

Abstract
The chaperone HSP70 promotes the survival of cells exposed to many different types of stresses, and is also potently anti-apoptotic. The major stress-induced form of this protein, HSP70–1, is overexpressed in a number of human cancers, yet is negligibly expressed in normal cells. Silencing of the gene encoding HSP70–1 (HSPA1A) is cytotoxic to transformed but not normal cells. Therefore, HSP70 is considered to be a promising cancer drug target, and there has been active interest in the identification and characterization of HSP70 inhibitors for cancer therapy. Because HSP70 behaves in a relatively non-specific manner in the control of protein folding, to date there are no reliably-identified “clients” of this protein, nor is there consensus as to what the phenotypic effects of HSP70 inhibitors are on a cancer cell. Here for the first time we compare three recently-identified HSP70 inhibitors, PES-Cl, MKT-077, and Ver-155008, for their ability to impact some of the known and reported functions of this chaperone; specifically, the ability to inhibit autophagy, to influence the level of HSP90 client proteins, to induce cell cycle arrest, and to inhibit the enzymatic activity of the anaphase-promoting complex/cyclosome (APC/C). We report that all three of these compounds can inhibit autophagy and cause reduced levels of HSP90 client proteins; however, only PES-Cl can inhibit the APC/C and induce G2/M arrest. Possible reasons for these differences, and the implications for the further development of these prototype compounds as anti-cancer agents, are discussed.
Keywords: phenylethynesulfonamide, PES, PES-Cl, Ver-155008, MKT-077, autophagy, cell cycle, anaphase promoting complex
Introduction
We previously identified the compound phenylethynesulfonamide (PES, also known as pifithrin mu) as one that binds specifically to HSP70, and disrupts the ability of this chaperone to interact with critical co-chaperones that bind to the carboxyl terminus of the substrate-binding domain of this protein.1 We showed that PES is cytotoxic to tumor cells but not non-transformed cells, and that silencing HSP70 significantly reduces the cytotoxicity of this compound.1 Further, we showed that PES can directly interact with recombinant HSP70, in a manner that is most consistent with a non-covalent association.2
We first became interested in PES as a possible cancer therapeutic when we discovered that this compound inhibits autophagy, using several different autophagy assays.1 This inhibition of autophagy likely occurs by virtue of the ability of PES to inhibit HSP70 at the lysosome, as there is a concomitant disruption of lysosome function that occurs following PES treatment. This disruption of lysosome function by an HSP70 inhibitor is consistent with the findings by others that HSP70 is required for lysosome integrity in cancer cells.3 We also showed that PES can also interact in some cancer cell lines with the constitutively expressed member of the HSP70 family, HSC70.4 With the knowledge that inhibiting both HSC70 and HSP70 leads to inhibition of HSP90 chaperone function,5 we investigated and then showed that incubation of cells with PES causes a reduction of HSP90 client proteins in the cell; this occurs due to sequestration of HSP90 client proteins into an insoluble fraction within the cell.4 Most recently, guided by data indicating that the activity of the anaphase promoting complex/cyclosome (APC/C) requires the function of an ATPase,6 we tested the hypothesis that PES and the related HSP70 inhibitor PES-Cl might inhibit the activity of APC/C in cell-free extracts. We showed that both PES and PES-Cl, but not the HSP90 inhibitor geldanamycin, inhibits the activity of the APC/C in cell-free assays.2 Consistent with this, we found that incubation with PES and PES-Cl causes cell cycle arrest in the G2/M phase of the cell cycle.2 The combined data support the premise that the HSP70 inhibitors PES and PES-Cl possess several notable anti-cancer activities. These include inhibition of autophagy, control of HSP90 client protein solubility, and inhibition of the APC/C.
Several groups have previously identified and characterized other HSP70 inhibitors (for a review see refs. 7–9). Two of these, VER-155008 and MKT-077, have been well-characterized and are commercially available. The HSP70 inhibitor VER-155008 has been co-crystallized with the HSC70/BAG-1 complex and shown to interact within the ATP-binding pocket of HSC70.10 Like PES, VER-155008 is preferentially cytotoxic to cancer cells but not normal cells, and reduces HSP90 client protein levels in tumor cells.11 The rhodacyanine dye derivative MKT-077 was first discovered as a compound that was cytotoxic to cancer cells but not normal cells, and later shown to bind to the mitochondrial HSP70 member HSPA9 (also called GRP75 or mortalin).12-14 More recently this compound was also found to bind to HSC70, and there is evidence that it can interact with and inhibit HSP70 as well.15 MKT-077 binds near the ATP binding site of HSP70 family members, and alters communication between the nucleotide binding domain and substrate binding domain of HSP70, resulting in impaired allostery between these domains.15 In sum, three different groups have identified three different HSP70 inhibitors, and all three inhibitors show cancer cell-selective cytotoxicity. But all three bind to different regions of HSP70 members (PES and PES-Cl to the substrate-binding domain, VER-155008 to the ATP binding site, and MKT-077 to an allosteric site near the ATP binding site), and may show different affinities for each member. Therefore, there exists the possibility that all three compounds impact different anti-cancer pathways in the cell. In this report, we compare the ability of these three compounds to reduce the viability of tumor cells, to inhibit autophagy, to influence the solubility and degradation of HSP90 client proteins, and to inhibit the APC/C. We find subtle differences in their mechanisms of action that may relate to their altered binding sites on HSP70. Notably, we also uncover a potentially novel mechanism for the inhibition of autophagy by these HSP70 inhibitors. Specifically, we find HSP70 reproducibly binds to the key autophagy protein Beclin-1, and that all three of these HSP70 inhibitors lead to sequestration of Beclin-1 into an insoluble fraction in the cell. These studies suggest an additional role for HSP70 in autophagy regulation. They should also serve as a useful foundation for the analysis of future derivatives and inhibitors of HSP70.
Results
All three HSP70 inhibitors are comparably cytotoxic to cancer cells and inhibit autophagy
All three HSP70 inhibitors employed here, PES-Cl, VER-155008, and MKT-077, have been previously shown to be cytotoxic to cancer cells; however, they have never been compared for cytotoxicity. Therefore, we first compared the cytotoxicity of PES-Cl, VER-155008, and MKT-077 in two different cancer cell lines, H1299 (a p53-null human lung adenocarcinoma) and A375 (a human melanoma line) using standard cytotoxicity and viability assays (MTT, as well as a dye exclusion assay, ViaCount). For this analysis, we also included the parent compound PES, which in our experience is slightly less cytotoxic than PES-Cl.2 This analysis revealed that all four compounds are cytotoxic to A375 cells, with IC50 values in the micromolar range; PES-Cl was slightly more effective, and VER-155008 slightly less effective, than the others (Fig. 1A). Nearly identical IC50 values were obtained for all four compounds in H1299 cells (data not shown). Similar data were obtained using the ViaCount assay, which like trypan blue assays measures cell count and cell viability via a dye-exclusion assay (Fig. S1A).

Figure 1. HSP70 inhibitors can inhibit autophagy and are cytotoxic to cancer cells. (A) Cell viability analysis (MTT) of A375 melanoma cell line treated with DMSO or the indicated doses of PES, PES-Cl, MKT-077, or VER-155008 for 48 h. The data depicted are representative of 3 independent experiments. Error bars indicate standard deviations. (B) Western blot analysis of apoptosis (cleaved lamin A, cleaved caspase 3) and autophagy (p62/SQSTM1, and LC3 I and II) markers in H1299 and A375 cell lines. Cells were treated with DMSO, 10 uM PES-Cl or MKT-077, or 20 uM VER-155008 for 24 h. Actin is indicated as a loading control. The presence of p62 monomers and oligomers is indicated. (C) Annexin V flow cytometric analysis was conducted on A375 cells treated with the indicated concentrations of PES-Cl, MKT-077, or VER-155008 for 24 h. The data depicted are the average results from 3 independent experiments. Error bars indicate standard deviations. (D) H1299 and A375 cells were treated with DMSO or 10 uM PES-Cl, 10 uM MKT-077, or 20 uM VER-155008 for 24 h. Whole cell lysates were then subjected to immunoblot analysis of HSP90 client proteins using the antibodies indicated. Actin is included as a loading control.
We next compared the ability of these compounds to induce apoptosis, using western blot analysis for cleaved lamin A and cleaved caspase-3, as well as Annexin V assays. These studies indicated that following 24 h incubation with each drug, both PES-Cl and VER-155008 demonstrated the ability to induce programmed cell death, as assessed by the accumulation of caspase-cleaved lamin A and cleaved caspase-3, particularly in A375 melanoma cells (Fig. 1B). Interestingly, using Annexin V assays, all three HSP70 inhibitors showed significant ability to induce the accumulation of Annexin V positive cells (Fig. 1C). The cause for the somewhat discrepant findings between Annexin V assays and cleaved lamin A and cleaved caspase-3 markers for the MKT-077 inhibitor is not known.
We next tested all three HSP70 inhibitors for the ability to inhibit autophagy, using western blot analysis for the autophagy-scaffolding protein p62/SQSTM1 (SQSTM1) and the autophagy marker LC3 II. Whereas the ability of PES-Cl to inhibit autophagy has been documented with several different autophagy assays, including electron microscopy,2 whether this activity is shared by all HSP70 inhibitors has never been shown. In these studies, we found that both PES-Cl and MKT-077 demonstrated significant ability to induce the accumulation of SQSTM1, which is normally degraded by autophagy (Fig. 1B). In the case of MKT-077, the ability to inhibit autophagy was quite pronounced, as evident by the increased levels of higher-order oligomeric forms of SQSTM1, along with the concomitant decreased level of the monomeric form of this protein (Fig. 1B). To confirm that we were witnessing autophagy inhibition by these compounds, we next analyzed LC3 II levels in cells treated with HSP70 inhibitors, in the presence and absence of NH4Cl, which freezes autophagic flux.16,17 Under conditions of halted flux, decreased levels of LC3 II in HSP70 inhibitor/NH4Cl-treated cells, compared with inhibitor alone, would support the conclusion that these compounds induce, rather than inhibit, autophagy. Instead, we found that incubation of H1299 cells with PES-Cl or MKT-077, in the presence or absence of NH4Cl to freeze autophagic flux, led to identical increases in LC3 II levels, supporting the conclusion that these HSP70 inhibitors inhibit LC3 II degradation, and therefore inhibit autophagy (Fig. S1B). The combined data support the premise that PES-Cl, and to a possibly greater extent MKT-077, can effectively inhibit autophagy. Interestingly, the compound VER-155008 showed a reproducible but markedly lesser ability to inhibit autophagy, compared with PES-Cl and MKT-077, yet showed significant ability to induce apoptosis; these data suggest that cell death induced by this HSP70 inhibitor is unlikely to be caused by autophagy inhibition (Fig. 1B).
All three HSP70 inhibitors cause altered levels of HSP90 client proteins
HSP70 is known to be an obligate co-chaperone for HSP90.5 Our group and others have shown that treatment of cells with HSP70 inhibitors causes HSP90 “client” proteins to be degraded or sequestered into an insoluble fraction in cells.4,11 To compare and contrast the ability of all three inhibitors to influence HSP90 client protein levels, we next analyzed the steady-state levels of three established HSP90 “client” proteins, CDK4, C-RAF, and HER2, following treatment with all three HSP70 inhibitors. We found that all three compounds demonstrated an ability to cause reduced levels of the HSP90 client proteins CDK4 and HER2 (Fig. 1D). Interestingly, these client proteins were affected differently in the two cell lines, H1299 and A375, suggesting cell-type specific differences (Fig. 1D). In sum, these combined data suggest that all three HSP70 inhibitors can reduce HSP90 chaperone function, but that there are subtle differences in the activity of these compounds in cancer cells, at least at the concentrations used.
Only the HSP70 inhibitor PES-Cl shows the ability to inhibit the APC/C in vitro
It has been known for some time that in cell extracts isolated from G2/M-arrested cells, the enzymatic activity of the anaphase promoting complex/cyclosome (APC/C) is poorly detectable unless exogenous ATP is added. Further, because non-hydrolyzable ATP is ineffective at activating APC/C activity in cell-free extracts, it was hypothesized that a chaperone ATPase was essential for APC/C activation in these assays.6 Based upon these data, we previously tested the ability of the HSP70 inhibitor PES-Cl, and the HSP90 inhibitor geldanamycin, to inhibit APC/C activity in vitro, as assessed by cyclin B1 degradation in cell-free extracts following ATP addition. Our data indicated that the HSP70 inhibitors PES and PES-Cl could effectively inhibit APC/C activity in vitro, whereas the HSP90 inhibitor geldanamycin was ineffective.2 Consistent with APC/C inhibition, we reported that tumor cells treated with the HSP70 inhibitors PES and PES-Cl undergo cell cycle arrest in the G2/M phase of the cell cycle.2
Because APC/C inhibition is a potentially novel and new activity of HSP70 inhibitors, we next compared the ability of the HSP70 inhibitors PES-Cl, MKT-077, and VER-155008 to inhibit APC/C activity in cell-free extracts, exactly as previously described.18 For these assays, identical amounts of HeLa cell extract purified from nocodazole-arrested G2/M cells are incubated with ATP to induce APC/C activity, in the absence and presence of PES-Cl, MKT-077, or VER-155008, or dilution vehicle (DMSO). These extracts are then followed for the ability to degrade endogenous cyclin B, a reliable marker of APC/C activity.18 This analysis reproducibly revealed that of the three HSP70 inhibitors, only PES-Cl was capable of inhibiting cyclin B degradation, and APC/C activity (Fig. 2A). We next analyzed the ability of these three compounds to induce G2/M cell cycle arrest using flow cytometry, as G2/M arrest would be the result of APC/C inhibition. This analysis revealed that of these three compounds, only PES-Cl was capable of causing G2/M arrest of tumor cells (Fig. 2B; Fig. S1C). These data indicate that, although these compounds share the ability to inhibit HSP70 function, they possess subtle differences in action that may be significant as they are further developed as anti-cancer agents.
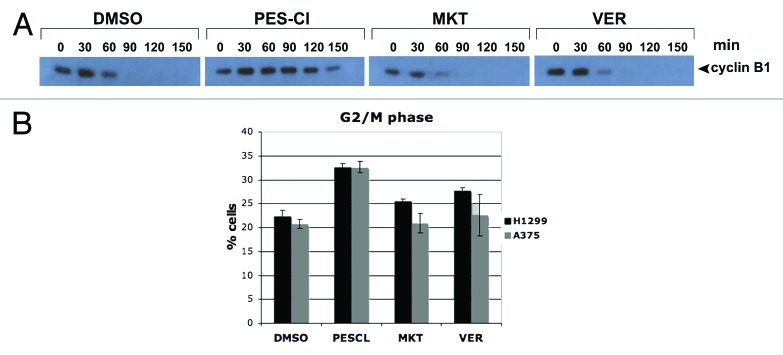
Figure 2. The HSP70 inhibitor PES-Cl inhibits APC/C function and causes G2/M arrest. (A) Western blot analysis of cyclin B1 level in cell-free extracts generated from synchronized HeLa cells treated with DMSO, 10 uM PES-CL, 10 uM MKT-077, or 20 uM VER-155008. The degradation of cyclin B1 by APC/C activity was analyzed at the time points indicated. The data depicted are representative of three independent experiments. (B) Cell cycle analysis of H1299 and A375 cells treated with DMSO, 10 uM PES-CL, 10 uM MKT-077, or 20 uM VER-155008 for 24 h followed by propidium iodide staining and flow cytometry. The graph shows the percentage of cells arrested in G2/M phase of the cell cycle after each treatment. Data are the average results from three independent experiments. Error bars mark standard error.
HSP70 inhibitors reduce Beclin-1 solubility
One issue that remains unresolved regarding these three HSP70 inhibitors is the mechanism whereby these compounds inhibit autophagy. Whereas it has been shown that HSP70 localizes to the lysosome membrane, and that silencing HSP70 causes lysosome dysfunction,3 whether or not HSP70 directly regulates key autophagy proteins has never been tested. Our finding that all three inhibitors tested here all inhibit autophagy afforded us an opportunity to address this question. Toward this question, we took note of a very recent publication indicating that HSP70 binds to the Vps34/Beclin-1 complex,19 which is a master regulatory complex for autophagy. Therefore, we focused on the possibility that either Vps34 or Beclin-1 might be client proteins of HSP70, and become sequestered in an insoluble, inactive fraction in cells following treatment with HSP70 inhibitors. Using techniques to separate the soluble from insoluble fractions in cells,4 we discovered that Beclin-1 moves from the soluble to the insoluble fraction in cells following treatment with all three autophagy inhibitors (Fig. 3A). As controls for this assay, we showed that the enzyme GAPDH did not move from the soluble to insoluble fraction, but that HSP70 itself did (Fig. 3B). Additionally, using immunoprecipitation–western blot analysis, we confirmed that immunoprecipitation with Beclin-1 antisera brought down HSP70 in the complex, and that this interaction was greatly enriched in cells treated with PES-Cl (Fig. 3C). These data recapitulate the findings of others that HSP70 binds to the Vps34/Beclin-1 complex,19 and support the premise that Beclin-1 is a novel “client” of HSP70. They also suggest that HSP70 inhibitors may impair autophagy, at least in part, by their ability to control the function of this critical autophagy regulator.

Figure 3. HSP70 inhibitors cause sequestration of Beclin-1 in an insoluble fraction in the cell. (A) Western analysis of Vps34, Beclin-1, and actin in H1299 cells treated with 10 uM PES-Cl or MKT-077 (MKT), or 20 uM VER-155008 (VER) for 24 h followed by fractionation of soluble and insoluble material and analysis by western blot using antibodies for the proteins indicated. Note that Beclin-1 is normally present in a soluble fraction in cells, but that after HSP70 inhibition, this protein changes to an insoluble fraction, indicative of sequestration and inactivation. (B) Western analysis of HSP70, GAPDH (control for soluble protein), and Histone H3 (control for insoluble protein) in H1299 cells treated with 10 uM PES-Cl or MKT-077 (MKT), or 20 uM VER-155008 (VER) for 24 h followed by fractionation of soluble and insoluble material and analysis by western blot using antibodies for the proteins indicated. (C) Immunoprecipitation–western blot analysis of the Beclin1/HSP70 complex in H1299 cells treated with DMSO or 10 uM PES-Cl for 24 h. Five hundred micrograms of lysate was immunoprecipitated with 0.5 ug of antisera to Beclin1, followed by SDS-PAGE, transfer, and western analysis using antisera to Vps34 and HSP70.
Discussion
Despite the fact that several HSP70 inhibitors have been identified and tested, to date there has not been a consensus on the phenotypic effects of HSP70 inhibition, nor are there universally-used assays for monitoring the activity or efficacy of these compounds. In this report we show that three different HSP70 inhibitors, all of which interact with different domains of this chaperone, and possibly also different allosteric states of this protein, are each cytotoxic to tumor cell lines, can all inhibit autophagy, and can also cause reduced levels of HSP90 client proteins. We posit that these three assays should be routinely used to monitor the effect of other HSP70 inhibitors and derivatives that are developed. We also propose that these compounds may manifest their cytotoxicity in tumor cells by virtue of these common activities.
Each of the three HSP70 inhibitors tested here-in binds to a different region of HSP70 family members. For example, the compound VER-155008 was identified as an ATP analog that overlaps with the ATP binding site of HSP70 and competes for ATP binding of HSP70.10 This compound likely inhibits HSP70 activity by keeping this protein in a state that has reduced affinity for substrate. Conversely, the compound MKT-077 binds near but not at the ATP binding site, and preferentially binds to the ADP-bound form of this enzyme.15 As the ADP-bound form has the highest affinity for substrate, it is possible that MKT-077 inhibits HSP70 activity by preventing the release of substrate and stalling the HSP70 folding cycle. Finally, we have previously shown that unlike MKT-077 and VER-155008, PES and PES-Cl interact with the substrate-binding domain of HSP70, and prevents the association of co-chaperones with this protein.1,2 Additionally, we also find that incubation with PES and PES-Cl causes HSP70 to form higher-order oligomeric states in the cell (Budina-Kolomets, unpublished observations). These differences in the binding site, and the effect of these compounds on HSP70 function, may underlie the biological/biochemical differences we see in their ability to inhibit the APC/C and induce G2/M arrest; this remains to be tested further. It is interesting to note, however, that G2/M arrest and mitotic abnormalities have been a consistent finding in cells with silenced or genetic knockout of HSP70.20,21
The mechanism whereby PES and PES-Cl inhibit APC/C activity is currently unknown. However, it is interesting to note that recent studies have shown that HSP70 family members, and the yeast ortholog Ssa1, have been repeatedly discovered in immunoprecipitates of CDK1 with cyclin B.22,23 Also, an HSP70 family member, HSP70–2, is a chaperone required for CDK1/cyclin B complex assembly, and proper cyclin B degradation during meiosis.24 The role that HSP70 plays in APC/C activity, and the mechanism whereby PES and PES-Cl inhibit this activity, will be important next questions to address.
Materials and Methods
Cells, antibodies, and reagents
H1299 and HeLa cells were obtained from the American Type Culture Collection and were used within six months of receipt; these were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS (Hyclone) and 100 units of penicillin/ streptomycin. A375 cells were obtained from the Herlyn laboratory (Wistar Institute) and authenticated by genotype analysis; these were maintained in RPMI supplemented with 5% FBS (Hyclone) and 100 units of penicillin/ streptomycin. For treatment with PES, PES-Cl, MKT-077, or VER-155008, stock solutions were made in DMSO and diluted in PBS; the final concentration of DMSO was less than 0.4%. Western analysis was performed using the following antibodies at supplier-recommended dilutions: p62/SQSTM1 (sc-28359, Santa Cruz Biotechnology), LC3 (NB100–2331, Novus), Her-2 (791–100, Vertana), CDK4 (sc-601, Santa Cruz), C-RAF (9422S, Cell Signaling), cyclin B (554177, PharMingen), actin (AC-15, Sigma), cleaved lamin A (2035S, Cell Signaling), Hsp70 (4873S, Cell Signaling), and cleaved caspase 3 (9661S, Cell Signaling). MTT assays were performed exactly as described.2 Separation of soluble and insoluble proteins from cells was performed exactly as described.4
Cell cycle, Annexin V, ViaCount assay; immunopreciptation–western blot analysis
Propidium iodide staining and cell cycle analysis, the ViaCount cell viability assay, and Annexin V staining were performed on the Guava EasyCyte System as per manufacturer protocols (Millipore). For IP-westerns, 500 ug of lysate was immunoprecipitated with 0.5 ug of antisera to Beclin1 (sc-10086, Santa Cruz Biotechnology), followed by SDS-PAGE, transfer, and western analysis using antisera to Vps34 (Z-RO16, Echelon Biosciences) and HSP70 (4873S, Cell Signaling).
Cell-free cyclin B degradation (APC/C) assay
For the APC/C activity assay, cell extracts were made as previously described.2,18 Degradation assay reactions contained 10 uL HeLa cell extract, 1 uL of 10X degradation reaction mixture (100 mM Tris-HCl [pH 7.6], 50 mM MgCl2, 10 mM DTT, 10 mg/ml ubiquitin, 100 mM phosphocreatine, 1 mg/ml creatine phosphokinase, and 5 mM ATP) and DMSO, 10 uM PES-CL, 10 uM MKT-077, or 20 uM VER-155008. Reactions were incubated at 30 °C and samples were withdrawn every 30 min for 3 h. The collected samples were immediately mixed with an equal volume 2ȕ SDS sample buffer and resolved by SDS-PAGE.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by NIH R01 CA139319 to Murphy M and by award number P30 CA010815 from the National Cancer Institute. This manuscript is dedicated to the fond memory of Patricia Lorenzo, PhD, a former member of the Molecular Oncogenesis study section and the University of Hawaii Cancer Center, who brightened science with her compassion, affability, and dedication. Her remarkable courage in the face of terminal cancer continues to be inspirational.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/26720
References
- 1.Leu JI, Pimkina J, Frank A, Murphy ME, George DL. A small molecule inhibitor of inducible heat shock protein 70. Mol Cell. 2009;36:15–27. doi: 10.1016/j.molcel.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balaburski GM, Leu JI, Beeharry N, Hayik S, Andrake MD, Zhang G, Herlyn M, Villanueva J, Dunbrack RL, Jr., Yen T, et al. A modified HSP70 inhibitor shows broad activity as an anticancer agent. Mol Cancer Res. 2013;11:219–29. doi: 10.1158/1541-7786.MCR-12-0547-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nylandsted J, Gyrd-Hansen M, Danielewicz A, Fehrenbacher N, Lademann U, Høyer-Hansen M, Weber E, Multhoff G, Rohde M, Jäättelä M. Heat shock protein 70 promotes cell survival by inhibiting lysosomal membrane permeabilization. J Exp Med. 2004;200:425–35. doi: 10.1084/jem.20040531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leu JI, Pimkina J, Pandey P, Murphy ME, George DL. HSP70 inhibition by the small-molecule 2-phenylethynesulfonamide impairs protein clearance pathways in tumor cells. Mol Cancer Res. 2011;9:936–47. doi: 10.1158/1541-7786.MCR-11-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Powers MV, Clarke PA, Workman P. Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer Cell. 2008;14:250–62. doi: 10.1016/j.ccr.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 6.Miniowitz-Shemtov S, Teichner A, Sitry-Shevah D, Hershko A. ATP is required for the release of the anaphase-promoting complex/cyclosome from inhibition by the mitotic checkpoint. Proc Natl Acad Sci U S A. 2010;107:5351–6. doi: 10.1073/pnas.1001875107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patury S, Miyata Y, Gestwicki JE. Pharmacological targeting of the Hsp70 chaperone. Curr Top Med Chem. 2009;9:1337–51. doi: 10.2174/156802609789895674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Powers MV, Jones K, Barillari C, Westwood I, van Montfort RL, Workman P. Targeting HSP70: the second potentially druggable heat shock protein and molecular chaperone? Cell Cycle. 2010;9:1542–50. doi: 10.4161/cc.9.8.11204. [DOI] [PubMed] [Google Scholar]
- 9.Murphy ME. The HSP70 family and cancer. Carcinogenesis. 2013;34:1181–8. doi: 10.1093/carcin/bgt111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williamson DS, Borgognoni J, Clay A, Daniels Z, Dokurno P, Drysdale MJ, Foloppe N, Francis GL, Graham CJ, Howes R, et al. Novel adenosine-derived inhibitors of 70 kDa heat shock protein, discovered through structure-based design. J Med Chem. 2009;52:1510–3. doi: 10.1021/jm801627a. [DOI] [PubMed] [Google Scholar]
- 11.Massey AJ, Williamson DS, Browne H, Murray JB, Dokurno P, Shaw T, Macias AT, Daniels Z, Geoffroy S, Dopson M, et al. A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon carcinoma cells. Cancer Chemother Pharmacol. 2010;66:535–45. doi: 10.1007/s00280-009-1194-3. [DOI] [PubMed] [Google Scholar]
- 12.Chiba Y, Kubota T, Watanabe M, Matsuzaki SW, Otani Y, Teramoto T, Matsumoto Y, Koya K, Kitajima M. MKT-077, localized lipophilic cation: antitumor activity against human tumor xenografts serially transplanted into nude mice. Anticancer Res. 1998;18(2A):1047–52. [PubMed] [Google Scholar]
- 13.Koren J, 3rd, Miyata Y, Kiray J, O’Leary JC, 3rd, Nguyen L, Guo J, Blair LJ, Li X, Jinwal UK, Cheng JQ, et al. Rhodacyanine derivative selectively targets cancer cells and overcomes tamoxifen resistance. PLoS One. 2012;7:e35566. doi: 10.1371/journal.pone.0035566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koya K, Li Y, Wang H, Ukai T, Tatsuta N, Kawakami M, Shishido, Chen LB. MKT-077, a novel rhodacyanine dye in clinical trials, exhibits anticarcinoma activity in preclinical studies based on selective mitochondrial accumulation. Cancer Res. 1996;56:538–43. [PubMed] [Google Scholar]
- 15.Rousaki A, Miyata Y, Jinwal UK, Dickey CA, Gestwicki JE, Zuiderweg ER. Allosteric drugs: the interaction of antitumor compound MKT-077 with human Hsp70 chaperones. J Mol Biol. 2011;411:614–32. doi: 10.1016/j.jmb.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Budina-Kolomets A, Hontz RD, Pimkina J, Murphy ME. A conserved domain in exon 2 coding for the human and murine ARF tumor suppressor protein is required for autophagy induction. Autophagy. 2013;9:9. doi: 10.4161/auto.25831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–75. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sudakin V, Chan GK, Yen TJ. Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J Cell Biol. 2001;154:925–36. doi: 10.1083/jcb.200102093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Y, Fiskus W, Yong B, Atadja P, Takahashi Y, Pandita TK, Wang HG, Bhalla KN. Acetylated hsp70 and KAP1-mediated Vps34 SUMOylation is required for autophagosome creation in autophagy. Proc Natl Acad Sci U S A. 2013;110:6841–6. doi: 10.1073/pnas.1217692110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rohde M, Daugaard M, Jensen MH, Helin K, Nylandsted J, Jäättelä M. Members of the heat-shock protein 70 family promote cancer cell growth by distinct mechanisms. Genes Dev. 2005;19:570–82. doi: 10.1101/gad.305405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hunt CR, Dix DJ, Sharma GG, Pandita RK, Gupta A, Funk M, Pandita TK. Genomic instability and enhanced radiosensitivity in Hsp70.1- and Hsp70.3-deficient mice. Mol Cell Biol. 2004;24:899–911. doi: 10.1128/MCB.24.2.899-911.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marteil G, Gagné JP, Borsuk E, Richard-Parpaillon L, Poirier GG, Kubiak JZ. Proteomics reveals a switch in CDK1-associated proteins upon M-phase exit during the Xenopus laevis oocyte to embryo transition. Int J Biochem Cell Biol. 2012;44:53–64. doi: 10.1016/j.biocel.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 23.Honey S, Schneider BL, Schieltz DM, Yates JR, Futcher B. A novel multiple affinity purification tag and its use in identification of proteins associated with a cyclin-CDK complex. Nucleic Acids Res. 2001;29:E24. doi: 10.1093/nar/29.4.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu D, Dix DJ, Eddy EM. HSP70-2 is required for CDC2 kinase activity in meiosis I of mouse spermatocytes. Development. 1997;124:3007–14. doi: 10.1242/dev.124.15.3007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.