

Abstract
Oriented Sample (OS) solid-state NMR spectroscopy can be used to determine the three-dimensional structures of membrane proteins in magnetically or mechanically aligned lipid bilayers. The bottleneck for applying this technique to larger and more challenging proteins is making resonance assignments, which is conventionally accomplished through the preparation of multiple selectively isotopically labeled samples and performing an analysis of residues in regular secondary structure based on Polarity Index Slant Angle (PISA) Wheels and Dipolar Waves. Here we report the complete resonance assignment of the full-length mercury transporter, MerF, an 81-residue protein, which is challenging because of overlapping PISA Wheel patterns from its two trans-membrane helices, by using a combination of solid-state NMR techniques that improve the spectral resolution and provide correlations between residues and resonances. These techniques include experiments that take advantage of the improved resolution of the MSHOT4-Pi4/Pi pulse sequence; the transfer of resonance assignments through frequency alignment of heteronuclear dipolar couplings, or through dipolar coupling correlated isotropic chemical shift analysis; 15N/15N dilute spin exchange experiments; and the use of the proton-evolved local field (PELF) experiment with isotropic shift analysis to assign the irregular terminal and loop regions of the protein, which is the major “blind spot” of the PISA Wheel/Dipolar Wave method.
Keywords: Solid-state NMR, membrane protein, aligned bilayers, dipolar coupling, chemical shift anisotropy, PISA Wheel, Dipolar Wave
Introduction
Oriented Sample (OS) solid-state NMR spectroscopy of stationary samples is an approach to determining the atomic resolution structures of biological macromolecules that can be aligned by their environment, such as proteins in virus particles or phospholipid bilayers (Opella et al. 2008; Murray et al. 2013; Opella 2013). When the macromolecules are aligned relative to the magnetic field the angle-dependent NMR observables, such as anisotropic chemical shifts and heteronuclear dipole-dipole couplings, converge to single line resonances (or doublets) (Opella and Waugh 1977). Signals are resolved since individual sites have different orientations relative to the direction of the field, and hence different resonance frequencies. Notably, resolution results from differences in orientation relative to the axis of alignment rather than differences in local environment.
The angles of N-H and C-H bonds, and chemical shift anisotropy vectors for each site of a membrane protein in liquid crystalline phospholipid bilayers can be measured relative to the axis of alignment with high accuracy and precision, and the three-dimensional structure of the protein can be determined using these angular restraints. However, the application of OS solid-state NMR to membrane proteins has been hindered by limitations in the methods for the assignment of resonances. Unlike solution NMR or magic angle spinning (MAS) solid-state NMR, systematic assignment methods that rely on a sequential “walk” along the backbone atoms are lacking in OS solid-state NMR because of the difficulty in dealing with the network of 13C-13C homonuclear dipolar couplings in uniformly 13C labeled samples. For a period of time, the main and only resonance assignment method was the analysis of Polarity Index Slant Angle (PISA) Wheels (Marassi and Opella 2000; Wang et al. 2000) and Dipolar Waves (Mesleh et al. 2002) that required multiple amino-acid-type selectively labeled/unlabeled spectra. This method was used in the several structures (De Angelis et al. 2006; Park et al. 2010b; Sharma et al. 2010; Marassi and Opella 2003; Park et al. 2003; Opella et al. 1999; Ketchem et al. 1993) recently determined by OS solid-state NMR of aligned, stationary samples. For the full-length mercury transporter MerF used as an example in this article, the two long transmembrane helices, which include between them more than 50 residues, are tilted at similar angles, and as a result, the spectra contain overlapped PISA Wheels, rendering the protein a challenging target for OS solid-state NMR structure determination. Without severe spectral overlap, the PISA wheel/Dipolar Wave method (Marassi and Opella 2003) works well for assigning resonances from residues in regular secondary structures but not with irregular regions of tertiary structures, such as those encountered in loops and structured terminal regions of membrane proteins. This is also an issue with MerF.
The methods that we combine for a more effective and comprehensive assignment strategy include: (1) The MSHOT4-Pi4/Pi pulse sequence (Lu et al. 2012; Lu and Opella 2013), which provides three-dimensional heteronuclear correlation (HETCOR)/separated local field (SLF) spectra of uniformly and selectively labeled samples. (2) The resonance assignment method of dipolar coupling correlated isotropic chemical shift (DCCICS) analysis (Lu et al. 2011), which was previously demonstrated on a smaller membrane protein. With selectively labeled samples, it complements the information obtained from PISA Wheel assignments. (3) Signals from the non-helical terminal and loop regions of the proteins have been difficult or impossible to assign with the PISA Wheel/Dipolar Wave approach. The proton-evolved local field (PELF) experiment (Schmidt-Rohr et al. 1994) improved by the MSHOT4-Pi4/Pi pulse sequence, efficiently resolves signals from these regions, and in combination with DCCICS analysis it provides a general method to assign signals from regions with irregular tertiary structure. (4) Resonances can be assigned with MAS solid-state NMR methods, and subsequently their recoupled 1H-15N heteronuclear dipolar couplings can be measured under rotational alignment (RA) condition (Park et al. 2010a; Das et al. 2012). These dipolar couplings and their assignments are scaled and transferred to SLF spectra acquired from selectively labeled and aligned, stationary samples, and enabling assignment of the latter spectra. (5) 15N/15N dilute spin exchange can be applied to selectively labeled samples to single out the adjacent pairs of same type of amino acid residues (Cross et al. 1983; Marassi et al. 1999a).
Materials and Methods
Sample preparation
The expression and purification of uniformly 15N-labeled full-length MerF has been described in detail (Lu et al. 2013). For the expression of amino acid type selectively 15N labeled MerF, a 15N-labeled amino acid (Cambridge Isotope Laboratories, www.isotope.com) was added to the growth media at a concentration of 0.1 g/l, while all the other 19 unlabeled amino acids were added at higher concentrations including 0.5 g/l Asp and Glu. The bacterial growth was generally shortened to < 3 hours after induction to minimize isotopic scrambling. In the last step of purification, size-exclusion chromatography on a Sephacryl S-200 column was followed by exhaustive dialysis to remove the sodium dodecyl sulfate (SDS) detergent. Alternatively, reverse-phase high-performance liquid chromatography (HPLC) (DeltaPak C4 Column, Waters, www.waters.com) was used (Lu et al. 2013). In both cases, the resulting pure polypeptides were lyophilized for long-term storage prior to preparing the NMR samples.
Magnetically aligned bicelle samples for OS solid-state NMR were prepared from a mixture of long-chain 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and short-chain 1,2-dihexanoyl-sn-glycero-3-phosphocholine (DHPC) lipids at q=3.2 (Sanders et al. 1994). In some samples, including the uniformly 15N-labeled sample used for the three-dimensional experiments, DMPC and DHPC were substituted by the non-hydrolysable ether-linked lipids 1,2-di-O-tetradecyl-sn-glycero-3-phosphocholine (14-O-PC) and 1,2-di-O-hexyl-sn-glycero-3-phosphocholine (6-O-PC) for long-term stability. All lipids were from Avanti Polar Lipids (www.avantilipids.com). The protocols for preparing the magnetically aligned bicelle samples for OS solid-state NMR have been described (De Angelis and Opella 2007; Sanders et al. 1994). Briefly, lyophilized MerF protein (in the range of 2 to 7 mg) was first solubilized in a 160μL solution containing 7mg DHPC, and then ∼35mg lyophilized DMPC was mixed into the solution. The resulting solution was repeatedly cycled between 42°C and 0°C with vortexing at both temperatures to improve sample mixing and facilitate the formation of bicelles. The pH was then adjusted to 6.0. 170μL of the solution was transferred to a 5 mm outside diameter flat-bottomed thin-wall glass tube (NE-RG5-T-15-FB, New Era, http://www.newera-spectro.com), which was sealed with a rubber plug. “Flipped” bicelle samples with the bilayer normals parallel to the field were prepared by the addition of ∼1.5mM YbCl3 to the already formed bicelles. Isotropic bicelle samples (q=0.1) for solution NMR were prepared by mixing ∼1mg MerF protein in 500μL solution containing 10% v/v D2O, 100mM 6-O-PC, 10mM 14-O-PC, 20mM MES and 1mM NaN3 at pH=6.0.
NMR spectroscopy
The solid-state NMR experiments were performed on a Bruker Avance 700 MHz spectrometer using a home-built 1H/15N double-resonance “low-E” probe with a strip shield (Wu et al. 2009). Sample temperature was maintained at 42°C and the recycle delay was 4-6 seconds. 15N chemical shift frequencies were referenced to the signal from solid ammonium sulfate, which was set to 26.8 ppm (De Angelis et al. 2006) and 1H chemical shift frequencies were referenced to the internal 1H2O resonance set to 4.7 ppm. The 1H carrier was set at 9 ppm for “unflipped” perpendicularly aligned bicelle samples and at 5 ppm for “flipped” parallel aligned bicelle samples. All two-dimensional SLF experiments performed on “unflipped” bicelle samples utilized the SAMPI4 pulse sequence (Nevzorov and Opella 2007). For “flipped” bicelle samples, both SAMPI4 and PISEMA (Wu et al. 1994) pulse sequences were used in order to cope with the larger dipolar couplings present in the samples. The three-dimensional HETCOR/SLF experiments and two-dimensional PELF experiments (Schmidt-Rohr et al. 1994) used the MSHOT-Pi4/Pi sequence (Lu et al. 2012; Lu and Opella 2013). All the experiments were performed with a radiofrequency field strength of ∼ 50 kHz in both channels, except that the 1H field strength was ∼70 kHz during 1H chemical shift evolution in two-dimensional HETCOR and three-dimensional HETCOR/SLF experiments and during the 1H-15N dipolar coupling dimension in PELF experiments. Spectra from the selectively 15N labeled samples were acquired with between 20 and 48 real points in the t1 dimension of PISEMA and SAMPI4 experiments, and the settings for the HETCOR/SLF and PELF experiments were similar to those previously described (Lu et al. 2012). For solution NMR experiments, the two-dimensional HSQC spectra were acquired at 50 °C on a 800 MHz spectrometer using a triple-resonance cryoprobe, and the chemical shifts are referenced to the 1H2O resonance defined as 4.534 ppm at 50 °C (Cavanagh et al. 1996). All the NMR data were processed using the program NMRPipe (Delaglio et al. 1995) and the spectra were assigned using the program Sparky (Goddard).
Results
Oriented Sample Solid-State NMR of Stationary Aligned Samples
Alignment of the nuclear spin interactions present at all protein sites relative to a fixed external axis is the essential physical principle of oriented sample solid-state NMR spectroscopy. The basic approach is best illustrated by aligned, stationary samples, which can be prepared in several ways. The most common ones are mechanical alignment on glass plates and magnetic alignment in bicelles with q > 2.5 (De Angelis and Opella 2007). It is also possible to align samples as drawn fibers (Opella and Waugh 1977), in narrow tubes (Chekmenev et al. 2006), and potentially other methods. Although mechanically aligned samples of protein-containing phospholipids on glass plates are the simplest to prepare, and provide the greatest flexibility in terms of choice of lipids, temperature, etc., magnetic alignment has the strong advantages of providing spectra with narrower line widths and having no uncertainty about the precise direction of alignment. Both mechanically and magnetically aligned samples can be ‘flipped’ between alignments with the bilayer normals parallel and perpendicular to the direction of the applied magnetic field. Bilayers aligned on glass plates have the additional advantage that they can be oriented at any angle with respect to the field by tilting the plates and/or the coil, which is useful in some specialized experiments (Park et al. 2006).
MerF in 14-O-PC/6-O-PC q=3.2 bicelles aligns spontaneously with the bilayer normals perpendicular to the direction of the field. Full-length MerF in bicelles forms particularly well-aligned samples, as judged by the line widths of individual resonances, and the overall resolution of the one-dimensional 15N NMR spectrum of a uniformly 15N labeled sample shown in Fig. 1. Two features in particular demonstrate that the sample is well aligned. The spectral region above 150 ppm is free of signal intensity, indicating the absence of residual powder pattern from unaligned or aggregated proteins and that the proteins are all undergoing fast uniaxial rotational diffusion. And the line widths of selected peaks are very narrow, approaching those of the best reported spectra, which required the use of Triton X-100 instead of DHPC as the “short chain” lipid and a small protein (Park and Opella 2010). The ability to prepare well-aligned samples lays the foundation for the subsequent steps of spectroscopy and resonance assignments of signals associated with individual residues. The frequencies of these resolved signals carry angular information that is the principal source of restraints for structure calculations. The frequencies are also the source of resolution in the spectra.
Fig. 1.
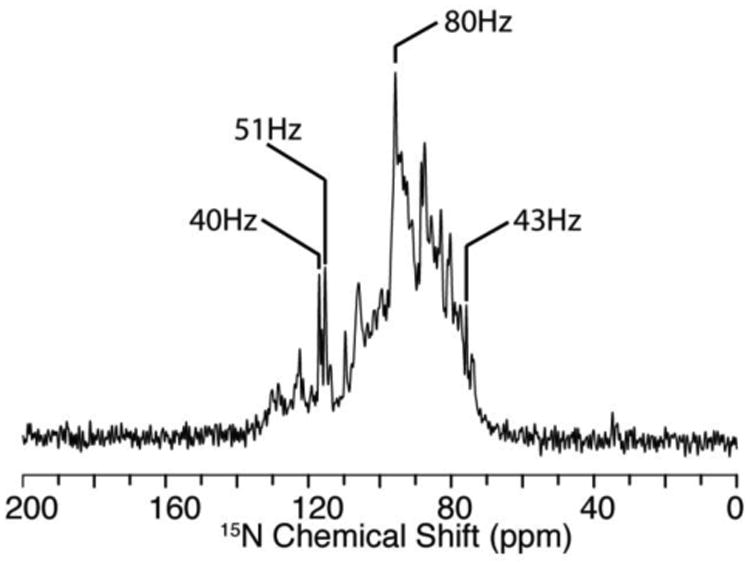
15N solid-state NMR spectrum of uniformly 15N labeled full-length MerF in q=3.2 14-O-PC/6-O-PC bicelles aligned with their normals perpendicular to the direction of the magnetic field. Line widths of selected resonances are marked in the figure.
The next task is to resolve and assign individual resonance signals in the spectrum. In previous examples of single trans-membrane or small two trans-membrane helical membrane proteins, this could be successfully accomplished with the combination of high-resolution two-dimensional SLF spectroscopy and the use of selective isotopic labeling. Since selectively labeled samples could be prepared for the majority of amino acids, signals from the rest could be identified by subtracting the spectra of selectively labeled samples from that of a uniformly labeled sample. With individual signals resolved and the confident assignment of their amino acid type, the resonance assignment could then be accomplished through the qualitative, geometric PISA Wheel (Marassi and Opella 2000; Wang et al. 2000) and Dipolar Wave analysis (Mesleh et al. 2002). This assignment method has been described in detail in earlier publications for those regions of proteins with large amounts of regular alpha helix (Marassi and Opella 2003) or beta sheet secondary structure (Marassi 2001), and has been recently optimized and automated in the AssignFit program (Tian et al. 2012). The method relies on the periodic geometry of α-helical residues that can be displayed as a helical wheel and therefore present themselves as the PISA Wheel in SLF spectra.
However, in the structure determination of larger and more challenging proteins, such as MerF, these methods alone are not sufficient to resolve and unambiguously assign signals from all residues. The first challenge is that two-dimensional SLF spectra do not fully resolve signals from uniformly labeled samples (Fig. 2A), or even in some selectively labeled samples when multiple copies of the same amino acid are present, such as the 14 leucines in MerF (Fig. 2D). The second challenge is the difficulty in unambiguously assigning signals with PISA Wheel analysis. Signals from residues in the two long trans-membrane helices produce overlapped PISA wheels in the crowded spectral region with 1H-15N dipolar couplings > 1.5 kHz. More critically, the third challenge is the assignment of the residues in the non-helical regions of the protein. Even for small membrane proteins, few irregularly structured residues could be readily assigned from the other signals in the selectively labeled samples. Since multiple sites are now present in this region and PISA Wheels and Dipolar Wave approaches are effective only for residues in regular secondary structures, an alternative assignment method needs to be developed.
Fig. 2.
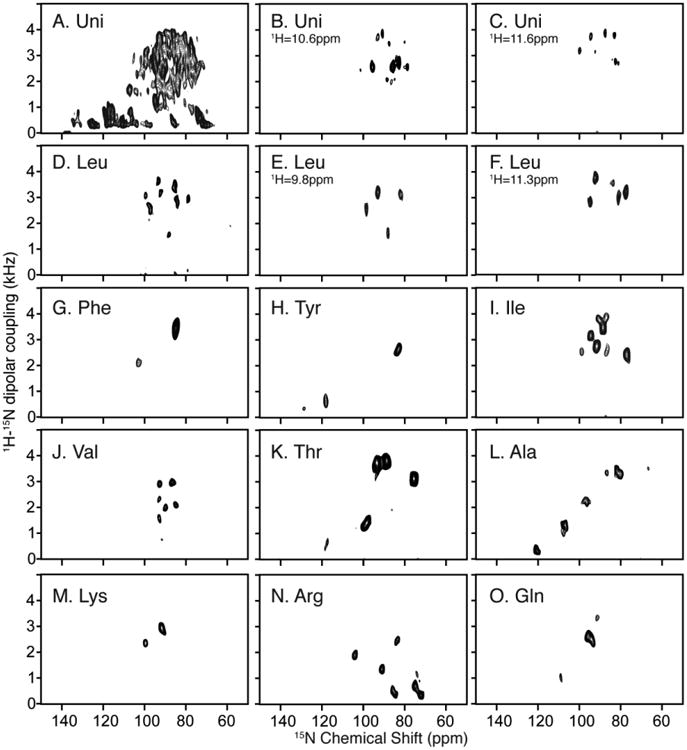
Two-dimensional SLF and three-dimensional HECTOR/SLF NMR spectra of uniformly and selectively 15N labeled samples of MerF in q=3.2 14-O-PC/6-O-PC magnetically aligned bilayers (q=3.2 bicelles). A. Two-dimensional SLF spectrum of uniformly 15N labeled MerF. B. and C. Two-dimensional planes at the designated 1H chemical shift frequencies from a three-dimensional HECTOR/SLF spectrum. D. Two-dimensional SLF spectrum of selectively 15N leucine labeled MerF. E. and F. Two-dimensional planes at the designated 1H chemical shift frequencies from a three-dimensional HECTOR/SLF spectrum. G-O. Two-dimensional SLF spectrum of other selectively labeled MerF samples. A-C are reprinted from (Lu et al. 2012). D and G-O are from (Howell 2007).
Here we show that the first challenge can be addressed by application of the MSHOT-Pi4/Pi pulse sequence, which improves the resolution in the 1H chemical shift dimension (Lu and Opella 2013). This enables three-dimensional HETCOR/SLF spectroscopy to be used effectively with superior resolving power to that observed in two-dimensional SLF spectroscopy without much loss of sensitivity. In the uniformly 15N labeled sample, except for 5 overlapped signals out of the total 64 observed amide signals, all are recognizable as distinct resonances in the three-dimensional HETCOR/SLF spectrum (Fig. 2B and 2C). This enables the few residues that cannot be selectively labeled during bacterial expression to be readily identified among the unassigned peaks after accounting for all of the successfully labeled amino acids. Additionally, all resonances assigned by relying on multiple spectra and experiments are now mapped onto a single spectrum, which provides the practical advantage of minimizing the effects of minor sample-to-sample variations.
Using stationary, aligned samples in the form of magnetically aligned bilayers (q=3.2 bicelles), spectra from multiple selectively 15N labeled samples are compared to that from a uniformly 15N labeled sample (Fig. 2). Although it is labor-intensive and time consuming to prepare multiple samples, these data provide a substantial amount of unambiguous information about the positions and assignments of resonances. Selectively 15N-labeled samples were prepared for Ala, Arg, Gln, Ile, Leu, Lys, Phe, Thr, Tyr and Val. As noted, several of the other amino acids, i.e., Asp, Gly and Ser, could not be labeled with high incorporation and selectivity. Nine of the ten samples provide well-resolved two-dimensional SLF spectra (Fig. 2G-2O). The leucine labeled sample is the exception; the 14 leucine resonances are not well resolved in two-dimensional spectra (Fig. 2D) but are fully resolved in a three-dimensional HETCOR/SLF spectrum as seen in the two examples of spectral planes (Fig. 2E and 2F).
Dipolar coupling correlated isotropic chemical shift analysis
For solution NMR and MAS solid-state NMR, both homonuclear and heteronuclear dipolar couplings are attenuated by motion, yielding isotropic chemical shift spectra and enabling the development of many magnetization transfer and sequential backbone assignment procedures. However, the ability to obtain angular information was lost until the introduction of methods to measure residual dipolar couplings (RDCs) in solution NMR of weakly aligned samples (Tolman et al. 1995) and dipolar recoupling in solid-state NMR (Griffin 1998). OS solid-state NMR spectra of aligned, stationary samples, on the other hand, retain all the anisotropic information from the beginning. However, it has a major drawback, which is the difficulty of sequential backbone assignments due to the presence of unaveraged 1H-1H and 13C-13C dipolar couplings. These limitations can be largely overcome by a strategy of making resonance assignment using established methods on isotropic NMR spectra first and subsequently transferring the information to OS solid-state NMR spectra.
In dipolar coupling correlated isotropic chemical shift (DCCICS) analysis (Lu et al. 2011), the 1H and 15N isotropic chemical shifts are transferred from solution NMR spectra on low q isotropic bicelles (Son et al. 2012), as demonstrated using the selectively 15N-Ile labeled sample of MerF shown in Fig. 3 and Table 1. The underlying principle of the method is that the 1H-15N dipolar coupling for the same pair of nuclei differs by exactly a factor of two between parallel (flipped) and perpendicular (unflipped) aligned protein-containing bicelles. As a result, a one-to-one correlation can be established between signals in SLF spectra of flipped and unflipped bicelles by aligning the appropriately scaled spectra. This enables two sets of anisotropic 15N and 1H chemical shifts to be measured for each residue, which are used to calculate the isotropic chemical shifts. These derived isotropic shifts can be compared with those observed in solution NMR spectra for regions of the protein that are not distorted by the presence of detergents or MAS solid-state NMR spectra to obtain their assignments. Notably, due to the broader line widths generally encountered in solid-state NMR, the isotropic chemical shift calculated in this manner have an error range, which was ∼1.5 ppm for the 15N chemical shift and ∼0.3 ppm for the 1H chemical shift in the previous well optimized case (Lu et al. 2011). The existence of this range, which can be improved through further spectroscopic development to improve resolution, currently relegates the DCCICS method to the role of assisting or confirming assignments rather than being able to make unique assignments on its own.
Fig. 3.
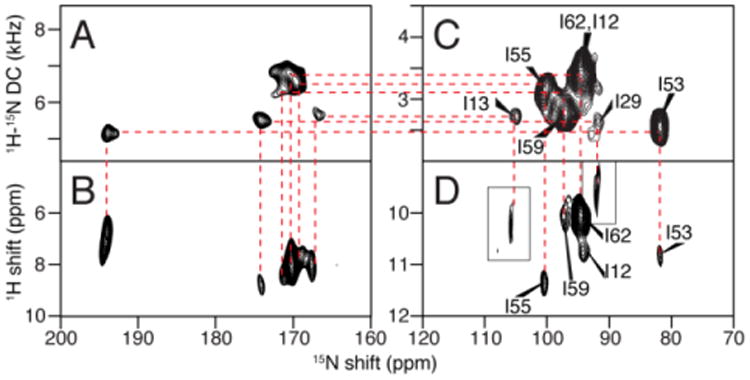
Example of the assignment of the isoleucine resonances of MerF by the method of heteronuclear dipolar coupling correlated isotropic chemical shift analysis. A., B., C., and D. are two-dimensional spectra of MerF in q=3.2 14-O-PC/6-O-PC bicelles. A. and B. are “flipped” with the bilayer normal parallel to the field; and C. and D. are “unflipped” with the bilayer normal perpendicular to the field. A. and C. are SLF spectra. B. and D. are HETCOR spectra. The assignment derived from combined DCCICS and PISA wheel analysis are labeled in spectra C and D. Insets in D are extracted from three-dimensional HETCOR/SLF spectrum of the same sample to occupy the missing peaks in two-dimensional spectrum due to selective magnetization transfer.
Table 1.
Summary of data for the assignment and isotropic chemical shift calculations of MerF isoleucine residues for DCCICS analysis.
| Residue assignment | Calculation from OS solid-state NMR | Measurement in solution NMR | The difference | ||
|---|---|---|---|---|---|
|
|
|||||
| 15N shift | 1H shift | 15N shift | 1H shift | ||
| 55 | 121.47 | 8.72 | 120.80 | 8.88 | 0.21 |
| 62 | 118.24 | 7.98 | 117.97 | 8.22 | 0.24 |
| 59 | 120.35 | 8.21 | 120.25 | 8.25 | 0.04 |
| 53 | 119.05 | 8.10 | 116.81 | 8.15 | 0.45 |
| 29 | 117.40 | 8.07 | 118.19 | 7.63 | 0.47 |
| 13 | 124.21 | 8.05 | 121.80 | 8.22 | 0.51 |
| 12 | 118.01 | 8.46 | 122.91 | 8.26 | 1.00 |
The assignment of the isoleucine resonances is shown here as an example of the DCCICS method (Fig. 3 and Table 1). The assignment starts at residue I55, which is well separated in the DCCICS analysis. I55 also further confirms the PISA Wheel predicted for the second trans-membrane helix of MerF (L48 to R64), which is anchored by the assigned F54 and Y60 signals. From the PISA Wheel fitting, the assignments of residues I53, I59 and I62, which are all in the second helix, can be obtained, and agree with the DCCICS analysis. Next, I13 can be assigned: from DCCICS the signal could be either I12 or I13, but the subsequent PISA wheel fitting shows that it can only be I13 in order to have I12 fit to one of the two isoleucine peaks left. I12 and I29 are then assigned to the last two isoleucine peaks. This assignment is further confirmed with the dipolar coupling frequency alignment method shown in the subsequent section “transfer of resonance assignments from rotational alignment solid-state NMR spectra”. The relatively large differences of residue I12 and I13 in DCCICS matching (Table 1) is likely due to the different membrane environments between low “q” isotropic bicelles and high “q” magnetically aligned bicelles. Because I12 and I13 are exposed to aqueous environment, they are susceptible to the variation in free detergent concentrations between the two samples. In contrast, the other isoleucine residues are likely to be in similar environments in both samples, because they are buried inside the membrane environment and are presumably in contact with long-chain 14-O-PC lipid in both samples (Lee et al. 2008).
Resonance assignment of loop and terminal region
It is possible to use DCCICS to assign the non-helical regions of solid-state NMR spectra from aligned samples. This is especially valuable for those residues that cannot be selectively labeled. The method relies on the MSHOT-Pi4 pulse sequence to improve the resolution of the PELF experiment. Previous DCCICS trials with SLF spectra resulted in incomplete assignments (data not shown) because the dipolar truncation effect (Gan 2000) of rotating-frame SLF experiments often render those signals with small 1H-15N dipolar couplings at the similar dipolar couplings of ∼0.3 kHz. As a result, one-to-one correlations cannot be easily established for signals from the same residues in spectra from flipped and unflipped bicelle samples. It also makes it difficult to resolve single signals with small heteronuclear dipolar couplings. Application of the improved PELF experiment addresses these issues, as shown in Fig. 4A and 4B.
Fig. 4.
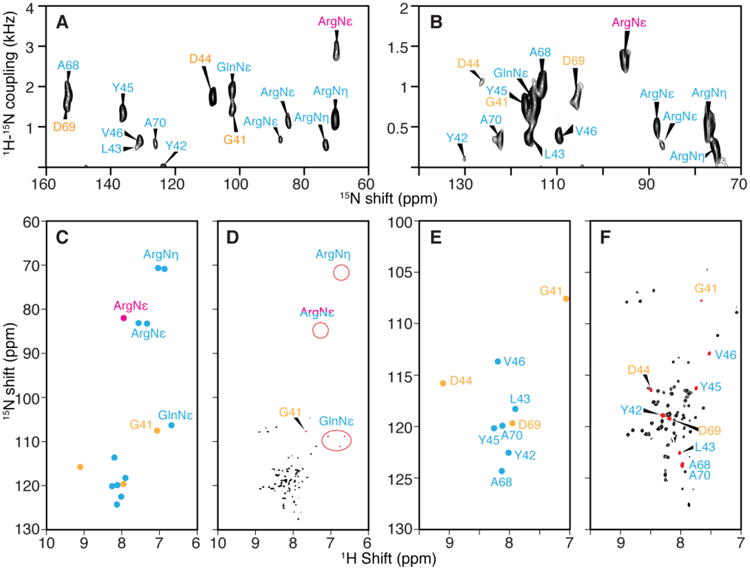
Resonance assignments of the terminal and loop regions of membrane proteins with irregular tertiary structure. PELF spectra of uniformly 15N labeled MerF in flipped (A.) and unflipped (B.) bicelles. C. 1H and 15N isotropic chemical shifts calculated from the spectra in panels A. and B. and three-dimensional HETCOR/SLF spectra. D. Solution HSQC NMR spectrum and the chemical shift of side chains. E. and F. Expansion of the backbone amide resonance region of the spectra in panels C. and D. Those resonances whose assignments can be determined from selectively labeled spectra are labeled in cyan color. In this group, one of the ArgNε peak (magenta color) is an example that DCCICS analysis is crucial for avoiding its mis-assignment as a backbone amide resonance. The remaining resonances that could not be selectively labeled (yellow color) are successfully assigned by DCCICS analysis. A is reprinted from (Lu et al. 2012) after adding resonance assignments.
An important advantage of the application of DCCICS is that side chain amide groups can be readily distinguished (Fig. 4C and 4D). These side chain signals, especially the GlnNε and the ArgNε with > 1 kHz dipolar couplings, can be easily misidentified as backbone amide signals. For example, after acquiring the SLF spectrum on selectively 15N-Arg labeled MerF sample (Fig. 2N), an ArgNε peak at 1.5 kHz dipolar coupling (labeled in magenta in Fig. 4) could be easily grouped together with the other two back bone amide peaks with large dipolar couplings (> 1 kHz). In this case, DCCICS analysis offers an unambiguous method of distinguishing backbone and side chain signals.
As shown in the preceding section, the currently existing error range of the DCCICS method makes it necessary to incorporate other information during resonance assignment. Here selective labeling with Ala, Leu, Tyr and Val was necessary to identify most of the signals and assignments (labeled in cyan in Fig. 4) in the terminal and loop regions of MerF, and their assignments were further verified in DCCICS analysis. After completing their assignment, DCCICS successfully provided additional assignments for the signals from D44, D69 and G41 (labeled yellow in Fig. 4), which could not be selectively labeled. The combination of DCCICS, selective labeling and the improved PELF pulse sequence is able to provide complete assignments for all residues in the terminal and loop regions of MerF that are visible in solid-state NMR.
Transfer of resonance assignments from rotational alignment solid-state NMR spectra
Besides the DCCICS method, a straightforward way to transfer assignments from isotropic NMR spectra to anisotropic OS solid-state NMR spectra is through the frequency alignment of 1H-15N dipolar couplings to MAS solid-state NMR spectra, and it is enabled by the recent finding that dipolar couplings measured by MAS solid-state NMR under rotationally alignment condition have the same frequencies as those observed in spectra of aligned, stationary samples in OS solid-state NMR after being scaled by a known factor (Park et al. 2010a). Fig. 5 shows three examples where the individual assignment of residues in OS solid-state NMR spectra on selectively labeled samples can be made by aligning their dipolar coupling frequencies to those measured in the previously assigned RA solid-state NMR spectra of MerF (Lu et al. 2013). The 1H-15N dipolar couplings measured from RA solid-state NMR spectra are equal to those measured from ‘“unflipped” bicelle samples by taking into account the established bicelle order parameter of ∼0.8 (Sanders et al. 1994) and the scaling factor of 0.5 due to the perpendicular alignment of the bilayer normals in the magnetic field.
Fig. 5.
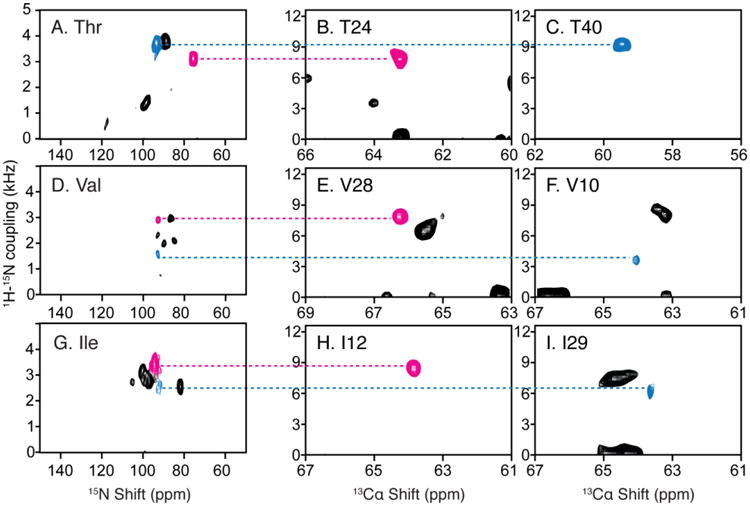
Resonance assignments of residues in OS solid-state NMR spectra utilizing the 1H-15N dipolar couplings measured in RA MAS solid-state NMR experiments. A. D. and G. Two-dimensional SLF spectrum of amino-acid-type selectively 15N labeled MerF in ‘unflipped’ DMPC/DHPC bicelles. B. C. E. F. H. and I. Two-dimensional SLF spectral planes extracted from a three-dimensional HnNCa experiment, of which the third dimension is 15N chemical shift (Lu et al. 2013). The scales during aligning the 1H-15N dipolar couplings are adjusted for the scaling factors of the bicelles vs. liposomes (0.8) and of the alignment perpendicular to the magnetic field vs. rotational alignment parallel to the bilayer normal (0.5).
T24, T40, V28 and V10 shown in Fig. 5 are all located near the end of a transmembrane helix, where PISA Wheel assignments often encounter difficulties because of ambiguity in determining the exact ending of the helix and possible deviations from an ideal helix in these regions. Here the assignment of these few key residues through dipolar coupling alignment in spectra of selectively labeled samples often provides a breakthrough in resolving the rest of the assignment of the specific amino acid type. The last example of I12 and I29 assignment further verifies the isoleucine assignment made with combined DCCICS and PISA wheel method (Fig. 3 and Table 1).
The dipolar coupling alignment method demonstrated above can be applied in a broader context before the full set of RA solid-state NMR spectra has been acquired and assigned. At least three three-dimensional spectra, HnNCa, HnNCo and HcCxCx, which require large amounts of signal averaging (Lu et al. 2013). In one scenario, resonance assignments may have already been obtained for a protein under MAS solid-state NMR experiments using conventional NCACX/NCOCX experiments (Pauli et al. 2001). In this case, only one additional experiment to recouple the dipolar coupling such as an HnNCa experiment (Lu et al. 2013), is needed to enable a direct transfer of resonance assignment to OS solid-state NMR spectra. In another scenario, partial resonance assignments of MAS spectra may be available from a combination of selective labeling/unlabeling procedures (Banigan et al. 2013); and in this case, a dipolar recoupling dimension can be integrated into one experiment to bring the assignment information to OS solid-state NMR.
Notably, the frequency alignment can also be used to transfer resonance assignment in the reverse direction from OS solid-state NMR spectra to MAS solid-state NMR spectra in the same manner. This is an attractive option because of the economical use of 15N-labeled amino acids and the high sensitivity of two-dimensional SLF experiments. Thus the amino-acid-type selective assignment information can be obtained efficiently in OS solid-state NMR, and subsequently to help resolve ambiguities encountered in assigning MAS solid-state NMR spectra.
Resonance assignment with 15N/15N dilute spin exchange
It is difficult to transfer magnetization along the protein backbone atoms in OS solid-state NMR in the absence of uniform 13C labeling. However, dilute spin exchange between proximate 15N sites of sequential residues has been shown to be effective assignment method in some cases, (Marassi et al. 1999b; Cross et al. 1983), and more recently, through several alternative pulse sequences (Nevzorov 2008; Xu et al. 2008; Traaseth et al. 2010; Tang et al. 2012). The application of dilute spin diffusion methodology to membrane proteins has been limited by the low sensitivity of the experiments (Knox et al. 2010).
To apply dilute spin diffusion experiments to larger membrane protein, it is essential to simplify the spectrum using methods such as amino acid type selective labeling. Fig. 6 demonstrates the application of two-dimensional dilute spin exchange (15N/15N) to selectively 15N-Leu labeled MerF. There are three pairs of vicinal leucine residues in the sequence, residues 7 and 8, residues 30 and 31, and residues 47 and 48. Both Mismatched Hartmann-Hahn (MMHH) spin diffusion (Nevzorov 2008) and proton-driven spin diffusion (Cross et al. 1983) have been used, and due to their different mechanisms, the intensities of cross peaks vary between the two types of spectra as expected. Two of the pairs, L7/8 and L47/48, are observed, while L30/31 cross peak cannot be observed because they have similar 15N chemical shifts and the cross peak is buried under the axial peaks. Notably, the intensity of cross peaks scales down according to the square of the labeling efficiency. This aggravates the contrast between high-intensity diagonal peaks and low-intensity cross peaks in the spectra.
Fig. 6.
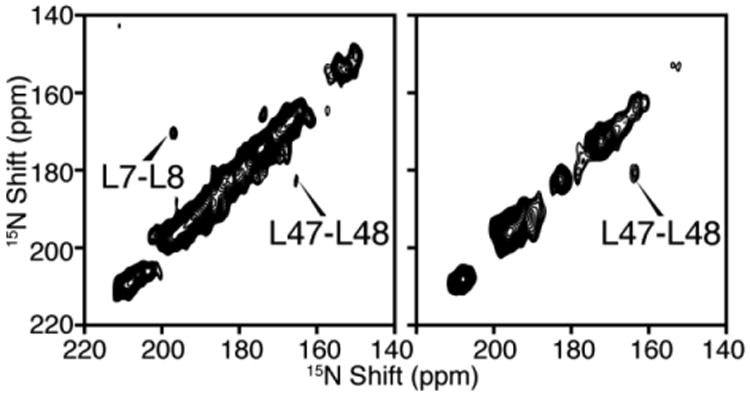
Two-dimensional 15N/15N dilute spin exchange spectra of selectively 15N-Leu labeled MerF in DMPC/DHPC bicelles aligned with their bilayer normal parallel to the magnetic field. A. Spectrum obtained using Mismatched Hartmann-Hahn (MMHH) spin diffusion (Nevzorov 2008). B. Spectrum obtained using Proton-driven Spin Diffusion (PDSD) (Cross et al. 1983).
Resonance frequencies of MerF protein
All the signals from the MerF residues that are immobile in the NMR time scale were assigned by this combinatorial set of resonance assignment methods described above. Signals from three N-terminal residues and 12 C-terminal residues are missing in OS solid-state NMR spectra of both uniformly and selectively 15N labeled samples, indicating that these residues are mobile. The resonance assignments and the anisotropic frequencies of the observed 64 residues are listed in Table 2.
Table 2.
The resonance assignment of 1H-15N dipolar coupling, anisotropic 1H chemical shift and anisotropic 15N chemical shift frequencies of MerF in perpendicularly magnetically aligned bicelles. The 1H-15N dipolar coupling of residues G41-V46 and A68-A70 are from PELF spectrum, and all other data are from three-dimensional HETCOR/SLF spectrum.
| Residue | 1H-15N DC (kHz) | 1H CS (ppm) | 15N CS (ppm) |
|---|---|---|---|
| K5 | 2502 | 10.23 | 100.05 |
| T6 | 3729 | 11.38 | 93.01 |
| L7 | 2633 | 10.53 | 99.27 |
| L8 | 3024 | 11.26 | 83.89 |
| R9 | 1650 | 9.11 | 106.85 |
| V10 | 1833 | 10.20 | 93.94 |
| S11 | 2673 | 11.29 | 81.12 |
| I12 | 3672 | 10.31 | 93.62 |
| I13 | 2934 | 10.21 | 105.44 |
| G14 | 2106 | 11.55 | 78.28 |
| T15 | 3910 | 11.00 | 93.15 |
| T16 | 1548 | 8.88 | 102.88 |
| L17 | 1575 | 10.09 | 90.50 |
| V18 | 2124 | 10.53 | 84.83 |
| A19 | 3413 | 11.35 | 87.94 |
| L20 | 2442 | 10.25 | 101.01 |
| S21 | 2110 | 10.14 | 88.62 |
| S22 | 3615 | 11.33 | 85.81 |
| F23 | 2476 | 10.42 | 101.41 |
| T24 | 2574 | 10.59 | 78.74 |
| V26 | 2088 | 10.64 | 88.61 |
| L27 | 3081 | 10.78 | 84.16 |
| V28 | 2865 | 10.61 | 91.48 |
| I29 | 2560 | 9.69 | 92.23 |
| L30 | 2757 | 11.65 | 82.60 |
| L31 | 3514 | 10.91 | 87.44 |
| G32 | 3313 | 11.20 | 85.03 |
| V33 | 2454 | 9.99 | 92.43 |
| V34 | 3076 | 11.46 | 86.38 |
| G35 | 3868 | 11.69 | 87.50 |
| L36 | 3033 | 11.59 | 96.26 |
| S37 | 2382 | 10.46 | 92.49 |
| A38 | 3738 | 11.35 | 82.87 |
| L39 | 3732 | 11.49 | 94.61 |
| T40 | 3766 | 10.67 | 92.96 |
| G41 | 769 | 6.82 | 117.15 |
| Y42 | 272 | 8.54 | 129.97 |
| L43 | 160 | 8.56 | 121.56 |
| D44 | 1066 | 10.33 | 127.70 |
| Y45 | 769 | 9.34 | 116.45 |
| V46 | 385 | 9.18 | 109.74 |
| L47 | 3181 | 10.08 | 95.36 |
| L48 | 3117 | 10.74 | 101.90 |
| A50 | 3519 | 10.88 | 80.27 |
| L51 | 3732 | 11.49 | 94.61 |
| A52 | 2559 | 10.63 | 95.68 |
| I53 | 2638 | 11.06 | 81.12 |
| F54 | 3468 | 11.19 | 87.31 |
| I55 | 3196 | 11.45 | 99.57 |
| G56 | 2515 | 11.18 | 76.88 |
| L57 | 3144 | 12.12 | 80.17 |
| T58 | 3904 | 10.73 | 90.82 |
| I59 | 2759 | 9.85 | 95.95 |
| Y60 | 2775 | 10.76 | 82.92 |
| A61 | 3519 | 10.88 | 80.27 |
| I62 | 3462 | 10.20 | 94.17 |
| Q63 | 2559 | 10.63 | 95.68 |
| R64 | 2575 | 10.46 | 85.74 |
| K65 | 2759 | 9.85 | 95.95 |
| R66 | 1650 | 9.11 | 106.85 |
| Q67 | 3247 | 10.79 | 90.66 |
| A68 | 1005 | 9.33 | 113.80 |
| D69 | 862 | 9.23 | 106.19 |
| A70 | 350 | 8.89 | 122.41 |
Discussion
A roadmap can be drawn to illustrate how these resonance assignment methods are closely connected and complementary to each other (Fig. 7). Five complementary resonance assignment methods are shown. PISA wheel, Dipolar Wave or AssignFit analysis is the conventional choice, and remains as the main resonance assignment method. Two other “information transfer” type of assignment methods, one introduced recently (Lu et al. 2011) and one in this article, bring in assignment information from isotropic NMR methods, including MAS solid-state NMR and solution NMR. The underlying motivation is to borrow the well-established assignment protocol in isotropic NMR methods and combine it with the advantage of OS solid-state NMR of preserving angular information of NMR observables. Two more categories of assignment method are listed, the spectroscopic method and sample preparation method. The former is an important focus of methodology development since the past few years. The latter is a category of many possible biochemical methods with site-directed mutagenesis and solid-phase peptide synthesis as two examples. The method is not demonstrated in this article but has been used in study of other membrane proteins (Kochendoerfer et al. 2004; Gayen et al. 2013). The strength of both methods are the low ambiguity, since a single known residue can be labeled or mutated. The disadvantages include the possibility of altering protein structure during mutagenesis, and the difficulties in synthesizing large hydrophobic polypeptides.
Fig. 7.
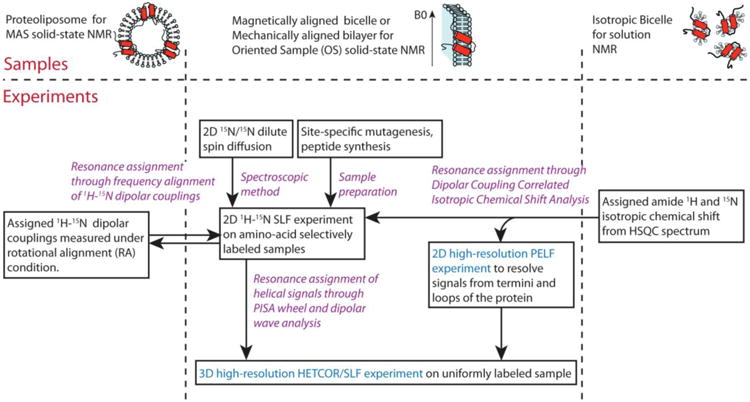
A roadmap for resonance assignments of membrane protein samples in OS solid-state NMR. Purple highlights the five complementary resonance assignment methods. Cyan highlights the experiments with significantly improved resolution by new pulse sequences.
It should be noted that the two “information transfer” methods (Fig. 7) bring assignments from the samples where membrane proteins are in slightly different lipid mimetic environments than the one used in OS solid-state NMR. Recently, examples from several membrane proteins have shown that the different detergent/lipid environment could alter the structures of the proteins (Zhou and Cross 2013; Zoonens et al. 2013; Cross et al. 2013; Zheng and Jia 2013; Raschle et al. 2010). Here the two sources of information, the proteoliposome and the isotropic bicelles, need to be treated separately. Firstly, the transfer can be performed relatively safely between samples of proteoliposome and magnetically aligned bicelle. Aligned bicelle has been established as a lipid bilayer environment similar to that of proteoliposome (Warschawski et al. 2011). Additionally, the high ratio of long-chain lipid to short-chain detergent in aligned bicelle samples reduces the free detergent concentration, making them similar to proteoliposome. An example is that the structures of the truncated form of MerF (MerFt) determined in liposome and aligned bicelle have also shown high degree of agreement (Das et al. 2012). On the other hand, the resonance assignment transfer between isotropic bicelle or detergent micelle and the high-q magnetically aligned bicelles deserves more carefulness. In favorable cases, the protein structure may stay the same (Franzin et al. 2007); but in some other cases such as MerFt in aligned bicelle versus in sodium dodecyl sulfate (SDS) micelle (Lu et al. 2013; Howell et al. 2005), the structure and dynamics can be largely different. In general, using the same set of lipid/detergent for aligned bicelle and isotropic bicelle can minimize the difference encountered in assignment transfer. Previously, DCCICS has been successfully applied to Pf1 coat protein between q=0 DHPC micelle and q=3.2 DMPC/DHPC bicelle (Lu et al. 2011). Here, we have transferred assignments between q=0.1 and q=3.2 14-O-PC/6-O-PC bicelle, and as discussed in previous section, residues I12 and I13 have experienced larger differences in chemical shifts, presumably due to the different environments.
In conclusion, the combination of a set of resonance assignment methods demonstrated in this article will likely find its greatest utility in assigning the spectra of larger membrane proteins by OS solid-state NMR. The assigned NMR observables contain angular restraints that can be directly used to calculate the three-dimensional structure of membrane proteins.
Acknowledgments
We thank Chris Grant and Albert Wu for assistance with instrumentation. The research was supported by Grants RO1GM099986 and P41EB002031 from the National Institutes of Health. It utilized the Biomedical Technology Resource for NMR Molecular Imaging of Proteins at the University of California, San Diego.
References
- Banigan J, Gayen A, Traaseth N. Combination of 15N reverse labeling and afterglow spectroscopy for assigning membrane protein spectra by magic-angle-spinning solid-state NMR: application to the multidrug resistance protein EmrE. J Biomol NMR. 2013;55(4):391–399. doi: 10.1007/s10858-013-9724-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh J, Fairbrother WJ, Palmer AG, III, Skelton NJ. Protein NMR spectroscopy: principles and practice. Academic Press; San Diego: 1996. [Google Scholar]
- Chekmenev EY, Gor'kov PL, Cross TA, Alaouie AM, Smirnov AI. Flow-Through Lipid Nanotube Arrays for Structure-Function Studies of Membrane Proteins by Solid-State NMR Spectroscopy. Biophys J. 2006;91(8):3076–3084. doi: 10.1529/biophysj.106.085191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross T, Murray D, Watts A. Helical membrane protein conformations and their environment. Eur Biophys J. 2013:1–25. doi: 10.1007/s00249-013-0925-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross TA, Frey MH, Opella SJ. Nitrogen-15 spin exchange in a protein. J Am Chem Soc. 1983;105(25):7471–7473. doi: 10.1021/ja00363a060. [DOI] [Google Scholar]
- Das BB, Nothnagel HJ, Lu GJ, Son WS, Tian Y, Marassi FM, Opella SJ. Structure Determination of a Membrane Protein in Proteoliposomes. J Am Chem Soc. 2012;134(4):2047–2056. doi: 10.1021/ja209464f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Angelis AA, Howell SC, Nevzorov AA, Opella SJ. Structure determination of a membrane protein with two trans-membrane helices in aligned phospholipid bicelles by solid-state NMR spectroscopy. J Am Chem Soc. 2006;128(37):12256–12267. doi: 10.1021/ja063640w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Angelis AA, Opella SJ. Bicelle samples for solid-state NMR of membrane proteins. Nature Protocols. 2007;2(10):2332–2338. doi: 10.1038/nprot.2007.329. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6(3):277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Franzin CM, Teriete P, Marassi FM. Structural similarity of a membrane protein in micelles and membranes. J Am Chem Soc. 2007;129(26):8078–8079. doi: 10.1021/ja0728371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Z. Spin Dynamics of Polarization Inversion Spin Exchange at the Magic Angle in Multiple Spin Systems. J Magn Reson. 2000;143(1):136–143. doi: 10.1006/jmre.1999.1971. [DOI] [PubMed] [Google Scholar]
- Gayen A, Banigan JR, Traaseth NJ. Ligand-Induced Conformational Changes of the Multidrug Resistance Transporter EmrE Probed by Oriented Solid-State NMR Spectroscopy. Angew Chem Int Ed. 2013;52(39):10321–10324. doi: 10.1002/anie.201303091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard TD, K DG. Sparky 3. University of California; San Francisco: [Google Scholar]
- Griffin RG. Dipolar recoupling in MAS spectra of biological solids. Nat Struct Biol. 1998;5:508–512. doi: 10.1038/749. [DOI] [PubMed] [Google Scholar]
- Howell SC. Dissertation. University of California; San Diego, California, USA: 2007. Application of nuclear magnetic resonance spectroscopy to the structure determination of the integral membrane proteins of the Mer operon. [Google Scholar]
- Howell SC, Mesleh MF, Opella SJ. NMR structure determination of a membrane protein with two transmembrane helices in micelles: MerF of the bacterial mercury detoxification system. Biochemistry. 2005;44(13):5196–5206. doi: 10.1021/bi048095v. [DOI] [PubMed] [Google Scholar]
- Ketchem R, Hu W, Cross T. Science. 5127. Vol. 261. New York, NY: 1993. High-resolution conformation of gramicidin A in a lipid bilayer by solid-state NMR; pp. 1457–1460. [DOI] [PubMed] [Google Scholar]
- Knox RW, Lu GJ, Opella SJ, Nevzorov AA. A Resonance Assignment Method for Oriented-Sample Solid-State NMR of Proteins. J Am Chem Soc. 2010;132(24):8255–8257. doi: 10.1021/ja102932n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochendoerfer GG, Jones DH, Lee S, Oblatt-Montal M, Opella SJ, Montal M. Functional characterization and NMR spectroscopy on full-length Vpu from HIV-1 prepared by total chemical synthesis. J Am Chem Soc. 2004;126(8):2439–2446. doi: 10.1021/ja038985i. [DOI] [PubMed] [Google Scholar]
- Lee D, Walter KFA, Brückner AK, Hilty C, Becker S, Griesinger C. Bilayer in Small Bicelles Revealed by Lipid-Protein Interactions Using NMR Spectroscopy. J Am Chem Soc. 2008;130(42):13822–13823. doi: 10.1021/ja803686p. [DOI] [PubMed] [Google Scholar]
- Lu GJ, Opella SJ. Motion-adapted pulse sequences for oriented sample (OS) solid-state NMR of biopolymers. J Chem Phys. 2013;139(8):084203. doi: 10.1063/1.4819331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu GJ, Park SH, Opella SJ. Improved 1H amide resonance line narrowing in oriented sample solid-state NMR of membrane proteins in phospholipid bilayers. J Magn Reson. 2012;220:54–61. doi: 10.1016/j.jmr.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu GJ, Son WS, Opella SJ. A general assignment method for oriented sample (OS) solid-state NMR of proteins based on the correlation of resonances through heteronuclear dipolar couplings in samples aligned parallel and perpendicular to the magnetic field. J Magn Reson. 2011;209(2):195–206. doi: 10.1016/j.jmr.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu GJ, Tian Y, Vora N, Marassi FM, Opella SJ. The Structure of the Mercury Transporter MerF in Phospholipid Bilayers: A Large Conformational Rearrangement Results From N-terminal Truncation. J Am Chem Soc. 2013;135(25):9299–9302. doi: 10.1021/ja4042115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marassi FM. A simple approach to membrane protein secondary structure and topology based on NMR spectroscopy. Biophys J. 2001;80(2):994–1003. doi: 10.1016/S0006-3495(01)76078-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marassi FM, Gesell JJ, Valente AP, Kim Y, Oblatt-Montal M, Montal M, Opella SJ. Dilute spin-exchange assignment of solid-state NMR spectra of oriented proteins: acetylcholine M2 in bilayers. J Biomol NMR. 1999a;14(2):141–148. doi: 10.1023/a:1008391823293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marassi FM, Gesell JJ, Valente AP, Kim Y, Oblatt-Montal M, Montal M, Opella SJ. Dilute spin-exchange assignment of solid-state NMR spectra of oriented proteins: acetylcholine M2 in bilayers. J Biomol NMR. 1999b;14(2):141–148. doi: 10.1023/a:1008391823293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marassi FM, Opella SJ. A solid-state NMR index of helical membrane protein structure and topology. J Magn Reson. 2000;144(1):150–155. doi: 10.1006/jmre.2000.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marassi FM, Opella SJ. Simultaneous assignment and structure determination of a membrane protein from NMR orientational restraints. Protein Sci. 2003;12(3):403–411. doi: 10.1110/ps.0211503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesleh MF, Veglia G, DeSilva TM, Marassi FM, Opella SJ. Dipolar Waves as NMR Maps of Protein Structure. J Am Chem Soc. 2002;124(16):4206–4207. doi: 10.1021/ja0178665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray DT, Das N, Cross TA. Solid State NMR Strategy for Characterizing Native Membrane Protein Structures. Acc Chem Res. 2013;46(9):2172–2181. doi: 10.1021/ar3003442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevzorov AA. Mismatched Hartmann-Hahn conditions cause proton-mediated intermolecular magnetization transfer between dilute low-spin nuclei in NMR of static solids. J Am Chem Soc. 2008;130(34):11282–11283. doi: 10.1021/ja804326b. [DOI] [PubMed] [Google Scholar]
- Nevzorov AA, Opella SJ. Selective averaging for high-resolution solid-state NMR spectroscopy of aligned samples. J Magn Reson. 2007;185(1):59–70. doi: 10.1016/j.jmr.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Opella SJ. Structure Determination of Membrane Proteins by Nuclear Magnetic Resonance Spectroscopy. Annu Rev Anal Chem. 2013;6(1):305–328. doi: 10.1146/annurev-anchem-062012-092631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opella SJ, Marassi FM, Gesell JJ, Valente AP, Kim Y, Oblatt-Montal M, Montal M. Structures of the M2 channel-lining segments from nicotinic acetylcholine and NMDA receptors by NMR spectroscopy. Nat Struct Biol. 1999;6(4):374–379. doi: 10.1038/7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opella SJ, Waugh JS. Two-dimensional 13C NMR of highly oriented polyethylene. J Chem Phys. 1977;66(11):4919–4924. [Google Scholar]
- Opella SJ, Zeri AC, Park SH. Structure, dynamics, and assembly of filamentous bacteriophages by nuclear magnetic resonance spectroscopy. Annu Rev Phys Chem. 2008;59:635–657. doi: 10.1146/annurev.physchem.58.032806.104640. [DOI] [PubMed] [Google Scholar]
- Park SH, Das BB, De Angelis AA, Scrima M, Opella SJ. Mechanically, Magnetically, and “Rotationally Aligned” Membrane Proteins in Phospholipid Bilayers Give Equivalent Angular Constraints for NMR Structure Determination. J Phys Chem B. 2010a;114(44):13995–14003. doi: 10.1021/jp106043w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SH, Marassi FM, Black D, Opella SJ. Structure and Dynamics of the Membrane-Bound Form of Pf1 Coat Protein: Implications of Structural Rearrangement for Virus Assembly. Biophys J. 2010b;99(5):1465–1474. doi: 10.1016/j.bpj.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SH, Mrse AA, Nevzorov AA, De Angelis AA, Opella SJ. Rotational diffusion of membrane proteins in aligned phospholipid bilayers by solid-state NMR spectroscopy. J Magn Reson. 2006;178(1):162–165. doi: 10.1016/j.jmr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Park SH, Mrse AA, Nevzorov AA, Mesleh MF, Oblatt-Montal M, Montal M, Opella SJ. Three-dimensional structure of the channel-forming trans-membrane domain of virus protein “u” (Vpu) from HIV-1. J Mol Biol. 2003;333(2):409–424. doi: 10.1016/j.jmb.2003.08.048. [DOI] [PubMed] [Google Scholar]
- Park SH, Opella SJ. Triton X-100 as the “Short-Chain Lipid” Improves the Magnetic Alignment and Stability of Membrane Proteins in Phosphatidylcholine Bilayers for Oriented-Sample Solid-State NMR Spectroscopy. J Am Chem Soc. 2010;132(36):12552–12553. doi: 10.1021/ja1055565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli J, Baldus M, van Rossum B, de Groot H, Oschkinat H. Backbone and Side-Chain 13C and 15N Signal Assignments of the α-Spectrin SH3 Domain by Magic Angle Spinning Solid-State NMR at 17.6 Tesla. ChemBioChem. 2001;2(4):272–281. doi: 10.1002/1439-7633(20010401)2:4<272∷aid-cbic272>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Raschle T, Hiller S, Etzkorn M, Wagner G. Nonmicellar systems for solution NMR spectroscopy of membrane proteins. Curr Opin Struct Biol. 2010;20(4):471–479. doi: 10.1016/j.sbi.2010.05.006. doi: http://dx.doi.org/10.1016/j.sbi.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders CR, Hare BJ, Howard KP, Prestegard JH. Magnetically-oriented phospholipid micelles as a tool for the study of membrane-associated molecules. Prog Nucl Magn Reson Spectrosc. 1994;26(Part 5):421–444. doi: 10.1016/0079-6565(94)80012-X. [DOI] [Google Scholar]
- Schmidt-Rohr K, Nanz D, Emsley L, Pines A. NMR Measurement of Resolved Heteronuclear Dipole Couplings in Liquid Crystals and Lipids. J Phys Chem. 1994;98(27):6668–6670. doi: 10.1021/j100078a002. [DOI] [Google Scholar]
- Sharma M, Yi M, Dong H, Qin H, Peterson E, Busath DD, Zhou HX, Cross TA. Science. 6003. Vol. 330. New York, NY: 2010. Insight into the Mechanism of the Influenza A Proton Channel from a Structure in a Lipid Bilayer; pp. 509–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son WS, Park SH, Nothnagel HJ, Lu GJ, Wang Y, Zhang H, Cook GA, Howell SC, Opella SJ. ‘q-Titration’ of long-chain and short-chain lipids differentiates between structured and mobile residues of membrane proteins studied in bicelles by solution NMR spectroscopy. J Magn Reson. 2012;214(0):111–118. doi: 10.1016/j.jmr.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Knox R, Nevzorov A. A spectroscopic assignment technique for membrane proteins reconstituted in magnetically aligned bicelles. J Biomol NMR. 2012;54(3):307–316. doi: 10.1007/s10858-012-9673-y. [DOI] [PubMed] [Google Scholar]
- Tian Y, Schwieters CD, Opella SJ, Marassi FM. AssignFit: A program for simultaneous assignment and structure refinement from solid-state NMR spectra. J Magn Reson. 2012;214:42–50. doi: 10.1016/j.jmr.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolman JR, Flanagan JM, Kennedy MA, Prestegard JH. Nuclear magnetic dipole interactions in field-oriented proteins: information for structure determination in solution. Proc Natl Acad Sci. 1995;92(20):9279–9283. doi: 10.1073/pnas.92.20.9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traaseth NJ, Gopinath T, Veglia G. On the Performance of Spin Diffusion NMR Techniques in Oriented Solids: Prospects for Resonance Assignments and Distance Measurements from Separated Local Field Experiments. J Phys Chem B. 2010;114(43):13872–13880. doi: 10.1021/jp105718r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Denny J, Tian C, Kim S, Mo Y, Kovacs F, Song Z, Nishimura K, Gan Z, Fu R, Quine JR, Cross TA. Imaging Membrane Protein Helical Wheels. J Magn Reson. 2000;144(1):162–167. doi: 10.1006/jmre.2000.2037. [DOI] [PubMed] [Google Scholar]
- Warschawski DE, Arnold AA, Beaugrand M, Gravel A, Chartrand E, Marcotte I. Choosing membrane mimetics for NMR structural studies of transmembrane proteins. Biochim Biophys Acta-Biomembr. 2011;1808(8):1957–1974. doi: 10.1016/j.bbamem.2011.03.016. [DOI] [PubMed] [Google Scholar]
- Wu CH, Grant CV, Cook GA, Park SH, Opella SJ. A strip-shield improves the efficiency of a solenoid coil in probes for high-field solid-state NMR of lossy biological samples. J Magn Reson. 2009;200(1):74–80. doi: 10.1016/j.jmr.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CH, Ramamoorthy A, Opella SJ. High-Resolution Heteronuclear Dipolar Solid-State NMR Spectroscopy. J Magn Reson, Ser A. 1994;109(2):270–272. doi: 10.1006/jmra.1994.1169. [DOI] [Google Scholar]
- Xu J, Struppe J, Ramamoorthy A. Two-dimensional homonuclear chemical shift correlation established by the cross-relaxation driven spin diffusion in solids. J Chem Phys. 2008;128(5):052308. doi: 10.1063/1.2826323. [DOI] [PubMed] [Google Scholar]
- Zheng J, Jia Z. Structural biology: Tiny enzyme uses context to succeed. Nature. 2013;497(7450):445–446. doi: 10.1038/nature12245. [DOI] [PubMed] [Google Scholar]
- Zhou HX, Cross TA. Influences of Membrane Mimetic Environments on Membrane Protein Structures. Annu Rev Biophys. 2013;42(1):361–392. doi: 10.1146/annurev-biophys-083012-130326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoonens M, Comer J, Masscheleyn S, Pebay-Peyroula E, Chipot C, Miroux B, Dehez F. Dangerous Liaisons between Detergents and Membrane Proteins. The Case of Mitochondrial Uncoupling Protein 2. J Am Chem Soc. 2013;135(40):15174–15182. doi: 10.1021/ja407424v. [DOI] [PubMed] [Google Scholar]