

Abstract
Circadian rhythms control metabolism and energy homeostasis, but the role of the skeletal muscle clock has never been explored. We generated conditional and inducible mouse lines with muscle-specific ablation of the core clock gene Bmal1. Skeletal muscles from these mice showed impaired insulin-stimulated glucose uptake with reduced protein levels of GLUT4, the insulin-dependent glucose transporter, and TBC1D1, a Rab-GTPase involved in GLUT4 translocation. Pyruvate dehydrogenase (PDH) activity was also reduced due to altered expression of circadian genes Pdk4 and Pdp1, coding for PDH kinase and phosphatase, respectively. PDH inhibition leads to reduced glucose oxidation and diversion of glycolytic intermediates to alternative metabolic pathways, as revealed by metabolome analysis. The impaired glucose metabolism induced by muscle-specific Bmal1 knockout suggests that a major physiological role of the muscle clock is to prepare for the transition from the rest/fasting phase to the active/feeding phase, when glucose becomes the predominant fuel for skeletal muscle.
Abbreviations: BSA, bovine serum albumin; 2-DG, 2-Deoxyglucose; GSEA, Gene Set Enrichment Analysis; imKO, inducible muscle-specific Bmal1 knockout; HK2, hexokinase 2; KHB, Krebs–Henseleit buffer; mKO, muscle-specific Bmal1 knockout; PDH, pyruvate dehydrogenase; PDK, PDH kinase; PDP, PDH phosphatase; SCN, suprachiasmatic nucleus; ZT, Zeitgeber time
Keywords: Bmal1, Circadian rhythms, Glucose metabolism, Glucose uptake, Skeletal muscle, Muscle insulin resistance
1. Introduction
Circadian rhythms and energy metabolism are inextricably linked. While many behavioral and physiological processes linked to energy metabolism show daily fluctuations under the control of an endogenous circadian clock and environmental cues, the circadian clock is itself influenced by feeding and activity rhythms, and ultimately metabolic state [1–3]. The regulation of body metabolism by normal circadian rhythms has important clinical implications, since chronic circadian misalignment, as occurs in shift work, is associated with a higher prevalence of insulin resistance, obesity, metabolic syndrome and diabetes [4–6].
The circadian timing system is composed of a central pacemaker located in the suprachiasmatic nucleus (SCN) of the hypothalamus and autonomous molecular oscillators present in all cells of the body. At the cellular level, the core of this 24-h rhythm generating oscillator is composed of molecular feedback loops [7]. In the major loop, the transcription factors BMAL1 and CLOCK bind as heterodimers to the promoters of Per and Cry genes inducing their transcription. Through negative feedback, PER and CRY proteins translocate to the nucleus and repress the CLOCK:BMAL1 complex. In a second regulatory loop, the expression of Bmal1 (Arntl) is controlled by CLOCK:BMAL1-dependent transcriptional activators of the ROR family, such as RORα, and repressors of the REV-ERB family, such as REV-ERBα. In addition to the core clock genes, all tissues contain a large number of oscillating genes, most of which are tissue-specific and regulated either directly by the core oscillator or by extrinsic circadian signals [7].
An important role for circadian clocks in the regulation of glucose metabolism is supported by genome-wide DNA-binding analyses and by ablation of clock genes. A time-resolved and genome-wide map of BMAL1 binding in mouse liver, derived from ChIP-seq analyses, has shown that carbohydrate metabolism is a major output of the circadian clock in this tissue [8]. Global inactivation of different clock genes leads to altered glucose metabolism: Clock mutant mice display hyperglycemia, obesity and metabolic syndrome [9]; Per1/Per2 double knockout mice and Cry1/Cr2 double knockout mice exhibit glucose intolerance [10,11]; Bmal1 null mice are also glucose intolerant, and show altered gluconeogenesis [10,12]. In addition, the normal circadian rhythm of increased insulin sensitivity during the active/feeding phase is completely abolished in global Bmal1 null mice [13].
Interpretation of whole body knockout studies is complicated by the fact that all tissues are affected, including both the central pacemaker and peripheral clocks. The hierarchical role of the central pacemaker was demonstrated by the finding that circadian rhythms of blood glucose, insulin and glucagon, as well as glucose tolerance, are all abolished by SCN lesion [14]. An equally important role for peripheral oscillators has been demonstrated by tissue-specific knockout of clock genes, with distinct effects depending on the tissue. Selective knockout of Bmal1 in the liver causes hypoglycemia [10], while ablation of the same gene in the endocrine pancreas leads to fasting hyperglycemia and severe glucose intolerance [15]. Local insulin sensitivity was found to be normal in mice lacking Bmal1 selectively in the liver or adipose tissue [10,16].
Although skeletal muscle is a primary site of insulin-dependent glucose disposal, the specific role the muscle clock plays in regulating glucose uptake and metabolism is not known. A daily rhythm in plasma glucose concentration has been described in both animals and humans, with a peak just before the start of the main activity period, reflecting fluctuations in glucose export, glucose uptake and insulin sensitivity [14]. However, it is not clear whether these circadian rhythms are controlled by peripheral clocks in muscle and adipose tissue, or by humoral or neural signals emanating from the SCN. A circadian rhythm of glucose uptake was also demonstrated in muscle cell cultures, but these daily fluctuations were observed both in basal and insulin-stimulated conditions [17]. To understand the physiological role the intrinsic muscle clock plays in glucose metabolism, we have generated two skeletal muscle-specific Bmal1 knockout models. We report here that muscle-specific Bmal1 ablation causes impaired insulin-dependent glucose uptake and reduced glucose oxidation in skeletal muscle, and we identify potential mechanisms involved in mediating these effects.
2. Materials and methods
2.1. Animals
Muscle-specific inactivation of Bmal1 (mKO) was obtained from the cross between a C57BL/6 mouse line with floxed Bmal1 [18] and a C57BL/6 mouse line carrying a Cre recombinase transgene under control of the Mlc1f promoter (Mlc1f–Cre) [19]. In the resulting mKO mice, the region coding for the BMAL1 basic helix–loop–helix DNA binding domain is excised. Cre-negative littermates were used as controls.
A second knockout model with inducible muscle-specific inactivation of Bmal1 (imKO) was obtained by crossing the Bmal1 floxed line with mice carrying an α-skeletal actin driven Cre recombinase fused to a mutated estrogen receptor [20], which can be activated by treatment with tamoxifen (i.p. 1 mg/day for 5 days). Cre negative littermates, also receiving tamoxifen treatment, were used as controls. All strains had been backcrossed a minimum of 6 times with C57BL/6 mice. Experimental protocols were reviewed and approved by the local Animal Care Committee, University of Padova. Animals were housed in a temperature-controlled room (22 °C) under a 12 h light–dark regimen, with lights on at ZT0 (6 am), lights off at ZT12 (6 pm), with standard chow diet (Mucedola, Settimo Milanese, Italy) and water provided ad libitum. All tissues were collected immediately after cervical dislocation at ZT0, 4, 8, 12, 16, and 20, snap frozen in liquid nitrogen and stored at −80 °C until subsequent use. To monitor locomotor activity levels, a telemetric sensor (PhysioTel, TA10TA-F20, Data Sciences, Inc.) was implanted i.p. under general anesthesia. Mice were allowed to recover for at least 2 weeks, and locomotor activity was recorded continuously for 12 days.
2.2. Immunofluorescence
Cryosections of fast and slow muscles were stained with monoclonal anti-myosin heavy chain (MyHC) antibodies produced in our lab [21], and now distributed by the Developmental Studies Hybridoma Bank (DSHB, University of Iowa): BA-D5 (IgG2b, supernatant, 1:100 dilution) specific for MyHC-I, SC-71 (IgG1, supernatant, 1:100 dilution) specific for MyHC-2A and BF-F3 (IgM, purified antibody, 1:100 dilution) specific for MyHC-2B. Type 2X fibers are not recognized by these antibodies, and so appear black. Three different secondary antibodies (Jackson ImmunoResearch) were used to selectively bind to each primary antibody: goat anti-mouse IgG1, conjugated with DyLight488 fluorophore (to bind to SC-71); goat anti-mouse IgG2b, conjugated with DyLight405 fluorophore (to bind to BA-D5); goat anti-mouse IgM, conjugated with DyLight549 fluorophore (to bind to BF-F3). Muscle sections, 8 µm thick, were incubated with M.O.M. IgG blocking solution (Vector) for 1 h at room temperature, then briefly washed twice with PBS for 2 min. A solution with all the primary antibodies in phosphate-buffered saline (PBS) containing 0.5% of bovine serum albumin (BSA) was then prepared, and sections were incubated for 1 h at 37 °C. After 3 washes (5 min each) with PBS, sections were incubated for 30 min at 37 °C with a solution with the three different secondary antibodies, diluted in PBS containing 0.5% of BSA and 5% of goat serum. After 3 washes with PBS (5 min each) and a brief rinse in water, sections were mounted with Elvanol. A control incubation with no primary antibodies was performed, and also control incubations with each primary antibody and non-specific secondary antibodies to exclude any possible cross-reaction. Pictures were collected with an epifluorescence Leica DM5000 B equipped with a Leica DFC 300 FX camera. Single-color images were merged to obtain a whole muscle reconstruction with Adobe Photoshop CS2 (Adobe Systems Inc.).
2.3. Electron microscopy
Muscles were fixed at resting length in 2.5% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4. During subsequent washing in phosphate buffer, small bundles of fibers were dissected from the superficial regions of the muscles and postfixed in 1% OsO4 in phosphate buffer and treated with uranyl acetate before dehydration and embedding in Epon. Ultrathin sections were stained with lead citrate.
2.4. Force measurements
In vivo force measurements were performed as described previously [22]. Briefly, mice were anesthetized and stainless steel electrode wires were placed on either side of the sciatic nerve. Torque production of the plantar flexors was measured using a muscle lever system (Model 305c; Aurora Scientific, Aurora ON, Canada). The force–frequency curves were determined by increasing the stimulation frequency in a stepwise manner, pausing for 30 s between stimuli to avoid effects due to fatigue. Muscle force was normalized for the weight of the gastrocnemius muscle.
2.5. RNA isolation and qPCR
Total RNA was isolated using TRIzol (Invitrogen) followed by cleanup with the RNeasy Mini Kit (Qiagen). RNA integrity was evaluated with the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA) and quantified with a NanoVue spectrophotometer (GE Healthcare Life Sciences, Baie d'Urfe, QC). Complementary DNA from each sample was generated from 0.8 μg of RNA reverse-transcribed with Invitrogen Superscript III reverse transcriptase. Primer sets were designed using Primer-BLAST (NCBI) and were each validated prior to use by gradient PCR and gel analysis to test for optimal annealing temperature, reaction efficiency and specificity. Duplicates of cDNA samples were then amplified on the 7900HT Fast Real-Time PCR System (Applied Biosystems) using the Fast SYBR Green RT-PCR kit (Applied Biosystems). Specificity of gene amplification was confirmed by analyzing the dissociation curve with SDS 2.4 software (Applied Biosystems). Analysis was performed using the standard curve method and all data were normalized relative to 36B4 expression and plotted in arbitrary units as mean±SEM. Primer sequences are available upon request.
2.6. Gene expression profiling and analyses
For gene expression profiling, 250 ng of RNA from tibialis anterior and soleus muscles was hybridized to Mouse Gene 1.0 ST Arrays (Affymetrix) using three biological replicates for each time point. Expression values were generated from fluorescence signals using the robust multi-array average procedure (RMA) [23]. Specifically, intensity levels have been background adjusted, normalized using quantile normalization, and log 2 expression values calculated using median polish summarization and Entrez custom chip definition files for mouse arrays (version 14.1.0) [24]. Differentially expressed genes were identified using the microarray Significant Profiles method coded in the maSigPro R package [25] with default parameters. Rhythmic genes that cycle with a 24-h period were identified using a Benjamini–Hochberg q-value<0.2 in the non-parametric algorithm JTK_Cycle [26]. Over-representation analysis was performed using Gene Set Enrichment Analysis software (GSEA; http://www.broadinstitute.org/gsea/index.jsp) [27] and gene sets from the KEGG database. Gene sets were defined as significantly enriched if the False Discovery Rate (FDR) was <0.25 when using Signal2Noise as metric and 10,000 permutations of phenotype labels. Expression data are available at Gene Expression Omnibus under the accession number GSE43071.
2.7. Global metabolic profiling
Metabolic profiling, peak identification, and curation were performed by Metabolon using described methods [28]. Briefly, the non-targeted metabolic profiling platform used by Metabolon combines three independent platforms: UHPLC/MS/MS optimized for basic species, UHPLC/MS/MS optimized for acidic species, and GC/MS. We examined 60 muscles (tibialis anterior) from distinct mice, corresponding to control and muscle-specific Bmal1 knockout, at 6 time points, 5 muscles per point. A total of 277 biochemicals were identified, but only sugar metabolites will be reported here (see Table S1).
2.8. CircadiOmics
All transcriptome and metabolome data are accessible through the CircadiOmics Website (http://circadiomics.igb.uci.edu), allowing users to search and view genes of interest and detected metabolites in Ctrl and Bmal1 mKO skeletal muscles, as well as their interactions and statistics. Importantly, gene expression and metabolite levels were derived from contralateral hindlimb muscles from the same animals, allowing for a more direct correlation of these metabolite–gene interactions. Networks and subnetworks can be explored and rearranged interactively, providing a comprehensive systems-level view of the circadian molecular pathways [29].
2.9. Comparative analysis of genomic sequences
For the identification of potential binding sites for clock genes, the genomic sequences for 6 eutherian species (about 400 bp upstream and 400 bp downstream of the relevant binding motif) were analyzed using the mVISTA tool (http://genome.lbl.gov/vista) and aligned with LAGAN. The conservation parameters were set as 70% of conservation identity and 100 bp minimum conservation window.
2.10. Glucose and insulin tolerance tests
Blood glucose was measured after an 8 h fast using a Glucocard G+ meter (Arkray Factory, Shiga, Japan). Glucose tolerance tests (GTT) and insulin tolerance tests (ITT) were performed at ZT12, coinciding with the hours near awakening and the normal physiological peak of glucose tolerance and insulin sensitivity [14]. Mice were fasted for 8 h before analysis during their normal rest/fasting phase. For GTT, mice were injected i.p. with glucose (2 g/kg body weight). Blood glucose was measured at 0, 10, 20, 30, 60 and 120 min via the tail vein. For ITT, mice were injected i.p. with human insulin (0.75 U/kg body weight; Sigma) and blood glucose was measured at −10, 0, 10, 20, 30, 60, and 90 min via the tail vein.
2.11. Insulin-stimulated glucose transport in isolated skeletal muscle
All experiments were performed between ZT8 and ZT12 in order to coincide with the hours immediately before and during awakening, the normal physiological peak of glucose tolerance and insulin sensitivity [14]. Mice were sacrificed by cervical dislocation and soleus muscles were rapidly and carefully removed. The method to measure glucose transport was similar to that described previously [30,31] with some modifications [32]. Muscles were pre-incubated for 40 min at 30 °C under gentle shaking (200 RPM) in 2 mL of a daily prepared, pre-gassed (1 h, 95% O2+5% CO2) modified Krebs–Henseleit buffer (KHB): 118 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 15 mM CO2–gassed NaHCO3, 1% fatty acid free bovine serum albumin (BSA), 5 mM glucose, and 10 mM HEPES. Thereafter, muscles were incubated an additional 20 min at 30 °C under gentle shaking (200 RPM) in 2 mL KHB+radiolabelled [3H]-2-Deoxyglucose (2-DG) (1.5 μCi/mL) with either 0, or for contralateral muscles, 2 mU/mL insulin (Sigma). Muscles were quickly removed, rinsed in sterile saline, and washed 3×5 min (100 RPM) in ice-cold KHB to clear extracellular space of 2-DG [33]. Muscles were then rinsed again in sterile saline, blotted with filter paper, weighed, and homogenized in 1 M KOH at 70 °C with periodic agitation for 15 min. Homogenates were centrifuged 10,000g at 4 °C for 5 min to clear insoluble components, and supernatants were pipetted into vials with 10 mL scintillation cocktail and [3H]-2-DG disintegrations per minute (dpm) for each vial were determined using a scintillation counter.
2.12. Glucose oxidation
All experiments were performed on mKO and control mice, and 24-h fasted control mice, between ZT8 and ZT12, again to coincide with the hours near awakening and the normal physiological peak of glucose tolerance and insulin sensitivity [14]. The method to measure glucose oxidation was similar to that previously described [34] with some modifications. Mice were sacrificed by cervical dislocation, and diaphragm muscles were rapidly and carefully removed, blotted, cut in half, weighed, and pre-incubated for 30 min at 37 °C under gentle shaking (200 RPM) in 0.5 mL of a daily prepared, pre-gassed (1 h, 95% O2+5% CO2) modified Krebs–Henseleit buffer (KHB): 118 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 15 mM CO2−gassed NaHCO3, 1% fatty acid free BSA, 5 mM glucose, and 10 mM HEPES. Thereafter, muscles were incubated an additional 90 min at 37 °C under gentle shaking (200 RPM) in 0.5 mL KHB+radiolabelled [1-14C]-glucose (0.5 μCi/mL). Reactions were terminated by addition of 100 μL 1 M perchloric acid, and the CO2 produced during the incubation was trapped in 100 μL NaOH that had been added to a small tube inside the reaction vial. Glucose oxidation rates were determined by measuring incorporation into 14CO2 by liquid scintillation counting.
2.13. PDHa activity
The activity of PDH in the active form (PDHa) was determined as previously described [35–37] after homogenization of 10–15 mg gastrocnemius muscle on ice, for 50 s using a glass homogenizer (Kontes, Vineland, NJ, USA) and quick-frozen in liquid nitrogen.
2.14. SDS-PAGE and western blotting
Muscle lysates were prepared as previously reported [38], protein concentration determined using the bicinchoninic acid assay (Pierce, Rockford, IL, USA) and SDS-PAGE performed using 10% hand-cast gels. Western blotting analysis was performed using specific antibodies to BMAL1 (kind gift from Dr. Charles Weitz), AKT1, AKT2 and HK2 (cell signaling), β Tubulin and Actin (Santa Cruz), phospho-AKT Ser473 and phospho-AKT Thr308 (Millipore), GLUT4 (Thermo Scientific), TBC1D1 (kind gift from Prof. Dr. Hadi Al-Hasani), PDH-E1α, phospho-PDH-E1α Ser300, and PDK4 (kind gifts from Prof. Grahame Hardie) and PDP1 (kind gift from Dr. Brian Robinson). The bands were visualized and quantified (Carestream Health, Rochester, NY, USA), then normalized by an internal standard [38].
2.15. Statistical analysis
All data are expressed as means±SEM unless otherwise stated. Statistical analysis was performed using unpaired Student's t-test or 2-way analysis of variant (ANOVA). When ANOVA revealed significant differences, further analysis was performed using Bonferroni's multiple comparison test. Differences between groups were considered statistically significant for p<0.05.
3. Results
3.1. Muscle-specific Bmal1 knockout mice
To determine the role of the muscle clock, we produced two mouse lines with skeletal muscle-specific knockout of Bmal1 (Arntl), an essential component of the core clock. The first line was a traditional tissue-specific knockout, for which we bred mice with a floxed Bmal1 allele (Bmal1fl/fl) [18] with mice carrying a Cre recombinase transgene under control of the Mlc1f promoter (Mlc1f–Cre), which is specifically active in skeletal muscle [19] (Figure S1A). The resulting muscle-specific knockout (hereafter referred to as mKO) mice showed a disrupted Bmal1 gene in skeletal muscles but not in heart and liver (Figure S1B) and markedly decreased levels of BMAL1 protein in skeletal muscle (Figure S1C). Bmal1 transcripts were reduced across the circadian cycle in both fast muscles (tibialis anterior, TA) and slow muscles (soleus, SOL) compared to control littermates (Bmal1fl/fl; Cre−/−), whereas Bmal1 levels and rhythmic expression were unchanged in cardiac muscle (Figure S1D). We also generated an inducible muscle-specific Bmal1 knockout line (hereafter referred to as imKO) by crossing floxed Bmal1 mice with HSA-Cre-ER mice, in which the human skeletal actin promoter controls a chimeric Cre recombinase fused with a mutated estrogen receptor that can be activated by tamoxifen [20] (Figure S1E). Two-month-old imKO mice and their control littermates were treated with tamoxifen daily for 5 days and examined at 1 or 5 months after tamoxifen treatment. Cre-mediated recombination of the Bmal1 gene was seen in skeletal muscles but not in the heart (Figure S1F). Bmal1 transcripts were reduced across the circadian cycle in skeletal muscles from imKO mice after tamoxifen treatment (Figure S1G), and BMAL1 protein levels were likewise markedly reduced (Figure S1H).
The phenotype of mKO mice was strikingly different from that of whole body Bmal1 knockout mice. Mice with whole body Bmal1 knockout were reported to stop growing around 16 weeks of age, display progressive arthropathy, sarcopenia and loss of fat tissue, and die between 26 and 52 weeks of age [39,40]. In contrast, mKO mice had a normal life span (Figure 1A) and growth curve (Figure 1B), with body weight being slightly increased compared to controls at advanced age. The weight of both fast and slow hindlimb muscles was significantly increased compared to controls (Figure 1C). In contrast, no significant difference was seen in the weight of epididymal fat pads (Figure 1D). The fiber type profile of mKO mice was similar to controls, with the slow soleus predominantly composed of type 1 and 2A fibers, with few type 2X fibers, and the fast extensor digitorum longus (EDL) predominantly composed of type 2B and 2X fibers, with few type 2A fibers (Figure 1E). However, quantitative analysis revealed a slight decrease in type 1 and a corresponding increase in type 2A fibers in soleus, and a very slight decrease in the relative proportion of type 2X fibers in EDL (Figure 1F). Muscle histology was apparently normal in mKO muscles even at advanced age, with no sign of fiber degeneration or regeneration or fibrosis (Figure 1G). Muscle force, as determined in vivo, showed a significant decrease when normalized for muscle mass (Figure 1H). A similar decrease in muscle force was previously reported for whole body Bmal1 knockout mice, which also show a disruption of sarcomere architecture, with loss of the regular disposition of thick and thin filaments [41]. However, electron microscopy revealed no significant change in muscle ultrastructure of mKO muscles: in particular, sarcomeres showed regular banding pattern and the hexagonal disposition of thick and thin filaments in cross-section was normal (Figure S2).
Figure 1.
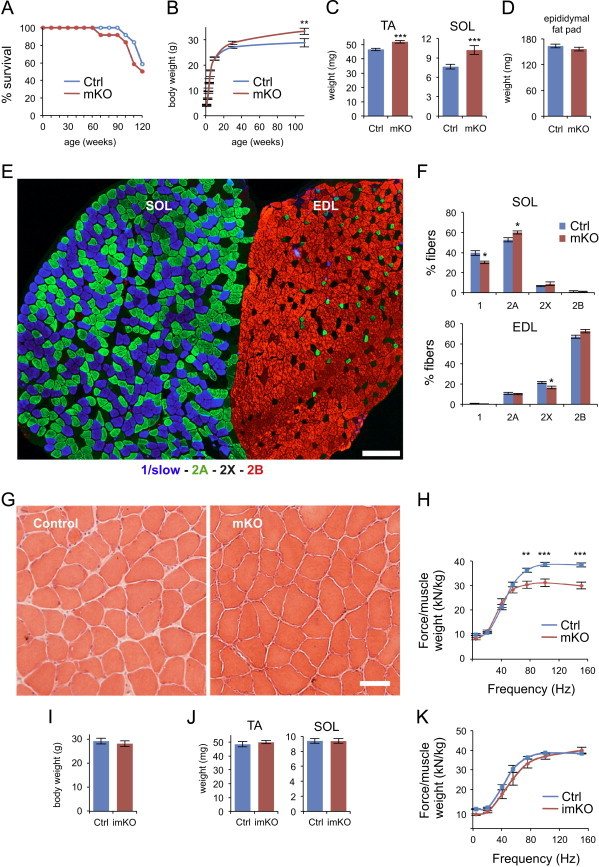
Characterization of skeletal muscle-specific Bmal1 knockout mice. (A) Survival curve of muscle-specific Bmal1 knockout mice (mKO) and control littermates (Ctrl) (n=12/group). (B) Body weight of control and mKO mice (mean±SEM; n=7–10/group/age; **p<0.01, 2-way ANOVA with the Bonferroni correction). (C) Weight of fast tibialis anterior (TA) and slow soleus (SOL) skeletal muscles from 5-month old mKO and control littermates (mean±SEM; n=36/group; ***p<0.001, Student's t-test). (D) Epididymal fat pad weight from 5-month old mKO and control littermates (mean±SEM; n=4/group). (E) Fiber type profile of slow soleus (SOL) and fast extensor digitorum longus (EDL) in transverse sections stained with anti-myosin antibodies specific for type-1/slow (blue), type-2A (green) and type 2B (red) myosin heavy chains. Type 2X fibers are unstained and appear black. Scale bar is 200 µμm. (F) Quantitation of the relative proportion of the different fiber types in SOL and EDL (mean±SEM, n=8 muscles per group with >90% of fibers measured; see methods; *p<0.05, Student's t-test). (G) Hematoxylin and eosin staining of transverse cryosections of SOL muscles from 26-month old mKO and control littermate. Scale bar is 50 µμm. (H) In vivo muscle force measurements performed on gastrocnemius muscles from 5-month old mKO and control littermates. Force normalized to muscle weight (mean±SEM, n=8–10 muscles per group; **p<0.01, ***p<0.001, 2-way ANOVA with Bonferroni correction). (I) Body weight of 7-month old inducible muscle-specific Bmal1 knockout mice (imKO, n=7) and control littermates (Ctrl, n=5) measured 5 months after tamoxifen injections (mean±SEM). (J) Weight of TA and SOL skeletal muscles from 7-month old imKO and control littermates measured 5 months after tamoxifen injections (mean±SEM; n=10–14/group). (K) In vivo muscle force measurements performed on gastrocnemius muscles from 7-month old imKO and control littermates 5 months after tamoxifen injection. Force normalized to muscle weight (mean±SEM, n=8–10 muscles per group).
The phenotype of imKO mice was similar to that of controls when examined at 1 or 5 months after tamoxifen treatment both with respect to body weight (Figure 1I) and muscle weight (Figure 1J). Muscle histology and fiber type profile were normal (not shown) and force production was not significantly different when compared to controls (Figure 1K). In contrast with global Bmal1 knockout, which results in loss of circadian rhythm of locomotor activity and reduced total activity levels [42], the circadian rhythm of locomotor activity was normal in muscle-specific Bmal1 knockout and imKO mice, but surprisingly activity levels were increased during the dark phase in mKO mice (Figure S3).
3.2. Clock gene expression and circadian transcriptome changes in mKO muscles
Bmal1-dependent genes, including Rev-erbα, Rev-erbβ, Dbp, Tef, Hlf, and Cry2, showed markedly reduced oscillation in TA muscles from mKO mice according to microarray analysis (Figure 2A) and qPCR of selected genes (Figure S4). Similar changes were detected in SOL muscles (Figure S5). Per1 expression was also reduced, but Per2 levels were only slightly affected. In contrast, Clock, Npas2 and E4bp4 (Nfil3) were all upregulated in mKO muscles. The Bmal1 paralog, Bmal2 (Arntl2/Mop9), which is downregulated in the liver of Bmal1 null mice [42] and can functionally replace Bmal1 [43], was found to be expressed at low levels without daily fluctuations in mouse skeletal muscle, and was unchanged in mKO muscles (Figure S6). The various responses of clock genes and clock-associated genes to Bmal1 inactivation were in agreement with previous reports for global and tissue-specific Bmal1 knockouts [16]. For example, the fact that Per2 oscillation was only mildly affected in mKO skeletal muscles likely reflects a role for oscillating systemic signals in the regulation of this gene, as was demonstrated in a liver-specific Bmal1 knockout [44,45].
Figure 2.
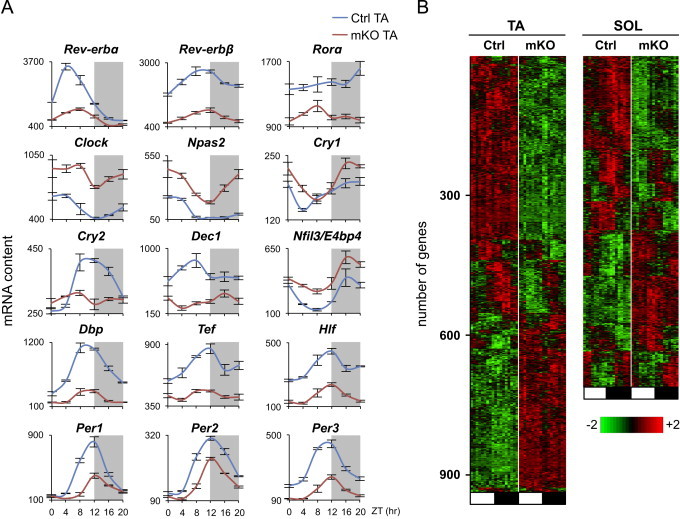
Altered gene expression caused by skeletal muscle-specific Bmal1 knockout. (A) Circadian expression profiles of core clock and clock-associated genes in TA muscles from control and mKO mice. Transcript levels were determined by microarray analysis and plotted as mean absolute expression levels (mean±SEM; n=3/group/timepoint). (B) Phase map of differentially expressed genes in control and mKO skeletal muscles (TA and SOL) identified by maSigPro (see Section 2). Muscles were collected at 4-h intervals throughout the light (ZT0-ZT8, white box) and dark phase (ZT12–ZT20, black box) and hybridized to Affymetrix arrays as described in the methods (n=3/group/timepoint).
Global gene expression was markedly altered in skeletal muscles from mKO mice, with 931 genes in TA and 706 in SOL identified as differentially regulated according to maSigPro analysis (Figure 2B). Out of the 684 (Ctrl TA) and 1359 (Ctrl SOL) 24-h cycling genes identified by the JTK_CYCLE algorithm [26], 487 genes in TA and 666 genes in SOL lost their 24-h oscillation in the absence of Bmal1, showing that a significant proportion of cycling genes in mouse skeletal muscles is controlled by the intrinsic muscle clock.
3.3. Impaired insulin-dependent glucose uptake in skeletal muscles from mKO and imKO mice
Global changes in muscle gene expression were further examined by Gene Set Enrichment Analysis (GSEA) [27] in order to identify significantly enriched signaling pathways altered by muscle-specific Bmal1 knockout. The “KEGG insulin signaling pathway”, comprising 132 genes coding for various components of the insulin-dependent signaling cascade, was among the top ranking gene sets altered in mKO muscles (p=0.024) in both TA (Figure S7A) and SOL muscles (Figure S7B). Transcriptome analysis indicated that the expression of a number of genes coding for major components of the insulin signaling pathway, including PI3K (p110), AKT1 and AKT2, were significantly up- or down-regulated in mKO muscles (Figure S8). Western blotting analyses showed that AKT2, the critical isoform in insulin-dependent glucose uptake, was also increased at the protein level in mKO muscles about 2-fold, whereas AKT1 was only slightly decreased (Figure S9).
To determine whether the gene expression changes shown by GSEA and Western blotting analysis lead to functional alterations in muscle insulin signaling, we measured rates of basal and insulin-stimulated glucose transport with [3H]-2-Deoxyglucose (2-DG) in isolated SOL muscles from mKO mice and their control littermates. Basal rates of 2-DG uptake were not different between control and mKO muscles, and the rate increased (+72%) in control muscle as expected in response to insulin. However, this increase was significantly reduced in mKO muscles (only +29%) (Figure 3A). Reduced sensitivity to insulin was even more striking in the inducible mKO model (imKO), in which no significant effect of insulin stimulation on glucose uptake was detected (Figure 3A). Next, we examined whether reduced muscle insulin sensitivity had consequences on global glucose homeostasis. Fasting blood glucose was unchanged in mKO mice both at ZT0 and ZT12 (Figure 3B). Glucose tolerance and insulin tolerance were likewise unchanged (Figure 3C and D), with similar results obtained in imKO mice (Figure S10).
Figure 3.
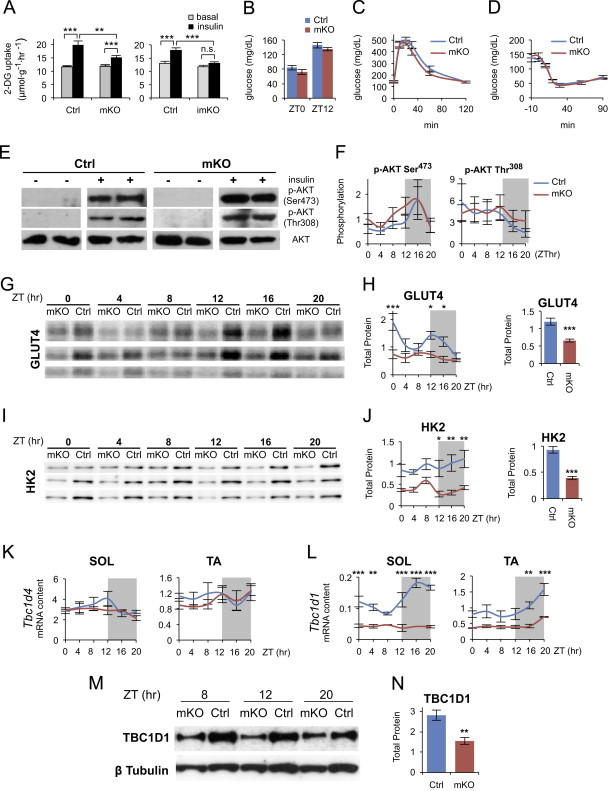
Insulin-dependent glucose uptake is impaired by muscle-specific Bmal1 deletion. (A) Insulin-stimulated [3H]-2-Deoxyglucose (2-DG) uptake by isolated SOL muscles from mKO (left panel) and imKO mice (right panel) versus respective controls (mean±SEM; n=15/group; **p<0.01, ***p<0.001, Student's t-test). (B) Fasting blood glucose levels at ZT0 (n=6/group) and ZT12 (n=10/group) from Ctrl and mKO mice (mean±SEM). (C) Glucose tolerance test performed at ZT12 in mKO mice versus control (mean±SEM; n=10/group). (D) Insulin tolerance test performed at ZT12 in mKO mice versus control (mean±SEM; n=8/group). (E) Basal (−) and insulin-induced (+) AKT Ser473 and Thr308 phosphorylation determined in isolated SOL muscles from mKO and Ctrl mice. (F) In situ diurnal AKT Ser473 phosphorylation (n=5/group/timepoint) and Thr308 phosphorylation (n=3/group/timepoint) in mKO and Ctrl gastrocnemius muscle quantified by densitometry after western blotting analysis (mean±SEM; arbitrary units; AKT Ser473 time effect F=2.88, p=0.02, 2-way ANOVA). (G) Western blots showing protein levels of GLUT4 across the day/night cycle in mKO and Ctrl gastrocnemius muscles from 36 different mice (n=3/group/timepoint). (H) Diurnal protein levels of GLUT4 quantified by densitometry after western blot analysis (mean±SEM; arbitrary units; n=5/group/timepoint; *p<0.05, ***p<0.001, 2-way ANOVA with Bonferroni correction, group effect F=32.2, p<0.0001; time effect F=4.45, p=0.002; group×time interaction F=2.64, p=0.03). Right panel shows mean 24-h GLUT4 protein levels (mean±SEM; arbitrary units; n=30/group; ***p<0.0001, Student's t-test). (I) Western blots showing protein levels of HK2 across the day/night cycle in mKO and Ctrl gastrocnemius muscles from 36 different mice (n=3/group/timepoint). (J) Diurnal protein levels of HK2 quantified by densitometry after western blot analysis (mean±SEM; arbitrary units; n=3/group/timepoint; *p<0.05, **p<0.01, 2-way ANOVA with the Bonferroni correction, group effect F=57.8, p<0.0001). Right panel shows mean 24-h HK2 protein levels (mean±SEM; arbitrary units; n=18/group; ***p<0.0001, Student's t-test). (K) and (L) Diurnal expression profiles of Tbc1d4 (K) and Tbc1d1 (L) transcripts in SOL and TA muscles from Ctrl and mKO mice detected by qPCR and plotted relative to 36B4 expression (mean±SEM; n=3/timepoint; **p<0.01, ***p<0.001, 2-way ANOVA with the Bonferroni correction). (M) Western blots showing TBC1D1 protein levels at three time points in mKO and Ctrl TA muscles. (N) Mean 24-h TBC1D1 protein levels in mKO and Ctrl TA muscles (mean±SEM; arbitrary units; n=4/group; **p<0.01, Student's t-test).
Muscle insulin resistance induced by high-fat-diet or obesity is associated with dysregulation of the initial steps of insulin-signaling cascade leading to decreased AKT activation. We therefore examined the effect of insulin on AKT phosphorylation in the same muscles used for the glucose uptake assay. As shown in Figure 3E, a similar increase in insulin-stimulated AKT phosphorylation at Ser473 and Thr308 was detected in mKO and control muscles. Likewise, no significant difference in AKT phosphorylation was observed in muscles from mKO and control mice examined throughout the day/night cycle (Figure 3F). Interestingly, the diurnal cycle in phosphorylation state of AKT Ser473 in control muscle, peaking in the middle of the active/feeding phase, was unchanged in mKO muscle.
Next, we focused on the final steps of the insulin-signaling cascade directly involved in glucose uptake. The expression of GLUT4, the insulin-dependent glucose transporter, showed only modest, but significantly decreased expression at the transcript level (group effect p<0.05, 2-way ANOVA; Figure S8). This change was even more evident at the protein level, with significant reductions particularly at ZT0, ZT12 and ZT16 (Figure 3G and H). Robust reductions in both transcript and protein levels were similarly observed for hexokinase 2 (HK2) (Figures S8 and 3I, J). AKT is known to control skeletal muscle glucose transport by phosphorylating the Rab-GTPase-activating proteins TBC1D4 (AS160) and TBC1D1, thus promoting the translocation of GLUT4 to the plasma membrane [46–48]. As shown in Figure 3K and L, Tbc1d4 transcript levels were essentially unchanged in mKO muscles, whereas Tbc1d1 transcripts were markedly downregulated. Based on the JTK_CYCLE algorithm, and in agreement with circadian gene expression profiles obtained from high-resolution DNA microarray experiments (http://bioinf.itmat.upenn.edu/circa/mouse), Tbc1d1 transcripts display a circadian rhythm in skeletal muscles with a peak during the active phase, and their oscillation is blunted in both slow and fast mKO muscles (Figure 3L). TBC1D1 protein levels were also markedly reduced across the day/night cycle in mKO muscles (Figure 3M and N). Tbc1d1 was previously identified as a circadian gene also in bone and atrial myocardium [49] and a comparative computational screen for conserved putative CLOCK:BMAL1-binding elements in the mouse and human genomes identified a potential cis-regulatory CLOCK:BMAL1 binding site within Tbc1d1 [50]. We observed that this 18 bp sequence is located in a non-coding region that is highly conserved among different mammalian species (Figure S11). Although a non-canonical E-Box, the core binding motif CACGGG was shown to efficiently bind the CLOCK:BMAL1 heterodimer in a screen for circadian transcription factor DNA binding activity [51]. Interestingly, Tbc1d1 knockout mice showed decreased insulin-dependent glucose uptake in isolated skeletal muscles, and GLUT4 protein abundance was similarly reduced by approximately 50% [52].
3.4. Altered glucose metabolism in mKO muscles
To gain further insight into the effect of muscle-specific Bmal1 KO on glucose metabolism, we examined the circadian metabolome of control and mKO muscles. Diurnal steady state levels of glycolytic intermediates were similar in mKO and control muscles, with a tendency for the initial metabolites of the glycolytic pathway, such as glucose-6-phosphate and fructose-6-phosphate, to show distinct diurnal fluctuations, with peak levels during the active/feeding phase and reduced levels during the rest/fasting phase (Figure 4A). In contrast, mKO muscles showed significantly increased levels of a wide range of metabolites from pathways linked to glycolysis, including the pentose phosphate pathway (xylulose-5P, ribose, ribulose), the polyol pathway (sorbitol, galactitol, fructose) and the glucuronic acid pathway (xylitol, xylulose) (Figure 4B and Table S1). Interestingly, many of these sugar metabolites increased especially or exclusively during the active/feeding phase, i.e. the time when glycolytic intermediates were increased in both Ctrl and mKO muscles. To test whether muscle glucose oxidation was impaired in mKO muscles, we measured in vitro [1-14C]-glucose oxidation rates in control and mKO muscles around ZT12, at the start of the active/feeding phase. Glucose oxidation rate was significantly decreased by 37% in muscles from mKO mice compared to control, similar to the 51% reduction observed in control muscles after a 24-h fast (Figure 5A).
Figure 4.
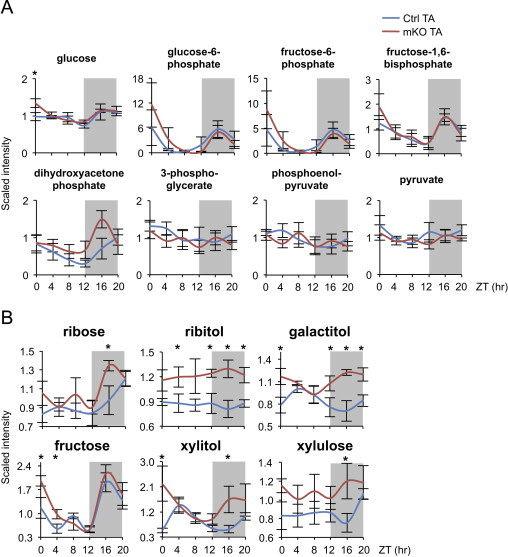
Altered sugar metabolites in skeletal muscles from Bmal1 mKO mice. (A) Diurnal variations of glycolytic metabolites in control (Ctrl) and mKO TA muscles (mean±SEM; n=5/group/timepoint; *p<0.05, repeated measures ANOVA). (B) Diurnal changes of other sugar metabolites in Ctrl and mKO TA muscles (mean±SEM; n=5/group/timepoint; *p<0.05, repeated measures ANOVA; see Table S1).
Figure 5.
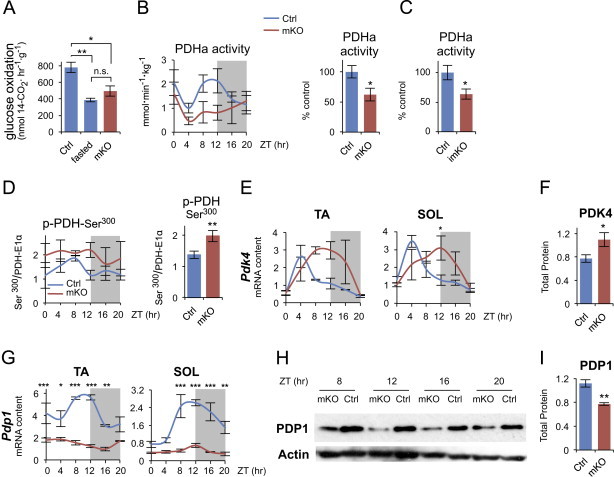
Altered glucose metabolism in skeletal muscles from Bmal1 mKO mice. (A) Glucose oxidation to CO2 measured in isolated diaphragms incubated with 5 mM [1-14C]-glucose at ZT12, at the light/dark transition (mean±SEM; n=5–10/group; *p<0.05, **p<0.01, Student's t-test). (B)–(D) Active pyruvate dehydrogenase (PDHa) activity and PDH phosphorylation in gastrocnemius muscles from Ctrl and mKO mice. (B) Diurnal pattern of PDHa activity. Data are expressed as mmoles/min/kg wet weight (mean±SEM; n=3/group/timepoint; 2-way ANOVA group effect F=6.66, p=0.016). Right panel shows mean 24-h PDHa activity in muscles from Ctrl and mKO mice (mean percent relative to control±SEM; n=18/group; *p<0.05, Student's t-test). (C) Mean 24-h PDHa activity in muscles from Ctrl (n=13) and imKO mice (n=16) (mean percent relative to control±SEM; *p<0.05, Student's t-test). (D) Diurnal PDH-E1α Ser300 phosphorylation quantified by densitometry after western blot analysis in muscles across the diurnal cycle (normalized to total PDH-E1α protein; mean±SEM; arbitrary units; n=3/group/timepoint; 2-way ANOVA group effect F=6.58, p=0.0173). Right panel shows mean 24-h PDH-E1α Ser300 phosphorylation in mKO and control muscles (mean±SEM; arbitrary units; n=18/group; **p<0.01, Student's t-test). (E) Diurnal gene expression profiles of PDH kinase 4 (Pdk4) in Ctrl and mKO TA and SOL muscles detected by qPCR and plotted relative to 36B4 expression (mean±SEM; n=3/group/timepoint; *p<0.05, 2-way ANOVA with the Bonferroni correction). (F) PDK4 protein levels in Ctrl and mKO muscles (gastrocnemius) immediately before and during the dark/active phase (average of values measured at ZT8, 12, 16 and 20) (mean±SEM; n=12; *p<0.05, Student's t-test). (G) Diurnal gene expression profiles of PDH phosphatase 1 (Pdp1) in Ctrl and mKO TA and SOL muscles detected by qPCR and plotted relative to 36B4 expression (mean±SEM; n=3/group/timepoint; *p<0.05, **p<0.01, ***p<0.001, 2-way ANOVA with the Bonferroni correction). (H) Western blots showing PDP1 protein levels, as measured immediately before and during the dark/active phase, in mKO and Ctrl gastrocnemius muscles. (I) PDP1 protein levels in mKO and Ctrl muscles (gastrocnemius) immediately before and during the dark/active phase (ZT8-20; mean±SEM; arbitrary units; n=4/group; **p<0.01, Student's t-test).
A crucial step in glucose oxidation is the irreversible oxidative decarboxylation of pyruvate to acetyl-CoA catalyzed by the pyruvate dehydrogenase (PDH) complex, a multisubunit mitochondrial complex. The PDH-E1α subunit catalyzes the rate-limiting step in the overall reaction, with the rate of conversion determined by the amount of PDH-E1α in the dephosphorylated active state (PDHa). We examined PDHa activity in muscles from control and mKO mice over 24 h and found that in control muscles, PDHa activity oscillated with a peak at ZT8-ZT12 (Figure 5B), immediately before and during the transition from the rest/fasting phase to the active/feeding phase. Similar diurnal fluctuations in PDHa activity were also reported in cardiac muscle [53]. PDHa activity was significantly decreased in Bmal1 mKO skeletal muscles, and the normal peak during the light/dark transition was completely abrogated (Figure 5B). PDHa activity was similarly decreased in muscles from inducible knockout (imKO) mice (Figure 5C).
Reduced PDHa activity suggests increased phosphorylation of the E1α subunit of the PDH complex. Indeed, while protein levels of PDH-E1α were unchanged in mKO muscles (Figure S12), Ser300 phosphorylation, a site that increases in skeletal muscle after fasting [54], was significantly increased in mKO muscles (Figure 5D). Reversible phosphorylation of PDH-E1α is controlled by PDH kinases (PDK) and PDH phosphatases (PDP). We therefore examined expression levels of Pdk4 and Pdp1, the genes coding for the major PDK and PDP isoforms expressed in skeletal muscle. Pdk4 was previously identified as a circadian gene in skeletal muscle [55] and in many other tissues [49]. As shown in Figure 5E, Pdk4 displayed normal peak expression during the fasting phase in control muscles, but showed a marked shift in expression phase in mKO muscles, persisting at high levels well into the feeding phase. A similar shift in Pdk4 expression was previously observed in muscles from a global Clock mutant [55]. Consistent results were seen in imKO mice examined 5 months after tamoxifen treatment (Figure S13), again with Pdk4 transcripts increased in imKO muscles at ZT12 compared to control littermates. PDK4 protein levels closely followed transcript levels, and were significantly increased in mKO muscles immediately before and during the active/feeding phase (Figure 5F). On the other hand, using JTK_CYCLE we identified Pdp1 as a 24-h cycling gene in both TA and SOL muscles, with a peak between ZT8 and ZT12, i.e. in the hours preceding the active/feeding phase. Pdp1 was also previously reported to be a circadian gene in the brain (listed as Ppm2c) [49]. As shown in Figure 5G, Pdp1 was markedly downregulated, and its oscillation completely abolished in muscles lacking Bmal1. Similar results were seen in muscles from the inducible knockout (imKO), with Pdp1 transcripts clearly reduced at all timepoints 5 months after tamoxifen (Figure S13). PDP1 protein levels were similarly reduced across the day/night cycle in mKO muscles (Figure 5H and I). Potential CLOCK:BMAL1-dependent E-boxes have not been described for either Pdk4 or Pdp1. However, Pdp1 (listed as Gene ID 381511) was previously identified among the top 100 genes with conserved putative D-box cis-regulatory elements [50]. The D-box transcriptional activators Dbp, Hlf and Tef are all direct targets of Bmal1, as is the D-Box transcriptional repressor E4bp4 (Nfil3) [8]. We found that this D-box sequence, located within the second intron of Pdp1, shows a high degree of conservation among many mammalian species in addition to human and mouse (Figure S14), suggesting Pdp1 may be an indirect Bmal1 target.
4. Discussion
Skeletal muscle is a major site of insulin-stimulated glucose disposal [56] and muscle insulin resistance has been considered to be one of the earliest factors in the pathogenesis of the metabolic syndrome [57]. The dissection of the mechanisms controlling muscle insulin sensitivity may therefore have important implications for understanding the causes of metabolic diseases. Circadian rhythms control body metabolism and their disruption is known to increase the risk for metabolic syndrome; however the relative roles of central and peripheral clocks remain to be established. Changes in skeletal muscle structure and function were described in a global Bmal1 knockout model [41]; however it is unclear whether these are direct consequences of muscle Bmal1 deficiency or secondary effects due to the disruption of the circadian pacemaker or other peripheral clocks. Global Bmal1 knockout causes not only loss of circadian rhythm of locomotor activity but also reduced total activity levels [42] and rescue experiments in which BMAL1 was constitutively expressed in skeletal muscle of Bmal1 null mice showed that muscle-specific rescue restored activity levels, though not circadian rhythmicity of activity [58]. This result was interpreted as indicating that BMAL1 function in muscle is important for activity. However, interpretation of this experiment is complicated by the fact that the Bmal1 transgene used for the rescue was controlled by an α-actin promoter expressed at constitutively high levels in skeletal muscle, without any circadian oscillation.
We show here that muscle-specific Bmal1 inactivation does not cause the dramatic changes seen in whole body Bmal1 knockout, such as progressive sarcopenia and premature aging. The phenotype of both knockout models examined here was essentially normal, except for a slight change in fiber type composition and decrease in muscle force in the mKO but not in the imKO line. Locomotor activity showed a normal circadian rhythm in both lines; however the total activity levels during the dark phase were significantly increased compared to control in the mKO but not in imKO line. The cause of these differences remains to be established and probably reflects changes taking place during development in the mKO model. Importantly, in spite of these differences, the effect on glucose metabolism was essentially similar in the two muscle-specific Bmal1 knockout lines.
The main result of this study is the finding that disruption of the intrinsic muscle clock causes muscle insulin resistance and altered muscle glucose metabolism. Insulin-dependent glucose uptake in isolated muscles was reduced by muscle-specific inactivation of Bmal1, induced either at early developmental stages (mKO) or in the adult (imKO). In contrast, no significant change in global glucose tolerance was observed in the absence of muscle Bmal1. Muscle insulin resistance without changes in global glucose tolerance was previously described in mice with muscle-specific knockout of the insulin receptor [30]. It is likely that other insulin-sensitive tissues, such as adipose tissue, compensate for reduced muscle insulin sensitivity at the whole body level [59].
What is the cause of the altered response to insulin in muscles from Bmal1 mKO mice? The canonical insulin signaling pathway is apparently not responsible for this change, since AKT phosphorylation, which is reduced in obesity and diabetes [60], was unchanged, and was similarly increased by insulin in muscles from control and Bmal1 mKO mice (Figure 3E). Other experimental conditions, such as lipid infusion [61–63], and skeletal muscle-specific genetic models, such as overexpression of PPARα [64] or inactivation of carnitine acetyltransferase [65], were also shown to cause muscle insulin resistance without affecting AKT phosphorylation. On the other hand, our results suggest that two major changes in muscle-specific Bmal1 knockout muscles may be linked to muscle insulin resistance and altered glucose metabolism: (1) reduced GLUT4 protein levels, with reduced expression and altered circadian oscillation of TBC1D1; (2) reduced PDH activity due to altered expression of Pdp1 and Pdk4, two circadian genes coding for the crucial PDH regulators (Figure 6).
Figure 6.
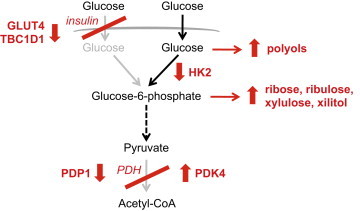
Glucose metabolism in skeletal muscles of Bmal1 mKO mice. In control mice glucose uptake is normally enhanced by insulin at the transition from the rest/fasting to the active/feeding phase, and PDH activity is increased by upregulation of PDP1 and downregulation of PDK4. In Bmal1 mKO mice, insulin-dependent glucose uptake is impaired due to decreased GLUT4 and TBC1D1 protein levels. As a result of reduced HK2 and PDHa activity, glucose metabolism is channeled to alternative pathways, including the polyol, pentose phosphate and glucuronic acid pathways.
In Bmal1 mKO muscles, GLUT4 protein levels were significantly reduced by 45% across the diurnal cycle compared to control levels (Figure 3G and H). This change is likely relevant, because skeletal muscle-specific GLUT4+/− mice, which have a 40–50% reduction in GLUT4 protein in skeletal muscles, also show a partial reduction in insulin-stimulated glucose uptake in skeletal muscles [66]. GLUT4 traffic to the plasma membrane is controlled by two Rab-GTPase-activating proteins: TBC1D1 and its paralog TBC1D4 (AS160) [67]. While Tbc1d4 expression was essentially unaffected by Bmal1 knockout, Tbc1d1 was strongly downregulated in mKO skeletal muscle (Figure 3G and H) resulting in reduced circadian TBC1D1 protein levels (Figure 3M). Interestingly, Tbc1d1 knockout mice were recently reported to show a 50% reduction in GLUT4 levels and impaired insulin-stimulated glucose uptake in skeletal muscle [52]. A similar effect was previously described in a recombinant mouse strain carrying a mutation of Tbc1d1 [48,68]. Our results therefore suggest that the loss of Bmal1 in skeletal muscle leads to reduced Tbc1d1 oscillation and expression levels, which in turn leads to reduced GLUT4 levels and insulin-stimulated glucose uptake, as previously suggested for the Tbc1d1 knockout model [52]. Interestingly, in both Tbc1d1 null mice and mKO mice GLUT4 protein was markedly reduced, whereas Glut4 mRNA levels were barely affected. In the case of Tbc1d1 null mice, it was suggested that GLUT4 protein changes are not due to an alteration of transcriptional regulation of the gene, but reflect a post-translational event possibly controlled by TBC1D1 [52]. One possibility is that TBC1D1 affects GLUT4 protein stability, but this remains to be established.
Glucose oxidation in Bmal1 mKO muscles is impaired by reduced PDH activity due to altered expression of two circadian genes, Pdp1 and Pdk4. During the rest/fasting phase, reduced insulin and contractile activity levels decrease glucose uptake and glycolytic flux in muscle cells, while promoting fatty acid β-oxidation, further inhibiting glucose oxidation by PDK4 activation and PDH inhibition [69]. During the active/feeding phase, skeletal muscle glucose uptake and oxidation are both stimulated by a rise in blood glucose and insulin secretion, in conjunction with increased locomotor activity. At this stage, PDP1, which is stimulated by contractile activity-dependent increase in intracellular Ca2+, dephosphorylates PDH-E1α, thus activating the PDH complex. Our results suggest that circadian PDH activity is driven by the intrinsic muscle clock, with PDHa normally activated in anticipation of the active/feeding phase by increased expression of Pdp1. In the absence of Bmal1, Pdp1 oscillation and expression is severely impaired, while Pdk4 levels and PDH phosphorylation remain high also during the active/feeding phase. PDH thus remains partially “locked” in a fasting state during the light/dark transition when it is normally activated. We suggest that as a result of the PDH block, glycolysis is partially uncoupled from glucose oxidation in Bmal1 KO muscles, and glycolytic flux in the context of PDH inhibition leads to the diversion of glycolytic intermediates to alternative metabolic fates, including the pentose phosphate and glucuronic acid pathways, with the shunting of glucose to the polyol pathway being favored by the concomitant decrease in HK2 levels (Figure 6).
In conclusion, we report here that a major physiological role of the muscle clock is to prepare muscle cells for the transition from the rest/fasting phase to the active/feeding phase, in anticipation of periodic fluctuations in fuel supply and demand. Reduced PDH activity and the concomitant block in insulin-stimulated glucose uptake are expected to impair the switch from lipid to glucose metabolism at the light/dark transition. Skeletal muscles with functional clocks are thus primed to choose “the right fuel at the right time“ [70], whereas muscle-specific Bmal1 inactivation and the consequent disruption of the muscle clock would cause a state of reduced metabolic flexibility [71], characterized by muscle insulin resistance and altered glucose metabolism.
Conflict of interest
None declared.
Acknowledgments
This work was supported by grants from the European Commission (Integrated Project MYOAGE no. 223576, ERC mitoCalcium, no. 294777 and ITN Program MUZIC); the Italian Space Agency (ASI, project OSMA); the Italian Ministries of Health (Ricerca Finalizzata) and of Education, University and Research (PRIN, FIRB), NIA (2P01AG025532-06A1), Cariparo Foundation (Padua), the Italian Association for Cancer Research (AIRC) and Telethon–Italy (GPP1005A and GGP11082). We thank Alexandra Chadt and Hadi Al-Hasani for the TBC1D1 antibody, Jessie Cameron and Brian Robinson for the PDP1 antibody, and Charles Weitz for the Bmal1 antibody. We are grateful to Rodolfo Costa, Charalambos Kyriacou, Antonio Vidal-Puig and Louis Hue for useful discussions, and to Gian Paolo Fadini for advice on insulin resistance tests. We also thank Lisa Agatea, Luisa Coletto and Judith Ceelen for technical assistance, and Brian Wittmann of Metabolon for help in the interpretation of metabolomics data.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Contributor Information
Kenneth A. Dyar, Email: kenneth.dyar@gmail.com.
Stefano Schiaffino, Email: stefano.schiaffino@unipd.it.
Appendix A. Supporting information
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.molmet.2013.10.005.
Appendix A. Supplementary materials
Supplementary Data
References
- 1.Asher G., Schibler U. Crosstalk between components of circadian and metabolic cycles in mammals. Cell Metabolism. 2011;13:125–137. doi: 10.1016/j.cmet.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 2.Green C.B., Takahashi J.S., Bass J. The meter of metabolism. Cell. 2008;134:728–742. doi: 10.1016/j.cell.2008.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang W., Ramsey K.M., Marcheva B., Bass J. Circadian rhythms, sleep, and metabolism. Journal of Clinical Investigation. 2011;121:2133–2141. doi: 10.1172/JCI46043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karlsson B., Knutsson A., Lindahl B. Is there an association between shift work and having a metabolic syndrome? Results from a population based study of 27,485 people. Occupational and Environmental Medicine. 2001;58:747–752. doi: 10.1136/oem.58.11.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buxton O.M., Cain S.W., O'Connor S.P., Porter J.H., Duffy J.F., Wang W. Adverse metabolic consequences in humans of prolonged sleep restriction combined with circadian disruption. Science Translational Medicine. 2012;4:129ra143. doi: 10.1126/scitranslmed.3003200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Esquirol Y., Bongard V., Ferrieres J., Verdier H., Perret B. Shiftwork and higher pancreatic secretion: early detection of an intermediate state of insulin resistance? Chronobiology International. 2012;29:1258–1266. doi: 10.3109/07420528.2012.719959. [DOI] [PubMed] [Google Scholar]
- 7.Dibner C., Schibler U., Albrecht U. The mammalian circadian timing system: organization and coordination of central and peripheral clocks. Annual Review of Physiology. 2010;72:517–549. doi: 10.1146/annurev-physiol-021909-135821. [DOI] [PubMed] [Google Scholar]
- 8.Rey G., Cesbron F., Rougemont J., Reinke H., Brunner M., Naef F. Genome-wide and phase-specific DNA-binding rhythms of BMAL1 control circadian output functions in mouse liver. PLoS Biology. 2011;9:e1000595. doi: 10.1371/journal.pbio.1000595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turek F.W., Joshu C., Kohsaka A., Lin E., Ivanova G., McDearmon E. Obesity and metabolic syndrome in circadian Clock mutant mice. Science. 2005;308:1043–1045. doi: 10.1126/science.1108750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lamia K.A., Storch K.F., Weitz C.J. Physiological significance of a peripheral tissue circadian clock. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:15172–15177. doi: 10.1073/pnas.0806717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lamia K.A., Papp S.J., Yu R.T., Barish G.D., Uhlenhaut N.H., Jonker J.W. Cryptochromes mediate rhythmic repression of the glucocorticoid receptor. Nature. 2011;480:552–556. doi: 10.1038/nature10700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rudic R.D., McNamara P., Curtis A.M., Boston R.C., Panda S., Hogenesch J.B. BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biology. 2004;2:e377. doi: 10.1371/journal.pbio.0020377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi S.Q., Ansari T.S., McGuinness O.P., Wasserman D.H., Johnson C.H. Circadian disruption leads to insulin resistance and obesity. Current Biology: CB. 2013;23:372–381. doi: 10.1016/j.cub.2013.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalsbeek A., Yi C.X., La Fleur S.E., Fliers E. The hypothalamic clock and its control of glucose homeostasis. Trends in Endocrinology and Metabolism. 2010;21:402–410. doi: 10.1016/j.tem.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 15.Marcheva B., Ramsey K.M., Buhr E.D., Kobayashi Y., Su H., Ko C.H. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. 2010;466:627–631. doi: 10.1038/nature09253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paschos G.K., Ibrahim S., Song W.L., Kunieda T., Grant G., Reyes T.M. Obesity in mice with adipocyte-specific deletion of clock component Arntl. Nature Medicine. 2012;18:1768–1777. doi: 10.1038/nm.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feneberg R., Lemmer B. Circadian rhythm of glucose uptake in cultures of skeletal muscle cells and adipocytes in Wistar-Kyoto, Wistar, Goto-Kakizaki, and spontaneously hypertensive rats. Chronobiology International. 2004;21:521–538. doi: 10.1081/cbi-200026958. [DOI] [PubMed] [Google Scholar]
- 18.Storch K.F., Paz C., Signorovitch J., Raviola E., Pawlyk B., Li T. Intrinsic circadian clock of the mammalian retina: importance for retinal processing of visual information. Cell. 2007;130:730–741. doi: 10.1016/j.cell.2007.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bothe G.W., Haspel J.A., Smith C.L., Wiener H.H., Burden S.J. Selective expression of Cre recombinase in skeletal muscle fibers. Genesis. 2000;26:165–166. [PubMed] [Google Scholar]
- 20.Schuler M., Ali F., Metzger E., Chambon P., Metzger D. Temporally controlled targeted somatic mutagenesis in skeletal muscles of the mouse. Genesis. 2005;41:165–170. doi: 10.1002/gene.20107. [DOI] [PubMed] [Google Scholar]
- 21.Schiaffino S., Gorza L., Sartore S., Saggin L., Ausoni S., Vianello M. Three myosin heavy chain isoforms in type 2 skeletal muscle fibres. Journal of Muscle Research and Cell Motility. 1989;10:197–205. doi: 10.1007/BF01739810. [DOI] [PubMed] [Google Scholar]
- 22.Blaauw B., Canato M., Agatea L., Toniolo L., Mammucari C., Masiero E. Inducible activation of Akt increases skeletal muscle mass and force without satellite cell activation. FASEB Journal: Official Publication of the Federation of American Societies for Experimental Biology. 2009;23:3896–3905. doi: 10.1096/fj.09-131870. [DOI] [PubMed] [Google Scholar]
- 23.Irizarry R.A., Hobbs B., Collin F., Beazer-Barclay Y.D., Antonellis K.J., Scherf U. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 24.Dai M., Wang P., Boyd A.D., Kostov G., Athey B., Jones E.G. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Research. 2005;33:e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Conesa A., Nueda M.J., Ferrer A., Talon M. maSigPro: a method to identify significantly differential expression profiles in time-course microarray experiments. Bioinformatics. 2006;22:1096–1102. doi: 10.1093/bioinformatics/btl056. [DOI] [PubMed] [Google Scholar]
- 26.Hughes M.E., Hogenesch J.B., Kornacker K. JTK_CYCLE: an efficient nonparametric algorithm for detecting rhythmic components in genome-scale data sets. Journal of Biological Rhythms. 2010;25:372–380. doi: 10.1177/0748730410379711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Eckel-Mahan K.L., Patel V.R., Mohney R.P., Vignola K.S., Baldi P., Sassone-Corsi P. Coordination of the transcriptome and metabolome by the circadian clock. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:5541–5546. doi: 10.1073/pnas.1118726109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patel V.R., Eckel-Mahan K., Sassone-Corsi P., Baldi P. CircadiOmics: integrating circadian genomics, transcriptomics, proteomics and metabolomics. Nature Methods. 2012;9:772–773. doi: 10.1038/nmeth.2111. [DOI] [PubMed] [Google Scholar]
- 30.Bruning J.C., Michael M.D., Winnay J.N., Hayashi T., Horsch D., Accili D. A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Molecular Cell. 1998;2:559–569. doi: 10.1016/s1097-2765(00)80155-0. [DOI] [PubMed] [Google Scholar]
- 31.Sakamoto K., McCarthy A., Smith D., Green K.A., Grahame Hardie D., Ashworth A. Deficiency of LKB1 in skeletal muscle prevents AMPK activation and glucose uptake during contraction. EMBO Journal. 2005;24:1810–1820. doi: 10.1038/sj.emboj.7600667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mackrell J.G., Cartee G.D. A novel method to measure glucose uptake and myosin heavy chain isoform expression of single fibers from rat skeletal muscle. Diabetes. 2012;61:995–1003. doi: 10.2337/db11-1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang C. Insulin-stimulated glucose uptake in rat diaphragm during postnatal development: lack of correlation with the number of insulin receptors and of intracellular glucose transporters. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:3621–3625. doi: 10.1073/pnas.82.11.3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jeoung N.H., Wu P., Joshi M.A., Jaskiewicz J., Bock C.B., Depaoli-Roach A.A. Role of pyruvate dehydrogenase kinase isoenzyme 4 (PDHK4) in glucose homoeostasis during starvation. Biochemical Journal. 2006;397:417–425. doi: 10.1042/BJ20060125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cederblad G., Carlin J.I., Constantin-Teodosiu D., Harper P., Hultman E. Radioisotopic assays of CoASH and carnitine and their acetylated forms in human skeletal muscle. Analytical Biochemistry. 1990;185:274–278. doi: 10.1016/0003-2697(90)90292-h. [DOI] [PubMed] [Google Scholar]
- 36.Constantin-Teodosiu D., Cederblad G., Hultman E. A sensitive radioisotopic assay of pyruvate dehydrogenase complex in human muscle tissue. Analytical Biochemistry. 1991;198:347–351. doi: 10.1016/0003-2697(91)90437-x. [DOI] [PubMed] [Google Scholar]
- 37.Putman C.T., Spriet L.L., Hultman E., Lindinger M.I., Lands L.C., McKelvie R.S. Pyruvate dehydrogenase activity and acetyl group accumulation during exercise after different diets. American Journal of Physiology. 1993;265:E752–E760. doi: 10.1152/ajpendo.1993.265.5.E752. [DOI] [PubMed] [Google Scholar]
- 38.Pilegaard H., Birk J.B., Sacchetti M., Mourtzakis M., Hardie D.G., Stewart G. PDH-E1alpha dephosphorylation and activation in human skeletal muscle during exercise: effect of intralipid infusion. Diabetes. 2006;55:3020–3027. doi: 10.2337/db06-0152. [DOI] [PubMed] [Google Scholar]
- 39.Bunger M.K., Walisser J.A., Sullivan R., Manley P.A., Moran S.M., Kalscheur V.L. Progressive arthropathy in mice with a targeted disruption of the Mop3/Bmal-1 locus. Genesis. 2005;41:122–132. doi: 10.1002/gene.20102. [DOI] [PubMed] [Google Scholar]
- 40.Kondratov R.V., Kondratova A.A., Gorbacheva V.Y., Vykhovanets O.V., Antoch M.P. Early aging and age-related pathologies in mice deficient in BMAL1, the core component of the circadian clock. Genes and Development. 2006;20:1868–1873. doi: 10.1101/gad.1432206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andrews J.L., Zhang X., McCarthy J.J., McDearmon E.L., Hornberger T.A., Russell B. CLOCK and BMAL1 regulate MyoD and are necessary for maintenance of skeletal muscle phenotype and function. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:19090–19095. doi: 10.1073/pnas.1014523107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bunger M.K., Wilsbacher L.D., Moran S.M., Clendenin C., Radcliffe L.A., Hogenesch J.B. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000;103:1009–1017. doi: 10.1016/s0092-8674(00)00205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi S., Hida A., McGuinness O.P., Wasserman D.H., Yamazaki S., Johnson C.H. Circadian clock gene Bmal1 is not essential; functional replacement with its paralog, Bmal2. Current Biology: CB. 2010;20:316–321. doi: 10.1016/j.cub.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buhr E.D., Yoo S.H., Takahashi J.S. Temperature as a universal resetting cue for mammalian circadian oscillators. Science. 2010;330:379–385. doi: 10.1126/science.1195262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kornmann B., Schaad O., Bujard H., Takahashi J.S., Schibler U. System-driven and oscillator-dependent circadian transcription in mice with a conditionally active liver clock. PLoS Biology. 2007;5:e34. doi: 10.1371/journal.pbio.0050034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.An D., Toyoda T., Taylor E.B., Yu H., Fujii N., Hirshman M.F. TBC1D1 regulates insulin- and contraction-induced glucose transport in mouse skeletal muscle. Diabetes. 2010;59:1358–1365. doi: 10.2337/db09-1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kramer H.F., Witczak C.A., Taylor E.B., Fujii N., Hirshman M.F., Goodyear L.J. AS160 regulates insulin- and contraction-stimulated glucose uptake in mouse skeletal muscle. Journal of Biological Chemistry. 2006;281:31478–31485. doi: 10.1074/jbc.M605461200. [DOI] [PubMed] [Google Scholar]
- 48.Szekeres F., Chadt A., Tom R.Z., Deshmukh A.S., Chibalin A.V., Bjornholm M. The Rab-GTPase-activating protein TBC1D1 regulates skeletal muscle glucose metabolism. American Journal of Physiology—Endocrinology and Metabolism. 2012;303:E524–E533. doi: 10.1152/ajpendo.00605.2011. [DOI] [PubMed] [Google Scholar]
- 49.Yan J., Wang H., Liu Y., Shao C. Analysis of gene regulatory networks in the mammalian circadian rhythm. PLoS Computational Biology. 2008;4:e1000193. doi: 10.1371/journal.pcbi.1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumaki Y., Ukai-Tadenuma M., Uno K.D., Nishio J., Masumoto K.H., Nagano M. Analysis and synthesis of high-amplitude Cis-elements in the mammalian circadian clock. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:14946–14951. doi: 10.1073/pnas.0802636105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reinke H., Saini C., Fleury-Olela F., Dibner C., Benjamin I.J., Schibler U. Differential display of DNA-binding proteins reveals heat-shock factor 1 as a circadian transcription factor. Genes and Development. 2008;22:331–345. doi: 10.1101/gad.453808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dokas J., Chadt A., Nolden T., Himmelbauer H., Zierath J.R., Joost H.G. Conventional knockout of Tbc1d1 in mice impairs insulin- and AICAR-stimulated glucose uptake in skeletal muscle. Endocrinology. 2013;154:3502–3514. doi: 10.1210/en.2012-2147. [DOI] [PubMed] [Google Scholar]
- 53.Bryson J.M., Cooney G.J., Wensley V.R., Blair S.C., Caterson I.D. Diurnal patterns of cardiac and hepatic pyruvate dehydrogenase complex activity in gold-thioglucose-obese mice. Biochemical Journal. 1993;295(Pt 3):731–734. doi: 10.1042/bj2950731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kiilerich K., Adser H., Jakobsen A.H., Pedersen P.A., Hardie D.G., Wojtaszewski J.F. PGC-1alpha increases PDH content but does not change acute PDH regulation in mouse skeletal muscle. American Journal of Physiology—Regulatory, Integrative and Comparative Physiology. 2010;299:R1350–R1359. doi: 10.1152/ajpregu.00400.2010. [DOI] [PubMed] [Google Scholar]
- 55.McCarthy J.J., Andrews J.L., McDearmon E.L., Campbell K.S., Barber B.K., Miller B.H. Identification of the circadian transcriptome in adult mouse skeletal muscle. Physiological Genomics. 2007;31:86–95. doi: 10.1152/physiolgenomics.00066.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.DeFronzo R.A., Jacot E., Jequier E., Maeder E., Wahren J., Felber J.P. The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes. 1981;30:1000–1007. doi: 10.2337/diab.30.12.1000. [DOI] [PubMed] [Google Scholar]
- 57.Rabol R., Petersen K.F., Dufour S., Flannery C., Shulman G.I. Reversal of muscle insulin resistance with exercise reduces postprandial hepatic de novo lipogenesis in insulin resistant individuals. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:13705–13709. doi: 10.1073/pnas.1110105108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McDearmon E.L., Patel K.N., Ko C.H., Walisser J.A., Schook A.C., Chong J.L. Dissecting the functions of the mammalian clock protein BMAL1 by tissue-specific rescue in mice. Science. 2006;314:1304–1308. doi: 10.1126/science.1132430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim J.K., Michael M.D., Previs S.F., Peroni O.D., Mauvais-Jarvis F., Neschen S. Redistribution of substrates to adipose tissue promotes obesity in mice with selective insulin resistance in muscle. Journal of Clinical Investigation. 2000;105:1791–1797. doi: 10.1172/JCI8305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Samuel V.T., Petersen K.F., Shulman G.I. Lipid-induced insulin resistance: unravelling the mechanism. Lancet. 2010;375:2267–2277. doi: 10.1016/S0140-6736(10)60408-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alkhateeb H., Chabowski A., Glatz J.F., Gurd B., Luiken J.J., Bonen A. Restoring AS160 phosphorylation rescues skeletal muscle insulin resistance and fatty acid oxidation while not reducing intramuscular lipids. American Journal of Physiology—Endocrinology and Metabolism. 2009;297:E1056–E1066. doi: 10.1152/ajpendo.90908.2008. [DOI] [PubMed] [Google Scholar]
- 62.Hoeg L.D., Sjoberg K.A., Jeppesen J., Jensen T.E., Frosig C., Birk J.B. Lipid-induced insulin resistance affects women less than men and is not accompanied by inflammation or impaired proximal insulin signaling. Diabetes. 2011;60:64–73. doi: 10.2337/db10-0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hoy A.J., Brandon A.E., Turner N., Watt M.J., Bruce C.R., Cooney G.J. Lipid and insulin infusion-induced skeletal muscle insulin resistance is likely due to metabolic feedback and not changes in IRS-1, Akt, or AS160 phosphorylation. American Journal of Physiology—Endocrinology and Metabolism. 2009;297:E67–E75. doi: 10.1152/ajpendo.90945.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Finck B.N., Bernal-Mizrachi C., Han D.H., Coleman T., Sambandam N., LaRiviere L.L. A potential link between muscle peroxisome proliferator- activated receptor-alpha signaling and obesity-related diabetes. Cell Metabolism. 2005;1:133–144. doi: 10.1016/j.cmet.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 65.Muoio D.M., Noland R.C., Kovalik J.P., Seiler S.E., Davies M.N., DeBalsi K.L. Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metabolism. 2012;15:764–777. doi: 10.1016/j.cmet.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zisman A., Peroni O.D., Abel E.D., Michael M.D., Mauvais-Jarvis F., Lowell B.B. Targeted disruption of the glucose transporter 4 selectively in muscle causes insulin resistance and glucose intolerance. Nature Medicine. 2000;6:924–928. doi: 10.1038/78693. [DOI] [PubMed] [Google Scholar]
- 67.Bogan J.S. Regulation of glucose transporter translocation in health and diabetes. Annual Review of Biochemistry. 2012;81:507–532. doi: 10.1146/annurev-biochem-060109-094246. [DOI] [PubMed] [Google Scholar]
- 68.Chadt A., Leicht K., Deshmukh A., Jiang L.Q., Scherneck S., Bernhardt U. Tbc1d1 mutation in lean mouse strain confers leanness and protects from diet-induced obesity. Nature Genetics. 2008;40:1354–1359. doi: 10.1038/ng.244. [DOI] [PubMed] [Google Scholar]
- 69.Hue L., Taegtmeyer H. The Randle cycle revisited: a new head for an old hat. American Journal of Physiology—Endocrinology and Metabolism. 2009;297:E578–E591. doi: 10.1152/ajpendo.00093.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Muoio D.M., Neufer P.D. Lipid-induced mitochondrial stress and insulin action in muscle. Cell Metabolism. 2012;15:595–605. doi: 10.1016/j.cmet.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kelley D.E., Goodpaster B., Wing R.R., Simoneau J.A. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. American Journal of Physiology. 1999;277:E1130–E1141. doi: 10.1152/ajpendo.1999.277.6.E1130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data