

Abstract
Adenosylcobalamin serves as a source of reactive free radicals that are generated by homolytic scission of the coenzyme’s cobalt-carbon bond. AdoCbl-dependent enzymes accelerate AdoCbl homolysis by ~1012-fold, but the mechanism by which this is accomplished remains unclear. We have combined experimental and computational approaches to gain molecular-level insight into this process for glutamate mutase. Two residues, glutamate-330 and lysine-326, form hydrogen bonds with the adenosyl group of the coenzyme. A series of mutations were introduced at these positions that impair the enzyme’s ability to catalyze coenzyme homolysis and tritium exchange with the substrate by 2 – 4 orders of magnitude. These mutations, together with the wild-type enzyme, were also characterized in silico by molecular dynamics simulations of the enzyme:AdoCbl:substrate with AdoCbl modeled in either the associated (Co-C bond formed) or the dissociated (adenosyl radical + CblII) state. The simulations reveal that the number of hydrogen bonds between the adenosyl group and the protein side-chains increases in the homolytically-dissociated state, with respect to the associated state, for both the wild-type and mutant enzymes. The mutations also cause a progressive increase in the mean distance between the 5′-carbon of the adenosyl radical and the abstractable hydrogen of the substrate. Interestingly, the distance between the 5′-carbon and substrate hydrogen, determined computationally, was found to inversely correlate with the logk for tritium exchange (r = 0.93) determined experimentally. Taken together, these results point to a dual role for these residues: they both stabilize the homolytic state through electrostatic interactions between the protein and the dissociated coenzyme, and correctly position the adenosyl radical to facilitate hydrogen abstraction from the substrate.
Introduction
Adenosylcobalamin (Coenzyme B12, AdoCbl, Figure 1) serves as a source of highly reactive carbon-based radicals that are “unmasked” by homolytic cleavage of the cofactor’s unique cobalt-carbon bond to yield a 5′-deoxyadenosine radical and cob(II)alamin.1–3 AdoCbl-dependent enzymes catalyze a variety of radical-mediated 1,2-rearrangement reactions4, 5 that are initiated by abstraction of a non-acidic hydrogen atom from the substrate by the adenosyl radical. The substrate radical generated in this process subsequently undergoes rearrangement to form a product radical, with the precise mechanism depending upon the nature of the substrate. In the final step, the product radical re-abstracts a hydrogen atom from Ado-H to form the product and regenerate the adenosyl radical, which then recombines with cob(II)alamin to regenerate AdoCbl (Figure 1).
Figure 1.

A: Structure of AdoCbl; B: Mechanistic scheme for the reaction catalyzed by glutamate mutase.
The unusual nature of the reactions catalyzed by AdoCbl-dependent enzymes has stimulated wide-ranging investigations into their mechanisms, including spectroscopic studies,6, 7 studies on model compounds8 and computational modeling.9–11 As a result, the role of the coenzyme as the source of free radicals, the identities of various radical intermediates, and the feasibility of hydrogen atom transfer steps and substrate-radical rearrangement steps are securely established. Pre-steady state kinetic measurements have shown that enzymes accelerate AdoCbl homolysis by ~1012 fold, and that homolysis and hydrogen abstraction are kinetically coupled steps so that the adenosyl radical is only transiently formed and never accumulates on the enzyme.12–14 Moreover, although the formation of organic radicals by this mechanism would be highly unfavorable in free solution, measurements on the enzymes indicate that the equilibrium constant for radical formation is close to 1, implying that the enzyme greatly stabilizes these radicals. However, the underlying mechanism by which these enzymes both accelerate AdoCbl homolysis and stabilize highly reactive free radical species remains poorly understood.
This study focuses on glutamate mutase, which catalyzes the unusual carbon skeleton rearrangement of L-glutamate to L-threo-3-methylaspartate.15–17 This enzyme has been the subject of extensive investigations by our laboratory and others. As a result, the identities of the reaction intermediates and the kinetics with which they are formed are well established for the wild-type enzyme.18–25 A high-resolution structure of glutamate mutase, crystallized in the presence of both AdoCbl and glutamate, provides an instructive starting point to investigate how the enzyme catalyzes homolysis of AdoCbl.26
Under the crystallization conditions, the enzyme underwent turnover, resulting in a clearly distinguishable mixture of glutamate and methylaspartate bound at the active site. The Co-C bond was cleaved during crystallization, revealing the ribose ring of the adenosyl moiety to exist in two distinct conformations that are related by a pseudo-rotation of the ribose ring. In one conformation, the 5′-carbon points toward the cobalt atom, with a Co-C distance of 3.2 Å and most likely reflects the conformation with the Co-C bond formed, we refer to this as the associated conformation. In the other, the 5′-carbon swings away from the cobalt atom positioning it close to the C-4 pro-S hydrogen of glutamate, which is abstracted during the reaction, we refer to this as the dissociated conformation. (Figure 2A).
Figure 2.

Structures of the associated (left) and dissociated (right) states of AdoCbl in glutamate mutase. A: Structures of wild-type enzyme determined crystallographically. B: Representative structures of wild-type enzyme obtained from MD simulations. C: Representative structures of the Glu330Asp mutant obtained from MD simulations. The substrate (glutamate) is shown in green and the 5′-carbon of adenosine is shown in black.
The ribose hydroxyl groups form hydrogen bonds with two protein side chains, Glu330 and Lys326, which differ significantly between the associated and dissociated conformers. In the associated conformer, hydrogen bonds are formed between the 2′-hydroxyl and the oxygen (O-28) of an acetamide group from the corrin ring, and between the 3′-hydroxyl and the amino group of Lys326 and carboxylate of Glu330. In the dissociated conformer, the ribose ring rotates about the glycosidic bond by ~ 25° and now forms hydrogen bonds exclusively between both the ribose 2′ and 3′-hydroxyl groups and the carboxylate of Glu330.
We have here combined both computational and experimental approaches to examine the effects of mutating Lys326 and Glu330 on the ability of the enzyme to catalyze homolysis of the Co-C bond and subsequent hydrogen abstraction from the substrate. This has allowed us to correlate changes in the structure of the active site, determined computationally, with changes in activity determined experimentally.
Experimental Procedures
Materials
AdoCbl was purchased from Sigma Chemical Co; L-[3,4-3H]-glutamic acid was purchased from Perkin Elmer; all other materials were purchased from commercial suppliers and were of the highest grade available. Site directed mutagenesis of the GlmES gene was performed by standard methods and the mutant enzymes were over-expressed and purified from recombinant E. coli as described previously.27
Enzymatic Synthesis of 5′-3H-AdoCbl
The exchange of tritium from glutamate into AdoCbl was performed by a modification of a previously reported method28. Reactions were performed in 10 mM Tris-Cl buffer, pH 8.0, containing 10% glycerol, 50 μM enzyme, 250 μM AdoCbl, 100 μM unlabeled L-glutamate, 75 μCi L-[3,4-3H]-glutamic acid (specific activity 184 Ci/mol at the exchangeable position) in a volume of 1.2 mL. Assays were performed under dim light in an anaerobic glove box to minimize exposure to oxygen and photolysis. Reactions were started by addition of 100 μL of substrate via a syringe and allowed to proceed at 37 °C. At various times, 100 μL aliquots were removed by syringe and quenched on ice by addition of 25 μL of 500 mM HCl. Samples were frozen in liquid nitrogen and stored at −20 °C prior to recovery of AdoCbl by reverse-phase HPLC.
Analysis of Radiolabeled AdoCbl
AdoCbl was purified by HPLC on a Vydac 201SP54 250 x 4.6 mm C18 reverse-phase column, 5 μm particle size, as described previously.28 The tritium content of AdoCbl was determined by liquid scintillation counting of the eluted peaks.
Equilibrium ultrafiltration measurements
AdoCbl (50 μM) was mixed with protein (50 μM) in 10 mM Tris-Cl buffer, 10% glycerol, pH 8.0, in a total volume of 500 μL. After incubation for 15 min at 37 °C, the sample was spin-filtered through a Microcon-30 filter until 75 μL had passed through the filter. The absorbance at 522 nm was recorded for the retentate and filterate. Kds were calculated assuming the filtrate absorbance represents the concentration of free AdoCbl and the retentate absorbance represents free + enzyme-bound AdoCbl.
Computational methods
The PDB structure of glutamate mutase ID 1I9C,26 was used for all simulations; mutant structures were generated using the PyMol molecular graphics system, version 1.3 Schrödinger, LLC. The Co-C bond is not formed in the crystal structure, however minimization using a forcefield including a term for the Co-C bond, followed by careful equilibration produced reasonable starting structures for simulation of the associated Co-C complex.
Histidine protonation states were determined by visual inspection of implied hydrogen bonds and pKas estimated using PROPKA29 (see supporting information). Some Asn and Gln residues were flipped to remove steric clashes as calculated using Molprobity.30 Where the calculated electron density in the crystal structure was indicative of more than one side-chain conformation, the conformation with the higher occupation number was chosen. In particular, there are two conformations of the adenosyl group, both of which have broken Co-C bonds. In only one of these conformations does the adenosyl group form hydrogen bonds with Glu330. This conformation was used as the starting structure for all simulations because it has the higher occupation number and because we believe it better resembles the dissociated state. The AmberTools 1.5 program ‘tleap’ was used to add hydrogen atoms. The protein was solvated in a water box with > 12 Å between the protein and the sides; sodium ions were added to neutralize the system.31
The AMBER ff99SB force field was used; parameters for modeling cobalamin and adenosine groups were adapted from previously published parameters derived and thoroughly tested against experimental structural data by other groups.32–34 The performance of the ff99SB force field has been shown to satisfactorily model hydrogen bonding, outperforming most semi-empirical QM/MM methods in reproducing some experimental properties such as NMR dipolar coupling constants.35–37
Charges for the cobalamin, adenosyl and glutamate substrate were calculated by the RESP fitting method at the HF/6-31G* level of theory using Gaussian09 and RED-IV via the RED server.38 Due to the known poor performance of Hartree-Fock in calculating properties of transition metals, the cobalamin charges close to the Co atom were refined by comparison with Mulliken charges calculated at the B3LYP/LACV3P* level with Jaguar 7.6, Schrödinger, LLC. The positions of water and ions were optimized with 100 steps of steepest descent followed by 900 steps of conjugate gradient minimization. The whole system was then similarly optimized with restraints of 5 kcal mol−1 Å2 on all α-carbons.
Energy minimizations were performed using the sander.MPI code in the AMBER11 package. Random velocities were assigned and the system was heated to 300 K by 50 ps Langevin dynamics (collision frequency of 5 ps−1) with α-Carbons restraints applied. 100 ps equilibration in the NPT ensemble at 300 K and 1 atm was performed with α-Carbon restraints present followed by gradual release of restraints over 50 ps of additional simulation. 18 ns production runs were performed using Langevin dynamics (collision frequency of 5 ps−1) at 300 K and 1 atm. All MD simulations were performed with a 2 fs time-step. The SHAKE algorithm was applied to constrain the length of all bonds involving hydrogen, allowing a timestep on a similar time scale to vibrations of bonds involving hydrogens to be used. Non-bonded interactions were not calculated between pairs of atoms separated by a distance greater than a cut-off of 10 Å. Periodic boundary conditions were used with a 130 × 100 × 110 Å orthorhombic box. Particle Mesh Ewald (PME) method was applied to treat long-range electrostatic interactions. All MD simulations were performed using the pmemd.mpi code in the AMBER11 package. Analysis of the MD simulations was performed using the cpptraj module of AmberTools 1.5.
Results
Glu330 is conserved in the structures of AdoCbl-dependent mutases,26, 39–41 indicating that this residue plays an important role in the mechanism of homolysis. Glu330 has been proposed to facilitate homolysis by stabilizing the dissociated state, either by forming stronger hydrogen bonding with the ribose ring or by forming an increased number of hydrogen bonds.22, 42 However, it could also be important for positioning the ribose ring to facilitate hydrogen abstraction from the substrate. Lys326 is not conserved, but is the only other side-chain to directly interact with the ribose group, suggesting it is also important for catalysis. To test these proposals, we constructed a series of enzyme variants at Lys326 and Glu330 and determined their effect on catalysis in vitro and on the structure and dynamics of the active site in silico. We then examined whether the experimentally determined changes in activity correlate with changes observed in various structural parameters derived computationally.
Five mutations were introduced into glutamate mutase by standard methods. These convert Glu330 to Gln, Asp and Ala and Lys326 to Gln and Met respectively. The Glu330Asp and Glu330Gln mutations are highly conservative: the former moves the carboxylate ~ 1 Å away from the ribose hydroxyl groups; the latter is almost isosteric with Glu and, while removing the charge, retains the ability to act as a hydrogen bond donor and acceptor. The Glu330Ala mutation was chosen as a “loss of function” mutation. In the case of Lys326, a mutation to Arg would generally be considered most conservative, but was not introduced in this case because modeling indicated the larger side-chain could not be accommodated without significant reorganization of the active site. Instead, the Lys326Met mutation was introduced as it mimics the steric bulk of lysine, but lacks its charge and potential for hydrogen bonding to the 2′-hydroxyl of ribose. The Lys326Gln mutation reduces the side-chain length and removes the positive charge, but the amide nitrogen retains the potential to act as a hydrogen bond donor to the 2′-hydroxyl group.
Activity of Mutants
The introduction of these mutations proved highly deleterious for activity. No activity could be detected for any of the mutant enzymes using the standard spectroscopic assay in which the formation of 3-methylaspartate is coupled to the formation of mesaconate through the action of 3-methylasparatase. From this we concluded that the mutant enzymes possess significantly less than 1 % of the wild-type activity. To establish that the lack of activity was not simply due to the inability of the mutant enzymes to bind AdoCbl, the affinity of the enzymes for AdoCbl was qualitatively assessed using equilibrium ultrafiltration. These measurements established that the mutations bound AdoCbl with similar, or slightly reduced, affinity to wild-type glutamate mutase. The Kd for AdoCbl of wild-type enzyme is 2 μM,27 whereas the activity measurements were conducted with 5 μM of mutant enzyme and up to 50 μM AdoCbl. It is therefore highly unlikely that the lack of activity results from the inability of the mutant enzymes to bind AdoCbl.
Spectral changes in cobalamin indicative Co-C bond cleavage
Upon homolysis, the electronic spectrum of AdoCbl undergoes extensive changes, characterized by an increase at 420 nm and decrease at 520 nm, that are associated with the conversion of 6-coordinate Co(III) to 5-coordinated Co(II). The u.v.-visible spectra of most AdoCbl enzymes, including glutamate mutase,43 when recorded during steady-state turnover, reflect this change and indicate that cob(II)alamin accumulates as an intermediate. However, consistent with the prediction that Glu330 and Lys326 stabilize the homolytic state, no changes to the spectra of the mutant enzymes were observed upon addition of substrate, indicating that Cbl(II), if formed, must be at very low concentrations.
During aerobic turnover, AdoCbl is irreversibly converted to hydroxocobalamin (CblOH), as a slow side reaction, through adventitious oxidation of Cbl(II). Thus, the formation of CblOH, which has a prominent peak at 340 nm, can be used as a proxy for Cbl(II) formation, even if Cbl(II) is only formed in low steady-state concentrations. CblOH formation was evident after several minutes upon incubation of wild-type holo-glutamate mutase with 50 mM L-glutamate in aerobic buffer at 37 °C. However, no CblOH could be detected after 24 h upon incubation of the mutant enzymes under similar conditions.
Tritium exchange between glutamate and AdoCbl
During the mechanism of AdoCbl-dependent isomerizations, the hydrogen abstracted from the substrate and the hydrogen atoms at 5′-position of AdoCbl can exchange positions, as shown in Figure 1. We exploited this fact to develop an extremely sensitive assay for AdoCbl cleavage and hydrogen transfer based on the exchange of tritium from glutamate into AdoCbl.
Measurements of deuterium and tritium kinetic isotope effects on wild-type glutamate mutase indicate that AdoCbl homolysis and hydrogen abstraction are kinetically coupled in the enzyme, and that these steps are partially rate-determining for protium and substantially rate-determining for tritium.12, 24 The failure to detect any significant concentration of Cbl(II) in the experiments noted above, implies that the mutations destabilize the dissociated coenzyme and thus recombination must be much faster than homolysis in the mutant enzymes. Under these conditions, provided that the exchange reaction does not approach equilibrium, the initial rate of tritium incorporation into AdoCbl reflects the rate of homolysis and hydrogen abstraction.
To measure tritium exchange, 50 μM enzyme, 250 μM AdoCbl, and 100 μM L-[3,4-3H]-glutamate, specific activity 184 μCi/μmol at the exchangeable position, were incubated in 10 mM Tris-Cl, pH 8.0, containing 10% glycerol, at 37 °C. All the mutant enzymes incorporated tritium into AdoCbl, with the reaction proceeding linearly for several hours (Figure 3). In all cases the specific activity of the 3H-AdoCbl formed remained less than 3% that of glutamate, so that the exchange of tritium back to glutamate remained negligible and thus the requirement that the measurement reflect the initial rate of tritium incorporation is met. The relative activities of the mutants span 2 orders of magnitude and may be summarized as Glu330Gln > Glu330Asp > Lys326Met > Glu330Ala > Lys326Gln. The rate constants for tritium exchange, kT, are summarized in Table 1. Interestingly, Glu330Asp is a more deleterious mutation than the Glu330Gln, and Lys326Met is far less deleterious than Glu330Ala. To facilitate comparisons with wild-type enzyme, for which the rate of homolysis and hydrogen transfer has been measured using pre-steady-state methods12, kT was corrected assuming a KIE of 20; a value that has been previously measured for the transfer of tritium between glutamate and AdoCbl24. Although this assumption may not be entirely valid, because the mutations may affect the value of the intrinsic KIE, we consider it very unlikely that changes in the KIE contribute significantly to the difference in the rates measured for the enzyme variants, which span 4 orders of magnitude. As an example, a recent study by Kohen and co-workers44 examining the effect of active site mutations designed to change the distance for hydride transfer between donor and acceptor atoms in dihydrofolate reductase found that intrinsic KIEs were increased by no more than 50 % at 37 °C in the mutant enzymes.
Figure 3.

Rates of tritium incorporation into AdoCbl by Glu330Gln (□), Glu330Asp (■), Glu330Ala (▲), Lys326Met (△) and Lys326Gln (○) mutant enzymes.
Table 1.
Apparent rate constants for tritium exchange (kT) and mean distance between adenosyl 5′-carbon and substrate-hydrogen (R(C5′-H)) for wild-type and mutant enzymes
| Mutant | kT (× 103 s−1) | log(kT) (relative to w.t.) | R(C5′-H) (Å) |
|---|---|---|---|
| Wild-type | 5000 ± 100* | 0 | 2.67 ± 0.18 |
| E330Q | 50 ± 2 | −2.0 | 2.85 ± 0.23 |
| E330D | 20 ± 1 | −2.4 | 3.27 ± 0.44 |
| K326M | 8.0 ± 0.1 | −2.8 | 3.30 ± 0.62 |
| E330A | 0.90 ± 0.04 | −3.7 | 3.61 ± 0.56 |
| K326Q | 0.70 ± 0.02 | −3.9 | 3.43 ± 0.39 |
Value calculated from data in reference 24
Molecular dynamics (MD) simulations
Further insight into the roles of Glu330 and Lys326 in controlling homolysis and hydrogen abstraction was provided by molecular dynamics simulations. Various factors have been proposed to explain how AdoCbl-dependent enzymes may catalyze Co-C bond cleavage. Computational studies point to electrostatic interactions as being important in stabilizing the dissociated state through increased hydrogen bonding with the ribosyl OH-groups.22, 42 The active site residues also likely play an important role in optimally positioning the 5′-carbon of the adenosyl radical for hydrogen abstraction from the substrate. To examine these possibilities, we performed molecular dynamics simulations on the wild-type and mutant enzymes, using the crystal structure of the enzyme:AdoCbl:substrate complex as a starting point26 and with AdoCbl modeled in either the associated (Co-C bond formed) or the dissociated (adenosyl radical + CblII) state.
No extensive changes in the peptide backbone were observed over the course of the simulations of the wild-type enzyme as indicated by a mean root mean squared deviation (RMSD) of backbone alpha carbons relative to the crystal structure of 1.3 Å for the dissociated state and 1.6 Å for the associated state. This shows that the protein structure is well modeled by the force field used and that the overall conformational state of the protein is similar in both the associated and dissociated states. The RMSDs of only the backbone alpha carbons of residues in the active site region (comprising all residues with at least one atom within 6Å of the adenosyl group) were also computed for the wild type and mutant enzymes. These fell within a narrow range of 1–1.6Å indicating that no large changes to the active site were introduced by any of the mutations. RMSD data is included in the supporting information, Table S3, together with overlays of wild type and mutant active site average structures, Figure S1.
The MD simulations were analyzed to calculate the mean distance, R(C5′-H), between the 5′-carbon of the adenosyl radical and the abstractable hydrogen of the substrate in the dissociated state. As shown in Figure 4, the distances extracted from the simulations were binned and fitted to a gamma distribution function (see supporting information for parameters associated with the fits), from which the mean distance, R(C5′-H), was calculated. The distances are tabulated in Table 1. The wild-type enzyme shows both the shortest mean distance, R(C5′-H) = 2.67 Å, and the narrowest distribution (standard deviation) of distances. For the most active mutant, Glu330Gln, R(C5′-H) increases to 2.85 Å and the distribution of inter-atomic distances is somewhat broader. For the least active mutants, Glu330Ala and Lys326Gln, R(C5′-H) increases to 3.61 Å and 3.43 Å respectively, and the distribution of inter-atomic distances becomes much broader. Interestingly for the Glu330Asp mutant, R(C5′-H) = 3.27 Å, is significantly longer than wild-type, which, may explain why, unexpectedly, it was less active than the Glu330Gln mutant. These results suggest that both residues are important for positioning the adenosyl radical optimally for hydrogen abstraction from the substrate.
Figure 4.

Gamma distribution plots of distances between the 5′-carbon of the adenosyl radical and the abstractable hydrogen of glutamate, R(C5′-H), for wild-type and mutant enzymes. Inset A representative fit of the binned data from MD simulations is shown for the E330Q mutant.
The MD simulations were also analyzed for the occurrence of hydrogen bonding configurations between the adenosyl-OH groups and Glu330 and Lys326. A hydrogen bond was considered to be present at any instantaneous configuration during the MD simulations if the distance between any donor – acceptor pair was < 3.5 Å and the donor-hydrogen-acceptor angle was greater than 135 degrees. Previous work has shown that simple geometric criteria for hydrogen bonding as used here yield good agreement with more elaborate energy-based criteria. The mean number of hydrogen bonds over the length of each simulation was calculated for both the associated and dissociated forms of the enzyme, and a similar analysis performed for the mutant enzymes. The data are summarized in Table 2. The data were further analyzed by calculating the mean hydrogen bond donor – acceptor distance from the simulations as a proxy for the strength of hydrogen bonding; this distance was obtained by averaging the distances over all structures where a hydrogen bond was considered present. (Table S4).
Table 2.
Hydrogen bond counts between adenosyl hydroxyl groups and side-chains at positions 326 and 330 in the coenzyme associated and dissociated configurations of glutamate mutase.
| Associated AdoCbl | Dissociated (Ado•+ Cbl(II)) | Δ H-bond (Dissociated – Associated) | |||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Ado - 330 | Ado - 326 | Total | Ado - 330 | Ado - 326 | Total | ||
| Wild-type | 0.90 | 0.26 | 1.16 | 2.03 | 0.41 | 2.44 | 1.28 |
| E330Q | 0.15 | 0.14 | 0.29 | 1.04 | 0.43 | 1.47 | 1.18 |
| E330D | 0.14 | 0.23 | 0.37 | 1.64 | 0.13 | 1.77 | 1.40 |
| K326M | 0.38 | 0.00 | 0.38 | 1.57 | 0.00 | 1.57 | 1.19 |
| E330A | 0.00 | 0.16 | 0.16 | 0.00 | 0.09 | 0.09 | −0.07 |
| K326Q | 0.06 | 0.57 | 0.63 | 1.53 | 0.15 | 1.68 | 1.05 |
With the exception of the Glu330Ala mutation, the number and strength of hydrogen bonds between the residue at position-330 and the ribose moiety is significantly greater in the dissociated state of AdoCbl than in the associated state. In the case of Glu330Ala, which cannot form hydrogen bonds, water molecules enter the active site to fill the void created by the mutation. The simulations indicate that, on average, the wild-type enzyme forms both more and shorter hydrogen bonds with the adenosyl-OH groups in both the associated and dissociated state than any of the mutant enzymes. This observation underscores the importance of the hydrogen bonds formed by this residue for catalysis.
Hydrogen bonding between Lys326 and adenosyl-OH groups appears less important. For the wild-type enzyme, the mean number of hydrogen bonds formed between Lys326 and adenosyl-OH groups during the simulations is fewer, and is not greatly different between the associated and dissociated states of AdoCbl. For the Lys326Met mutant no hydrogen bonding is possible at this position, but for the other mutants there is no obvious relationship between the mutation and the number or strength of hydrogen bonds formed to ribose at this position. However, mutation of Lys326 does appear to impair the ability of Glu330 to form hydrogen bonds with the ribose OH groups and, as discussed below, the 5′-carbon is much less well-positioned for hydrogen abstraction. The low activity of the Lys326Met and Lys326Gln mutants suggests that Lys326 plays an important role in positioning Glu330 to effectively hydrogen bond with the ribose group.
Correlations between simulation and experiment
To provide further support that the changes to the structure and dynamics of the active site identified in the simulations were indeed contributing to the experimentally measured deceases in enzyme activity, we examined the data for correlations between computationally derived parameters and experimentally measured tritium exchange rate, kT. Interestingly, -logkT exhibited a good linear correlation with R(C5′-H), r = 0.93, r2 = 0.86 (Figure 5), pointing to the important role that these residues play in positioning the adenosyl radical relative to substrate. Changes in hydrogen bonding patterns between mutants (either in the associated or dissociated state) showed no clear relationship with the activity of the mutant enzyme. We do not interpret this to mean that changes in hydrogen bonding interactions are unimportant for the reduced activities of the mutants. Rather any clear correlation between changes in hydrogen bonding interactions and mutant activity is masked because the activities are also affected by changes in the positioning of the adenosyl radical in the active site due to mutation.
Figure 5.
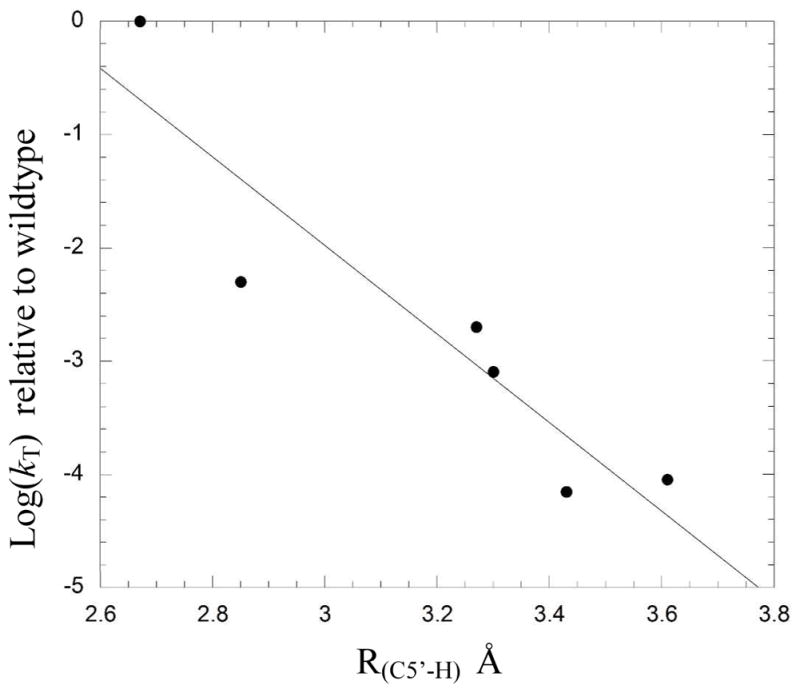
Correlation between R(C5′-H) determined from MD simulations and logkT determined by experiment.
Discussion
The fundamental principles by which AdoCbl-dependent enzymes accelerate coenzyme homolysis and stabilize the resulting reactive free radical intermediates remain the most poorly understood aspect of these enzymes. In this work, we sought to gain insight into the mechanism of radical generation in AdoCbl-dependent enzymes by a coordinated approach employing both computation and experiment.
The crystal structure of glutamate mutase suggested that Glu330 and Lys326 might play an important role in catalyzing homolysis of the coenzyme glutamate mutase. The results reported here demonstrate that this is indeed the case. By developing a highly sensitive assay for homolysis and hydrogen abstraction, we were able measure the activities of mutant enzymes that are slower by 2 – 4 orders of magnitude than wild-type enzyme. This wide range of activities allowed us to examine whether changes in the structure and dynamics of the active site identified by molecular dynamics simulations correlated with changes in activity. This strategy has provided a powerful approach to gain new atomic-level insights into reaction mechanism that would have been unavailable from either experimental observation or computation alone.
Experiments on glutamate mutase and other AdoCbl enzymes have shown that transfer of hydrogen between substrate and coenzyme involves tunneling and is the rate-determining step in the reaction.20, 45–47 As such, the position of the adenosyl radical with respect to the substrate is expected to be critical for efficient catalysis, but to this point experimental evidence to support this statement has been lacking. The simulations point to the important role that Lys326 and Glu330 play in positioning the transiently-formed adenosyl radical for hydrogen abstraction. R(C5′-H) for wild-type glutamate mutase is both shorter and exhibits a narrower standard deviation than any of the mutants, suggesting that it is optimized for reaction. Significantly, the correlation between -logkT, which reflects the activation energy for the transfer of tritium between substrate and coenzyme, 6and R(C5′-H) computed from MD simulations provides strong support for the idea that optimizing the distance between the 5′-carbon and the abstractable substrate hydrogen is vital for efficient catalysis in glutamate mutase and other AdoCbl enzymes.
The formation of stronger electrostatic and/or hydrogen bonding interactions between coenzyme and protein in the dissociated state of AdoCbl has been identified as one potential mechanism by which the enzyme promotes homolysis of the coenzyme.22, 42 Our results support this hypothesis, but show that this is not the only factor in efficient catalysis. Our MD simulations do not quantify the catalytic contribution that hydrogen bonding makes, but they do indicate that for wild-type glutamate mutase, hydrogen bonding is significantly increased between Glu330 and ribose in the dissociated state. The mutant enzymes also show increased numbers of hydrogen bonds in the dissociated state relative to the associated state (Table 2), although they form fewer hydrogen bonds in total. However, changes in electrostatic interactions between the coenzyme and the protein in the dissociated state appear insufficient to fully explain the difference in activity between wild-type and mutant enzymes.
This is illustrated by the Glu330Asp mutant, which in the simulations forms well-defined hydrogen bonds with the ribose hydroxyl groups in the dissociated state, but is less active than the Glu330Gln mutant, even though the neutral glutamine side chain would be expected to form weaker hydrogen bonds with the ribose ring than the negatively charged carboxylate side chain. Our analysis rationalizes this trend by showing that hydrogen bonding between ribose and the shorter aspartate chain constrains the 5′-carbon so that it is, on average, 0.42 Å further from the substrate than the glutamine mutant (Figure 2). Interestingly, the same activity trend was observed in ornithine aminomutase and methylmalonyl-CoA mutase when the equivalent glutamate residues were mutated to glutamine and aspartate.41
In conclusion, our results provide evidence that both electrostatic interactions between the enzyme and the ribosyl moiety of the coenzyme and precise positioning of the adenosyl radical by active site residues are important factors in catalysis by AdoCbl-dependent enzymes. This work represents the first study to examine in atomistic detail the structural features of the active site that underpin these catalytic principles. None of the mutant enzymes accumulate detectable amounts of cob(II)alamin during catalysis, indicating that they do not stabilize the dissociated state as well as the wild-type. We propose that two factors contribute to the high levels of coenzyme homolysis observed in AdoCbl enzymes. First, favorable electrostatic interactions, as previously proposed by Warshel and co-workers,42 between the dissociated coenzyme and protein are necessary to offset part of the enthalpic cost of radical generation. Second, precise positioning of the transiently-formed adenosyl radical by the enzyme, a feature that has not been widely discussed previously, facilitates the rapid transfer of hydrogen from the substrate to coenzyme, to generate the substrate radical that is further stabilized by electronic effects.
Supplementary Material
Acknowledgments
This research was supported by NIH grant GM 093088 to E.N.G.M. A.J.M. is an EPSRC Leadership Fellow (grant number EP/G007705/1).
Abbreviations used are
- AdoCbl
Adenosylcobalamin
- CblOH
hydroxycobalamin
- Cbl(II)
cob(II)alamin
Footnotes
Supporting Information Available
Protonation states for histidine residues used in MD simulations; mean hydrogen bonding distances and distance distribution plots obtained from MD simulations and parameters used to fit the plots. This information is available via the internet at http://pubs.acs.org.
References
- 1.Marsh ENG, Patterson DP, Li L. Adenosyl radical: reagent and catalyst in enzyme reactions. ChemBioChem. 2010;11:604–621. doi: 10.1002/cbic.200900777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Matthews RG. Cobalamin- and Corrinoid-Dependent Enzymes, In. Met Ions Life Sci. 2009:53–114. doi: 10.1039/BK9781847559159-00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frey PA, Hegerman AD, Reed GH. Free radical mechanisms in enzymology. Chem Rev. 2006;106:3302–3316. doi: 10.1021/cr050292s. [DOI] [PubMed] [Google Scholar]
- 4.Toraya T. Radical catalysis in coenzyme B12-dependent isomerization (eliminating) reactions. Chem Rev. 2003;103:2095–2127. doi: 10.1021/cr020428b. [DOI] [PubMed] [Google Scholar]
- 5.Banerjee R. Radical carbon skeleton rearrangements: Catalysis by coenzyme B12-dependent mutases. Chem Rev. 2003;103:2083–2094. doi: 10.1021/cr0204395. [DOI] [PubMed] [Google Scholar]
- 6.Brunold TC, Conrad KS, Liptak MD, Park K. Spectroscopically validated density functional theory studies of the B12 cofactors and their interactions with enzyme active sites. Coord Chem Rev. 2009;253:779–794. [Google Scholar]
- 7.Robertson WD, Wang M, Warncke K. Characterization of Protein Contributions to Cobalt-Carbon Bond Cleavage Catalysis in Adenosylcobalamin-Dependent Ethanolamine Ammonia-Lyase by using Photolysis in the Ternary Complex. J Am Chem Soc. 2011;133:6968–6977. doi: 10.1021/ja107052p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown KL. Chemistry and enzymology of vitamin B12. Chem Rev. 2005;105:2075–2149. doi: 10.1021/cr030720z. [DOI] [PubMed] [Google Scholar]
- 9.Sandala GM, Smith DM, Radom L. Modeling the Reactions Catalyzed by Coenzyme B-12-Dependent Enzymes. Acc Chem Res. 2010;43:642–651. doi: 10.1021/ar900260c. [DOI] [PubMed] [Google Scholar]
- 10.Marsh ENG, Melendez GDR. Adenosylcobalamin enzymes: Theory and experiment begin to converge. Biochim Biophys Acta, Proteins Proteomics. 2012;1824:1154–1164. doi: 10.1016/j.bbapap.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jensen KP, Ryde U. Cobalamins uncovered by modern electronic structure calculations. Coord Chem Rev. 2009;253:769–778. [Google Scholar]
- 12.Marsh ENG, Ballou DP. Coupling of cobalt-carbon bond homolysis and hydrogen atom abstraction in adenosylcobalamin-dependent glutamate mutase. Biochemistry. 1998;37:11864–11872. doi: 10.1021/bi980512e. [DOI] [PubMed] [Google Scholar]
- 13.Padmakumar R, Banerjee R. Evidence that cobalt-carbon bond homolysis is coupled to hydrogen atom abstraction from substrate in methylmalonyl-CoA mutase. Biochemistry. 1997;36:3713–3718. doi: 10.1021/bi962503g. [DOI] [PubMed] [Google Scholar]
- 14.Bandarian V, Reed GH. Isotope effects in the transient phases of the reaction catalyzed by ethanolamine ammonia-lyase: Determination of the number of exchangeable hydrogens in the enzyme-cofactor complex. Biochemistry. 2000;39:12069–12075. doi: 10.1021/bi001014k. [DOI] [PubMed] [Google Scholar]
- 15.Marsh ENG. Insights into the mechanisms of adenosylcobalamin (coenzyme B-12)-dependent enzymes from rapid chemical quench experiments. Biochem Soc Trans. 2009;37:336–342. doi: 10.1042/BST0370336. [DOI] [PubMed] [Google Scholar]
- 16.Gruber K, Kratky C. Coenzyme B-12 dependent glutamate mutase. Curr Opin Chem Biol. 2002;6:598–603. doi: 10.1016/s1367-5931(02)00368-x. [DOI] [PubMed] [Google Scholar]
- 17.Marsh ENG. Coenzyme B12-dependent glutamate mutase. Bioorg Chem. 2000;28:176–189. doi: 10.1006/bioo.2000.1168. [DOI] [PubMed] [Google Scholar]
- 18.Rommel JB, Kaestner J. The Fragmentation-Recombination Mechanism of the Enzyme Glutamate Mutase Studied by QM/MM Simulations. J Am Chem Soc. 2011;133:10195–10203. doi: 10.1021/ja202312d. [DOI] [PubMed] [Google Scholar]
- 19.Yoon M, Kalli A, Lee HY, Hakensson K, Marsh ENG. Intrinsic Deuterium Kinetic Isotope Effects in Glutamate Mutase Measured by an Intramolecular Competition Experiment. Angew Chem. 2007;46:8455–8459. doi: 10.1002/anie.200702448. [DOI] [PubMed] [Google Scholar]
- 20.Cheng MC, Marsh ENG. Evidence for coupled motion and hydrogen tunneling of the reaction catalyzed by glutamate mutase. Biochemistry. 2007;46:883–889. doi: 10.1021/bi0616908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sandala GM, Smith DM, Marsh ENG, Radom L. Towards an Improved Understanding of the Glutamate Mutase System. J Am Chem Soc. 2007;129:1623–1633. doi: 10.1021/ja066432c. [DOI] [PubMed] [Google Scholar]
- 22.Jensen KP, Ryde U. How the Co-C bond is cleaved in coenzyme B-12 enzymes: A theoretical study. J Am Chem Soc. 2005;127:9117–9128. doi: 10.1021/ja050744i. [DOI] [PubMed] [Google Scholar]
- 23.Chih HW, Marsh ENG. Mechanism of glutamate mutase: identification and kinetic competence of acrylate and glycyl radical as intermediates in the rearrangment of glutamate to methylaspartate. J Am Chem Soc. 2000;122:10732–10733. [Google Scholar]
- 24.Chih H-W, Marsh ENG. Tritium partitioning and isotope effects in adenosylcobalamin-dependent glutamate mutase. Biochemistry. 2001;40:13060–13067. doi: 10.1021/bi011298o. [DOI] [PubMed] [Google Scholar]
- 25.Bothe H, Darley DJ, Albracht SP, Gerfen GJ, Golding BT, Buckel W. Identification of the 4-glutamyl radical as an intermediate in the carbon skeleton rearrangement catalyzed by coenzyme B12-dependent glutamate mutase from Clostridium cochlearium. Biochemistry. 1998;37:4105–4113. doi: 10.1021/bi971393q. [DOI] [PubMed] [Google Scholar]
- 26.Gruber K, Reitzer R, Kratky C. Radical shuttling in a protein: Ribose pseudorotation controls alkyl-radical transfer in the coenzyme B12 dependent enzyme glutamate mutase. Angew Chem, Int Ed. 2001;40:3377–3380. doi: 10.1002/1521-3773(20010917)40:18<3377::aid-anie3377>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 27.Chen HP, Marsh ENG. Adenosylcobalamin-dependent glutamate mutase: examination of substrate and coenzyme binding in an engineered fusion protein possessing simplified subunit structure and kinetic properties. Biochemistry. 1997;36:14939–14945. doi: 10.1021/bi971374g. [DOI] [PubMed] [Google Scholar]
- 28.Cheng M-C, Marsh ENG. Pre-steady state measurement of intrinsic secondary tritium isotope effects associated with the homolysis of adenosylcobalamin and the formation of 5′-deoxyadensosine in glutamate mutase. Biochemistry. 2004;43:2155–2158. doi: 10.1021/bi036122w. [DOI] [PubMed] [Google Scholar]
- 29.Rostkowski M, Olsson MH, Søndergaard CR, Jensen JH. Graphical analysis of pH-dependent properties of proteins predicted using PROPKA. Bmc Struct Biol. 2011;11 doi: 10.1186/1472-6807-11-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen VB, Arendall WB, Head JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2009;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown KL, Marques HM. Molecular modeling of the mechanochemical triggering mechanism for catalysis of carbon-cobalt bond homolysis in coenzyme B12. J Inorg Biochem. 2001;83:121–132. doi: 10.1016/s0162-0134(00)00188-4. [DOI] [PubMed] [Google Scholar]
- 33.Walker RC, de Souza MM, Mercer IP, Gould IR, Klug DR. Large and Fast Relaxations inside a Protein: Calculation and Measurement of Reorganization Energies in Alcohol Dehydrogenase. J Phys Chem B. 2002:11658–11665. [Google Scholar]
- 34.Pavelites JJ, Gao J, Bash PA, Mackerell AD. A molecular mechanics force field for NAD+ NADH, and the pyrophosphate groups of nucleotides. J Comput Chem. 1997;18:221–239. [Google Scholar]
- 35.Seabra GM, Walker RC, Roitberg AE. Are Current Semiempirical Methods Better Than Force Fields? A Study from the Thermodynamics Perspective. J Phys Chem A. 2009;113:11938–11948. doi: 10.1021/jp903474v. [DOI] [PubMed] [Google Scholar]
- 36.Lange AF, van der Spoel D, de Groot BL. Scrutinizing Molecular Mechanics Force Fields on the Submicrosecond Timescale with NMR Data. Biophys J. 2010;99:647–655. doi: 10.1016/j.bpj.2010.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lindorff-Larsen K, Maragakis P, Piana S, Eastwood MP, Dror RO, Shaw DE. Systematic Validation of Protein Force Fields against Experimental Data. PLoS ONE. 2012;7:e32131. doi: 10.1371/journal.pone.0032131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vanquelef E, Simon S, Marquant G, Garcia E, Klimerak G, Delepine JC, Cieplak P, Dupradeau FY. R.E.D. Server: a web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments. Nucleic Acids Res. 2011;39:W511–W517. doi: 10.1093/nar/gkr288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wolthers KR, Levy C, Scrutton NS, Leys D. Large-scale Domain Dynamics and Adenosylcobalamin Reorientation Orchestrate Radical Catalysis in Ornithine 4,5-Aminomutase. J Biol Chem. 2010;285:13942–13950. doi: 10.1074/jbc.M109.068908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mancia F, Keep NH, Nakagawa A, Leadlay PF, McSweeney S, Rasmussen B, Bosecke P, Diat O, Evans PR. How coenzyme B12 radicals are generated: the crystal structure of methylmalonyl-coenzyme A mutase at 2 A resolution. Structure. 1996;4:339–350. doi: 10.1016/s0969-2126(96)00037-8. [DOI] [PubMed] [Google Scholar]
- 41.Makins C, Pickering AV, Mariani C, Wolthers KR. Mutagenesis of a Conserved Glutamate Reveals the Contribution of Electrostatic Energy to Adenosylcobalamin Co-C Bond Homolysis in Ornithine 4,5-Aminomutase and Methylmalonyl-CoA Mutase. Biochemistry. 2013;52:878–888. doi: 10.1021/bi3012719. [DOI] [PubMed] [Google Scholar]
- 42.Sharma PK, Chu ZT, Olsson MHM, Warshel A. A new paradigm for electrostatic catalysis of radical reactions in vitamin B12 enzymes. Proc Natl Acad Sci U S A. 2007;104:9661–9666. doi: 10.1073/pnas.0702238104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen HP, Marsh ENG. How enzymes control the reactivity of adenosylcobalamin: effect on coenzyme binding and catalysis of mutations in the conserved histidine-aspartate pair of glutamate mutase. Biochemistry. 1997;36:7884–7889. doi: 10.1021/bi970169y. [DOI] [PubMed] [Google Scholar]
- 44.Stojkovic V, Perissinotti LL, Willmer D, Benkovic SJ, Kohen A. Effects of the Donor-Acceptor Distance and Dynamics on Hydride Tunneling in the Dihydrofolate Reductase Catalyzed Reaction. J Am Chem Soc. 2012;134:1738–1745. doi: 10.1021/ja209425w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoon M, Song H, Hakansson K, Marsh ENG. Intrinsic deuterium kinetic isotope effects in glutamate mutase measured by an intramolecular competition experiment. Biochemistry. 2010;49:3168–3173. [Google Scholar]
- 46.Dybala-Defratyka A, Paneth P, Banerjee R, Truhlar DG. Coupling of hydrogenic tunneling to active site motion in the hydrogen radical transfer catalyzed by a coenzyme B12 mutase. Proc Natl Acad Sci U S A. 2007;104:10774–10779. doi: 10.1073/pnas.0702188104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chowdhury S, Banerjee R. Evidence for quantum mechanical tunneling in the coupled cobalt-carbon bond homolysis-substrate radical generation reaction catalyzed by methylmalonyl-CoA mutase. J Am Chem Soc. 2000;122:5417–5418. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.