

Abstract
Dendritic cells (DCs) function as professional antigen presenting cells and are critical for linking innate immune responses to the induction of adaptive immunity. Many current cancer DC vaccine strategies rely on differentiating DCs, feeding them tumor antigens ex vivo, and infusing them into patients. Importantly, this strategy relies on prior knowledge of suitable “tumor-specific” antigens to prime an effective anti-tumor response. DCs express a variety of receptors specific for the Fc region of immunoglobulins, and antigen uptake via Fc receptors is highly efficient and facilitates antigen presentation to T cells. Therefore, we hypothesized that expression of the mouse IgG1 Fc region on the surface of tumors would enhance tumor cell uptake by DCs and other myeloid cells and promote the induction of anti-tumor T cell responses. To test this, we engineered a murine lymphoma cell line expressing surface IgG1 Fc and discovered that such tumor cells were taken up rapidly by DCs, leading to enhanced cross-presentation of tumor-derived antigen to CD8+ T cells. IgG1-Fc tumors failed to grow in vivo and prophylactic vaccination of mice with IgG1-Fc tumors resulted in rejection of unmanipulated tumor cells. Furthermore, IgG1-Fc tumor cells were able to slow the growth of an unmanipulated primary tumor when used as a therapeutic tumor vaccine. Our data demonstrate that engagement of Fc receptors by tumors expressing the Fc region of IgG1 is a viable strategy to induce efficient and protective anti-tumor CD8+ T cell responses without prior knowledge of tumor-specific antigens.
Keywords: Fc receptors, IgG1, dendritic cells, cross-presentation, CD8 T cell priming, cancer vaccine, MHC Class I
Introduction
Current anti-cancer treatments are composed of various combinations of surgery, radiotherapy, chemotherapy and molecularly-targeted therapies. The efficacy of many of these therapies is limited by their toxicity and inability to eliminate all tumor cells.1 Despite extensive progress in modifying tumor-specific T cells2 and advances in dendritic cell therapy,3 cancer immunotherapy is still viewed as a complex and confounding therapeutic. This comes as no surprise, considering the number of mechanisms by which tumors bypass immune checkpoints,4 and thus immune-mediated clearance.
Antigen-presenting dendritic cells (DCs) form a critical link between the innate and adaptive immune systems. When naïve DCs encounter pathogens, they recognize microbial products leading to upregulation of surface major histocompatibility complex (MHC) molecules, costimulatory molecules and production of inflammatory cytokines, such as IL-6, IL-12, and type I interferons.5 Mature DCs then migrate to draining lymph nodes where they present antigen and prime CD4 and CD8 T cells.5 A number of current cancer immunotherapy strategies rely on differentiating CD34+ peripheral blood stem cells or monocytes into DCs ex vivo, pulsing them with tumor antigen and infusing them into patients with the hope of inducing effective CD4 and CD8 T cell responses against tumors.3 This approach has had measurable clinical success,6 but a number of factors may limit its efficacy. First, the many subsets of DCs in vivo differ broadly in their capacity to activate T cells.7 Second, ex vivo manipulated DCs display altered patterns of expression of adhesion molecules and chemokine receptors, which may affect their ability to efficiently migrate to lymphoid organs and prime naïve T cells against the tumor antigen.8 Third, injected DCs have a short half-life in vivo and, without persistent antigen presentation, the magnitude of activation and differentiation of T cells could be variable depending on the quality of the injected DCs.9,10 Finally, and perhaps most importantly, infusion of tumor-antigen loaded DCs into patients requires prior knowledge of which tumor-specific antigens or peptides induce effective anti-tumor immunity.9
T cell responses to infection are driven largely by pattern recognition receptor (PRR)-mediated detection of conserved pathogen associated molecular patterns (PAMPs) by DCs.5 As tumors are autologous, they inherently lack many of the patterns that would elicit a productive immune response to infection/microbial non-self.11 However, a number of phagocytic and endocytic receptors, including Fc receptors, scavenger receptors and mannose receptors, could potentially be exploited to target tumors to dendritic cells.3,12,13 Such targeting is likely to enhance uptake of tumor cells by DCs and lead to the presentation of tumor-derived antigens on MHC molecules.14 Concomitant activation of PRRs could then provide additional signals aiding induction of optimal effector responses against tumor cells.13
Four classes of IgG Fc receptors (FcγR) are expressed widely on cells of both the myeloid and lymphoid lineages, and impart effector functions to IgG subclasses.15 Of these, FcγRIIB and FcγRIII predominantly bind to IgG1, the dominant IgG isotype found in mouse serum.15 FcγRIII is an activating Fc receptor found broadly on the surface of myeloid cells and is the only IgG receptor expressed by NK cells.15,16 FcγRIIB, an inhibitory IgG receptor, is the only IgG Fc receptor expressed by B cells. It is also expressed on a variety of myeloid cells, but is not expressed by NK cells.16 On NK cells and myeloid cells, FcγRIII is known to potently mediate antibody-dependent cell-mediated cytotoxicity (ADCC) through binding to IgG1 immune complexes, a process negatively regulated on myeloid cells by concurrent signals through FcγRIIB.16-18
Antibodies targeting cell-surface antigens expressed by tumors have shown great promise in eliminating cancer cells.17,19-22 Part of the efficacy of therapeutic anti-tumor antibodies may be through ADCC.17 It has also been suggested that these antibodies may induce CTL responses by targeting tumors to dendritic cells.23-25 Indeed, Fc receptor-mediated uptake of antigen-antibody complexes triggers highly efficient presentation of Fc-targeted antigens and induction of T cell responses.18,26-29
In all nucleated cells, cytosolic antigens are presented on MHC class I (MHC-I) molecules. In specialized cells capable of phagocytosis, such as macrophages, endocytosed antigens are presented largely on MHC-II molecules. In contrast, DCs possess the unique ability to cross-present endocytosed antigen to CD8+ T cells via MHC-I. Cross-presentation is critical for the initiation of CD8 T cell responses to intracellular pathogens that do not infect DCs directly.7 Targeting antigens to Fc receptors on DCs also leads to very efficient priming of CD8 T cells.29-31 We hypothesized that targeted recognition of tumor cells by dendritic cells/myeloid cells via murine IgG1 Fc, the least inflammatory mouse IgG isotype, would promote efficient CTL responses while minimizing inflammatory side effects. In this study, we show that tumor cells expressing the Fc portion of murine IgG1 enhance the cross-presentation of a model antigen, and trigger a potent anti-tumor immune response in vitro and in vivo.
Results
Engineering IgG1 Fc-tagged tumor cells
To direct trafficking of tumor cells and their antigens to Fc receptors on dendritic cells, we expressed the Fc region of IgG1 on the surface of the tumor cell line EG7. The CH2 and CH3 domains (residues 237 to 430) of murine IgG1 Fc can be efficiently expressed on the cell surface in reverse orientation by fusing IgG1 Fc with the transmembrane domain of transferrin receptor.32,33 This chimeric protein approach was previously exploited to propagate pseudorabies virus with the Fc portion incorporated into its viral envelope. This modified virus was then used for immunization studies.33 We cloned the murine chimeric IgG1 Fc-transferrin fusion into a retroviral vector (MSCV 2.2) (Fig. 1A) and transduced EG7 cells34 (the murine lymphoma cell line EL4 that expresses the model antigen ovalbumin). The resulting Fc-transferrin expressing EG7 cells are hereafter referred to as EG7-Fc. A control cell line was constructed by transducing EG7 cells with an MSCV vector lacking the IgG1 Fc insert (EG7- Empty vector, EG7-EV). Both EG7 and EL4 cells form aggressive tumors when injected subcutaneously in mice and ultimately result in lethality in 1–2 mo. The pMIG vector MSCV 2.2 contains an IRES sequence followed by GFP. Thus, both transduced cell lines express GFP, but surface Fc expression was seen only in cells transfected with the IgG1-Fc containing vector (Fig. 1B). Polyclonal cultures of transduced cells were derived from sorted cells that expressed high levels of GFP. We found no difference in the doubling time or 3H-thymidine incorporation rate in these two modified cell lines, suggesting that retroviral modification did not alter the growth kinetics of the transduced cells (Fig. S1).

Figure 1. Creation of IgG1 Fc expressing tumor cells. (A) A construct expressing IgG1 Fc‐transferrin transmembrane region was subcloned into a retroviral vector expressing IRES‐GFP. EG7 cells were transduced with retro virus expressing either GFP containing empty vector (EG7-EV) or the IgG1 Fc construct EG7-Fc). (B) Lower panels show staining for GFP (X axis) and IgG1 (Y axis) in sorted cells that express just the empty vector (left panel) or the mTR-Fc construct (Right panel).
IgG1-Fc expressing tumors enhance dendritic cell cross-presentation
We hypothesized that engagement of Fc receptors on dendritic cells by tumor cells that expressed IgG1-Fc would enhance processing of tumor-specific and tumor-associated antigens and their presentation to CD4 and CD8 T lymphocytes. To test our hypothesis, we incubated bone marrow-derived dendritic cells35 (Fig. 2A and B) or DCs derived from the spleen after Flt3L injection36 (Fig. 2C and D), with live EG7-Fc or EG7-EV for 12 h. DCs were then purified to > 99.9% purity via fluorescence activated cell sorting (FACS) and incubated with OVA-specific CD8 and CD4 T cells from OT-I and OT-II TCR transgenic (Tg) animals, respectively.37,38 CD8 T cells from OT-I TCR Tg mice co-cultured with DCs that were pre-incubated with EG7-Fc tumor cells showed significantly greater proliferation compared with OT-I T cells co-cultured with DCs pre-incubated with control EG7-EV cells (Fig. 2A and C). Surprisingly, CD4 OT-II lymphocytes co-cultured with DCs pre-incubated with either cell line showed limited proliferation (Fig. 2B and D). We also found that DCs that were pre-incubated with EG7-Fc tumor cells induced greater IFN-γ, TNF-α and Granzyme B production by CD8 T cells, and these cells were able to kill target cells more efficiently than EG7-EV induced CD8+ cells (Fig. 3). Importantly, incubation of DCs with the FcγRII and FcγRIII blocking antibody 2.4G2 prior to co-culture with tumor cell lines was sufficient to blunt the enhancement of CD8 priming seen after incubation with EG7-Fc tumor cells (Fig. 4A).

Figure 2. Dendritic cells exposed to EG7-Fc bearing tumors induce robust activation of antigen specific CD8 T cells. (A, B) GM‐CSF derived BMDCS and (C, D) Flt3‐ligand derived splenic DCs were co‐incubated with either EG7-EV cells or EG7-Fc cells for a period of 12‐16 h. DCs were then sorted on a flow cytometer and were then plated at different concentrations in the presence of either OT‐I (A, C) or OT‐II (B, D) T cells. After 3 d of culturing, T cell proliferation was measured by 3H thymidine incorporation. Ovalbumin (10 μg/ml) pulsed DCs were used as a control to measure both CD8 (OT‐I) and CD4 (OT‐II) T cell proliferation. The data are representative of five independent experiments.
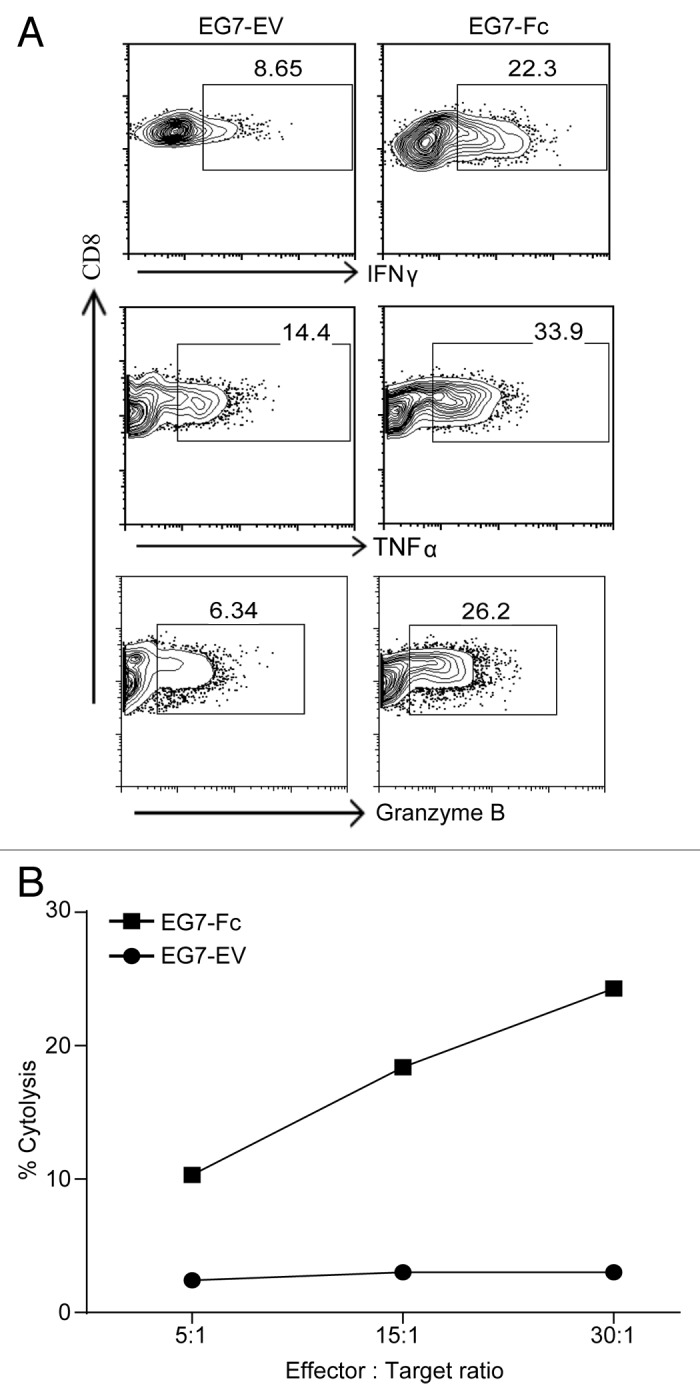
Figure 3. Dendritic cells exposed to EG7-Fc bearing tumors prime functionally superior CD8 T cells. GM‐CSF derived BMDCS were co‐incubated with either EG7-EV cells or EG7-Fc cells for a period of 12‐16 h. DCs were then sorted on a flow cytometer and incubated at 1:10 ratio with OT‐I T cells. After 3 d of culturing, CD8 T cells were assessed for intracellular IFN-gamma, TNF-α and Granzyme B (A). CD8 T cells were also incubated with target cells (EG7) at the indicated effector:target ratio for a period of 12 h to measure cytotoxicity (B). The data are representative of two independent experiments.

Figure 4. Enhanced CD8 T cell activation can be inhibited by blocking Fc receptors and enhanced by the addition of TLR3 agonist. (A) GM‐CSF BMDCs were pre‐incubated with a control antibody or a blocking antibody to Fc receptors (clone 2.4G2) before being cultured with either EG7-EV or EG7‐Fc cells. After overnight culture, DCs were sorted and plated at different concentrations before addition of OT‐I T cells. T cell proliferation was measured after 72 h as described previously. B‐C, Experiments performed as previously with the addition of 10 µM poly I:C to tumor‐BMDC cultures 8 h prior to incubation with OT‐I cells (B) and OT‐II cells (C). The data are representative of three independent experiments.
The observation that targeting of tumors to Fc receptors enhances the priming of CD8, but not CD4 T cells, argues that the expression of the Fc portion of IgG1 on tumor cells enhances cross-presentation of tumor cell-derived antigens, but does not enhance presentation of tumor-derived antigens by MHC class-II. Notably, no significant differences were observed in the cell surface expression of the DC activation molecules CD40 and CD86 after 12 h of culture with tumor cells (Fig. S2), suggesting that the enhanced cross-priming we observed was unlikely to be dependent on these costimulatory molecules. Therefore, we next tested whether we could further enhance T cell responses to Fc-targeted tumor antigens by activating DCs exposed to EG7-Fc using TLR ligands. Because the TLR3 ligand poly IC is approved for use in humans and has been shown to be an effective adjuvant in vivo,39 we decided to test its ability to influence CD8 T cell responses induced by DCs incubated with tumor cells. Stimulation of DCs with poly I:C greatly enhanced cross-presentation of EG7-Fc derived antigens to CD8 T cells (Fig. 4B and C). In contrast, addition of poly I:C did not enhance CD4 T cell priming under similar conditions. Taken together, these findings suggest that effective cross-presentation and CD8 T cell activation can be induced by simply targeting tumor cargo to Fc receptors on DCs, and that concomitant activation of TLR3 can further enhance these CD8 T cell responses.
IgG1-Fc expressing tumors interact longer with BMDCs
We hypothesized that IgG1-Fc expression on tumor cells would prolong interaction time with DCs and thereby enhance antigen uptake. To test this possibility, we used live cell imaging to observe the interactions between these two cell types in a 4 h culture. As our tumor cell lines expressed GFP, they were easily differentiated from BMDCs using fluorescence and bright field channels. Tumor/DC interaction events were defined as contacts between the two cell types that were initiated during the 4 h culture and ceased during the 4 h culture. The average duration of these interactions was found to be nearly 10-fold longer with EG7-Fc compared with EG7-EV (Fig. 5). These extended interaction times could potentially lead to enhanced uptake of IgG1-Fc expressing tumor cells by DCs, and result in increased class I presentation and CD8 priming.
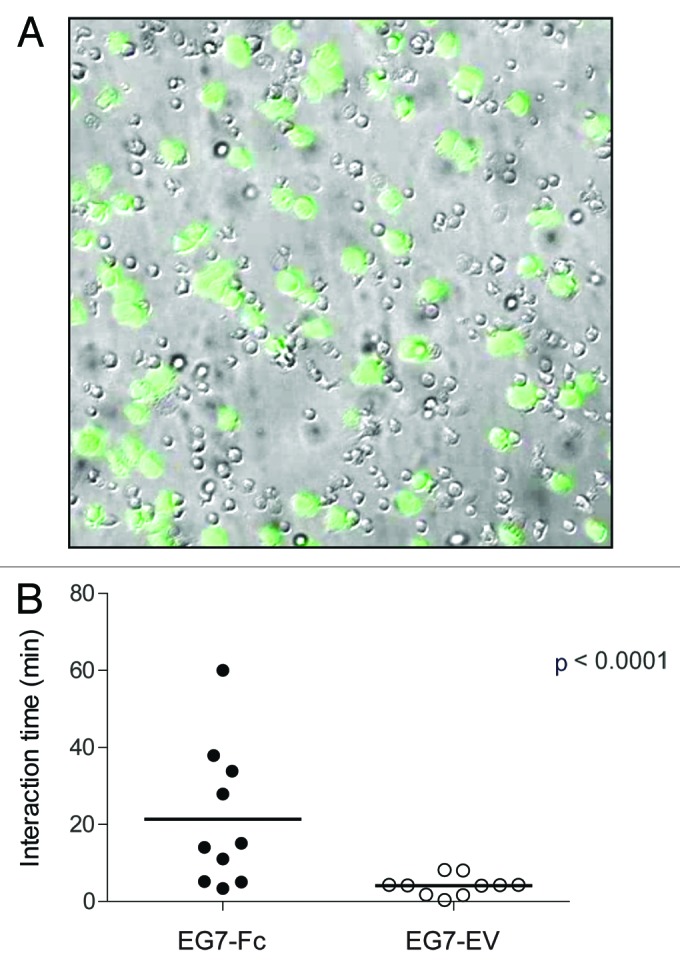
Figure 5. DC-Tumor interaction time is prolonged when tumors express IgG1-Fc. (A) A representative bright field and fluorescence image overlay from a 4 h time-lapsed imaging experiment with DCs co-cultured with GFP expressing cancer cells (green) is given. (B) Interaction times of DCs with tumor cells were analyzed as described in the materials and methods. The data are representative of two independent experiments and p values were determined by two-tailed unpaired t-test.
IgG1-Fc tumors exhibit decreased growth in vivo and stimulate increased anti-tumor CD8 T cell responses
Having observed that tumors expressing IgG1-Fc were able to enhance cross-priming of CD8 T cells in vitro, we wanted to explore the growth and survival of these tumors in vivo. We challenged 15 mice with 500 000 live EG7-Fc or EG7-EV tumor cells injected subcutaneously in the flank. We then euthanized animals on days 7, 14, and 30 (n = 5 mice for each time point) and resected all visible tumors. By day 14, the average weight of EG7-Fc tumors was significantly lower than the average weight of EG7-EV tumors (p < 0.05 unpaired t-test). By day 30, no visible tumors were apparent in mice challenged with EG7-Fc tumors, while all control tumors formed large subcutaneous masses (Fig. 6A). To examine the immune response to these tumors, cells collected from draining lymph nodes from tumor bearing mice on day 7 were incubated with purified BMDCs that had been fed tumor cells for 12–16 h prior to incubation with T cells. CD8 T cells from the draining lymph nodes from EG7-Fc tumor-bearing mice showed higher proliferative responses compared with those from EG7-EV tumor bearing mice (Fig. 6B).
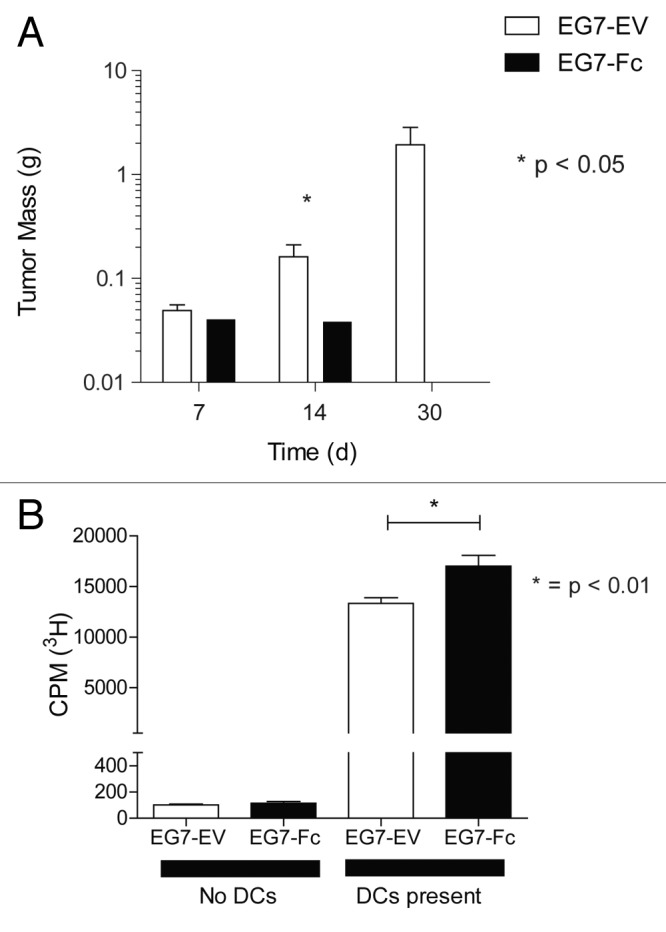
Figure 6. EG7-Fc tumor cells fail to grow in vivo and induce higher CD8 T cell responses. (A) Groups of 15 mice were implanted subcutaneously with 5 × 105 tumor cells in the flanks. Five mice from each group were sacrificed on days 7, 14 and 21 and tumors were excised and weighed to measure growth. Mice that received empty vector expressing tumor cells grew large tumors by day 30, however mice that received mTR‐Fc cells failed to grow detectable tumors at day 30. Both groups had palpable and measurable tumors at days 7 and 14. (B) Draining lymph nodes (inguinal) were harvested and pooled from 5 mice for each group. CD8+ T cells were purified using negative selection and allowed to proliferate on BMDCs that had been cocultured with tumor cells for 12 h prior and purified by FACS. After 48 h of culture, CD8 T cell proliferation was measured by 3H-thymidine incorporation. The data are representative of three independent experiments and p values were determined by two-tailed unpaired t-test.
Vaccination with inactivated IgG1-Fc tumors protects against subsequent challenge with tumor
The use of ovalbumin-expressing tumors in the above-described studies allowed us to precisely determine the effects of IgG1-Fc on antigen presentation. The potential power of this approach, however, is that it can effectively induce anti-tumor responses without prior knowledge of tumor-specific antigens. Therefore, in vivo studies using unmanipulated tumors are essential to determine the potential therapeutic utility. To understand if IgG1-Fc expressing tumors induce a memory CD8 response to tumor-specific or tumor-associated antigens in vivo, we tested if treatment of mice with EG7-Fc tumor cells would protect the mice against development of a tumor when challenged with unmanipulated tumor cells (EG7). To ensure that the EG7-Fc and EG7-EV cells used for vaccination would not form primary tumors in vivo, we treated these cells with mitomycin C, a chemotherapeutic agent that is toxic to tumor cell lines, prior to immunization. We established that mitomycin C treatment was sufficient to completely abolish replication as measured by 3H-thymidine incorporation (Fig. S1). We treated mice with 5 × 105 mitomycin C inactivated tumor cells (n = 5 each group) as a primary vaccine. Twelve days later we challenged mice with 5 × 105 EG7 cells in the contra-lateral flank and followed tumor growth by measuring tumor size on days 12, 14, 17, 21, and 25. Mice immunized with mitomycin C treated EG7-Fc expressing cells were less likely to develop measurable tumors than mice immunized with EG7-EV tumor cells (Fig. 7A). These data suggest that IgG1 Fc expressing tumor cells can induce an adaptive immune response that is long-lasting and can prevent growth of an unmanipulated parent tumor cell at a later time point. Taken together, these data suggest that this may be a highly effective approach for prophylactic cancer vaccination.

Figure 7. EG7-Fc tumors are functional both as a prophylactic inactivated cell vaccine and as a therapeutic live cell vaccine. (A) Groups of 5 mice were administered either EG7-EV or EG7‐Fc cells (5 × 105, mitomycin C treated) as vaccines in the left flanks. After 14 d, both groups received unmanipulated live EG7 cells subcutaneously in the right flank. Mice were followed longitudinally and tumor volumes were assessed by using Vernier’s Calipers. (B‒C) Mice were injected with 5 × 105 live unmanipulated EG7 tumors into the left flank. Mice (n = 15 each group) were then treated with 5 × 105 of live tumor (EG7-EV or EG7-Fc) or vehicle in the right flank on day 1, 2, 4, and 10. Mice treated with EG7-Fc expressing tumors had significantly smaller primary tumors in the left flank by day 21 than animals treated with vehicle (p < 0.05, independent t-test) while animals treated with EG7-EV did not show any diminution in tumor size compared with the vehicle. Panel (B) shows the raw data, panel C shows mean tumor volume of each group. The data are representative of two independent experiments.
IgG1-Fc tumors are effective as therapeutic whole cell tumor vaccines
To evaluate the efficacy of EG7-Fc as a therapeutic approach to treating established tumors, we implanted unmanipulated EG7 cells on day 0 and subsequently injected mice with live EG7-Fc or EG7-EV tumor cells in the contra lateral flank on days 1, 2, 4, and 10. This strategy was designed to approximate vaccination following surgical removal of a primary tumor where a small number of replicating cells can serve as a source of relapse. The sizes of the primary tumors were measured on day 7, 10, 14, 16, 18, and 21 in a blinded fashion. Mice treated with EG7-Fc had significantly smaller primary tumors by day 18 (vehicle) and day 21 (Empty Vector) (n = 15 mice each group) (Fig. 7B and C). In addition, injection of Fc-bearing tumors did not lead to the development of secondary tumors, while mice that received non-Fc bearing tumor cells developed several secondary tumors, consistent with our earlier data. These data argue that the immune response generated by Fc-expressing tumors has the ability to halt or reverse the growth of a previously established parental tumor.
Discussion
The lack of effective presentation of tumor specific or tumor-associated antigens to the immune system continues to be a major obstacle in tumor immunotherapy. Known barriers to effective antitumor immune responses include the immunosuppressive tumor microenvironment, lack of cross-presentation of tumor antigen, and blunted effector responses.4 We present here an approach that targets genetically modified tumors to DCs through transgenic expression of the Fc fragment of IgG1 on the tumor cell surface. Consequently, DC uptake of IgG1-Fc bearing tumors leads to cross-priming of CD8 T cells. In vivo, this approach proved beneficial in promoting shrinkage of pre-existing tumors in mice that were therapeutically “vaccinated” with IgG1-Fc bearing tumor cells. This approach circumvents the requirement for prior knowledge of the tumor antigens that can lead to effective CD8 T cell activation and could have therapeutic potential for a broad spectrum of human cancers.
Polymorphisms in Fc gamma receptors have been associated with an improved clinical response to targeted tumor-associated monoclonal antibodies, suggesting that interactions between the Fc portion of the antibodies and their receptors are important mediators of antitumor responses.40 This, coupled with the evidence that Fc-FcR interactions are important for the uptake, internalization and presentation of antigen to CTLs,31,41 makes enhancing the cross-presentation of tumor antigen an attractive strategy for improving anti-tumor immunity. Furthermore, it is rational to believe that enhanced cross-presentation may be able to diminish tolerance to tumor antigens as one study found that antibody-mediated cross-presentation of antigen can break T cell tolerance in a mouse model of type 1 diabetes.42 Our observation that expression of IgG1-Fc on the surface of tumor cells was able to enhance cross-presentation of tumor-specific antigens and produce measurable clinical efficacy in tumor clearance suggest that this is an immunotherapeutic strategy that is functionally achievable in vivo.
In contrast to a previous study using immune complexes to deliver antigen for cross-presentation,43 our findings suggest that Fc engagement and enhanced cross-priming is not associated with overt DC maturation, as measured by upregulation of CD40 and CD86 (Fig. S2) and secretion of pro-inflammatory cytokines such as IL-6 and IL-12 (Fig. S3). One possible explanation for this difference is that cross-presentation induced by immune complexes is qualitatively different from cross-presentation induced by cell-associated Fc. Our data showing that cross-priming by BMDCs can be further augmented by ligation of TLR3 suggest that DCs simultaneously activated via PRRs may induce quantitatively higher responses. Further experiments are needed to determine whether combining cell-associated Fc engagement with TLR ligation can influence the quality of CD8 T cell responses against tumor antigens in vivo.
Dendritic cells have been shown to capture antigen from virally-infected cells and cross-present them to CTLs in a process called nibbling.44 Our data argue that prolonged dendritic cell-tumor cell interactions result in enhanced cross-presentation and cross-priming. Further experiments are needed to determine whether the prolonged interactions between DCs and FcR-expressing tumor cells result in a quantitative difference in the number of MHC class I molecules loaded with antigen, or whether some other mechanism may be responsible for the enhanced cross-priming of CTLs we observed. Importantly, targeting of the tumor cells via Fc receptors enhanced cross-presentation by both bone marrow derived myeloid DCs and a heterogeneous population of DCs obtained after FLT3 ligand administration in vivo, suggesting the utility of this approach in humans may not be limited to targeting of Fc-expressing tumors to specific DC subsets.45 It remains to be seen, however, whether DCs are the primary cells that acquire and cross-present tumor antigens in vivo following Fc-expressing tumor cell vaccination. Notably, it has been suggested previously that macrophages are the primary cell type that cross-presents tumor antigen and primes CD8 T cells in vivo.46,47
A surprising outcome of our experiments is that Fc receptor-mediated targeting of tumor cells to DCs in vitro does not appear to enhance MHC class II-mediated antigen presentation. Similar findings have been reported by a recent study where enhanced cancer cell phagocytosis by macrophages using an IgG1 anti-CD47 antibody led to increased priming of CD8 T cells but not of CD4 T cells.47 One possible explanation for this observation is that CD8 T cells have much lower requirements for costimulation compared with CD4 T cells.48,49 Thus, our results may be explained by the fact that we see no upregulation of costimulatory molecules by DCs incubated with EG7-Fc cells; however, stimulation of DCs using poly I:C also failed to enhance CD4 T cell activation, while CD8 responses were significantly increased. These data imply that antigenic cargo is handled very differently when targeted via the Fc receptors and suggest the possibility that cross-presentation of antigens on MHC class I is a preferred pathway when DCs take up cells expressing the Fc portion of IgG1. This could have additional benefits in vivo since treatment with Fc-bearing tumors presumably would not induce unwanted CD4 T cell responses against self-antigens that may result in inflammation or auto-immunity. Nonetheless, the modest CD4 T cell responses that are generated in response to Fc-bearing tumors are clearly sufficient to provide the necessary help to CD8 T cells for generation of memory, as both prophylactic and therapeutic approaches using EG7-Fc are highly effective.50 It is also possible that CD4 help in the context of cross-presentation is not necessary for CD8 responses, which could make this approach very attractive for generating long-lasting anti-tumor CD8 responses. Because our work involves use of ovalbumin-expressing tumor cells, further investigation is needed to determine if this approach will work to induce CD8 responses against native tumor-derived antigens.
A number of approaches have been taken to improve tumor immunogenicity via the genetic modification of tumors. In a manner similar to our approach, one group induced ectopic surface expression of a chimeric protein of IgG2a and CD98 on murine melanoma cells.51 IgG2a is thought to be more effective at promoting ADCC than IgG1. Notably, these investigators did not observe a significant survival benefit of ectopic IgG2a expression in vivo.
Remarkably, recently established immunotherapies using antibodies that block endogenous immunoregulatory pathways have resulted in cures of tumors that were resistant to conventional treatments.4 Not all patients, however, respond to these therapies, and their immune-mediated side effects can be debilitating. Therefore, there are three major obstacles to effective tumor immunotherapy. First, anti-tumor immunity must be generated. Second, the immunosuppressive conditions in the tumor stroma must be alleviated. Third, the immunopathological side effects of broader therapeutics, such as CTLA4-targeted agents, must be mitigated. The approach described here addresses the first obstacle and suggests the third obstacle can be minimized. Specifically, vaccination of patients with their own tumors that have been modified to express the Fc region of IgG1 on their surface may initiate adaptive immune responses to their primary tumor and have therapeutic value as a tumor vaccine. Further, relapse in patients harboring residual minimal disease after appropriate conventional therapies may be prevented by the presence of circulating memory anti-tumor lymphocytes. These experiments thus provide a foundation for the development of an effective whole-cell therapeutic cancer vaccine strategy. Further work will determine if this strategy, alone or in combination with other potentially cooperating therapeutics, represents a promising therapeutic avenue toward improving outcomes for cancer patients.
Materials and Methods
Mice
OT-I and OT-II mice were obtained from Jackson Laboratories. Control C57BL/6 mice were obtained from the UT Southwestern mouse breeding core facility. Mice were maintained in specific pathogen-free conditions. Mice were used between 6 and 12 wk of age.
Ethics statement
All mouse experiments were done as per protocols approved by the University of Texas Southwestern (UTSW) Medical Center’s Institutional Animal Care and Use Committee (IACUC) and in direct accordance with US. Public Health Service policy and the Animal Welfare Act. Appropriate sedatives, anesthetics and analgesics, as approved by the committee were used to ensure minimal pain and suffering to the animals.
Cell lines and DCs
EG7 cells (ATCC) and murine primary cells were cultured in complete RPMI- 1640 supplemented with 10% FCS, 100 U/ml penicillin, 100 µg/ml streptomycin, 2 mM L-glutamine, 10 mM HEPES, and 1 mM sodium pyruvate (all from Hyclone). BMDCs were generated from bone marrow progenitors. Cells were harvested from femurs and iliac bones of WT mice, cultured for 5 d in complete RPMI-1640 supplemented with 5% FCS, 100 U/ml penicillin, 100 g/ml streptomycin, 2 mM L-glutamine, 10 mM HEPES, and 1 mM sodium pyruvate (all from Hyclone) and GM-CSF. Media was replenished on day 2 and day 4 of culture. Splenic FLT3 ligand induced DCs were obtained as described previously.52
Reagents and antibodies
Allophyocyanin (APC) labeled anti-CD11c, phycoerythrin (PE) labeled anti-Thy 1.2, anti-Mouse IgG1 biotin and Streptavidin APC (all from Biolegend) were used for staining of cells for flow cytometry analysis.
Retroviral transduction
Retrovirus was prepared from 293T cells (ATCC) transfected with MSCV 2.2, VSV-g (Clontech) and pcl-ECO (Imgenex) and VSV-g expressing plasmids using PEI transfection reagent (Polysciences Inc). Virus was harvested from 293T cultures after 24 h of transfection and centrifuged with EG7 cells for 90 min at 24°C at 1200 RPM. Transformed cells were grown as above in 10% FCS containing RPMI and repeatedly sorted for high expression of GFP using FACS on a MoFLo cell sorter (Beckman Coulter).
Purification of T cells
Spleens and lymph nodes were harvested from 8- to 12-week-old mice. CD4+ and CD8+ T cells were purified from the spleens by negative selection as previously described.53
T cell activation assays
BMDCs were prepared as above and cultured with modified EG7 for 12 h. Cells were stained for CD11c APC and Thy 1.2 PE as above and sorted for positive expression of CD11c and the absence of Thy 1.2. Purified DCs were cultured with purified T cells from OT-I and OT-II animals as above at various ratios of DCs to T cells for 2 d at 37 °C in round bottom 96-well plates. Proliferation of T cells was determined by incorporation of (3H) thymidine for the last 12–16 h of the culture (Perkin Elmer). For blocking experiments DCs were treated with 2.4G2 antibody (BD Biosciences) at a concentration of 1 μg/mL for 30 min prior to the incubation with tumor for ~12 h.
Intracellular staining
BMDCs were cultured (1:1) with EG7-Fc or EG7-EV for 12 hrs. Cells were stained for CD11c and Thy1.2 and sorted for CD11c positive and Thy1.2 negative population by FACS. Naive OT-I cells were cultured with purified CD11c positive BMDCs (1:5 ratio) for 2 d at 37 °C in 48 well plate. Primed OT-I cells were stimulated with 50 ng/ml phorbol myristate acetate (PMA) and 1 μM ionomycin in the presence of 1 μmg/ml brefeldin A for 5 h, followed by surface staining, fixed with 4% paraformaldehyde, permeabilized with 0.3% saponin, and stained for intracellular cytokines. The stained cells were analyzed with a FACSCalibur flow cytometer (BD Biosciences). Data were analyzed with FlowJo software (Tree Star).
T cell cytotoxicity assays
OT-I T cells were primed with EG7-EV or EG7-Fc fed BMDCs for 72 h and used as effector cells. EG7-EV and EL4 cells mixed 1:1 were used as target cells and cultured with (5:1, 15:1, and 30:1 effector/target ratios) and without effector cells for 12 h. The percentage of remaining EG7-EV cells (GFP+) were then measured by flow cytometry. Antigen specific cytolysis were calculated with the following formula: %Cytolysis = (%EG7effector-%EG7no effector)/%EG7no effector.
Tumor implantation experiments
For engraftment studies, 5 × 105 modified EG7 cells were implanted subcutaneously into the inguinal region of mice. Tumors were measured 2‒3 times per week by calipers and mice with tumors greater than 2.5 cm in any one dimension were sacrificed. Tumors were also quantified by mass at the time of death. Tumor volume was calculated, as described before, using a standard formula for estimation of volume based on two dimensional caliper measurements.54 For tumor vaccine experiments, modified EG7 cells were treated with 50 µg/ml of mitomycin C in PBS for 5 h at 37 °C. Cells were washed 4 times with 10% FCS in PBS, counted and 5 × 105 treated cells were injected into mice. Draining lymph nodes were harvested from tumor bearing mice, purified by negative selection and allowed to proliferate on BMDCs fed tumor as above. Therapeutic vaccine experiments were performed as above with 5 × 105 cells used in initial tumor implantation and for vaccine dose. Measurements were performed by a member of the lab (T.B.) who was blinded to therapy for the entire duration of the experiment.
Live cell imaging
A pDV Deltavision deconvolution microscope equipped with a 20× Olympus objective, Cool Snap HQ2 camera, and FITC filters was used for all imaging experiments. The time-lapsed imaging was controlled with Deltavision SoftWoRx software. A single brightfield and fluorescent image was acquired every 10 s for 4 h at 37 °C. Images were processed and interaction times were analyzed in ImageJ (NIH). Tumor cell interactions that initiated and commenced within experiment duration were analyzed. Interaction was defined as when the DC showed membrane spreading across the cancer cell surface or membrane projections that continually sampled the cancer cell surface.
Supplementary Material
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/mabs/article/27052
Acknowledgments
This study was performed by funding from biomedical research endowment to Pasare C from UT Southwestern Medical center. Furlan S was supported by funding from Children’s Cancer Fund (Dallas, TX). The authors thank Dr James Forman for critical reading of the manuscript.
Disclosure of Potential Conflicts of Interest
No potential conflict of interest was disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/27052
References
- 1.Wagle N, Emery C, Berger MF, Davis MJ, Sawyer A, Pochanard P, Kehoe SM, Johannessen CM, Macconaill LE, Hahn WC, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29:3085–96. doi: 10.1200/JCO.2010.33.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg SA, Restifo NP, Yang JC, Morgan RA, Dudley ME. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12:265–77. doi: 10.1038/nrc3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327:291–5. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, et al. IMPACT Study Investigators Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–22. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 7.Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12:557–69. doi: 10.1038/nri3254. [DOI] [PubMed] [Google Scholar]
- 8.Topalian SL, Weiner GJ, Pardoll DM. Cancer immunotherapy comes of age. J Clin Oncol. 2011;29:4828–36. doi: 10.1200/JCO.2011.38.0899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schuler G, Schuler-Thurner B, Steinman RM. The use of dendritic cells in cancer immunotherapy. Curr Opin Immunol. 2003;15:138–47. doi: 10.1016/S0952-7915(03)00015-3. [DOI] [PubMed] [Google Scholar]
- 10.Bousso P, Robey E. Dynamics of CD8+ T cell priming by dendritic cells in intact lymph nodes. Nat Immunol. 2003;4:579–85. doi: 10.1038/ni928. [DOI] [PubMed] [Google Scholar]
- 11.Janeway CA., Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54:1–13. doi: 10.1101/SQB.1989.054.01.003. [DOI] [PubMed] [Google Scholar]
- 12.Flinsenberg TW, Compeer EB, Koning D, Klein M, Amelung FJ, van Baarle D, Boelens JJ, Boes M. Fcγ receptor antigen targeting potentiates cross-presentation by human blood and lymphoid tissue BDCA-3+ dendritic cells. Blood. 2012;120:5163–72. doi: 10.1182/blood-2012-06-434498. [DOI] [PubMed] [Google Scholar]
- 13.Cruz LJ, Rueda F, Cordobilla B, Simón L, Hosta L, Albericio F, Domingo JC. Targeting nanosystems to human DCs via Fc receptor as an effective strategy to deliver antigen for immunotherapy. Mol Pharm. 2011;8:104–16. doi: 10.1021/mp100178k. [DOI] [PubMed] [Google Scholar]
- 14.Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol. 2012;30:1–22. doi: 10.1146/annurev-immunol-100311-102839. [DOI] [PubMed] [Google Scholar]
- 15.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008;8:34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 16.Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol. 2001;19:275–90. doi: 10.1146/annurev.immunol.19.1.275. [DOI] [PubMed] [Google Scholar]
- 17.Nimmerjahn F, Ravetch JV. Antibodies, Fc receptors and cancer. Curr Opin Immunol. 2007;19:239–45. doi: 10.1016/j.coi.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 18.Desai DD, Harbers SO, Flores M, Colonna L, Downie MP, Bergtold A, Jung S, Clynes R. Fc gamma receptor IIB on dendritic cells enforces peripheral tolerance by inhibiting effector T cell responses. J Immunol. 2007;178:6217–26. doi: 10.4049/jimmunol.178.10.6217. [DOI] [PubMed] [Google Scholar]
- 19.Boross P, Leusen JH. Mechanisms of action of CD20 antibodies. Am J Cancer Res. 2012;2:676–90. [PMC free article] [PubMed] [Google Scholar]
- 20.Pokrass MJ, Liu MF, Lindorfer MA, Taylor RP. Activation of complement by monoclonal antibodies that target cell-associated β₂-microglobulin: implications for cancer immunotherapy. Mol Immunol. 2013;56:549–60. doi: 10.1016/j.molimm.2013.05.242. [DOI] [PubMed] [Google Scholar]
- 21.Boross P, Lohse S, Nederend M, Jansen JH, van Tetering G, Dechant M, Peipp M, Royle L, Liew LP, Boon L, et al. IgA EGFR antibodies mediate tumour killing in vivo. EMBO Mol Med. 2013;5:1213–26. doi: 10.1002/emmm.201201929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scott AM, Wolchok JD, Old LJ. Antibody therapy of cancer. Nat Rev Cancer. 2012;12:278–87. doi: 10.1038/nrc3236. [DOI] [PubMed] [Google Scholar]
- 23.Signorino E, Brusa D, Granata R, Malavasi F, Ferrone S, Matera L. Contribution of dendritic cells’ FcgammaRI and FcgammaRIII to cross-presentation of tumor cells opsonized with the anti-MHC class I monoclonal antibodies. Cancer Biol Ther. 2007;6:1932–7. doi: 10.4161/cbt.6.12.4973. [DOI] [PubMed] [Google Scholar]
- 24.Dhodapkar KM, Krasovsky J, Williamson B, Dhodapkar MV. Antitumor monoclonal antibodies enhance cross-presentation ofcCellular antigens and the generation of myeloma-specific killer T cells by dendritic cells. J Exp Med. 2002;195:125–33. doi: 10.1084/jem.20011097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weiner LM, Dhodapkar MV, Ferrone S. Monoclonal antibodies for cancer immunotherapy. Lancet. 2009;373:1033–40. doi: 10.1016/S0140-6736(09)60251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rafiq K, Bergtold A, Clynes R. Immune complex-mediated antigen presentation induces tumor immunity. J Clin Invest. 2002;110:71–9. doi: 10.1172/JCI15640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harbers SO, Crocker A, Catalano G, D’Agati V, Jung S, Desai DD, Clynes R. Antibody-enhanced cross-presentation of self antigen breaks T cell tolerance. J Clin Invest. 2007;117:1361–9. doi: 10.1172/JCI29470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Getahun A, Dahlström J, Wernersson S, Heyman B. IgG2a-mediated enhancement of antibody and T cell responses and its relation to inhibitory and activating Fc gamma receptors. J Immunol. 2004;172:5269–76. doi: 10.4049/jimmunol.172.9.5269. [DOI] [PubMed] [Google Scholar]
- 29.Regnault A, Lankar D, Lacabanne V, Rodriguez A, Théry C, Rescigno M, Saito T, Verbeek S, Bonnerot C, Ricciardi-Castagnoli P, et al. Fcgamma receptor-mediated induction of dendritic cell maturation and major histocompatibility complex class I-restricted antigen presentation after immune complex internalization. J Exp Med. 1999;189:371–80. doi: 10.1084/jem.189.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amigorena S. Fc gamma receptors and cross-presentation in dendritic cells. J Exp Med. 2002;195:F1–3. doi: 10.1084/jem.20011925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.den Haan JM, Bevan MJ. Constitutive versus activation-dependent cross-presentation of immune complexes by CD8(+) and CD8(-) dendritic cells in vivo. J Exp Med. 2002;196:817–27. doi: 10.1084/jem.20020295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stabila PF, Wong SC, Kaplan FA, Tao W. Cell surface expression of a human IgG Fc chimera activates macrophages through Fc receptors. Nat Biotechnol. 1998;16:1357–60. doi: 10.1038/4339. [DOI] [PubMed] [Google Scholar]
- 33.Takashima Y, Tsukamoto M, Ota H, Matsumoto Y, Hayashi Y, Otsuka H. Immunization with pseudorabies virus harboring Fc domain of IgG makes a contribution to protection of mice from lethal challenge. Vaccine. 2005;23:3775–82. doi: 10.1016/j.vaccine.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 34.Moore MW, Carbone FR, Bevan MJ. Introduction of soluble protein into the class I pathway of antigen processing and presentation. Cell. 1988;54:777–85. doi: 10.1016/S0092-8674(88)91043-4. [DOI] [PubMed] [Google Scholar]
- 35.Mayordomo JI, Zorina T, Storkus WJ, Zitvogel L, Celluzzi C, Falo LD, Melief CJ, Ildstad ST, Kast WM, Deleo AB, et al. Bone marrow-derived dendritic cells pulsed with synthetic tumour peptides elicit protective and therapeutic antitumour immunity. Nat Med. 1995;1:1297–302. doi: 10.1038/nm1295-1297. [DOI] [PubMed] [Google Scholar]
- 36.Mach N, Gillessen S, Wilson SB, Sheehan C, Mihm M, Dranoff G. Differences in dendritic cells stimulated in vivo by tumors engineered to secrete granulocyte-macrophage colony-stimulating factor or Flt3-ligand. Cancer Res. 2000;60:3239–46. [PubMed] [Google Scholar]
- 37.Clarke SR, Barnden M, Kurts C, Carbone FR, Miller JF, Heath WR. Characterization of the ovalbumin-specific TCR transgenic line OT-I: MHC elements for positive and negative selection. Immunol Cell Biol. 2000;78:110–7. doi: 10.1046/j.1440-1711.2000.00889.x. [DOI] [PubMed] [Google Scholar]
- 38.Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76:34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- 39.Longhi MP, Trumpfheller C, Idoyaga J, Caskey M, Matos I, Kluger C, Salazar AM, Colonna M, Steinman RM. Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J Exp Med. 2009;206:1589–602. doi: 10.1084/jem.20090247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferris RL, Jaffee EM, Ferrone S. Tumor antigen-targeted, monoclonal antibody-based immunotherapy: clinical response, cellular immunity, and immunoescape. J Clin Oncol. 2010;28:4390–9. doi: 10.1200/JCO.2009.27.6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dhodapkar KM, Krasovsky J, Williamson B, Dhodapkar MV. Antitumor monoclonal antibodies enhance cross-presentation ofcCellular antigens and the generation of myeloma-specific killer T cells by dendritic cells. J Exp Med. 2002;195:125–33. doi: 10.1084/jem.20011097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harbers SO, Crocker A, Catalano G, D’Agati V, Jung S, Desai DD, Clynes R. Antibody-enhanced cross-presentation of self antigen breaks T cell tolerance. J Clin Invest. 2007;117:1361–9. doi: 10.1172/JCI29470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Regnault A, Lankar D, Lacabanne V, Rodriguez A, Théry C, Rescigno M, Saito T, Verbeek S, Bonnerot C, Ricciardi-Castagnoli P, et al. Fcgamma receptor-mediated induction of dendritic cell maturation and major histocompatibility complex class I-restricted antigen presentation after immune complex internalization. J Exp Med. 1999;189:371–80. doi: 10.1084/jem.189.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harshyne LA, Watkins SC, Gambotto A, Barratt-Boyes SM. Dendritic cells acquire antigens from live cells for cross-presentation to CTL. J Immunol. 2001;166:3717–23. doi: 10.4049/jimmunol.166.6.3717. [DOI] [PubMed] [Google Scholar]
- 45.Nierkens S, Tel J, Janssen E, Adema GJ. Antigen cross-presentation by dendritic cell subsets: one general or all sergeants? Trends Immunol. 2013;34:361–70. doi: 10.1016/j.it.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Asano K, Nabeyama A, Miyake Y, Qiu CH, Kurita A, Tomura M, Kanagawa O, Fujii S, Tanaka M. CD169-positive macrophages dominate antitumor immunity by crosspresenting dead cell-associated antigens. Immunity. 2011;34:85–95. doi: 10.1016/j.immuni.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 47.Tseng D, Volkmer JP, Willingham SB, Contreras-Trujillo H, Fathman JW, Fernhoff NB, Seita J, Inlay MA, Weiskopf K, Miyanishi M, et al. Anti-CD47 antibody-mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc Natl Acad Sci U S A. 2013;110:11103–8. doi: 10.1073/pnas.1305569110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pardigon N, Bercovici N, Calbo S, Santos-Lima EC, Liblau R, Kourilsky P, Abastado JP. Role of co-stimulation in CD8+ T cell activation. Int Immunol. 1998;10:619–30. doi: 10.1093/intimm/10.5.619. [DOI] [PubMed] [Google Scholar]
- 49.Shahinian A, Pfeffer K, Lee KP, Kündig TM, Kishihara K, Wakeham A, Kawai K, Ohashi PS, Thompson CB, Mak TW. Differential T cell costimulatory requirements in CD28-deficient mice. Science. 1993;261:609–12. doi: 10.1126/science.7688139. [DOI] [PubMed] [Google Scholar]
- 50.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–6. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- 51.Riddle DS, Sanz L, Chong H, Thompson J, Vile RG. Tumor cell surface display of immunoglobulin heavy chain Fc by gene transfer as a means to mimic antibody therapy. Hum Gene Ther. 2005;16:830–44. doi: 10.1089/hum.2005.16.830. [DOI] [PubMed] [Google Scholar]
- 52.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–6. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 53.Pasare C, Medzhitov R. Toll-dependent control mechanisms of CD4 T cell activation. Immunity. 2004;21:733–41. doi: 10.1016/j.immuni.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 54.Euhus DM, Hudd C, LaRegina MC, Johnson FE. Tumor measurement in the nude mouse. J Surg Oncol. 1986;31:229–34. doi: 10.1002/jso.2930310402. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.