

Abstract
Alzheimer's disease (AD) is a slowly progressing disorder in which pathophysiological abnormalities, detectable in vivo by biomarkers, precede overt clinical symptoms by many years to decades. Five AD biomarkers are sufficiently validated to have been incorporated into clinical diagnostic criteria and commonly used in therapeutic trials. Current AD biomarkers fall into 2 categories: biomarkers of amyloid-β plaques and of tau-related neurodegeneration. Three of the 5 are imaging measures and two are cerebrospinal fluid analytes. AD biomarkers do not evolve in an identical manner but rather in a sequential but temporally overlapping manner. Models of the temporal evolution of AD biomarkers can take the form of plots of biomarker severity (degree of abnormality) vs. time. In this review we discuss several time-dependent models of AD which take into consideration varying age of onset (early vs. late) and the influence of aging and co-occurring brain pathologies that commonly arise in the elderly.
Search terms: Alzheimer's disease, Alzheimer's biomarkers, amyloid imaging, Alzheimer's imaging, Alzheimer's modeling, PET AND Alzheimer's, MRI AND Alzheimer's
Pathological Features of Alzheimer's disease
Two well-known abnormal protein aggregates characterize Alzheimer's disease (AD) pathologically (Hyman et al., 2012). The hallmark of amyloid-β (Aβ) deposits in AD are neuritic plaques and cerebral amyloid angiopathy (CAA). Neuritic plaques are extra cellular and consist of a dense central fibrillar Aβ core with inflammatory cells and dystrophic neurites in its periphery. CAA is also extracellular and consists of fibrillar Aβ deposited in the wall of arterioles in both the leptomeninges and penetrating vessels (Johnson et al., 2007). The second major proteinopathy is aggregated tau protein which are intracellular aggregates of hyper phosphorylated tau in the form of neurofibrillary tangles (NFT). NFTs follow a stereotypic topographic progression pattern first appearing in the brainstem and transentorhinal area then progressing to the hippocampus, to paralimbic and adjacent medial-basal temporal cortex, to cortical association areas, and last to primary sensory-motor and visual areas (Braak and Braak, 1991). Another important pathological feature is neurodegeneration which maps onto NFT distribution topographically (not onto Aβ amyloid distribution) and is characterized macroscopically as atrophy and microscopically as loss of neurons and neuronal processes (Braak and Braak, 1994; Terry et al., 1991).
Clinical-autopsy correlation studies demonstrate a much tighter correlation between NFT and cognitive impairment than between amyloid and cognitive impairment (Bennett et al., 2004; Dickson et al., 1995; Gomez-Isla et al., 1997; Ingelsson et al., 2004). The tightest correlation between cognitive impairment and various autopsy features of AD though seems to be with neurodegeneration (Savva et al., 2009), specifically synapse loss (Terry et al., 1991).
Approximately 30% of cognitively normal elderly subjects have sufficient pathology to meet criteria for AD at autopsy (Knopman et al., 2003; Price and Morris, 1999); however, absence of cognitive symptoms is rarely seen in individuals with severe NFT burden.
Heterogeneity of brain pathology in older adults
The accumulation of brain pathologies seems to be a nearly inevitable consequence of aging; very few elderly individuals have no findings at autopsy and the older the individual the more this holds true (Nelson et al., 2011). The common age-associated brain pathologies are AD (plaques and tangles), medial temporal tangles without plaques, ischemic cerebro-vascular disease (which includes not only macroscopic cortical and subcortical infarctions but also microinfarctions, which seem to be particularly important, and ischemic demyelination), hippocampal sclerosis, alpha synuclein deposits (Lewy bodies), TDP43 inclusions, and agyrophyllic grains (Markesbery et al., 2006; Schneider et al., 2009; Sonnen et al., 2011; White, 2009). These same pathologies are also found in subjects who were cognitively normal at the time of death albeit generally at lower overall pathological burdens than in demented subjects. The most common finding at autopsy in elderly persons who meet clinical and pathological criteria for AD dementia is mixed pathology – plaques and tangles plus one or more of the non-AD pathologies above. Throughout this paper therefore AD is discussed from 2 perspectives: early onset which is a reasonable model of “pure AD”; and late onset which usually consists of AD plus one or more co-occurring pathological processes.
AD pathogenesis: early onset vs. late onset AD
Early Onset AD
AD is commonly categorized clinically as either early onset (onset of clinical symptoms before age 65) or late onset. Early onset AD is uncommon, accounting for a few percent of cases at most. A proportion of early onset AD cases occur in individuals with autosomal dominant mutations in one of three genes; the amyloid precursor protein gene on chromosome 21, the presenilin-1 gene on chromosome 14, or the presenilin-2 gene on chromosome 1 (Goate et al., 1991; Levy-Lahad et al., 1995; Sherrington et al., 1995; St George-Hyslop et al., 1987). Along with Down syndrome, all known autosomal dominant mutations leading to AD influence processing of the amyloid precursor protein (APP) in a manner resulting in 1) increased production of Aβ42 or all Aβ species or 2) increased aggregation/decreased clearance of Aβ (Holtzman et al., 2011). These result in increasing the accumulation of oligomeric and fibrillar Aβ (Scheuner et al., 1996).
It is notable that primary tauopathies lead to forms of fronto-temporal lobar degeneration, cortico-basal degeneration and progressive supranuclear palsy, but never to pathological AD (i.e. not to Aβ accumulation). Animal models in which human amyloid precursor protein with or without presenilin mutations have been inserted into the mouse genome recapitulate the Aβ-linked aspects seen in autosomal dominate AD including increased CSF tau (Maia et al., 2013). Transgenic mouse models of Aβ amyloidosis and cellular evidence indicates that Aβ exacerbates tauopathy (Lewis et al., 2001; Oddo et al., 2004) (perhaps by kindling the auto propagation of NFTs (Clavaguera et al., 2009; de Calignon et al., 2012; Frost et al., 2009; Guo and Lee, 2011)), not the reverse (Oddo et al., 2003)}. The available genetic evidence points to Aβ over production of all Aβ species (APP duplication, APP Swedish mutation), relative Aβ42 versus Aβ40 overproduction (N-terminal APP mutations/presenilin mutations), or enhanced aggregation/decreased clearance (APP mutations within the Aβ domain) as causative in autosomal dominate AD (Holtzman et al., 2011). This has led to the amyloid cascade hypothesis which asserts that dysfunction in the Aβ pathway is the initiating event in the disease (Glenner and Wong, 1984; Hardy and Selkoe, 2002). Soluble Aβ rather than insoluble Aβ is felt to initiate the disease cascade (Hardy, 2009). A simple mechanistic diagram of the amyloid cascade hypothesis is shown below:
Over production/aggregation of Aβ42 ➔ tauopathy ➔ neurodegeneration ➔ clinical symptoms
Late Onset AD
Late onset AD accounts for the overwhelming majority of cases. While most autosomal dominant AD is believed to usually be caused by over-production and subsequent aggregation of Aβ42 (a more fibrillogenic form of Aβ) from the beginning of life, late onset AD may most often be a disease of inadequate Aβ clearance again leading to increased aggregation and accumulation (Mawuenyega et al., 2010). While deterministic genetic mutations for sporadic AD have not been found, genetics nonetheless plays a very important role in risk. The ε4 allele of the apolipoprotein E (APOE) gene is the major known genetic risk factor (Roses, 1994). The ε4 allele of APOE increases the risk of developing AD and also lowers the mean age at onset of the disease in a dose-dependent fashion (Corder et al., 1993; Strittmatter et al., 1993). The major mechanism by which APOE ε4 contributes to AD pathogenesis appears to be by modulating the aggregation and clearance of Aβ peptide (Castellano et al., 2011; Kim et al., 2009) leading to increased deposition (Morris et al., 2010; Vemuri et al., 2010) which implicates this pathway in causation of late onset AD. However, APOE is also implicated in many functions other than Aβ trafficking that may contribute to AD pathogenesis, including regulating brain lipid metabolism, neuronal repair, synaptic function, inflammation, and mitochondrial function (Bu, 2009; Mahley and Huang, 2009). The effect of any specific genetic variant other than APOE4 at the population level is minor. Risk variants including those of CLU, CR1, PICALM, CD33, APP, A673T, ABCA7, EPHA1, TREM 2, SORL1, and BIN1 do point to Aβ but also to alternative pathways including immunologic, inflammatory, neurotropic, endocytosis, cholesterol metabolism, ubiquitination, and tau (Bertram et al., 2008; Griciuc et al., 2013; Guerreiro et al., 2013; Harold et al., 2009; Hollingworth et al., 2011; Jonsson et al., 2013; Lambert et al., 2009; Naj et al., 2011; Seshadri et al., 2010). Thus from a genetics standpoint sporadic AD is complex.
Late onset AD is also more complex than early onset from a pathological perspective. As noted above late onset AD typically occurs along with other age related pathologies. In fact the contribution of pathologies such as hippocampal sclerosis and cerebrovascular disease to dementia seems to increase relative to AD pathology with advancing age beyond 85 years (Nelson et al., 2011). A further complicating factor is the fact that subcortical and medial temporal tauopathy exists at autopsy very commonly by middle age in individuals who have no plaques (Braak and Braak, 1997; Braak and Del Tredici, 2011; Haroutunian et al., 1999; Price and Morris, 1999). For example in autopsies numbering in the thousands, Braak and Braak (Braak and Braak, 1997) find that by the late 70s nearly all individuals (97%) have some tauopathy, while only 17% have Aβ amyloid deposits. The fact that brainstem and medial temporal lobe tau commonly precedes amyloid plaques has led some to suggest that a different pathogenic model should exist for late onset vs. early onset AD.
Role of Aβ in pathogenesis of AD
The initiating event in the molecular cascade that eventually leads to clinical and pathological AD has been controversial for decades. The amyloid cascade hypothesis (Glenner and Wong, 1984; Hardy and Selkoe, 2002) assumes serial causal events initiated by abnormal Aβ production/aggregation. As discussed above, this seems to be a reasonable pathogenic model of early onset AD. An alternative position is that tau hyper-phosphorylation and Aβ elevation are independently arising pathophysiological processes that interact with pathogenic synergy (Duyckaerts, 2011; Duyckaerts et al., 1997; Mesulam, 1999; Small and Duff, 2008) which is particularly germane to late onset AD (Chetelat, 2013; Desikan et al., 2011; Jack et al., 2013a; Knopman et al., 2013a, b). A sequence of pathological events proposed by Price and Morris (Price and Morris, 1999) for late onset AD seems to best explain the fact that while small amounts of medial temporal tauopathy often precede amyloid plaque formation, Aβ seems to drive the progression of the disease. Price and Morris (Price and Morris, 1999) propose that tauopathy develops first, but is confined to subcortical and medial temporal limbic areas, is clinically benign and best thought of as a feature of typical aging. Neocortical Aβ deposits develop later, independently from medial temporal tauopathy. By unknown mechanism(s) distant Aβ aggregation transforms the medial temporal tauopathy leading to spread from medial temporal limbic areas to widespread extension throughout the neocortex. The proposed relationship between cognition and amyloid in late onset AD is not predominantly due to direct neurotoxicity of aggregated Aβ, but rather to its role in aiding the propagation of an antecedent tau related neurodegeneration throughout a topographically characteristic susceptible network (Raj et al., 2012; Seeley et al., 2009). It is tau related neurodegeneration that is ultimately responsible for clinical symptoms (a position reinforced by the poor association between the topographic distribution of amyloid and clinical phenotype in atypical forms of AD (Rabinovici et al., 2008; Wolk et al., 2012)). This view does not conflict with the amyloid cascade hypothesis in that it acknowledges the central role of Aβ in disease pathogenesis. As outlined in a recent review (Jack et al., 2013a) and in the section on AD pathogenesis, while tauopathy and Aβothapy may arise independently, evidence suggests that Aβ drives tau pathology not the reverse. This view also does not imply that therapeutic interventions targeting Aβ in late onset disease are misguided given the essential role Aβ plays in pathogenesis in late onset (as well as early onset) AD.
AD biomarkers
A biomarker is a physiological, biochemical, or anatomic parameter that can be objectively measured as an indicator of normal biologic processes, pathological processes, or responses to a therapeutic intervention. At present five AD biomarkers are well enough established to be used in clinical trials and in modern diagnostic criteria (Albert et al., 2011; Dubois et al., 2010; Jack et al., 2011a; McKhann et al., 2011; Sperling et al., 2011a). Two of these are cerebrospinal fluid (CSF) proteins and three are brain imaging measures. These five biomarkers fall into two major mechanistic categories: First are measures of Aβ deposition: These are CSF Aβ42 which decreases with increasing amyloid plaque load (Bouwman et al., 2009; Fagan et al., 2007; Mattsson et al., 2009; Shaw et al., 2009; Visser et al., 2009), and positron emission tomography (PET) amyloid imaging (Drzezga, 2010; Klunk et al., 2004; Nordberg et al., 2013; Rodrigue et al., 2012; Rowe et al., 2010; Villemagne et al., 2011) (Fig 1a). These two biomarkers of amyloid are highly correlated when measured in the same individuals (Fagan et al., 2006; Jagust et al., 2009; Tolboom et al., 2009; Weigand et al., 2011). The validity of these biomarkers of amyloid plaque deposition has been established by autopsy correlation studies (Clark et al., 2011; Fleisher et al., 2011; Ikonomovic et al., 2008; Sojkova et al., 2011; Strozyk et al., 2003; Tapiola et al., 2009).
Figure 1. Voxel based comparisons of amyloid PET (Fig 1a), FDG PET (Fig 1b) and structural MRI (Fig 1c) illustrate differences between subjects with Alzheimer's disease (n=50) and cognitively normal elderly (n=50).

AD and cognitively normal elderly subjects were from the Mayo Clinic and were matched on age and gender. Fig 1a, amyloid PET maps (thresholded at FWE, P < 0.001 without partial volume correction) illustrate greater retention of Pittsburgh Compound B (PIB) in AD vs cognitively normal elderly in most brain areas; primary sensory motor, visual and the medial temporal lobe are spared. Fig 1b, FDG PET maps (thresholded at FWE, P < 0.001 without partial volume correction) illustrate decreased FDG uptake in the basal temporal, lateral temporal – parietal, lateral pre-frontal, and posterior cingulate-precuneus in AD compared to cognitively normal elderly. This spatial pattern constitutes an “AD-signature” in FDG PET. Fig 1c, structural MRI maps (thresholded at FWE, P < 0.05) illustrate grey matter loss in the medial, basal and lateral temporal, lateral parietal, occipital, insula and precuneus in AD compared to cognitively normal elderly. This spatial pattern constitutes an “AD-signature” in structural MRI. All voxel wise comparisons generated with SPM5. 3D displays by Brain Net Viewer (http://www.nitrc.org/projects/bnv/). The color bar scale indicates the t-test differences between the groups.
The second major AD biomarker category is neurodegeneration, defined as progressive loss of neurons and their processes with a corresponding progressive impairment in neuronal function. The 3 major AD neurodegnerative biomarkers are increased levels of CSF total (t-tau) and phosphorylated (p-tau) tau (Fagan et al., 2009; Mattsson et al., 2009; Shaw et al., 2009; Visser et al., 2009), atrophy on structural MRI (Desikan et al., 2009; Dickerson and Wolk, 2012; Hua et al., 2008; Morra et al., 2008; Morra et al., 2009; Vemuri et al., 2009), and hypo metabolism on FDG PET (Jagust et al., 2010). The validity of these biomarkers of neurodegeneration due to AD is supported by autopsy correlation studies. A caveat is that direct tissue-to-image correlation can be established between tissue stains and in vivo or ex vivo imaging findings, whereas this correlation is more indirect for CSF analytes. Elevations in t-tau (Tapiola et al., 2009) and p-tau (Buerger et al., 2006; Tapiola et al., 2009) correlate with neurofibrillary tangle (NFT) burden at autopsy. Atrophy on MRI correlates with neuron loss (Bobinski et al., 2000; Zarow et al., 2005), Braak NFT stage (Jack et al., 2002; Vemuri et al., 2008; Whitwell et al., 2008), and tau immunostaining burden (Whitwell et al., 2008), and does not correlate well with Aβ load measured by immunohistochemistry (Josephs et al., 2008). Thus MRI is a measure of tau associated neurodegeneration. Antemortem FDG hypometabolism is also correlated to NFT burden and not to plaque burden at autopsy (DeCarli et al., 1992).
An important distinction between biomarkers of Aβ deposition and biomarkers of neurodegeneration concerns their specificity for AD pathophysiology. CSF Aβ42 and amyloid PET are specific for fibrillar Aβ amyloid deposition (in the extracellular space or in vessel walls). In contrast, neurodegenerative biomarkers are not always specific for neurodegeneration due to AD (Jack et al., 2002). In subjects with AD, both FDG PET and MRI follow a modalityspecific topology that is characteristic of AD (Figs 1b and c). An FDG PET or MRI study is transformed into a quantitative biomarker by extracting and summing values from AD-signature regions (as illustrated in Figs 1b and c) using an anatomic atlas that is spatially registered to the subject's imaging study (Senjem et al., 2005). However, atrophy and FDG hypo metabolism caused by non-AD etiologies can overlap spatially with these AD-like topographic patterns. Non-AD etiologies associated with atrophy, FDG hypo metabolism or both that commonly occur in elderly people include cerebro vascular disease, Lewy body disease, hippocampal sclerosis, non-AD degenerative disorders such as Lewy body disease, primary tauopathies, lobar degeneration, Creutzfelt Jakob disease, and perhaps TDP43. A feature of most automated image analysis algorithms that contribute to the non-specificity of MRI and FDG is that the algorithms typically interrogate only a modality-specific AD-like topographic pattern and do not examine areas outside of this predefined topology. Elevation of CSF tau (particularly ptau) seems to be more specific for AD – i.e. it is not elevated in primary tauopathies – however total tau is elevated in Creutzfelt Jakob disease, head trauma and acute stroke. Thus the interpretation of these neurodegenerative biomarkers in empiric studies of elderly subjects is confounded by overlap with co-occurring pathologies that ideally should be distinguished from AD. The importance of this will become evident when discussing modeling of biomarker evolution.
Other potential biomarkers of AD exist at various stages of evaluation. These include other CSF analytes such as VILIP-1 (Tarawneh et al., 2011) diffusion MRI, perfusion MRI, functional MRI (both resting state or task free, and task activation MRI), and tau PET ligands. These are either very new or have not yet demonstrated the test re-test precision or the diagnostic efficacy to be considered a major biomarker. This may change with further development and testing.
Temporal evolution of AD biomarkers
Models of the temporal evolution of AD biomarkers can take the form of plots of biomarker severity (degree of abnormality) vs. time (Fig 2). For imaging biomarkers (Fig 1), movement up the y axis in Fig 2 corresponds to both topographic progression as well as increasing severity in brain areas that are already involved. This type of plot illustrates two key types of relationships in a single display. First is the shape of each biomarker curve as a function of time (i.e. functional form). Biomarkers could increase linearly with time, exponentially, sigmoidally, etc (Fjell et al., 2013b). Different shapes carry different implications concerning the biological mechanisms driving the observed changes in a biomarker with time. The second key relationship concerns the temporal ordering of biomarkers with respect to each other and to the evolution of clinical symptoms. Modeling biomarker trajectories as curves that are time-shifted relative to one another reflects the notion that the different underlying pathophysiologic processes do not evolve simultaneously at identical rates but rather in a temporally ordered manner. A model of biomarker evolution with time depends on both the biology underlying biomarker changes and the sensitivity and specificity of the biomarkers to the biological signals that each responds to. We emphasize that biology (the order or functional form of pathophysiological events) and technology (ability to detect events) are inseparable aspects of the application of biomarkers for human use. For example, it is entirely possible that a highly sensitive biomarker of a later occurring pathological process may become abnormal before a relatively insensitive biomarker of an earlier pathological process.
Figure 2. Temporal evolution of biomarkers.
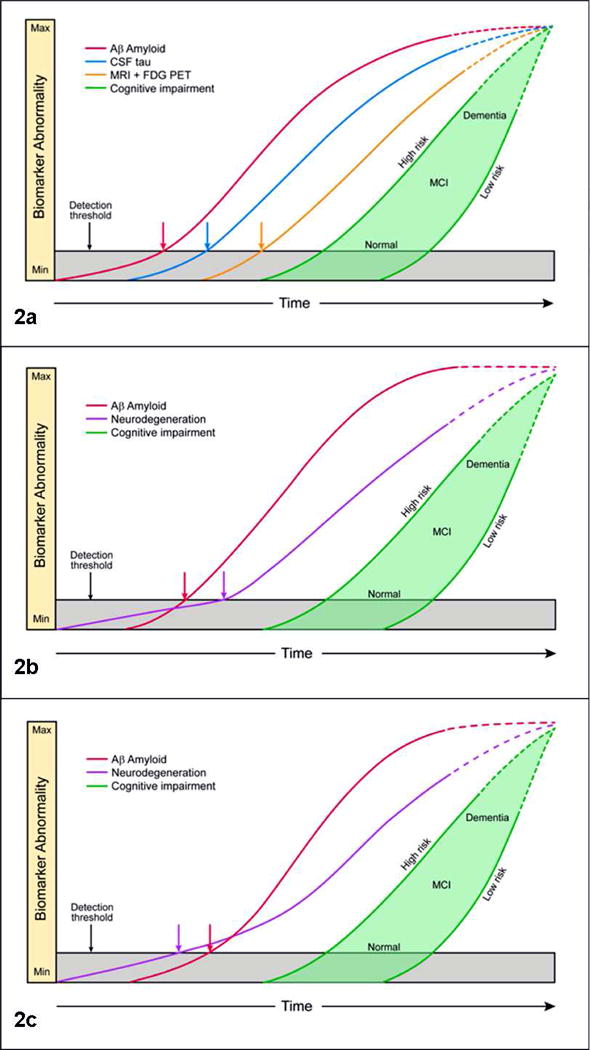
Fig 2a, biomarker model of pure AD. The horizontal axis is time and the vertical axis severity of biomarker abnormality from completely normal (min) to abnormal (max). The threshold for biomarker detection of pathophysiology is denoted by a horizontal line. The grey area denotes the zone in which abnormal pathophysiology lies below the biomarker detection threshold. Amyloid biomarkers become abnomral first, followed by CSF tau, followed by FDH PET and MRI. Cognitive impairment (green filled area) is the last event in the progression of the disease. A range of cognitive responses are possible that depend on the individual's risk profile. The cognitive response curve is shifted to the left for those with low cognitive reserve and to the right for those with high cognitive reserve. At a given point on the disease time line, a person with high cognitive reserve can be cognitively normal while a low cognitive response person with the same biomarker profile can be impaired. All biomarker curves (as well as cognitive impairment) are configured as sigmoids, but the curves have a progressively steeper slope in the right-hand tail for later-changing biomarkers. The right hand tails of the curves are dashed indicating that biomarker trajectories in end stage dementia are unknown at this time. MCI = mild cognitive impairment. Fig 2b, amyloid-first biomarker model of late onset AD where comorbid pathologies are likely. Figure labels are as in Fig 2a, except here the CSF tau, structural MRI and FDG PET are grouped under the generic neurodegenerative label. This reflects the fact that neurodegenerative biomarkers can be non-specific and that the proportional contribution of various possible etiologies to neurodegeneration cannot be known in vivo. We assume that in elderly individuals tauopathy and in many individuals non-AD neurodegenerative pathologies arise first but lie beneath the detection threshold of biomarkers of neurodegeneration. Aβopathy arises later independently and is the first AD biomarker to become positive. The neurodegeneration curve has a shallow slope initially, but the slope steepens after the onset of amyloidosis. Fig 2c, neurodegeneration-first biomarker model of late onset AD where comorbid pathologies are likely. Figure labels are as in Fig 2a, except here one or more neurodegenerative biomarkers become abnormal first reflecting the onset of tauopathy and in some individuals other neurodegenerative pathologies prior to amyloid. Observing the onset of an abnormal neurodegenerative biomarker prior to an abnormal amyloid biomarker does not alter the role of amyloid as an inducer of neurodegeneration. This is indicated by showing the neurodegeneration curve with a shallow slope initially which steepens after the onset of amyloidosis.
Initial models of the temporal evolution of AD biomarkers were based on observations in elderly subjects spanning the cognitive continuum (Jack et al., 2010a; Jack et al., 2008; Jack et al., 2009; Mormino et al., 2009; Perrin et al., 2009) and were supported by earlier autopsy data (Ingelsson et al., 2004). These initial models proposed that amyloid biomarkers become abnormal first, then biomarkers of tau related neurodegeneration, followed last by overt clinical symptoms. One model took the additional step of proposing not only an ordering scheme but also that the general shape or functional form for biomarker evolution was sigmoidal with time (Jack et al., 2010a). Following publication of these initial biomarker models subsequent empiric studies in both elderly individuals (Buchhave et al., 2012; Forster et al., 2012; Jack et al., 2011b; Jack et al., 2010b; Landau et al., 2012; Lo et al., 2011; Villemagne et al., 2013; Villemagne et al., 2011) and in carriers of autosomal dominant mutations (Bateman et al., 2012; Reiman et al., 2012) found that ordering of biomarkers was generally consistent with that proposed in the models. In addition, empirical data has confirmed that at least one class of biomarkers – amyloid – does follow a sigmoidal shape with time (Caroli and Frisoni, 2010; Fleisher et al., 2012; Jack et al., 2012b; Jack et al., 2013b; Villain et al., 2012; Villemagne et al., 2013). Given that late stage AD subjects have not been thoroughly studied with biomarkers to date, it is not surprising that convincing evidence for plateauing (i.e. sigmoidal shape) has only been found for the earliest changing class of AD biomarkers (amyloid).
The discussion in the previous sections though suggests that different models might exist for early onset vs. late onset AD primarily because cognitive and neurodegenerative biomarker abnormalities due to non –AD pathologies and to aging all may precede or co-occur with AD pathology in the elderly. We propose that 3 different sets of biomarker models should be considered, each of which culminates in the common clinical phenotype of AD dementia.
Pure AD biomarker model – amyloid first
The concept of pathologically pure AD (i.e. in the absence of non-AD age related pathologies) seems appropriate with onset early in life when non-AD age related pathologies are not highly prevalent. This is usually the case in autosomal dominant AD and many APOE4 homozygotes. A model of the temporal ordering of the 5 major AD biomarkers for early onset AD is outlined below and is illustrated in Fig 2a. The temporal ordering scheme in this biomarker model of “pure” AD matches the sequence of molecular events proposed in the amyloid cascade hypothesis (Glenner and Wong, 1984; Hardy and Selkoe, 2002).
Aβ 42 over production and aggregation/decreased clearance leads to amyloid plaque formation. CSF Aβ 42 and amyloid PET are the first biomarkers to become abnormal.
Aβopathy induces a medial temporal tauopathy and promotes its spread into the neocortex. CSF tau then becomes abnormal. In some cases, Aβopathy also induces synuclein aggregation as well.
Tauopathy leads to detectable abnormalities in imaging biomarkers of neurodegeneration -structural MRI and FDG PET.
Clinical symptoms follow the MRI and FDG PET abnormalities (Reed et al., 2010; Vemuri et al., 2011).
In the early to mid-clinically symptomatic phases of the disease amyloid biomarkers approach a plateau while MRI, FDG PET and tau continue to increase.
In the end stage of the disease, we hypothesize that all biomarkers approach a plateau, however this is conjecture. Patients can live for many years after they are no longer able to participate in research studies. In this end stage, patients may survive for prolonged periods unresponsive and bed ridden with little external evidence of further disease progression. Systematic studies of biomarker progression in this stage of the disease have not been done and are difficult to perform. Hence the right hand portions of all biomarker curves in Figure 2 are drawn as dotted lines, indicating lack of knowledge about the terminal phase of the disease.
Late onset AD biomarker models
Modeling late onset AD differs from early onset AD in several important ways. First, AD pathophysiology commonly co-exists with age related non-AD pathologies discussed above (Schneider et al., 2007; Schneider et al., 2009). Second neurodegenerative changes may occur as a function of aging in the absence of any specific disease process, akin to well documented agerelated declines in other organ systems like muscle mass or renal perfusion (Fjell et al., 2013a; Jagust, 2013). Brain systems particularly vulnerable to aging seem to be those sub serving attention/executive function and declarative memory (Jagust, 2013). A candidate substrate for age related declines in in cognitive function, brain volumes and FDG PET uptake in the absence of specific pathologies is synapse loss (Jagust, 2013; Yeoman et al., 2012). Thus unlike early onset AD, late onset AD commonly exists on a background of neurodegenerative changes due to aging, non-AD pathologies, or both (Jack et al., 2010b). Third, a phenomenon that has been recognized by a number of investigators is that biomarkers of neurodegeneration (especially structural MRI and FDG PET) are not specific for AD and may show AD-like patterns of abnormality due to non-AD conditions (Dickerson and Wolk, 2012; Fjell et al., 2013a; Jack et al., 2002; Jack et al., 2013a; Jack et al., 2010a; Jack et al., 2012a; Jack et al., 2010b; Jagust, 2013; Raz et al., 2007; Wirth et al., 2013a; Wirth et al., 2013b). A corollary to this is that validated biomarkers that are specific for non-AD pathologies are lacking. Therefore in any given person, we cannot know the precise pathological etiologies underlying an abnormal structural MRI or FDG PET scan nor the proportional contribution of various possible etiologies to neurodegeneration (Jack et al., 2010b). Realistic interpretation of neurodegenerative biomarkers in the elderly should acknowledge that many possible etiologies and proportional mixes of etiologies may exist for an abnormal neurodegenerative biomarker finding. The reason modeling of AD biomarkers seems to work empirically in the elderly is because AD is the dominant pathology leading to dementia.
While considerable evidence, described earlier under “temporal evolution of AD biomarkers”, supports an amyloid-first biomarker model of late onset AD, evidence has also appeared that supports the idea that cognitive decline and neurodegenerative biomarker abnormalities might precede abnormal amyloid biomarkers in some elderly individuals who later develop AD. This evidence falls into 3 categories. First, autopsy studies indicate that by late 50s 2/3 of the population and by the late 70s essentially everyone has some degree of medial temporal tauopathy very often without amyloid plaques (Braak and Braak, 1997). Therefore tauopathy ought to precede amyloid in many individuals who eventually develop late onset AD (Braak and Braak, 1997; Haroutunian et al., 1999; Price and Morris, 1999). While we currently lack in vivo data on the topographic staging of tau akin to Braak staging (Braak and Braak, 1997), this will likely be remedied in the future by new PET tau ligands (Chien et al., 2013), (Fodero-Tavoletti et al., 2011; Maruyama et al., 2013). Second, FDG PET and functional MRI studies have identified AD-like imaging abnormalities in cognitively normal APOE4 carriers (compared to non-carriers) who were either too young to be amyloid positive (Filippini et al., 2009; Reiman et al., 2004) or who were documented to be amyloid negative (Jagust and Landau, 2012; Sheline et al., 2010). These APOE4 carriers are much more likely to develop AD in the future than non-carriers. While these studies are suggestive they do not prove that neurodegenerative abnormalities can precede amyloidosis in individuals who do develop incident pre-clinical AD. That requires longitudinal documentation of incident amyloid positivity. To that end, a longitudinal study has recently proved proof that MRI and FDG PET can be abnormal prior to documented indicant amyloid positivity in the elderly (Jack et al., 2013c). This study (Jack et al., 2013c) has demonstrated empirically that both “amyloid-first” and “neurodegeneration-first” biomarker profile pathways to pre-clinical AD exist. We therefore describe both amyloid first and neurodegeneration first models of late onset AD below.
The sequence of molecular and biomarker events in our proposed amyloid-first model of late onset AD is illustrated in Fig 2b and outlined below.
Neurodegeneration due to tauopathy, non-AD pathologies, aging or a combination first appears in the brainstem and medial temporal lobe but lies beneath the detection threshold of biomarkers of neurodegeneration.
Aβ deposition arises later and progresses independently in neocortical association areas. Aβ biomarkers (CSF Aβ42 and amyloid PET) then become positive.
Aβopathy transforms slowly progressing medial temporal tauopathy into an aggressive process and induces its spread into the neocortex (Clavaguera et al., 2009; de Calignon et al., 2012; Frost et al., 2009; Guo and Lee, 2011; Price and Morris, 1999). One or more neurodegenerative biomarkers then become abnormal. Because in any older subject we cannot know if AD pathology exits in isolation or is accompanied by co-existing neurodegenerative processes, we group all neurodegeneration biomarkers together under a single generic neurodegeneration heading in this model without attempting to distinguish tau from non-tau related etiologies (which is different from the model of early onset AD where CSF tau and MRI/PET are treated distinctly because non-AD pathology is unlikely to be present). The accelerating effect of amyloid on neurodegeneration is illustrated in the neurodegeneration curve which has a shallow slope initially, but the slope steepens after the onset of amyloidosis.
Clinical symptoms follow the onset of abnormalities in neurodegeneration biomarkers. Subjects who are at high risk of cognitive impairment from AD are shown in Figure 2 with a cognitive response curve that is shifted to the left in time (Jack et al., 2013a; Jack et al., 2010a). Such high-risk subjects may harbor more significant co-morbid pathologies, more genetic risk alleles, or have low cognitive reserve (Reed et al., 2010; Rentz et al., 2010; Roe et al., 2008; Vemuri et al., 2011). In contrast, low-risk subjects with fewer co-morbid brain pathologies, a protective genetic profile and high cognitive reserve can co-exist with substantial AD pathophysiology and still maintain normal cognitive function. Thus cognitive response in Figure 2 is illustrated as a zone with low and high risk borders (Jack et al., 2013a). The variance in cognitive response (i.e. the range on the y axis projection) decreases as end stage dementia is approached which reflects the fact that there is relatively little variation in the end stage clinical phenotype.
The sequence of molecular and biomarker events in our proposed neurodegeneration-first model of late onset AD is illustrated in Fig 2c and outlined below.
Age related neurodegeneration due to medial temporal tauopathy, non-AD pathologies, aging or a mixture occur first. CSF tau, MRI, FDG PET or any combination of these become abnormal and are denoted as a single generic neurodegeneration biomarker curve.
Aβ deposition arises independently in neocortical association areas. CSF Aβ42 and amyloid PET become abnormal.
Aβopathy transforms tau related neurodegeneration into an aggressive process and induces its spread throughout the neocortex (Clavaguera et al., 2009; de Calignon et al., 2012; Frost et al., 2009; Guo and Lee, 2011; Price and Morris, 1999). As outlined in the section on the role of Aβ in pathogenesis of AD, evidence that neurodegenerative imaging measures can become abnormal before amyloid biomarkers does not negate the central role of Aβ in late onset AD where Aβ is envisioned as an accelerator of tau related neurodegeneration (rather than an initiator of tau related neurodegeneration as in early onset AD).
Neurodegenerative biomarker abnormalities then accelerate.
Clinical symptoms follow the progression of neurodegenerative biomarker abnormalities.
Conclusions
AD is a slowly evolving disorder in which pathophysiological abnormalities precede overt clinical symptoms by many years to decades. The only in vivo window into the disease in its long pre-clinical phase is biomarkers which, unlike autopsy, can be sampled repeatedly in individual subjects over time. A thorough understanding of the disease hinges on developing accurate, comprehensive models of AD biomarker evolution. Designing interventional strategies that target the right molecular pathways at an appropriate stage in the disease depends on accurate models of AD biomarker evolution (Fox et al., 2005; Frisoni and Delacourte, 2009; Rinne et al., 2010; Sperling et al., 2011b). This will lead to interventions that may be complex but are correctly tailored to the individual patient – i.e. individualized medicine. Finally, evolving diagnostic criteria (Albert et al., 2011; Dubois et al., 2010; Jack et al., 2012a; McKhann et al., 2011; Sperling et al., 2011a) must incorporate AD biomarkers, but cannot proceed effectively without accurate models of those biomarkers.
In Brief Paragraph.
We intend to familiarize readers with the major biomarkers of Alzheimer's disease and relate them to specific pathophysiological processes. We also outline a framework for temporal evolution of AD biomarkers in the context of both early and late onset disease.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Clifford R Jack, Jr, Email: jack.clifford@mayo.edu.
David M Holtzman, Email: holtzman@neuro.wustl.edu.
References
- Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, Snyder PJ, Phelps CH. The diagnosis of mild cognitive impairment due to Alzheimer's disease: Recommendations from the National Institute on Aging and Alzheimer's Association Workgroup. Alzheimers Dement. 2011;7:270–279. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer's Disease. The New England journal of medicine. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE. Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol. 2004;61:378–384. doi: 10.1001/archneur.61.3.378. [DOI] [PubMed] [Google Scholar]
- Bertram L, Lange C, Mullin K, Parkinson M, Hsiao M, Hogan MF, Schjeide BM, Hooli B, Divito J, Ionita I, Jiang H, Laird N, Moscarillo T, Ohlsen KL, Elliott K, Wang X, Hu-Lince D, Ryder M, Murphy A, Wagner SL, Blacker D, Becker KD, Tanzi RE. Genome-wide association analysis reveals putative Alzheimer's disease susceptibility loci in addition to APOE. American journal of human genetics. 2008;83:623–632. doi: 10.1016/j.ajhg.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobinski M, de Leon MJ, Wegiel J, Desanti S, Convit A, Saint Louis LA, Rusinek H, Wisniewski HM. The histological validation of post mortem magnetic resonance imaging-determined hippocampal volume in Alzheimer's disease. Neuroscience. 2000;95:721–725. doi: 10.1016/s0306-4522(99)00476-5. [DOI] [PubMed] [Google Scholar]
- Bouwman FH, Schoonenboom NS, Verwey NA, van Elk EJ, Kok A, Blankenstein MA, Scheltens P, van der Flier WM. CSF biomarker levels in early and late onset Alzheimer's disease. Neurobiol Aging. 2009;30:1895–1901. doi: 10.1016/j.neurobiolaging.2008.02.007. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Morphological criteria for the recognition of Alzheimer's disease and the distribution pattern of cortical changes related to this disorder. Neurobiol Aging. 1994;15:355–356. doi: 10.1016/0197-4580(94)90032-9. discussion 379-380. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18:351–357. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K. The pathological process underlying Alzheimer's disease in individuals under thirty. Acta Neuropathol. 2011;121:171–181. doi: 10.1007/s00401-010-0789-4. [DOI] [PubMed] [Google Scholar]
- Bu G. Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10:333–344. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchhave P, Minthon L, Zetterberg H, Wallin AK, Blennow K, Hansson O. Cerebrospinal Fluid Levels of beta-Amyloid 1-42, but Not of Tau, Are Fully Changed Already 5 to 10 Years Before the Onset of Alzheimer Dementia. Arch Gen Psychiatry. 2012;69:98–106. doi: 10.1001/archgenpsychiatry.2011.155. [DOI] [PubMed] [Google Scholar]
- Buerger K, Ewers M, Pirttila T, Zinkowski R, Alafuzoff I, Teipel SJ, DeBernardis J, Kerkman D, McCulloch C, Soininen H, Hampel H. CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer's disease. Brain. 2006;129:3035–3041. doi: 10.1093/brain/awl269. [DOI] [PubMed] [Google Scholar]
- Caroli A, Frisoni GB. The dynamics of Alzheimer's disease biomarkers in the Alzheimer's Disease Neuroimaging Initiative cohort. Neurobiol Aging. 2010;31:1263–1274. doi: 10.1016/j.neurobiolaging.2010.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ, Holtzman DM. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med. 2011;3:89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chetelat G. Alzheimer disease: Abeta-independent processes-rethinking preclinical AD. Nature reviews. Neurology. 2013;9:123–124. doi: 10.1038/nrneurol.2013.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien DT, Bahri S, Szardenings AK, Walsh JC, Mu F, Su MY, Shankle WR, Elizarov A, Kolb HC. Early Clinical PET Imaging Results with the Novel PHF-Tau Radioligand [F-18]-T807. Journal of Alzheimer's disease : JAD. 2013;34:457–468. doi: 10.3233/JAD-122059. [DOI] [PubMed] [Google Scholar]
- Clark CM, Schneider JA, Bedell BJ, Beach TG, Bilker WB, Mintun MA, Pontecorvo MJ, Hefti F, Carpenter AP, Flitter ML, Krautkramer MJ, Kung HF, Coleman RE, Doraiswamy PM, Fleisher AS, Sabbagh MN, Sadowsky CH, Reiman PEM, Zehntner SP, Skovronsky DM. Use of Florbetapir-PET for Imaging BAmyloid Pathology. JAMA: The Journal of the American Medical Association. 2011;305:275–283. doi: 10.1001/jama.2010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, Jucker M, Goedert M, Tolnay M. Transmission and spreading of tauopathy in transgenic mouse brain. Nature cell biology. 2009;11:909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- de Calignon A, Polydoro M, Suarez-Calvet M, William C, Adamowicz DH, Kopeikina KJ, Pitstick R, Sahara N, Ashe KH, Carlson GA, Spires-Jones TL, Hyman BT. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. 2012;73:685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCarli CS, Atack JR, Ball MJ, Kaye JA, Grady CL, Fewster P, Schapiro MB, Rapoport SI. Postmortem regional neurofibrillary tangle densities, but not senile plaque densities, are related to regional cerebral metabolic rates for glucose during life in Alzheimer's disease. Neurodegeneration. 1992;1:113–121. [Google Scholar]
- Desikan RS, Cabral HJ, Hess CP, Dillon WP, Glastonbury CM, Weiner MW, Schmansky NJ, Greve DN, Salat DH, Buckner RL, Fischl B. Automated MRI measures identify individuals with mild cognitive impairment and Alzheimer's disease. Brain. 2009;132:2048–2057. doi: 10.1093/brain/awp123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desikan RS, McEvoy LK, Thompson WK, Holland D, Roddey JC, Blennow K, Aisen PS, Brewer JB, Hyman BT, Dale AM. Amyloid-beta associated volume loss occurs only in the presence of phospho-tau. Ann Neurol. 2011;70:657–661. doi: 10.1002/ana.22509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson BC, Wolk DA. MRI cortical thickness biomarker predicts AD-like CSF and cognitive decline in normal adults. Neurology. 2012;78:84–90. doi: 10.1212/WNL.0b013e31823efc6c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, Crystal HA, Bevona C, Honer W, Vincent I, Davies P. Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol Aging. 1995;16:285–298. doi: 10.1016/0197-4580(95)00013-5. discussion 298-304. [DOI] [PubMed] [Google Scholar]
- Drzezga A. Amyloid-plaque imaging in early and differential diagnosis of dementia. Ann Nucl Med. 2010;24:55–66. doi: 10.1007/s12149-009-0330-9. [DOI] [PubMed] [Google Scholar]
- Dubois B, Feldman HH, Jacova C, Cummings JL, Dekosky ST, Barberger-Gateau P, Delacourte A, Frisoni G, Fox NC, Galasko D, Gauthier S, Hampel H, Jicha GA, Meguro K, O'Brien J, Pasquier F, Robert P, Rossor M, Salloway S, Sarazin M, de Souza LC, Stern Y, Visser PJ, Scheltens P. Revising the definition of Alzheimer's disease: a new lexicon. Lancet Neurol. 2010;9:1118–1127. doi: 10.1016/S1474-4422(10)70223-4. [DOI] [PubMed] [Google Scholar]
- Duyckaerts C. Tau pathology in children and young adults: can you still be unconditionally baptist? Acta Neuropathol. 2011;121:145–147. doi: 10.1007/s00401-010-0794-7. [DOI] [PubMed] [Google Scholar]
- Duyckaerts C, Uchihara T, Seilhean D, He Y, Hauw JJ. Dissociation of Alzheimer type pathology in a disconnected piece of cortex. Acta Neuropathol. 1997;93:501–507. doi: 10.1007/s004010050645. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Head D, Shah AR, Marcus D, Mintun M, Morris JC, Holtzman DM. Decreased cerebrospinal fluid Abeta(42) correlates with brain atrophy in cognitively normal elderly. Ann Neurol. 2009;65:176–183. doi: 10.1002/ana.21559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA, DeKosky ST, Morris JC, Holtzman DM. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006;59:512–519. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 2007;64:343–349. doi: 10.1001/archneur.64.3.noc60123. [DOI] [PubMed] [Google Scholar]
- Filippini N, MacIntosh BJ, Hough MG, Goodwin GM, Frisoni GB, Smith SM, Matthews PM, Beckmann CF, Mackay CE. Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc Natl Acad Sci U S A. 2009;106:7209–7214. doi: 10.1073/pnas.0811879106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, McEvoy L, Holland D, Dale AM, Walhovd KB. Brain changes in older adults at very low risk for Alzheimer's disease. J Neurosci. 2013a;33:8237–8242. doi: 10.1523/JNEUROSCI.5506-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Westlye LT, Grydeland H, Amlien I, Espeseth T, Reinvang I, Raz N, Holland D, Dale AM, Walhovd KB. Critical ages in the life course of the adult brain: nonlinear subcortical aging. Neurobiol Aging. 2013b;34:2239–2247. doi: 10.1016/j.neurobiolaging.2013.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleisher AS, Chen K, Liu X, Roontiva A, Thiyyagura P, Ayutyanont N, Joshi AD, Clark CM, Mintun MA, Pontecorvo MJ, Doraiswamy PM, Johnson KA, Skovronsky DM, Reiman EM. Using positron emission tomography and florbetapir F18 to image cortical amyloid in patients with mild cognitive impairment or dementia due to Alzheimer disease. Arch Neurol. 2011;68:1404–1411. doi: 10.1001/archneurol.2011.150. [DOI] [PubMed] [Google Scholar]
- Fleisher AS, Chen K, Quiroz YT, Jakimovich LJ, Gomez MG, Langois CM, Langbaum JB, Ayutyanont N, Roontiva A, Thiyyagura P, Lee W, Mo H, Lopez L, Moreno S, Acosta-Baena N, Giraldo M, Garcia G, Reiman RA, Huentelman MJ, Kosik KS, Tariot PN, Lopera F, Reiman EM. Florbetapir PET analysis of amyloid-beta deposition in the presenilin 1 E280A autosomal dominant Alzheimer's disease kindred: a cross-sectional study. Lancet neurology. 2012;11:1057–1065. doi: 10.1016/S1474-4422(12)70227-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodero-Tavoletti MT, Okamura N, Furumoto S, Mulligan RS, Connor AR, McLean CA, Cao D, Rigopoulos A, Cartwright GA, O'Keefe G, Gong S, Adlard PA, Barnham KJ, Rowe CC, Masters CL, Kudo Y, Cappai R, Yanai K, Villemagne VL. 18F-THK523: a novel in vivo tau imaging ligand for Alzheimer's disease. Brain. 2011;134:1089–1100. doi: 10.1093/brain/awr038. [DOI] [PubMed] [Google Scholar]
- Forster S, Grimmer T, Miederer I, Henriksen G, Yousefi BH, Graner P, Wester HJ, Forstl H, Kurz A, Dickerson BC, Bartenstein P, Drzezga A. Regional expansion of hypometabolism in Alzheimer's disease follows amyloid deposition with temporal delay. Biological psychiatry. 2012;71:792–797. doi: 10.1016/j.biopsych.2011.04.023. [DOI] [PubMed] [Google Scholar]
- Fox NC, Black RS, Gilman S, Rossor MN, Griffith SG, Jenkins L, Koller M. Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology. 2005;64:1563–1572. doi: 10.1212/01.WNL.0000159743.08996.99. [DOI] [PubMed] [Google Scholar]
- Frisoni GB, Delacourte A. Neuroimaging outcomes in clinical trials in Alzheimer's disease. J Nutr Health Aging. 2009;13:209–212. doi: 10.1007/s12603-009-0060-7. [DOI] [PubMed] [Google Scholar]
- Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. The Journal of biological chemistry. 2009;284:12845–12852. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenner GG, Wong CW. Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122:1131–1135. doi: 10.1016/0006-291x(84)91209-9. [DOI] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, Hollister R, West H, Mui S, Growdon JH, Petersen RC, Parisi JE, Hyman BT. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, Tanzi RE. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78:631–643. doi: 10.1016/j.neuron.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton A, Hardy J. TREM2 variants in Alzheimer's disease. The New England journal of medicine. 2013;368:117–127. doi: 10.1056/NEJMoa1211851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JL, Lee VM. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. The Journal of biological chemistry. 2011;286:15317–15331. doi: 10.1074/jbc.M110.209296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J. The amyloid hypothesis for Alzheimer's disease: a critical reappraisal. J Neurochem. 2009;110:1129–1134. doi: 10.1111/j.1471-4159.2009.06181.x. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan AR, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schurmann B, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frolich L, Hampel H, Hull M, Rujescu D, Goate AM, Kauwe JS, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin A, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton AB, Guerreiro R, Muhleisen TW, Nothen MM, Moebus S, Jockel KH, Klopp N, Wichmann HE, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans PA, O'Donovan M, Owen MJ, Williams J. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haroutunian V, Purohit DP, Perl DP, Marin D, Khan K, Lantz M, Davis KL, Mohs RC. Neurofibrillary tangles in nondemented elderly subjects and mild Alzheimer disease. Archives of neurology. 1999;56:713–718. doi: 10.1001/archneur.56.6.713. [DOI] [PubMed] [Google Scholar]
- Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, Dowzell K, Jones N, Stretton A, Thomas C, Richards A, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nature Genetics. 2011;43:429–435. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Morris JC, Goate AM. Alzheimer's disease the challenge of the second century. Sci Transl Med. 2011;3:77sr71. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua X, Leow AD, Lee S, Klunder AD, Toga AW, Lepore N, Chou YY, Brun C, Chiang MC, Barysheva M, Jack CR, Jr, Bernstein MA, Britson PJ, Ward CP, Whitwell JL, Borowski B, Fleisher AS, Fox NC, Boyes RG, Barnes J, Harvey D, Kornak J, Schuff N, Boreta L, Alexander GE, Weiner MW, Thompson PM, Alzheimer's Disease Neuroimaging, I 3D characterization of brain atrophy in Alzheimer's disease and mild cognitive impairment using tensor-based morphometry. Neuroimage. 2008;41:19–34. doi: 10.1016/j.neuroimage.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Thies B, Trojanowski JQ, Vinters HV, Montine TJ. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomovic MD, Klunk WE, Abrahamson EE, Mathis CA, Price JC, Tsopelas ND, Lopresti BJ, Ziolko S, Bi W, Paljug WR, Debnath ML, Hope CE, Isanski BA, Hamilton RL, DeKosky ST. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer's disease. Brain. 2008;131:1630–1645. doi: 10.1093/brain/awn016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingelsson M, Fukumoto H, Newell KL, Growdon JH, Hedley-Whyte ET, Frosch MP, Albert MS, Hyman BT, Irizarry MC. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–931. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- Jack CR, Jr, Albert MS, Knopman DS, McKhann GM, Sperling RA, Carillo M, Thies W, Phelps CH. Introduction to the recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011a;7:257–262. doi: 10.1016/j.jalz.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Dickson DW, Parisi JE, Xu YC, Cha RH, O'Brien PC, Edland SD, Smith GE, Boeve BF, Tangalos EG, Kokmen E, Petersen RC. Antemortem MRI findings correlate with hippocampal neuropathology in typical aging and dementia. Neurology. 2002;58:750–757. doi: 10.1212/wnl.58.5.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Shaw LM, Vemuri P, Wiste HJ, Weigand SD, Lesnick T, Pankratz VS, Donohue M, Trojanowski JQ. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet neurology. 2013a;12:207–216. doi: 10.1016/S1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010a;9:119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Knopman DS, Weigand SD, Wiste HJ, vemuri P, Lowe V, Kantarci K, Gunter JL, senjem ML, Ivnik RJ, Roberts R, rocca WA, Boeve BF, Petersen RC. An operational approach to NIA-AA crtiteria for preclinical Alzheimer's disease. Ann Neurol. 2012a;71:765–775. doi: 10.1002/ana.22628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Lowe VJ, Senjem ML, Weigand SD, Kemp BJ, Shiung MM, Knopman DS, Boeve BF, Klunk WE, Mathis CA, Petersen RC. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain. 2008;131:665–680. doi: 10.1093/brain/awm336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, Shiung MM, Gunter JL, Boeve BF, Kemp BJ, Weiner M, Petersen RC. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain. 2009;132:1355–1365. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Vemuri P, Wiste HJ, Weigand SD, Aisen PS, Trojanowski JQ, Shaw LM, Bernstein MA, Petersen RC, Weiner MW, Knopman DS. Evidence for Ordering of Alzheimer Disease Biomarkers. Arch Neurol. 2011b;68:1526–1535. doi: 10.1001/archneurol.2011.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Vemuri P, Wiste HJ, Weigand SD, Lesnick TG, Lowe V, Kantarci K, Bernstein MA, Senjem ML, Gunter JL, Boeve BF, Trojanowski JQ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Knopman DS. Shapes of the Trajectories of 5 Major Biomarkers of Alzheimer Disease. Arch Neurol. 2012b;69:856–867. doi: 10.1001/archneurol.2011.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Wiste HJ, Lesnick TG, Weigand SD, Knopman DS, Vemuri P, Pankratz VS, Senjem ML, Gunter JL, Mielke MM, Lowe VJ, Boeve BF, Petersen RC. Brain beta-amyloid load approaches a plateau. Neurology. 2013b;80:890–896. doi: 10.1212/WNL.0b013e3182840bbe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Wiste HJ, Vemuri P, Weigand SD, Senjem ML, Zeng G, Bernstein MA, Gunter JL, Pankratz VS, Aisen PS, Weiner MW, Petersen RC, Shaw LM, Trojanowski JQ, Knopman DS. Brain beta-amyloid measure and magnetic resonance imaging atophy both predict time-to-progression from mild cognitive impairment to Alzheimer's disease. Brain. 2010b;133:3336–3348. doi: 10.1093/brain/awq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Wiste HJ, Weigand SD, Knopman DS, Lowe V, Vemuri P, Mielke MM, Jones DT, Senjem ML, Gunter JL, Gregg BE, Pankratz VS, Petersen RC. Amyloid-first and neurodegeneration-first profiles characterize incident amyloid PET positivity. Neurology. 2013c doi: 10.1212/01.wnl.0000435556.21319.e4. Epub 10/18/2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust W. Vulnerable neural systems and the borderland of brain aging and neurodegeneration. Neuron. 2013;77:219–234. doi: 10.1016/j.neuron.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust WJ, Bandy D, Chen K, Foster NL, Landau SM, Mathis CA, Price JC, Reiman EM, Skovronsky D, Koeppe RA. The Alzheimer's Disease Neuroimaging Initiative positron emission tomography core. Alzheimers Dement. 2010;6:221–229. doi: 10.1016/j.jalz.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust WJ, Landau SM. Apolipoprotein E, Not Fibrillar beta-Amyloid, Reduces Cerebral Glucose Metabolism in Normal Aging. J Neurosci. 2012;32:18227–18233. doi: 10.1523/JNEUROSCI.3266-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust WJ, Landau SM, Shaw LM, Trojanowski JQ, Koeppe RA, Reiman EM, Foster NL, Petersen RC, Weiner MW, Price JC, Mathis CA. Relationships between biomarkers in aging and dementia. Neurology. 2009;73:1193–1199. doi: 10.1212/WNL.0b013e3181bc010c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA, Gregas M, Becker JA, Kinnecom C, Salat DH, Moran EK, Smith EE, Rosand J, Rentz DM, Klunk WE, Mathis CA, Price JC, Dekosky ST, Fischman AJ, Greenberg SM. Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann Neurol. 2007;62:229–234. doi: 10.1002/ana.21164. [DOI] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, Rujescu D, Hampel H, Giegling I, Andreassen OA, Engedal K, Ulstein I, Djurovic S, Ibrahim-Verbaas C, Hofman A, Ikram MA, van Duijn CM, Thorsteinsdottir U, Kong A, Stefansson K. Variant of TREM2 associated with the risk of Alzheimer's disease. The New England journal of medicine. 2013;368:107–116. doi: 10.1056/NEJMoa1211103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephs KA, Whitwell JL, Ahmed Z, Shiung MM, Weigand SD, Knopman DS, Boeve BF, Parisi JE, Petersen RC, Dickson DW, Jack CR., Jr Betaamyloid burden is not associated with rates of brain atrophy. Ann Neurol. 2008;63:204–212. doi: 10.1002/ana.21223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer's disease. Neuron. 2009;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Langstrom B. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Knopman DS, Jack CR, Jr, Wiste HJ, Weigand SD, Vemuri P, Lowe VJ, Kantarci K, Gunter JL, Senjem ML, Mielke MM, Roberts RO, Boeve BF, Petersen RC. Brain injury biomarkers are not dependent on beta-amyloid in normal elderly. Ann Neurol. 2013a;73:472–480. doi: 10.1002/ana.23816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopman DS, Jack CR, Jr, Wiste HJ, Weigand SD, Vemuri P, Lowe VJ, Kantarci K, Gunter JL, Senjem ML, Mielke MM, Roberts RO, Boeve BF, Petersen RC. Selective worsening of brain injury biomarker abnormalities in cognitively normal elderly persons with beta-amyloidosis. JAMA Neurol. 2013b;70:1030–1038. doi: 10.1001/jamaneurol.2013.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopman DS, Parisi JE, Salviati A, Floriach-Robert M, Boeve BF, Ivnik RJ, Smith GE, Dickson DW, Johnson KA, Petersen LE, McDonald WC, Braak H, Petersen RC. Neuropathology of cognitively normal elderly. J Neuropathol Exp Neurol. 2003;62:1087–1095. doi: 10.1093/jnen/62.11.1087. [DOI] [PubMed] [Google Scholar]
- Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fievet N, Barberger-Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A, Helisalmi S, Porcellini E, Hanon O, de Pancorbo MM, Lendon C, Dufouil C, Jaillard C, Leveillard T, Alvarez V, Bosco P, Mancuso M, Panza F, Nacmias B, Bossu P, Piccardi P, Annoni G, Seripa D, Galimberti D, Hannequin D, Licastro F, Soininen H, Ritchie K, Blanche H, Dartigues JF, Tzourio C, Gut I, Van Broeckhoven C, Alperovitch A, Lathrop M, Amouyel P. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- Landau SM, Mintun MA, Joshi AD, Koeppe RA, Petersen RC, Aisen PS, Weiner MW, Jagust WJ. Amyloid deposition, hypometabolism, and longitutdinal cognitive decline. Ann Neurol. 2012;72:578–586. doi: 10.1002/ana.23650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- Lo RY, Hubbard AE, Shaw LM, Trojanowski JQ, Petersen RC, Aisen PS, Weiner MW, Jagust WJ. Longitudinal Change of Biomarkers in Cognitive Decline. Arch Neurol. 2011;68:1257–1266. doi: 10.1001/archneurol.2011.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW, Huang Y. Alzheimer disease: multiple causes, multiple effects of apolipoprotein E4, and multiple therapeutic approaches. Ann Neurol. 2009;65:623–625. doi: 10.1002/ana.21736. [DOI] [PubMed] [Google Scholar]
- Maia LF, Kaeser SA, Reichwald J, Hruscha M, Martus P, Staufenbiel M, Jucker M. Changes in Amyloid-beta and Tau in the Cerebrospinal Fluid of Transgenic Mice Overexpressing Amyloid Precursor Protein. Sci Transl Med. 2013;5:194re192. doi: 10.1126/scitranslmed.3006446. [DOI] [PubMed] [Google Scholar]
- Markesbery WR, Schmitt FA, Kryscio RJ, Davis DG, Smith CD, Wekstein DR. Neuropathologic substrate of mild cognitive impairment. Arch Neurol. 2006;63:38–46. doi: 10.1001/archneur.63.1.38. [DOI] [PubMed] [Google Scholar]
- Maruyama M, Shimada H, Suhara T, Shinotoh H, Ji B, Maeda J, Zhang MR, Trojanowski JQ, Lee VM, Ono M, Masamoto K, Takano H, Sahara N, Iwata N, Okamura N, Furumoto S, Kudo Y, Chang Q, Saido TC, Takashima A, Lewis J, Jang MK, Aoki I, Ito H, Higuchi M. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron. 2013;79:1094–1108. doi: 10.1016/j.neuron.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattsson N, Zetterberg H, Hansson O, Andreasen N, Parnetti L, Jonsson M, Herukka SK, van der Flier WM, Blankenstein MA, Ewers M, Rich K, Kaiser E, Verbeek M, Tsolaki M, Mulugeta E, Rosen E, Aarsland D, Visser PJ, Schroder J, Marcusson J, de Leon M, Hampel H, Scheltens P, Pirttila T, Wallin A, Jonhagen ME, Minthon L, Winblad B, Blennow K. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. Jama. 2009;302:385–393. doi: 10.1001/jama.2009.1064. [DOI] [PubMed] [Google Scholar]
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science. 2010;330:1774. doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morric JC, Rosser MN, Scheltens P, Thies W, Weintraub S, Phelps CH. The diagnosis of dementia due to Alzheimer's disease: Recommendations from the National Institute on Aging and the Alzheimer's Assocation Workgroup. Alzheimers Dement. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesulam MM. Neuroplasticity failure in Alzheimer's disease: bridging the gap between plaques and tangles. Neuron. 1999;24:521–529. doi: 10.1016/s0896-6273(00)81109-5. [DOI] [PubMed] [Google Scholar]
- Mormino EC, Kluth JT, Madison CM, Rabinovici GD, Baker SL, Miller BL, Koeppe RA, Mathis CA, Weiner MW, Jagust WJ. Episodic memory loss is related to hippocampal-mediated beta-amyloid deposition in elderly subjects. Brain. 2009;132:1310–1323. doi: 10.1093/brain/awn320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morra JH, Tu Z, Apostolova LG, Green AE, Avedissian C, Madsen SK, Parikshak N, Hua X, Toga AW, Jack CR, Jr, Weiner MW, Thompson PM. Validation of a fully automated 3D hippocampal segmentation method using subjects with Alzheimer's disease mild cognitive impairment, and elderly controls. Neuroimage. 2008;43:59–68. doi: 10.1016/j.neuroimage.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morra JH, Tu Z, Apostolova LG, Green AE, Avedissian C, Madsen SK, Parikshak N, Toga AW, Jack CR, Jr, Schuff N, Weiner MW, Thompson PM. Automated mapping of hippocampal atrophy in 1-year repeat MRI data from 490 subjects with Alzheimer's disease, mild cognitive impairment, and elderly controls. Neuroimage. 2009;45:S3–15. doi: 10.1016/j.neuroimage.2008.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, Mintun MA. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67:122–131. doi: 10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Duxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, De Jager PL. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nature Genetics. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PT, Head E, Schmitt FA, Davis PR, Neltner JH, Jicha GA, Abner EL, Smith CD, Van Eldik LJ, Kryscio RJ, Scheff SW. Alzheimer's disease is not “brain aging”: neuropathological, genetic, and epidemiological human studies. Acta Neuropathol. 2011;121:571–587. doi: 10.1007/s00401-011-0826-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordberg A, Carter SF, Rinne J, Drzezga A, Brooks DJ, Vandenberghe R, Perani D, Forsberg A, Langstrom B, Scheinin N, Karrasch M, Nagren K, Grimmer T, Miederer I, Edison P, Okello A, Van Laere K, Nelissen N, Vandenbulcke M, Garibotto V, Almkvist O, Kalbe E, Hinz R, Herholz K. A European multicentre PET study of fibrillar amyloid in Alzheimer's disease. European journal of nuclear medicine and molecular imaging. 2013;40:104–114. doi: 10.1007/s00259-012-2237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer's disease. Neurobiol Aging. 2003;24:1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Perrin RJ, Fagan AM, Holtzman DM. Multimodal techniques for diagnosis and prognosis of Alzheimer's disease. Nature. 2009;461:916–922. doi: 10.1038/nature08538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann Neurol. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Rabinovici GD, Jagust WJ, Furst AJ, Ogar JM, Racine CA, Mormino EC, O'Neil JP, Lal RA, Dronkers NF, Miller BL, Gorno-Tempini ML. Abeta amyloid and glucose metabolism in three variants of primary progressive aphasia. Ann Neurol. 2008;64:388–401. doi: 10.1002/ana.21451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Kuceyeski A, Weiner M. A network diffusion model of disease progression in dementia. Neuron. 2012;73:1204–1215. doi: 10.1016/j.neuron.2011.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raz N, Rodrigue KM, Kennedy KM, Acker JD. Vascular health and longitudinal changes in brain and cognition in middle-aged and older adults. Neuropsychology. 2007;21:149–157. doi: 10.1037/0894-4105.21.2.149. [DOI] [PubMed] [Google Scholar]
- Reed BR, Mungas D, Farias ST, Harvey D, Beckett L, Widaman K, Hinton L, Decarli C. Measuring cognitive reserve based on the decomposition of episodic memory variance. Brain. 2010;133:2196–2209. doi: 10.1093/brain/awq154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Functional brain abnormalities in young adults at genetic risk for lateonset Alzheimer's dementia. Proc Natl Acad Sci U S A. 2004;101:284–289. doi: 10.1073/pnas.2635903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Quiroz YT, Fleisher AS, Chen K, Velez-Pardo C, Jimenez-Del-Rio M, Fagan AM, Shah AR, Alvarez S, Arbelaez A, Giraldo M, Acosta-Baena N, Sperling RA, Dickerson B, Stern CE, Tirado V, Munoz C, Reiman RA, Huentelman MJ, Alexander GE, Langbaum JB, Kosik KS, Tariot PN, Lopera F. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer's disease in the presenilin 1 E280A kindred: a case-control study. Lancet neurology. 2012;11:1048–1056. doi: 10.1016/S1474-4422(12)70228-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentz DM, Locascio JJ, Becker JA, Moran EK, Eng E, Buckner RL, Sperling RA, Johnson KA. Cognition, reserve, and amyloid deposition in normal aging. Ann Neurol. 2010;67:353–364. doi: 10.1002/ana.21904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinne JO, Brooks DJ, Rossor MN, Fox NC, Bullock R, Klunk WE, Mathis CA, Blennow K, Barakos J, Okello AA, Rodriguez Martinez de Liano S, Liu E, Koller M, Gregg KM, Schenk D, Black R, Grundman M. 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer's disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 2010;9:363–372. doi: 10.1016/S1474-4422(10)70043-0. [DOI] [PubMed] [Google Scholar]
- Rodrigue KM, Kennedy KM, Devous MD, Sr, Rieck JR, Hebrank AC, Diaz-Arrastia R, Mathews D, Park DC. beta-Amyloid burden in healthy aging: regional distribution and cognitive consequences. Neurology. 2012;78:387–395. doi: 10.1212/WNL.0b013e318245d295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe CM, Mintun MA, D'Angelo G, Xiong C, Grant EA, Morris JC. Alzheimer disease and cognitive reserve: variation of education effect with carbon 11-labeled Pittsburgh Compound B uptake. Arch Neurol. 2008;65:1467–1471. doi: 10.1001/archneur.65.11.1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roses AD. Apolipoprotein E affects the rate of Alzheimer disease expression: betaamyloid burden is a secondary consequence dependent on APOE genotype and duration of disease. J Neuropathol Exp Neurol. 1994;53:429–437. doi: 10.1097/00005072-199409000-00002. [DOI] [PubMed] [Google Scholar]
- Rowe CC, Ellis KA, Rimajova M, Bourgeat P, Pike KE, Jones G, Fripp J, Tochon-Danguy H, Morandeau L, O'Keefe G, Price R, Raniga P, Robins P, Acosta O, Lenzo N, Szoeke C, Salvado O, Head R, Martins R, Masters CL, Ames D, Villemagne VL. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging. 2010;31:1275–1283. doi: 10.1016/j.neurobiolaging.2010.04.007. [DOI] [PubMed] [Google Scholar]
- Savva GM, Wharton SB, Ince PG, Forster G, Matthews FE, Brayne C. Age, neuropathology, and dementia. N Engl J Med. 2009;360:2302–2309. doi: 10.1056/NEJMoa0806142. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69:2197–2204. doi: 10.1212/01.wnl.0000271090.28148.24. [DOI] [PubMed] [Google Scholar]
- Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol. 2009;66:200–208. doi: 10.1002/ana.21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron. 2009;62:42–52. doi: 10.1016/j.neuron.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senjem ML, Gunter JL, Shiung MM, Petersen RC, Jack CR., Jr Comparison of different methodological implementations of voxel-based morphometry in neurodegenerative disease. Neuroimage. 2005;26:600–608. doi: 10.1016/j.neuroimage.2005.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, Bis JC, Smith AV, Carassquillo MM, Lambert JC, Harold D, Schrijvers EM, Ramirez-Lorca R, Debette S, Longstreth WT, Jr, Janssens AC, Pankratz VS, Dartigues JF, Hollingworth P, Aspelund T, Hernandez I, Beiser A, Kuller LH, Koudstaal PJ, Dickson DW, Tzourio C, Abraham R, Antunez C, Du Y, Rotter JI, Aulchenko YS, Harris TB, Petersen RC, Berr C, Owen MJ, Lopez-Arrieta J, Varadarajan BN, Becker JT, Rivadeneira F, Nalls MA, Graff-Radford NR, Campion D, Auerbach S, Rice K, Hofman A, Jonsson PV, Schmidt H, Lathrop M, Mosley TH, Au R, Psaty BM, Uitterlinden AG, Farrer LA, Lumley T, Ruiz A, Williams J, Amouyel P, Younkin SG, Wolf PA, Launer LJ, Lopez OL, van Duijn CM, Breteler MM. Genome-wide analysis of genetic loci associated with Alzheimer disease. Jama. 2010;303:1832–1840. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, Dean R, Siemers E, Potter W, Lee VM, Trojanowski JQ. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403–413. doi: 10.1002/ana.21610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline YI, Morris JC, Snyder AZ, Price JL, Yan Z, D'Angelo G, Liu C, Dixit S, Benzinger T, Fagan A, Goate A, Mintun MA. APOE4 allele disrupts resting state fMRI connectivity in the absence of amyloid plaques or decreased CSF Abeta42. J Neurosci. 2010;30:17035–17040. doi: 10.1523/JNEUROSCI.3987-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Small SA, Duff K. Linking Abeta and tau in late-onset Alzheimer's disease: a dual pathway hypothesis. Neuron. 2008;60:534–542. doi: 10.1016/j.neuron.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sojkova J, Driscoll I, Iacono D, Zhou Y, Codispoti KE, Kraut MA, Ferrucci L, Pletnikova O, Mathis CA, Klunk WE, O'Brien RJ, Wong DF, Troncoso JC, Resnick SM. In vivo fibrillar beta-amyloid detected using [11C]PiB positron emission tomography and neuropathologic assessment in older adults. Arch Neurol. 2011;68:232–240. doi: 10.1001/archneurol.2010.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnen JA, Santa Cruz K, Hemmy LS, Woltjer R, Leverenz JB, Montine KS, Jack CR, Kaye J, Lim K, Larson EB, White L, Montine TJ. Ecology of the aging human brain. Arch Neurol. 2011;68:1049–1056. doi: 10.1001/archneurol.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Jr, Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carillo MC, Thies W, Morrison-Bogorad M, Wagster MV, Phelps CH. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Assocation workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011a;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Jack CR, Jr, Aisen PS. Testing the right target and right drug at the right stage. Sci Transl Med. 2011b;3:111cm133. doi: 10.1126/scitranslmed.3002609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St George-Hyslop PH, Tanzi RE, Polinsky RJ, Haines JL, Nee L, Watkins PC, Myers RH, Feldman RG, Pollen D, Drachman D, et al. The genetic defect causing familial Alzheimer's disease maps on chromosome 21. Science. 1987;235:885–890. doi: 10.1126/science.2880399. [DOI] [PubMed] [Google Scholar]
- Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, Schmechel D, Saunders AM, Goldgaber D, Roses AD. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:8098–8102. doi: 10.1073/pnas.90.17.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strozyk D, Blennow K, White LR, Launer LJ. CSF Abeta 42 levels correlate with amyloid-neuropathology in a population-based autopsy study. Neurology. 2003;60:652–656. doi: 10.1212/01.wnl.0000046581.81650.d0. [DOI] [PubMed] [Google Scholar]
- Tapiola T, Alafuzoff I, Herukka SK, Parkkinen L, Hartikainen P, Soininen H, Pirttila T. Cerebrospinal fluid {beta}-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch Neurol. 2009;66:382–389. doi: 10.1001/archneurol.2008.596. [DOI] [PubMed] [Google Scholar]
- Tarawneh R, D'Angelo G, Macy E, Xiong C, Carter D, Cairns NJ, Fagan AM, Head D, Mintun MA, Ladenson JH, Lee JM, Morris JC, Holtzman DM. Visinin-like protein-1: diagnostic and prognostic biomarker in Alzheimer disease. Ann Neurol. 2011;70:274–285. doi: 10.1002/ana.22448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Tolboom N, van der Flier WM, Yaqub M, Boellaard R, Verwey NA, Blankenstein MA, Windhorst AD, Scheltens P, Lammertsma AA, van Berckel BN. Relationship of cerebrospinal fluid markers to 11C-PiB and 18F-FDDNP binding. J Nucl Med. 2009;50:1464–1470. doi: 10.2967/jnumed.109.064360. [DOI] [PubMed] [Google Scholar]
- Vemuri P, Weigand SD, Przybelski SA, Knopman DS, Smith GE, Trojanowski JQ, Shaw LM, Decarli CS, Carmichael O, Bernstein MA, Aisen PS, Weiner M, Petersen RC, Jack CR., Jr Cognitive reserve and Alzheimer's disease biomarkers are independent determinants of cognition. Brain. 2011;134:1479–1492. doi: 10.1093/brain/awr049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri P, Whitwell JL, Kantarci K, Josephs KA, Parisi JE, Shiung MS, Knopman DS, Boeve BF, Petersen RC, Dickson DW, Jack CR., Jr Antemortem MRI based STructural Abnormality iNDex (STAND)-scores correlate with postmortem Braak neurofibrillary tangle stage. Neuroimage. 2008;42:559–567. doi: 10.1016/j.neuroimage.2008.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri P, Wiste HJ, Weigand SD, Knopman DS, Shaw LM, Trojanowski JQ, Aisen PS, Weiner M, Petersen RC, Jack CR., Jr Effect of apolipoprotein E on biomarkers of amyloid load and neuronal pathology in Alzheimer disease. Ann Neurol. 2010;67:308–316. doi: 10.1002/ana.21953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri P, Wiste HJ, Weigand SD, Shaw LM, Trojanowski JQ, Weiner MW, Knopman DS, Petersen RC, Jack CR., Jr MRI and CSF biomarkers in normal, MCI, and AD subjects: Diagnostic discrimination and cognitive correlations. Neurology. 2009;73:287–293. doi: 10.1212/WNL.0b013e3181af79e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villain N, Chetelat G, Grassiot B, Bourgeat P, Jones G, Ellis KA, Ames D, Martins RN, Eustache F, Salvado O, Masters CL, Rowe CC, Villemagne VL. Regional dynamics of amyloid-beta deposition in healthy elderly, mild cognitive impairment and Alzheimer's disease: a voxelwise PiB-PET longitudinal study. Brain : a journal of neurology. 2012;135:2126–2139. doi: 10.1093/brain/aws125. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, Szoeke C, Macaulay SL, Martins R, Maruff P, Ames D, Rowe CC, Masters CL. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet neurology. 2013;12:357–367. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Pike KE, Chetelat G, Ellis KA, Mulligan RS, Bourgeat P, Ackermann U, Jones G, Szoeke C, Salvado O, Martins R, O'Keefe G, Mathis CA, Klunk WE, Ames D, Masters CL, Rowe CC. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann Neurol. 2011;69:181–192. doi: 10.1002/ana.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser PJ, Verhey F, Knol DL, Scheltens P, Wahlund LO, Freund-Levi Y, Tsolaki M, Minthon L, Wallin AK, Hampel H, Burger K, Pirttila T, Soininen H, Rikkert MO, Verbeek MM, Spiru L, Blennow K. Prevalence and prognostic value of CSF markers of Alzheimer's disease pathology in patients with subjective cognitive impairment or mild cognitive impairment in the DESCRIPA study: a prospective cohort study. Lancet Neurol. 2009;8:619–627. doi: 10.1016/S1474-4422(09)70139-5. [DOI] [PubMed] [Google Scholar]
- Weigand SD, Vemuri P, Wiste HJ, Senjem ML, Pankratz VS, Aisen PS, Weiner MW, Petersen RC, Shaw LM, Trojanowski JQ, Knopman DS, Jack CR, Jr, Initiative, A.s.D.N Transforming cerebrospinal fluid Abeta42 measures into calculated Pittsburgh compound B units of brain Abeta amyloid. Alzheimers Dement. 2011;7:133–141. doi: 10.1016/j.jalz.2010.08.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White L. Brain lesions at autopsy in older Japanese-American men as related to cognitive impairment and dementia in the final years of life: a summary report from the Honolulu-Asia aging study. J Alzheimers Dis. 2009;18:713–725. doi: 10.3233/JAD-2009-1178. [DOI] [PubMed] [Google Scholar]
- Whitwell JL, Josephs KA, Murray ME, Kantarci K, Przybelski SA, Weigand SD, Vemuri P, Senjem ML, Parisi JE, Knopman DS, Boeve BF, Petersen RC, Dickson DW, Jack CR., Jr MRI correlates of neurofibrillary tangle pathology at autopsy: a voxel-based morphometry study. Neurology. 2008;71:743–749. doi: 10.1212/01.wnl.0000324924.91351.7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth M, Madison CM, Rabinovici GD, Oh H, Landau SM, Jagust WJ. Alzheimer's disease neurodegenerative biomarkers are associated with decreased cognitive function but not beta-amyloid in cognitively normal older individuals. J Neurosci. 2013a;33:5553–5563. doi: 10.1523/JNEUROSCI.4409-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth M, Villeneuve S, Haase CM, Madison CM, Oh H, Landau SM, Rabinovici GD, Jagust WJ. Associations Between Alzheimer Disease Biomarkers, Neurodegeneration, and Cognition in Cognitively Normal Older People. JAMA Neurol. 2013b doi: 10.1001/jamaneurol.2013.4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolk DA, Price JC, Madeira C, Saxton JA, Snitz BE, Lopez OL, Mathis CA, Klunk WE, Dekosky ST. Amyloid imaging in dementias with atypical presentation. Alzheimers Dement. 2012;8:389–398. doi: 10.1016/j.jalz.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeoman M, Scutt G, Faragher R. Insights into CNS ageing from animal models of senescence. Nat Rev Neurosci. 2012;13:435–445. doi: 10.1038/nrn3230. [DOI] [PubMed] [Google Scholar]
- Zarow C, Vinters HV, Ellis WG, Weiner MW, Mungas D, White L, Chui HC. Correlates of hippocampal neuron number in Alzheimer's disease and ischemic vascular dementia. Ann Neurol. 2005;57:896–903. doi: 10.1002/ana.20503. [DOI] [PMC free article] [PubMed] [Google Scholar]