

Abstract
Chromosomes must be highly compacted and organized within cells, but how this is achieved in vivo remains poorly understood. We report the use of Hi-C to map the structure of bacterial chromosomes. Analysis of Hi-C data and polymer modeling indicates that the Caulobacter crescentus chromosome consists of multiple, largely independent spatial domains likely comprised of supercoiled plectonemes arrayed into a bottlebrush-like fiber. These domains are stable throughout the cell cycle and re-established concomitantly with DNA replication. We provide evidence that domain boundaries are established by highly-expressed genes and the formation of plectoneme-free regions, whereas the histone-like protein HU and SMC promote short-range compaction and the cohesion of chromosomal arms, respectively. Collectively, our results reveal general principles for the organization and structure of chromosomes in vivo.
In all organisms chromosomal DNA must be compacted nearly three orders of magnitude to fit within the limited volume of a cell. Chromosomes must adopt structures that are compatible with critical cellular processes such as transcription, DNA replication, and chromosome segregation. While bacterial chromosomes are probably highly organized within cells (1–6), the resolution of previous studies is limited. For eukaryotes, chromosome conformation capture coupled with deep-sequencing, or Hi-C, has enabled higher resolution studies of chromosome structure in vivo (7, 8). These studies have suggested that interphase chromosomes are organized into a series of topological or structural domains <1 Mb in size (8–11), but the factors that create, maintain, and influence these domains is presently unknown.
To study the organization of bacterial chromosomes with high-resolution, we used Hi-C on Caulobacter cells (fig. S1–S2). We performed Hi-C on swarmer cells that each contain a single circular and unreplicated chromosome. To analyze our Hi-C data, we divided the genome into 10-kilobase (kb) bins with interaction frequencies for each restriction fragment assigned to corresponding bins. We visualized interactions as a heat map where each matrix position, mij, reflects the relative frequency of interactions between loci in bins i and j. For a description of data processing, normalization, reproducibility, and comparability to a previous 5C study (4), see the SOM (figs. S3–S6).
The swarmer cell interaction matrix contains two prominent diagonals (Fig. 1A). The main diagonal reflects high frequency interactions between loci on the same chromosomal arm. The other, less prominent diagonal captures lower frequency inter-arm contacts, i.e. those between loci on one chromosomal arm and those on the opposite arm of the circular genome. These locus pairs are separated by substantial distances in the primary genome sequence, but the Hi-C data indicates they are often physically adjacent and capable of interacting. This overall pattern, also seen by 5C (4), is consistent with the Caulobacter chromosome adopting an elongated structure with the single origin anchored at one pole and the two chromosome arms running the length of the cell in close proximity.
Fig. 1. Partitioning of the Caulobacter chromosome into chromosomal interaction domains (CIDs).

(A) Normalized NcoI Hi-C contact map for Caulobacter swarmer cells displaying contact frequencies for pairs of 10-kb bins across the genome. Axes indicate the genome position of each bin. Inset: simplified genomic map showing the origin of replication (ori) and terminus (ter) along with the right (black) and left (grey) chromosomal arms. (B) Hi-C contact map for one arm of the chromosome rotated 45° clockwise with directional preference plots below. Left- and right-ward preferences are shown as green and red bars, respectively. CIDs are outlined in yellow and numbered. Highly-expressed genes at CID boundaries are listed (hypothetical genes designated by GenBank ID). (C) Polymer chromosome model indicating polarly-anchored origin (magenta), chromosome backbone (black), and plectonemes (grey, with every 10th plectoneme on one arm in a color). (D) Comparison of experimental and simulated Hi-C contact maps, indicating that plectoneme-free regions (PFRs) can account for CIDs.
Further inspection of the Hi-C interaction matrix revealed highly self-interacting regions, or chromosomal interaction domains (CIDs), of the genome that appear as squares along the main diagonal (Fig. 1A) or as triangles if rotating the contact map 45° clockwise (Fig. 1B, S7–S8). Loci within a CID interact preferentially with other loci within the same CID compared to other CIDs. Loci at the border of each CID strongly favor interactions with loci on their left- or right-hand side but not both, whereas loci in the middle of a CID show high levels of interaction with loci to both sides. The Hi-C matrix exhibited variability in boundary sharpness and some nested domains (Fig. 1B, S8–S9). This hierarchical organization resembles the so-called topologically-associated domains (TADs) previously observed in eukaryotic Hi-C data (8–11).
To systematically map the boundaries of CIDs, we generated plots of directional preference as a function of genome position (Fig. 1B, S7–S9). There were 23 CIDs, ranging in length from 30 to 420 kb (table S1). CIDs were not artefacts of restriction site or sequencing read densities (fig. S10), and were independently verified using a recombination-based assay for interaction frequencies (fig. S11). The CIDs identified must be present in most cells, as Hi-C reflects interactions in a population of cells. Individual cells could have other, perhaps transient, domains.
CID boundaries were enriched for highly-expressed genes (p=7.7×10−5, Fisher's exact test, fig. S10). Of the 23 CID boundaries, 17 contained one or more highly-transcribed genes (Fig. 1B, S8,S10). We hypothesized that high gene expression unwinds the DNA duplex and creates plectoneme-free regions (PFRs), which form barriers between CIDs. These PFRs likely prevent the diffusion of supercoils and physically separate CIDs, thereby decreasing the contact probabilities of loci in different domains, as also suggested in Salmonella (1).
To better understand the 3D organization of the Caulobacter chromosome, we developed a detailed polymer model (see SOM, figs. S12–S15). The chromosome was modeled as a circular polymer comprising a dense array of plectonemes that have no sequence specificity and are stochastic in length and location (Fig. 1C, S12). We generated an equilibrium ensemble of chromosome conformations, simulated the Hi-C procedure on 25,000 modeled chromosomes, and compared the resulting data to experimental Hi-C data. By systematically varying model parameters, we identified values that provided the best fit to the observed Hi-C contact frequencies (fig. S13–S14).
Our model reflects two broad levels of chromosomal organization. On one level, the DNA is arranged into a fiber of ~300 plectonemes separated by small spacers, resembling a bottlebrush. At a higher level, the bottlebrush fiber forms a circular chromosome tethered at the pole by an origin-proximal region with chromosomal arms in close proximity down the long-axis of the cell. We also used the model to examine the effects of PFRs on interactions between loci. A single PFR of ~2 kb created a space of ~100–200 nm between flanking loci. This spacer reduced contacts between neighboring plectonemes and prevented diffusion of supercoils through the PFR in the simulations, recapitulating a CID boundary (Fig. 1D, S16). We then introduced PFRs into the chromosome model at the locations of the 20 most highly-expressed genes. Simulated Hi-C data generated a pattern of CIDs that resembled those observed experimentally (Fig. 1D, S17).
To probe the role of gene expression in chromosome structure, we performed Hi-C on swarmer cells treated for 30 minutes with rifampicin (rif), an inhibitor of transcription elongation (12). The interaction matrix for rif-treated cells was globally similar to that of untreated cells, indicating that the overall shape of the chromosome was unperturbed (Fig. 2A–B). However, CID boundaries were severely disrupted in rif-treated cells leading to a nearly domain-free organization (Fig. 2B, S18–19). Simulations of rif-treated chromosomes, performed by removing PFRs, also produced domain-free contact maps (fig. S19–21).
Fig. 2. Effect of inhibiting transcription on CID boundaries.

Normalized BglII Hi-C contact maps for (A) untreated and (B) rifampicin-treated swarmer cells. (C) Hi-C contact maps for wild-type, ΔrsaA, and ΔrsaA + van::PrsaA-rsaA. Only the region of the genome containing the van locus (dashed line) is shown.
We also moved the highly-expressed gene rsaA to the vanA locus, a poorly expressed region of the genome (fig. S22). The native vanA locus normally resides within a CID, but insertion of rsaA generated a sharp new CID boundary at this position in the genome (Fig. 2C). Relocating rsaA to the xylX locus, ~1.7 Mb from the vanA locus, also created a new CID boundary at this location (fig. S23). We conclude that highly-expressed genes play a direct role in defining chromosomal domain boundaries.
We also used Hi-C to probe the effect of inhibiting supercoiling on chromosomal organization. Swarmer cells were incubated for 30 minutes with a sublethal dose of novobiocin (fig. S24) and then subjected to Hi-C analysis (Fig. 3A–B, S18). Novobiocin, which inhibits DNA gyrase and negative supercoiling (13), significantly reduced the frequency of interactions in the 20–200 kb range while modestly increasing interactions in the 200–800 kb range relative to the untreated wild type (fig. S25). Additionally, novobiocin reduced the sharpness and positions of CID boundaries (Fig. 3B, S18). The decrease in interaction frequencies in the 20–200 kb range does not result from the emergence of a subpopulation of cells with gross defects in chromosome organization (fig. S26).
Fig. 3. Contribution of supercoiling, HU, and SMC to chromosome organization.
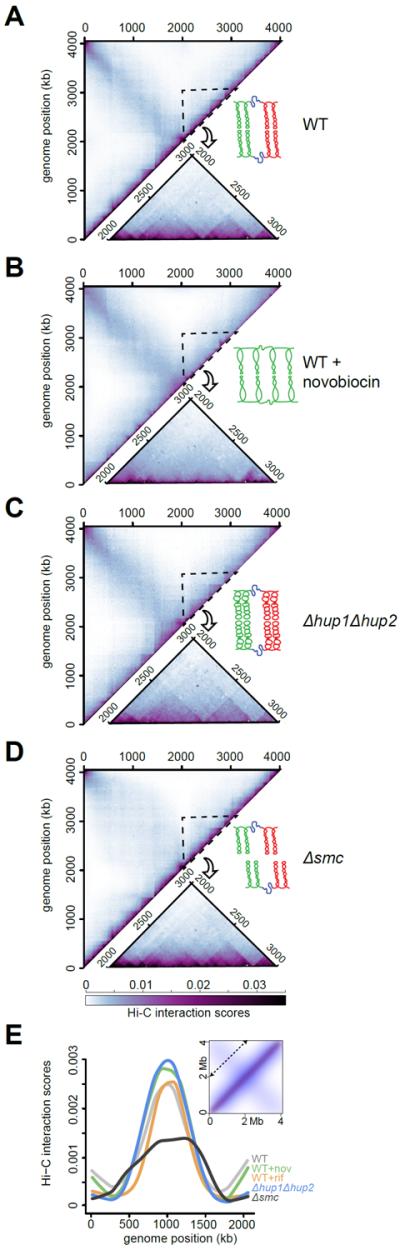
Normalized BglII Hi-C contact maps for (A) wild type, (B) novobiocin-treated wild type, (C) Δhup1Δhup2, and (D) Δsmc swarmer cells. Only the upper left regions of the symmetric Hi-C maps are shown. A region from 2–3 Mb is enlarged and shown below each map. A cartoon summarizing the effects of each perturbation is shown with neighboring domains of plectonemic DNA in red and green and plectoneme-free regions in blue. (E) Hi-C scores for the diagonal (indicated in inset) of each contact map.
To model the effect of novobiocin, we increased the spacing between duplexes in a plectoneme monomer and increased the average spacing between plectonemes five-fold. Subsequent simulations reproduced the partial loss of CID boundaries and changes in contact frequencies observed (fig. S19–20, S27). Taken together, our results demonstrate that supercoiling is (i) critical to genome compaction in the 20–200 kb range and (ii) helps establish CIDs in vivo.
To investigate the role of nucleoid-associated proteins in chromosomal organization, we focused on the histone-like proteins HU1 and HU2 (14, 15). We isolated swarmer cells from a Δhup1Δhup2 strain and performed Hi-C. The interaction matrix for Δhup1Δhup2 was grossly similar to that of wild-type cells (Fig. 3C). The correlation between directional preferences for each bin along the chromosome of wild type and Δhup1Δhup2 cells was high (r=0.73, p<10−15) indicating that CID boundaries were retained, although they became less pronounced in the absence of HU1 and HU2 (fig. S28). The contact probability plot for Δhup1Δhup2 cells revealed a significant decrease in short-range contacts, up to ~100 kb, compared to the wild type (fig. S25). These changes suggest that deleting HU may disrupt interactions within and between neighboring plectonemes without affecting inter-plectoneme spacing, which is critical for producing CID boundaries (Fig. 3C, S20). We modeled the effect of deleting HU by increasing the spacing between duplexes within a plectoneme; simulations based on this model recapitulated the Hi-C data (fig. S29). These analyses indicate that HU facilitates local, short-range compaction of the genome, possibly through the packing and stabilization of plectonemic DNA.
SMC (structural maintenance of chromosomes) homologs are found in all domains of life, and can form ring-like structures that facilitate chromosome cohesion or compaction (16–18). Hi-C analysis of Caulobacter Δsmc swarmer cells showed a clear drop in the frequency of inter-chromosomal arm interactions (Fig. 3D–E). Concomitantly, loci typically interacted with a wider range of loci on the opposite chromosomal arm compared to wild-type cells. In contrast, the frequencies of intra-arm interactions (fig. S25) and CID boundaries (fig. S28) were largely unaffected in the absence of SMC. Although many bacterial SMC proteins may compact DNA (19, 20), our data suggest that Caulobacter SMC contributes significantly to the cohesion of chromosome arms in swarmer cells.
To study chromosome organization changes during cell-cycle progression, we performed Hi-C on synchronized cells collected at regular intervals during the cell cycle (Fig. 4). After replication initiation in Caulobacter, one origin remains near the stalked pole while the other moves to the opposite pole (6). As the cell cycle progressed, the Hi-C contact maps indicated progressively more interactions between origin- and terminus-proximal loci (Figs. 4, S30), particularly at the 30 and 45 minute time-points when the newly translocated origin is close to the polarly-localized terminus (Fig. 4). However, the frequency of these interactions was still nearly 16-fold less than an average interaction between loci within 100 kb. This difference implies that the translocating chromosome is largely insulated from the anchored chromosome despite their physical proximity, which is likely to be advantageous to the segregation process by preventing the entanglement of chromosomes destined for different daughter cells. Also, the CIDs identified in swarmer cells (Fig. 1A–B) remained intact throughout the cell cycle (Fig. 4) indicating that CIDs must get re-established concurrently with, or shortly after, DNA replication (fig. S31–32), which may also help newly-replicated chromosomes from becoming entangled.
Fig. 4. Dynamics of chromosomal organization during cell cycle progression.

Plots show a section of the Hi-C interaction map for the cell-cycle time points indicated.
CIDs are reminiscent of the topologically-associated domains (TADs) documented in eukaryotes (9–11, 21) suggesting that domains on a 100-kb length-scale are a fundamental unit of chromosome structure in all organisms. Like TADs, Caulobacter CIDs often appear as nested domains, which may be related to the Mb-scale macrodomains identified in E. coli (3, 5). The identification of CIDs indicates that many domain barriers in Caulobacter are relatively fixed; however, within each CID there could be additional barriers that arise and dissipate, perhaps stochastically, as suggested in Salmonella and E. coli (1, 22), and CID boundaries may change in different growth conditions. Importantly, although chromosomal domains have now been documented in several organisms, the factors determining domain boundaries had been unclear. Although the DNA-binding proteins HU and SMC contribute to chromosome organization, they do not significantly affect CIDs. Instead, our work points to supercoiling and highly-expressed genes as critical determinants of domain formation in bacteria, and we suspect similar mechanisms contribute to creating TADs in higher organisms where boundaries may also be enriched for highly-expressed genes (9).
Supplementary Material
Acknowledgements
Hi-C data were deposited in GEO (GSE45966). We thank M. Umbarger for help optimizing Hi-C, and G. Fudenberg, A. Goloborodko, M. Hu, and T. Maxwell for discussions. M.T.L. is an Early Career Scientist of Howard Hughes Medical Institute. T.B.K.L is a postdoctoral fellow of the Life Sciences Research Foundation. This work was supported by NIH grants to M.T.L. (R01GM082899) and L.A.M. (U54CA143874).
References
- 1.Booker BM, Deng S, Higgins NP. Mol Microbiol. 2010;78:1348. doi: 10.1111/j.1365-2958.2010.07394.x. [DOI] [PubMed] [Google Scholar]
- 2.Fisher JK, et al. Cell. 2013;153:882. doi: 10.1016/j.cell.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Niki H, Yamaichi Y, Hiraga S. Genes Dev. 2000;14:212. [PMC free article] [PubMed] [Google Scholar]
- 4.Umbarger MA, et al. Mol Cell. 2011;44:252. doi: 10.1016/j.molcel.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valens M, Penaud S, Rossignol M, Cornet F, Boccard F. EMBO J. 2004;23:4330. doi: 10.1038/sj.emboj.7600434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Viollier PH, et al. Proc. Natl. Acad. Sci. U.S.A. 2004;101:9257. doi: 10.1073/pnas.0402606101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dekker J, Rippe K, Dekker M, Kleckner N. Science. 2002;295:1306. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 8.Lieberman-Aiden E, et al. Science. 2009;326:289. [Google Scholar]
- 9.Dixon JR, et al. Nature. 2012;485:376. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nora EP, et al. Nature. 2012;485:381. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sexton T, et al. Cell. 2012;148:458. doi: 10.1016/j.cell.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 12.Campbell EA, et al. Cell. 2001;104:901. doi: 10.1016/s0092-8674(01)00286-0. [DOI] [PubMed] [Google Scholar]
- 13.Gellert M, O'Dea MH, Itoh T, Tomizawa J. Proc. Natl. Acad. Sci. U.S.A. 1976;73:4474. doi: 10.1073/pnas.73.12.4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dillon S, Dorman CJ. Nat Rev Microbiol. 2009;8:185. doi: 10.1038/nrmicro2261. [DOI] [PubMed] [Google Scholar]
- 15.Guo F, Adhya S. Proc. Natl. Acad. Sci. U.S.A. 2007;104:4309. doi: 10.1073/pnas.0611686104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Badrinarayanan A, Reyes-Lamothe R, Uphoff S, Leake MC, Sherratt DJ. Science. 2012;338:528. doi: 10.1126/science.1227126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hirano T. Nat Rev Mol Cell Biol. 2006;7:311. doi: 10.1038/nrm1909. [DOI] [PubMed] [Google Scholar]
- 18.Schwartz MA, Shapiro L. Molecular Microbiology. 2011;82:1359. doi: 10.1111/j.1365-2958.2011.07836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graumann PL, Knust T. Chromosome Res. 2009;17:265. doi: 10.1007/s10577-008-9014-x. [DOI] [PubMed] [Google Scholar]
- 20.Volkov A, Mascarenhas J, Andrei-Selmer C, Ulrich HD, Graumann PL. Mol Cell Biol. 2003;23:5638. doi: 10.1128/MCB.23.16.5638-5650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hou C, Li L, Qin ZS, Corces VG. Mol Cell. 2012;48:471. doi: 10.1016/j.molcel.2012.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Postow L, Hardy CD, Arsuaga J, Cozzarelli NR. Genes Dev. 2004;18:1766. doi: 10.1101/gad.1207504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jensen R, Shapiro L. Proc. Natl. Acad. Sci. U.S.A. 1999;96:10661. doi: 10.1073/pnas.96.19.10661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Imakaev M, et al. Nat Methods. 2012;9:999. doi: 10.1038/nmeth.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hong S, et al. Proc. Natl. Acad. Sci. U.S.A. 2012;110:1674. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.