM reduced l-Glutathione in 50 mM Tris-HCl, pH 8.0).
Acyl-CoA in vitro binding assay
Recombinant His-Atg37ΔC (WT and acbs mutant) and GST-Atg30 were purified, as described above. Then they were mixed at a final concentration of 0.07 µM with increasing concentrations of [14C]palmitoyl-CoA (NEC555010UC; PerkinElmer) or [14C]oleoyl-CoA (NEC651A010UC; PerkinElmer) ranging from 1.5 µM to 9.0 µM in 100 µl of binding buffer (10 mM potassium phosphate, pH 7.4). The reaction mixture was incubated for 30 min at 30°C. Tubes were kept on ice for 10 min and mixed with 600 µl of ice-cold 50% slurry of lipidex-1000 (6008301; PerkinElmer) in binding buffer to bind unbound acyl-CoA. The samples were incubated on ice for 10 min and centrifuged at 12,000 g for 5 min at 4°C, and 200 µl of supernatant (bound acyl-CoA) was taken for analysis of radioactivity counts. Assays were performed in triplicate with protein samples and blanks at each concentration of acyl-CoA. Radioactivity counts of blanks were subtracted from the counts of samples.
Protein in vitro binding assay
Recombinant His-Atg37ΔC (WT and acbs mutant), GST-Pex3ΔN, GST-Atg30, and GST were purified as described above. For the Atg37ΔC and Pex3ΔN binding assay, we mixed 56 pmol of His-Atg37ΔC (0.28 µM) and 14 pmol of GST-Pex3ΔN or GST (0.07 µM) in 200 µl. For the Atg37ΔC and Atg30 binding assay, we mixed 56 pmol of His-Atg37ΔC (0.28 µM) and 28 pmol of GST-Atg30 or GST (0.14 µM) in 200 µl. The reaction was performed in the binding buffer (20 mM Tris-Cl, pH 7.5, 150 mM NaCl, and 0.05% vol/vol NP-40) for 1 h at 4°C. The binding buffer also contained 0, 36, or 360 µM palmitoyl-CoA (870716P; Avanti Polar Lipids, Inc.). Then, 50 µl of reaction volume was taken as input and 150 µl was incubated overnight at 4°C with 50 µl of washed Glutathione Sepharose 4B media (17-0756-01; GE Healthcare) to bind GST-tagged proteins. The next day, 50 µl of the supernatant was used as the unbound fraction. GST-tagged proteins bound to Glutathione Sepharose beads were washed three times with binding buffer, each time with a 5-min rotation at 4°C. Bound proteins were eluted with 150 µl of 2× SDS loading buffer and boiled for 5 min. Input and unbound samples were mixed separately with 6× SDS loading buffer and boiled for 5 min. Samples were analyzed by immunoblotting.
Protein interactions in Y2H system
We used the GAL4-based Matchmaker Y2H system (Takara Bio Inc.). Full-length or truncated ORFs were inserted in activation domain (pGAD-GH) or DNA binding domain (pGBT9) vectors as indicated in the figures.
Immunoprecipitation of proteins
150 OD of cells were induced in methanol medium for 4 h (HA IPs). Alternatively, cells were grown in methanol medium for 16 h and transferred to glucose medium for 0.5 h (25 or 150 OD of cells were taken for mCherry or HA IPs, respectively). Cells were pelleted and resuspended in 0.25 (mCherry IPs) or 1 (HA IPs) ml of IP buffer (0.5% vol/vol NP-40, 50 mM Hepes-KOH, pH 7.4, 50 mM NaCl, 1 mM EDTA, 10% vol/vol glycerol, 10 mM NaF, and 1 mM protease inhibitors). Protease inhibitors used were: PMSF, leupeptin, aprotinin, and a protease inhibitor cocktail. Then, 125 (mCherry IPs) or 250 (HA IPs) µl of glass beads were added and the mixture was frozen for 1 h at −80°C. To break the cells, samples were thawed on ice followed by 10 cycles of vortexing (2 min at maximum speed followed by 2 min on ice). Cell debris was removed by centrifugation at 21,000 g for 10 min. For mCherry IPs, cell lysates were incubated for 1 h at 4°C with 50 µl of washed GammaBind G Sepharose (17-0885-02; GE Healthcare) to preclear the lysate of proteins that bind Sepharose nonspecifically. 10 µl of precleared lysate or 25 µl of lysate was taken as the input sample, diluted 20 or 8 times, mixed with 2× or 6× SDS loading buffer, and boiled for 5 min for mCherry or HA IPs, respectively. For HA IPs, the rest of the cell lysate was incubated 4 h at 4°C with 50 µl of washed EZview Red Anti-HA Affinity Gel (E6779; Sigma-Aldrich) to bind HA-tagged protein. For mCherry IPs, the remaining 240 µl of precleared lysate was incubated overnight at 4°C with 10 µl of α-DsRed polyclonal antibody (632496; Takara Bio Inc.). To pull down antibody with mCherry-Atg8, 50 µl of washed Sepharose was added to the lysate and incubated for 1 h at 4°C. For all IPs, the beads were washed three times with IP buffer. For mCherry IPs, bound proteins were eluted with 50 µl of 2× SDS loading buffer and boiled for 5 min. For HA IPs, bound proteins were eluted with 200 µl of 0.1 M glycine-HCl, pH 2.5, and neutralized with a 2 M Tris base. Then, input and elution fractions were mixed separately with 6× SDS-loading buffer and boiled for 5 min. Samples were analyzed by immunoblotting.
Immunoprecipitation of Atg30-TAP was performed as described previously (Farré et al., 2008). In brief, cells were grown in methanol medium for 16–18 h and transferred to glucose medium without nitrogen for 0.5 h. For extraction, 50 OD of cells were washed in Dulbecco’s PBS (BRL 14190; Gibco), lysed as above with 250 µl of glass beads in 1 ml of IP buffer (1% wt/vol CHAPS, 50 mM Hepes-KOH, pH 7.2, 150 mM NaCl, 1 mM EDTA, 10% vol/vol glycerol, 50 mM NaF, and 1 mM protease inhibitors, as above), and centrifuged at 500 g for 10 min. The supernatant was incubated at 4°C for 30 min with rotation to solubilize membrane proteins and centrifuged at 21,000 g for 10 min. Cleared cell lysate was incubated 1 h at 4°C with 100 µl of washed human IgG-agarose (A6284; Sigma-Aldrich). Then, beads were washed five times with IP buffer, mixed with 100 µl of 2× SDS-loading buffer, and boiled for 5 min. Samples were analyzed by immunoblotting with input and IP corresponding to 0.2 and 7.5 OD of cells per lane, respectively.
Phosphatase treatment
10 OD of cells were induced in methanol medium for 4 h. Cells were pelleted and resuspended in 200 µl of IP buffer, which contained 500 mM NaCl and 50 mM NaF. Then, 100 µl of glass beads were added and the mixture was frozen for 1 h at −80°C. Cell breakage and removal of debris was as above. The cell lysates were mixed with 40 µl of 6× SDS-loading buffer and boiled for 5 min. Then, 10 µl of boiled lysates were added to 40 ml of potato acid phosphatase (PAP) buffer (50 mM MES, pH 6.0, 1 mM DTT, and 1 mM protease inhibitors as above) containing 1 U of PAP (P3752; Sigma-Aldrich) and incubated for 2 h at 30°C. The reaction mixtures were boiled for 5 min with 10 µl of 6× SDS loading buffer and analyzed by immunoblotting.
Cellular fractionation and carbonate treatment
375 OD of cells were induced in methanol medium for 4 h. Cells were pelleted and washed twice with water. Cells were resuspended in 4 ml of Zymolyase buffer (0.5 M KCl, 5 mM MOPS-KOH, pH 7.2, and 10 mM Na2SO3) with 0.5 mg Zymolyase 100T per gram of cells. Cells were incubated for 30 min at 30°C, 80 rpm, and spheroplasts were pelleted at 2,200 g for 8 min at 4°C. Spheroplasts were washed with 3 ml of homogenization buffer (5 mM MES-KOH, pH 5.5, and 1 M sorbitol) per gram of cells. 20 firm strokes were applied in a Dounce homogenizer to break the spheroplasts in the presence of ice-cold homogenization buffer containing protease (1 mM PMSF, leupeptin, aprotinin, and protease inhibitor cocktail) and phosphatase (50 mM NaF) inhibitors. The unbroken spheroplasts, cell debris, and nuclei were removed by two sequential centrifugations at 1,000 g for 10 min at 4°C. After the second centrifugation the supernatant was considered to be the postnuclear supernatant (PNS). Centrifugation of the PNS at 27,000 g (Optima Max-E, Beckman Coulter) generated supernatant (27S) and pellet (27P). The 27P was resuspended in salt (1 M NaCl), carbonate (100 mM Na2CO3 in 10 mM Tris-HCl, pH 11.5), or detergent (2% Triton X-100 in 1 M NaCl) and incubated for 30 min at 4°C with rotation. Samples were centrifuged at 200,000 g (Optima Max-E, Beckman Coulter) to generate high-speed supernatant (S) and pellet (P), which were analyzed by immunoblotting.
Protease protection
The 27P fraction was isolated from the PNS of cells induced in methanol medium for 4 h, as described above. However, for protease protection analysis, homogenization buffer did not contain PMSF and NaF. The 27P was resuspended in ice-cold homogenization buffer with leupeptin, aprotinin, and protease inhibitor cocktail to a final concentration of 1 µg/µl. Proteinase K (160 µg) and trypsin (80 µg) were added to 400 µg of the pellet fraction in the presence or absence of 0.5% Triton X-100. Aliquots were taken after incubation at RT for the indicated times. 12.5% TCA was added to terminate reactions. Samples were analyzed by immunoblotting.
Fluorescence microscopy
0.1 ml of cell culture was collected and kept on ice before observation. For slides, 5 µl of cell culture was mixed with 5 µl of 1% low-melt agarose kept at 37°C. Images were captured at RT using a motorized fluorescence microscope (Axioskop 2 MOT) with a Plan-Apochromat 100×/1.40 NA oil differential interference contrast (DIC) objective lens and monochrome digital camera (AxioCam MRm; all from Carl Zeiss). Optimal exposition times were automatically applied to capture images. Z-stack images (25 slices, 0.255 µm apart) were used to prove Atg37-GFP localization in the middle of the peroxisome cluster. All images were acquired and processed using the AxioVision software (Carl Zeiss), versions 4.7.1 and 4.8.2, respectively. Additionally, we used version 10.0 of Photoshop CS3 Extended (Adobe).
Biochemical studies of pexophagy
The AOX plate assay was performed as described previously (Stasyk et al., 2008). In brief, cells were replica plated twice: from YPD to methanol medium for 2 d and from methanol to glucose or ethanol medium for 14 or 17 h, respectively. Then, the glucose and ethanol plates were overlaid with 10 ml of the AOX activity assay mixture (50 mM Tris-HCl, pH 8.0, 0.5 mg/ml 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) or ABTS), 1 mg/ml digitonin, 0.3% (wt/vol) low melt agarose, 6 IU/ml peroxidase, and 1% (vol/vol) methanol). The last two components were added to the mixture after it was boiled and cooled to 37°C. The mixture was allowed to set on top of the plates and the plates were incubated at 30°C until the biomass of pexophagy mutants was stained green due to a high residual activity of the peroxisomal AOX.
For pexophagy assays in liquid culture, yeast cells were pregrown in YPD, as above. Cells were washed twice with YNB solution and transferred to 25 ml of methanol medium with a starting OD600 of 0.3 for 15 h. Then, cells were washed twice with YNB solution and transferred to 25 ml of ethanol medium without nitrogen (SE-N) at an OD600 of 2. At each time-point, 1 ml culture samples were taken and TCA precipitated, as described previously (Baerends et al., 2000). Samples were analyzed by immunoblotting.
MIPA and pexophagosome formation
Endogenously expressed GFP-Atg8 and mCherry-Atg8 or GFP-Atg26 expressed from the promoter of the GAPDH gene were used as the phagophore markers. Cells expressing mCherry-Atg8 were grown in methanol medium and cells with GFP-Atg8 or GFP-Atg26 were grown in methanol medium with 2.5 µg/ml of FM 4-64 (T-3166; Invitrogen) for 15 h, transferred to glucose or ethanol medium for 1 h, and analyzed by fluorescence microscopy.
Cvt and autophagy assays in yeast cells
The prApe1 maturation and GFP-Atg8 processing assays were used as described previously (Nazarko et al., 2009). In brief, for both assays, cells were pregrown twice in YPD, washed twice with YNB solution, and transferred to glucose medium at an OD600 of 0.03–0.1 for 13–14 h. Samples corresponding to 1 OD600 of cells were taken to study prApe1 maturation in growth conditions. To study prApe1 maturation and GFP-Atg8 processing under starvation conditions, cells were washed and transferred to the glucose medium without nitrogen (SD-N). Samples corresponding to 1 OD600 of cells were taken after 6 h to study prApe1 maturation in starvation conditions. 1-ml culture samples were used to study GFP-Atg8 processing during a 0–4-h time-course in SD-N.
To examine cell viability under nitrogen starvation conditions, cells were grown in YPD medium, as described above. Cells were washed twice with YNB solution and transferred to 50 ml of glucose medium with a starting OD600 of 0.1 for 15 h. Then, cells were washed twice with YNB solution and transferred to 50 ml of fresh SD-N at an OD600 of 1. At each time point, 0.1-ml culture samples were taken, diluted, and plated on YPD plates in triplicate. Colonies were counted after 2–3 d of incubation at 37°C.
Human cells and transfection
Human cervical carcinoma cell line, HeLa (DSM ACC 57), and human neuronally committed teratocarcinoma cell line, Ntera-2 (American Type Culture Collection CRL-1973), were maintained in DMEM medium supplemented with 10% FBS and antibiotics in 5% CO2 at 37°C. For gene knockdown, we used the ON-TARGETplus SMARTpool series of siRNAs from Thermo Fisher Scientific: siATG7 (L-020112-00-0005), siATG16L1 (L-021033-01-0005), and siACBD5 (L-016183-02-0005). These products represent gene-specific pools of four individual siRNAs per gene with minimized off-target effects. As a negative control, we used the ON-TARGETplus Non-targeting Pool (D-001810-10-05; Thermo Fisher Scientific). For transfection of siRNA, cells were seeded onto coverslips in 6-well plates. The next day, cells were transfected with 24 pmol of siRNA and 4 µl of Lipofectamine RNAiMAX (13778030; Invitrogen) in a total volume of 400 µl of the serum-free medium, and grown for 3 d. siRNA efficiency was examined by immunoblotting and/or immunofluorescence microscopy with rabbit anti-ATG7 (2054-1; Epitomics), anti-ATG16L1 (HPA012577; Sigma-Aldrich), or anti-ACBD5 (HPA011861; Sigma-Aldrich) antibodies. Transfection of HeLa and Ntera-2 cells with mRFP-SKL (provided by J.C. Farré, University of California, San Diego, La Jolla, CA ; pJCF395), mRFP-EGFP-SKL (pAT003), pEGFPc1-Htt-Q72 (provided by D. Rubinsztein, University of Cambridge, Cambridge, England, UK; Furlong et al., 2000), or mTagRFP-mWasabi-LC3 (provided by J. Lin, Peking University, Peking, China; Zhou et al., 2012) plasmid DNA was performed using X-tremeGENE 9 DNA transfection reagent (06365787001; Roche) 1 d after the transfection with siRNA. Primary fibroblasts of patients with PBD (provided by S. Ferdinandusse, University of Amsterdam, Amsterdam, Netherlands; Ebberink et al., 2010) were grown as described above. Cotransfection of siRNA and plasmid DNA was performed using Basic Fibroblasts Nucleofector kit (VPI-1002; Lonza). Fibroblasts were analyzed for intensity of mRFP-SKL 4 d after transfection with siRNA.
Tandem fluorochrome pexophagy assay
This assay is a variation of a recently published method (Deosaran et al., 2013) and is based on the differential stability of mRFP and EGFP variants under acidic conditions. HeLa and Ntera-2 cells were transfected with siRNAs and mRFP-EGFP-SKL plasmid, as described above. 2 d after the first transfection, HeLa cells were cultured in the presence of lysosomal inhibitors, 120 µM leupeptin (l-2023; Sigma-Aldrich) and 2 µM E-64 (ALX-260-007; Enzo Life Sciences), for another 24 h. After incubation, cells were washed in PBS and fixed by 4% PFA in PBS for 15 min at RT. Images were captured and processed, as described above. The number of HeLa cells with mRFP-labeled autolysosomes due to lysosomal delivery of the peroxisomal reporter, mRFP-EGFP-SKL, was counted and presented as a percentage. At least 50 cells per treatment were counted in two independent experiments. Alternatively, the mRFP and EGFP intensities per cell in Ntera-2 cells were automatically quantified and presented as a ratio.
Automated image analysis of peroxisome abundance
HeLa cells were transfected with siRNAs, and primary fibroblasts were transfected with siRNAs and mRFP-SKL plasmid, as described above. After incubation, cells were washed in PBS and fixed by 4% PFA in PBS for 15 min at RT. For measurement of catalase intensity, fixed cells were permeabilized using 0.1% vol/vol Triton X-100 in PBS, blocked using 1% wt/vol BSA and 0.1% vol/vol Triton X-100 in PBS, and stained for 1 h with rabbit anti-catalase antibody (219010; EMD Millipore). Then, cells were washed and stained for 1 h with peroxidase-conjugated goat anti–rabbit secondary antibody (172-1019; Bio-Rad Laboratories). Nuclei were stained with DAPI (10236276001; Roche). Images were captured and processed, as described above. The same optimized exposition times and linear display settings were applied to all images in a series. Automated image analysis was performed using the CellProfiler software (Carpenter et al., 2006) and self-made analysis pipelines. In short, nuclei served as seed regions to identify cytoplasm using a threshold method for the readout channel. Fluorescence intensity in at least 10 individual fields of view was analyzed on a single cell level using the measurement module of CellProfiler. The mRFP-SKL intensity measurement in primary fibroblasts was performed after manual cropping in at least 20 individual images per treatment.
Aggrephagy assay of mutant huntingtin
HeLa cells were transfected with siRNAs and EGFP-Htt-Q72 plasmid, as described above. 2 d after the first transfection, cells were treated overnight with 20 µM MG132 (C2211; Sigma-Aldrich) to inhibit proteasomal degradation. In the morning, cells were washed in PBS and fixed by 4% PFA in PBS for 15 min at RT. Images were captured and processed, as described above. The number of cells with EGFP-Htt-Q72 aggregates was counted and presented as a percentage. At least 50 cells per treatment were counted in two independent experiments.
LC3-positive vesicle formation
HeLa cells were transfected with siRNAs and mTagRFP-mWasabi-LC3 plasmid, as described above. Transfection efficiency of the plasmid was assessed visually (>60% and identical for all wells). 3 d after siRNA transfection, cells were starved for 3 h in EBSS medium (14155-063; Life Technologies) in the presence of 20 µM chloroquine (tlrl-chq; InvivoGen), which was added to neutralize lysosomal pH and visualize both autophagosomes and autolysosomes (Klionsky et al., 2012). Cells were permeabilized with 0.025% digitonin in PBS for 5 min to wash out cytosolic LC3 protein and enrich for the vesicle-associated LC3 (Kaminskyy et al., 2011). Then cells were washed in PBS and fixed by 4% PFA in PBS for 15 min at RT. Images were captured and processed, as described above. The number of cells with vesicle-bound LC3 was counted and presented as a percentage of 75–200 cells per treatment.
LC3 lipidation and SQSTM1 degradation
HeLa cells were transfected with siRNAs, as described above. 3 d after siRNA transfection, cells were lysed, and levels of SQSTM1, LC3-I, and LC3-II were analyzed by immunoblotting with rabbit anti-SQSTM1 (PM045; MBL International Corporation) and mouse anti-LC3 (0231-100/LC3-5F10; NanoTools) antibodies. β-Actin served as a loading control and was detected with mouse anti–β-Actin (A5441; Sigma-Aldrich) antibody.
Statistical analysis
Statistical testing was performed using a two-tailed Student’s t test (unpaired). The critical significance level α was set to 5% (*, P < 0.05). Quantitations of data are presented as the mean, with error bars representing SD.
Online supplemental material
Fig. S1 shows supplemental data on Atg37 modification, interaction, and the role in phagophore formation. Fig. S2 shows that Atg37 binds Pex3, but not Atg8, under pexophagy conditions. Fig. S3 shows a multiple sequence alignment of Atg37 orthologues. Fig. S4 shows that ATG37/ACBD5 localizes to peroxisomes in human cells. Fig. S5 shows supplemental data on the role of ATG37/ACBD5 in pexophagy. Table S1 lists P. pastoris strains and plasmids used in this study. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201307050/DC1.
Supplementary Material
Acknowledgments
We thank S.J. Field and W.F. Loomis for critical reading of the manuscript; J.C. Farré for providing strains, plasmids, and helpful advice; N. Rayapuram for purification of Pex3-TAP; S. Ferdinandusse for patient fibroblasts; J. Lin for the mTagRFP-mWasabi-LC3 plasmid; D. Rubinsztein for the pEGFPc1-Htt-Q72 plasmid; and S.P. Briggs and J.R. Yates III for help with mass spectroscopy.
This research was supported by National Institutes of Health grants DK094843 (to T.Y. Nazarko), GM069373 (to S. Subramani), and GM085764 through the San Diego Center for Systems Biology (to A. Till and S. Subramani). A. Till was also supported by Deutsche Forschungsgemeinschaft fellowships Ti/640 1-1 and Ti/640 2-1.
The authors declare no competing financial interests.
Footnotes
Abbreviations used in this paper:
- 30BS
- Atg30 binding site
- ACBD5
- acyl-CoA–binding domain containing protein 5
- ACBS
- acyl-CoA binding site
- AD
- Alzheimer’s disease
- AIM
- Atg8 family–interacting motif
- AOX
- alcohol oxidase
- Atg
- autophagy-related
- Cvt
- cytoplasm-to-vacuole targeting
- EBSS
- Earle’s Balanced Salt Solution
- LC3
- microtubule-associated protein 1 light chain 3
- MIPA
- micropexophagic apparatus
- PAP
- potato acid phosphatase
- PBD
- peroxisome biogenesis disorder
- PMP
- peroxisomal membrane protein
- PNS
- postnuclear supernatant
- prApe1
- precursor of aminopeptidase I
- RPC
- receptor protein complex
- SKL
- Ser-Lys-Leu
- TMD
- transmembrane domain
- WT
- wild type
- Y2H
- yeast two-hybrid
References
- Abu-Safieh L., Alrashed M., Anazi S., Alkuraya H., Khan A.O., Al-Owain M., Al-Zahrani J., Al-Abdi L., Hashem M., Al-Tarimi S., et al. 2013. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 23:236–247 10.1101/gr.144105.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baerends R.J., Faber K.N., Kram A.M., Kiel J.A., van der Klei I.J., Veenhuis M. 2000. A stretch of positively charged amino acids at the N terminus of Hansenula polymorpha Pex3p is involved in incorporation of the protein into the peroxisomal membrane. J. Biol. Chem. 275:9986–9995 10.1074/jbc.275.14.9986 [DOI] [PubMed] [Google Scholar]
- Bjørkøy G., Lamark T., Brech A., Outzen H., Perander M., Overvatn A., Stenmark H., Johansen T. 2005. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171:603–614 10.1083/jcb.200507002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter A.E., Jones T.R., Lamprecht M.R., Clarke C., Kang I.H., Friman O., Guertin D.A., Chang J.H., Lindquist R.A., Moffat J., et al. 2006. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 7:R100 10.1186/gb-2006-7-10-r100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Sawada O., Kohno H., Le Y.Z., Subauste C., Maeda T., Maeda A. 2013. Autophagy protects the retina from light-induced degeneration. J. Biol. Chem. 288:7506–7518 10.1074/jbc.M112.439935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregg J.M., Russell K.A. 1998. Transformation. Methods Mol. Biol. 103:27–39 [DOI] [PubMed] [Google Scholar]
- Deosaran E., Larsen K.B., Hua R., Sargent G., Wang Y., Kim S., Lamark T., Jauregui M., Law K., Lippincott-Schwartz J., et al. 2013. NBR1 acts as an autophagy receptor for peroxisomes. J. Cell Sci. 126:939–952 10.1242/jcs.114819 [DOI] [PubMed] [Google Scholar]
- Ebberink M.S., Csanyi B., Chong W.K., Denis S., Sharp P., Mooijer P.A., Dekker C.J., Spooner C., Ngu L.H., De Sousa C., et al. 2010. Identification of an unusual variant peroxisome biogenesis disorder caused by mutations in the PEX16 gene. J. Med. Genet. 47:608–615 10.1136/jmg.2009.074302 [DOI] [PubMed] [Google Scholar]
- Ebberink M.S., Koster J., Visser G., Spronsen F., Stolte-Dijkstra I., Smit G.P., Fock J.M., Kemp S., Wanders R.J., Waterham H.R. 2012. A novel defect of peroxisome division due to a homozygous non-sense mutation in the PEX11β gene. J. Med. Genet. 49:307–313 10.1136/jmedgenet-2012-100778 [DOI] [PubMed] [Google Scholar]
- Farré J.C., Vidal J., Subramani S. 2007. A cytoplasm to vacuole targeting pathway in P. pastoris. Autophagy. 3:230–234 [DOI] [PubMed] [Google Scholar]
- Farré J.C., Manjithaya R., Mathewson R.D., Subramani S. 2008. PpAtg30 tags peroxisomes for turnover by selective autophagy. Dev. Cell. 14:365–376 10.1016/j.devcel.2007.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farré J.C., Mathewson R.D., Manjithaya R., Subramani S. 2010. Roles of Pichia pastoris Uvrag in vacuolar protein sorting and the phosphatidylinositol 3-kinase complex in phagophore elongation in autophagy pathways. Autophagy. 6:86–99 10.4161/auto.6.1.10535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farré J.C., Burkenroad A., Burnett S.F., Subramani S. 2013. Phosphorylation of mitophagy and pexophagy receptors coordinates their interaction with Atg8 and Atg11. EMBO Rep. 14:441–449 10.1038/embor.2013.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlong R.A., Narain Y., Rankin J., Wyttenbach A., Rubinsztein D.C. 2000. Alpha-synuclein overexpression promotes aggregation of mutant huntingtin. Biochem. J. 346:577–581 10.1042/0264-6021:3460577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Birmingham C.L., Shahnazari S., Shiu J., Zheng Y.T., Smith A.C., Campellone K.G., Heo W.D., Gruenheid S., Meyer T., et al. 2011. Antibacterial autophagy occurs at PI(3)P-enriched domains of the endoplasmic reticulum and requires Rab1 GTPase. Autophagy. 7:17–26 10.4161/auto.7.1.13840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inestrosa N.C., Carvajal F.J., Zolezzi J.M., Tapia-Rojas C., Serrano F., Karmelic D., Toledo E.M., Toro A., Toro J., Santos M.J. 2013. Peroxisome proliferators reduce spatial memory impairment, synaptic failure, and neurodegeneration in brains of a double transgenic mice model of Alzheimer’s disease. J. Alzheimers Dis. 33:941–959 [DOI] [PubMed] [Google Scholar]
- Iwata J., Ezaki J., Komatsu M., Yokota S., Ueno T., Tanida I., Chiba T., Tanaka K., Kominami E. 2006. Excess peroxisomes are degraded by autophagic machinery in mammals. J. Biol. Chem. 281:4035–4041 10.1074/jbc.M512283200 [DOI] [PubMed] [Google Scholar]
- Johnson M.A., Snyder W.B., Cereghino J.L., Veenhuis M., Subramani S., Cregg J.M. 2001. Pichia pastoris Pex14p, a phosphorylated peroxisomal membrane protein, is part of a PTS-receptor docking complex and interacts with many peroxins. Yeast. 18:621–641 10.1002/yea.711 [DOI] [PubMed] [Google Scholar]
- Kalish J.E., Keller G.A., Morrell J.C., Mihalik S.J., Smith B., Cregg J.M., Gould S.J. 1996. Characterization of a novel component of the peroxisomal protein import apparatus using fluorescent peroxisomal proteins. EMBO J. 15:3275–3285 [PMC free article] [PubMed] [Google Scholar]
- Kaminskyy V., Abdi A., Zhivotovsky B. 2011. A quantitative assay for the monitoring of autophagosome accumulation in different phases of the cell cycle. Autophagy. 7:83–90 10.4161/auto.7.1.13893 [DOI] [PubMed] [Google Scholar]
- Kikuchi M., Hatano N., Yokota S., Shimozawa N., Imanaka T., Taniguchi H. 2004. Proteomic analysis of rat liver peroxisome: presence of peroxisome-specific isozyme of Lon protease. J. Biol. Chem. 279:421–428 10.1074/jbc.M305623200 [DOI] [PubMed] [Google Scholar]
- Kirkin V., Lamark T., Sou Y.S., Bjørkøy G., Nunn J.L., Bruun J.A., Shvets E., McEwan D.G., Clausen T.H., Wild P., et al. 2009. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol. Cell. 33:505–516 10.1016/j.molcel.2009.01.020 [DOI] [PubMed] [Google Scholar]
- Klionsky D.J., Abdalla F.C., Abeliovich H., Abraham R.T., Acevedo-Arozena A., Adeli K., Agholme L., Agnello M., Agostinis P., Aguirre-Ghiso J.A., et al. 2012. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 8:445–544 10.4161/auto.19496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komduur J.A., Veenhuis M., Kiel J.A. 2003. The Hansenula polymorpha PDD7 gene is essential for macropexophagy and microautophagy. FEMS Yeast Res. 3:27–34 [DOI] [PubMed] [Google Scholar]
- Kou J., Kovacs G.G., Höftberger R., Kulik W., Brodde A., Forss-Petter S., Hönigschnabl S., Gleiss A., Brügger B., Wanders R., et al. 2011. Peroxisomal alterations in Alzheimer’s disease. Acta Neuropathol. 122:271–283 10.1007/s00401-011-0836-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft C., Reggiori F., Peter M. 2009. Selective types of autophagy in yeast. Biochim. Biophys. Acta. 1793:1404–1412 10.1016/j.bbamcr.2009.02.006 [DOI] [PubMed] [Google Scholar]
- Kuo T.C., Chen C.T., Baron D., Onder T.T., Loewer S., Almeida S., Weismann C.M., Xu P., Houghton J.M., Gao F.B., et al. 2011. Midbody accumulation through evasion of autophagy contributes to cellular reprogramming and tumorigenicity. Nat. Cell Biol. 13:1214–1223 10.1038/ncb2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardakheh F.K., Yekezare M., Machesky L.M., Heath J.K. 2009. Spred2 interaction with the late endosomal protein NBR1 down-regulates fibroblast growth factor receptor signaling. J. Cell Biol. 187:265–277 10.1083/jcb.200905118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardakheh F.K., Auciello G., Dafforn T.R., Rappoport J.Z., Heath J.K. 2010. Nbr1 is a novel inhibitor of ligand-mediated receptor tyrosine kinase degradation. Mol. Cell. Biol. 30:5672–5685 10.1128/MCB.00878-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijaljica D., Nazarko T.Y., Brumell J.H., Huang W.P., Komatsu M., Prescott M., Simonsen A., Yamamoto A., Zhang H., Klionsky D.J., Devenish R.J. 2012. Receptor protein complexes are in control of autophagy. Autophagy. 8:1701–1705 10.4161/auto.21332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motley A.M., Nuttall J.M., Hettema E.H. 2012. Pex3-anchored Atg36 tags peroxisomes for degradation in Saccharomyces cerevisiae. EMBO J. 31:2852–2868 10.1038/emboj.2012.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukaiyama H., Oku M., Baba M., Samizo T., Hammond A.T., Glick B.S., Kato N., Sakai Y. 2002. Paz2 and 13 other PAZ gene products regulate vacuolar engulfment of peroxisomes during micropexophagy. Genes Cells. 7:75–90 10.1046/j.1356-9597.2001.00499.x [DOI] [PubMed] [Google Scholar]
- Mukaiyama H., Baba M., Osumi M., Aoyagi S., Kato N., Ohsumi Y., Sakai Y. 2004. Modification of a ubiquitin-like protein Paz2 conducted micropexophagy through formation of a novel membrane structure. Mol. Biol. Cell. 15:58–70 10.1091/mbc.E03-05-0340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatogawa H., Suzuki K., Kamada Y., Ohsumi Y. 2009. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat. Rev. Mol. Cell Biol. 10:458–467 10.1038/nrm2708 [DOI] [PubMed] [Google Scholar]
- Nazarko T.Y., Farré J.C., Subramani S. 2009. Peroxisome size provides insights into the function of autophagy-related proteins. Mol. Biol. Cell. 20:3828–3839 10.1091/mbc.E09-03-0221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordgren M., Wang B., Apanasets O., Fransen M. 2013. Peroxisome degradation in mammals: mechanisms of action, recent advances, and perspectives. Front Physiol. 4:145 10.3389/fphys.2013.00145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oku M., Warnecke D., Noda T., Müller F., Heinz E., Mukaiyama H., Kato N., Sakai Y. 2003. Peroxisome degradation requires catalytically active sterol glucosyltransferase with a GRAM domain. EMBO J. 22:3231–3241 10.1093/emboj/cdg331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti A.C., Zybailov B., Washburn M.P. 2004. Principles and applications of multidimensional protein identification technology. Expert Rev. Proteomics. 1:275–282 10.1586/14789450.1.3.275 [DOI] [PubMed] [Google Scholar]
- Pippucci T., Savoia A., Perrotta S., Pujol-Moix N., Noris P., Castegnaro G., Pecci A., Gnan C., Punzo F., Marconi C., et al. 2011. Mutations in the 5′ UTR of ANKRD26, the ankirin repeat domain 26 gene, cause an autosomal-dominant form of inherited thrombocytopenia, THC2. Am. J. Hum. Genet. 88:115–120 10.1016/j.ajhg.2010.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punzo F., Mientjes E.J., Rohe C.F., Scianguetta S., Amendola G., Oostra B.A., Bertoli-Avella A.M., Perrotta S. 2010. A mutation in the acyl-coenzyme A binding domain-containing protein 5 gene (ACBD5) identified in autosomal dominant thrombocytopenia. J. Thromb. Haemost. 8:2085–2087 10.1111/j.1538-7836.2010.03979.x [DOI] [PubMed] [Google Scholar]
- Santos M.J., Quintanilla R.A., Toro A., Grandy R., Dinamarca M.C., Godoy J.A., Inestrosa N.C. 2005. Peroxisomal proliferation protects from beta-amyloid neurodegeneration. J. Biol. Chem. 280:41057–41068 10.1074/jbc.M505160200 [DOI] [PubMed] [Google Scholar]
- Schrader M., Bonekamp N.A., Islinger M. 2012. Fission and proliferation of peroxisomes. Biochim. Biophys. Acta. 1822:1343–1357 10.1016/j.bbadis.2011.12.014 [DOI] [PubMed] [Google Scholar]
- Snyder W.B., Faber K.N., Wenzel T.J., Koller A., Lüers G.H., Rangell L., Keller G.A., Subramani S. 1999. Pex19p interacts with Pex3p and Pex10p and is essential for peroxisome biogenesis in Pichia pastoris. Mol. Biol. Cell. 10:1745–1761 10.1091/mbc.10.6.1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stasyk O.V., Nazarko T.Y., Sibirny A.A. 2008. Methods of plate pexophagy monitoring and positive selection for ATG gene cloning in yeasts. Methods Enzymol. 451:229–239 [DOI] [PubMed] [Google Scholar]
- Strømhaug P.E., Bevan A., Dunn W.A., Jr 2001. GSA11 encodes a unique 208-kDa protein required for pexophagy and autophagy in Pichia pastoris. J. Biol. Chem. 276:42422–42435 10.1074/jbc.M104087200 [DOI] [PubMed] [Google Scholar]
- Tanida I., Ueno T., Kominami E. 2004. LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 36:2503–2518 10.1016/j.biocel.2004.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till A., Lakhani R., Burnett S.F., Subramani S. 2012. Pexophagy: the selective degradation of peroxisomes. Int. J. Cell Biol. 2012:512721 10.1155/2012/512721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titorenko V.I., Terlecky S.R. 2011. Peroxisome metabolism and cellular aging. Traffic. 12:252–259 10.1111/j.1600-0854.2010.01144.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttle D.L., Dunn W.A., Jr 1995. Divergent modes of autophagy in the methylotrophic yeast Pichia pastoris. J. Cell Sci. 108:25–35 [DOI] [PubMed] [Google Scholar]
- Wanders R.J., Ferdinandusse S., Brites P., Kemp S. 2010. Peroxisomes, lipid metabolism and lipotoxicity. Biochim. Biophys. Acta. 1801:272–280 10.1016/j.bbalip.2010.01.001 [DOI] [PubMed] [Google Scholar]
- Wiemer E.A., Lüers G.H., Faber K.N., Wenzel T., Veenhuis M., Subramani S. 1996. Isolation and characterization of Pas2p, a peroxisomal membrane protein essential for peroxisome biogenesis in the methylotrophic yeast Pichia pastoris. J. Biol. Chem. 271:18973–18980 10.1074/jbc.271.31.18973 [DOI] [PubMed] [Google Scholar]
- Wiese S., Gronemeyer T., Ofman R., Kunze M., Grou C.P., Almeida J.A., Eisenacher M., Stephan C., Hayen H., Schollenberger L., et al. 2007. Proteomics characterization of mouse kidney peroxisomes by tandem mass spectrometry and protein correlation profiling. Mol. Cell. Proteomics. 6:2045–2057 10.1074/mcp.M700169-MCP200 [DOI] [PubMed] [Google Scholar]
- Yang Z., Klionsky D.J. 2009. An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. 335:1–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Klionsky D.J. 2010. Mammalian autophagy: core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 22:124–131 10.1016/j.ceb.2009.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Léon S., Subramani S. 2006. Two independent pathways traffic the intraperoxisomal peroxin PpPex8p into peroxisomes: mechanism and evolutionary implications. Mol. Biol. Cell. 17:690–699 10.1091/mbc.E05-08-0758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C., Zhong W., Zhou J., Sheng F., Fang Z., Wei Y., Chen Y., Deng X., Xia B., Lin J. 2012. Monitoring autophagic flux by an improved tandem fluorescent-tagged LC3 (mTagRFP-mWasabi-LC3) reveals that high-dose rapamycin impairs autophagic flux in cancer cells. Autophagy. 8:1215–1226 10.4161/auto.20284 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.