

Abstract
We examined the effect of interleukin-17 (IL-17) on the expression of Toll-like receptors (TLRs) in fibroblast-like synoviocytes (FLS) from patients with rheumatoid arthritis (RA) and osteoarthritis (OA). We investigated the region downstream of IL-17 for TLR expression. We also investigated the downstream signals responsible for the effect of IL-17 in TLR expression. Levels of IL-17 protein in the serum and synovial fluid of RA and OA patients were measured by ELISA. The IL-17 mRNA expression in peripheral blood mononuclear cells and synovial fluid mononuclear cells was measured by RT-PCR. RA and OA FLS were incubated with IL-17 and/or IL-23 for 24 hr. To block the signal transducer and activator of transcription 3 (STAT3) pathway, FLS were treated with S3I-201 before incubation with IL-17 and IL-23. Synovial tissue samples from RA and OA patients were stained with antibodies to IL-17, TLR2, TLR3, TLR4, STAT3 and phospho-STAT3. Levels of IL-17 protein were higher in the serum and synovial fluid from RA patients compared with those from OA patients. The IL-17 mRNA expression in synovial fluid monocytes was also higher in RA than in OA patients. Immunohistochemical staining showed greater expression of IL-17, TLR2, TLR3 and TLR4 in synovial samples from RA compared with OA patients. Interleukin-17 increased the expression of TLR2, TLR3 and TLR4 in RA FLS; IL-23 augmented the IL-17-induced expression of TLR2, TLR3 and TLR4 in RA FLS. Blocking STAT3 with S3I-201 reduced IL-17-induced TLR3 expression in RA FLS. Our results suggest that IL-17 is a major cytokine in pathogenesis on RA. The IL-17 influences the innate immune system by increasing the synovial expression of TLR2, TLR3 and TLR4. We may control TLR3 expression via the STAT3 pathway in RA FLS.
Keywords: human, interleukin-17, rheumatoid arthritis, signal transducer and activator of transcription 3, synovial fibroblasts, Toll-like receptors
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease involving progressive articular damage caused by inflammatory cells and synoviocytes. The RA synovial lining cells comprise activated fibroblast-like synoviocytes (FLS) and macrophages. The RA FLS are considered key cells that mediate the destruction of cartilage and bone in the affected joints.1 The synovial membranes are thickened and hyperplastic in RA.2 Synovial activation is driven by both pro-inflammatory cytokines and cytokine-independent pathways, including endogenous retroviral elements and Toll-like receptors (TLRs).2 These pathways are connected by a complex network of autocrine and paracrine acting factors.
After interleukin-17 (IL-17) and T helper 17 (Th17) cells were detected in the immune system, some investigators showed a relationship between IL-17 and synovial inflammation in RA. Interleukin-17 became another key cytokine in RA, and the abundance of Th17 cells in RA synovial fluid has been shown.3 High IL-17 level is detected in serum and synovial fluid of RA and autoimmune arthritis models.3–7 The IL-17 is linked to other inflammatory cytokines such as IL-1, tumour necrosis factor-α (TNF-α) and IL-23 p19.7
The RA FLS are part of the innate immune system, and they express pattern-recognition receptors such as TLRs. Some studies have reported higher TLR2, TLR3 and TLR4 expression in RA FLS.1,8–10 The innate and adaptive immune systems are connected by many cytokines and intracellular molecules. Stimulation by the TLR pathway induces the production of pro-inflammatory cytokines such as TNF-α, IL-1, IL-6, IL-17 and IL-23, and induces osteoclastogenesis by receptor activator of nuclear factor-κB ligand (RANKL) or matrix metalloproteinase 1 (MMP-1), MMP-3 and MMP-13.7–10 Activation of TLRs also drives regulatory mechanisms such as regulatory T cell activation and IL-10 secretion.11–15
In a recent study, we found that IL-17 increases the synovial expression of TLR2, TLR4 and TLR9 in a collagen-induced arthritis (CIA) model.4 This was the first report that IL-17, an inflammatory cytokine, up-regulates TLR expression. The TLR stimulation induces the production and gene activation of diverse cytokines including both pro-inflammatory and anti-inflammatory cytokines, depending on the cell type and environment. Interestingly, IL-17 induced by Th17 cells or TLR activation up-regulates the TLR expression in the FLS of CIA mice, suggesting that IL-17 provides another pathway of inflammatory amplification. The aim of our current study was to confirm our observations of the role of IL-17 in TLR-induced up-regulation in human RA FLS. First, we confirmed that TLRs are expressed in human RA FLS. Second, we investigated whether IL-17 increases the synovial expression of TLRs in CIA and RA FLS. We found that, in the CIA synovium, IL-1β and IL-6 are involved in the IL-17-induced aggravation of arthritis and TLR expression. Finally, we studied how IL-17 controls the expression of TLRs in human RA FLS.
Materials and methods
Patients
Five patients with RA fulfilling the 1987 revised criteria of the American College of Rheumatology (formerly the American Rheumatism Association) were enrolled in this study. Synovial tissues were isolated from five patients with RA (mean age 56·6 ± 4·7 years, range 32–70 years) undergoing total knee replacement surgery. These tissues were compared with tissues from four age-matched and sex-matched patients with osteoarthritis (OA) and one healthy control subject. Informed consent was obtained from each participant, and the experimental protocol was approved by the Catholic University of Korea Human Research Ethics Committee.
Isolation and culture of FLS
Synoviocytes were isolated by enzymatic digestion of synovial tissue specimens obtained from patients with RA and patients with OA undergoing total joint replacement surgery. The tissue samples were minced into 2–3 mm pieces and treated for 4 hr with 4 mg/ml type II collagenase (Worthington, Freehold, NJ) in Dulbecco's modified Eagle's medium (DMEM) at 37° in 5% CO2. Dissociated cells were centrifuged at 500 g, resuspended in DMEM supplemented with 10% fetal calf serum, 2 mm l-glutamine, 100 units/ml penicillin and 100 ng/ml streptomycin, plated in 75-cm2 flasks, and incubated overnight. The non-adherent cells were then removed, and the adherent cells were cultivated in DMEM supplemented with 10% fetal calf serum. Synoviocytes from passages 4–8 were used in each experiment. The cells were morphologically homogeneous and exhibited the appearance of synovial fibroblasts, with typical bipolar configuration under inverse microscopy. The purity of cells was tested by flow cytometric analysis using phycoerythrin-conjugated anti-CD14 (PharMingen, San Diego, CA) and fluorescein isothiocyanate-conjugated anti-CD3 or anti-Thy-1 (CD90) monoclonal antibodies (PharMingen). At passage 4, most cells (> 98%) expressed the surface markers for fibroblasts (CD90+), whereas < 3% of the cells were CD14+ and CD3+. In the experiments conducted to determine the effects of IL-17 and its associated molecules on TLR2, TLR3 and TLR4, the FLS were washed in DMEM and then incubated for an additional 48 hr in serum-free DMEM supplemented with insulin–transferrin–selenium A (ITSA; Gibco, Grand Island, NY). The medium was exchanged with fresh DMEM-ITSA, IL-17 was then added, and the cells were incubated for 24 hr.
Isolation of peripheral blood mononuclear cells and synovial fluid mononuclear cells
Peripheral blood mononuclear cells (PBMC) and synovial fluid mononuclear cells (SFMC) were prepared in heparinized tubes by Ficoll–Hypaque density gradient centrifugation. The PBMC and SFMC were treated with RNA Zol-B (Biotecx, Houston, TX) to isolate the RNA.
Reverse transcriptase–PCR for TLR2, TLR3 and TLR4
Total mRNA was extracted from the FLS using RNAzol-B according to the manufacturer's instructions. Reverse transcription of 2 μg of total mRNA was performed at 42° using the Superscript reverse transcription system (Takara, Shiga, Japan). The following primers were used for each molecule: TLR2: 5′-GCC AAA GTC TTG ATT GAT TGG-3′ (sense) and 5′-TTG AAG TTC TCC AGC TCC TG-3′ (antisense); TLR3: 5′-GAT CTG TCT CAT AAT GGC TTG-3′ (sense) and 5′-GAC AGA TTC CGA ATG CTT GTG-3′ (antisense); TLR4: 5′-TGG ATA CGT TTC CTT ATA AG-3′ (sense) and 5′-GAA ATG GAG GCA CCC CTT C-3′ (antisense); and β-actin: 5′-GGA CTT CGA GCA AGA GAT GG-3′ (sense) and 5′-TGT GTT GGG GTA CAG GTC TTT G-3′ (antisense). The reactions were processed in a DNA thermal cycler (PerkinElmer Cetus, Wellesley, MA) through 30 cycles of 30 seconds of denaturation at 95° and 30 seconds of annealing at 60°, followed by 30 seconds of elongation at 72°. The results are expressed as the ratio of product to β-actin product.
Western blot
The RA and OA FLS lysates were prepared from approximately 1 × 105 cells by homogenization in lysis buffer and were centrifuged (19 000 g, 15 min). The protein concentration in the supernatant was determined using the Bradford method (Bio-Rad, Hercules, CA). Protein samples were separated by 10% SDS–PAGE, and transferred to a nitrocellulose membrane (Amersham Pharmacia Biotech, Uppsala, Sweden). For Western blotting, the membrane was pre-incubated with 0·5% skimmed milk in TTBS buffer (0·1% Tween-20 in Tris-buffered saline) at room temperature for 2 hr. Primary antibodies to TLR2 (Santa Cruz Biotechnology, Dallas, TX), TLR3 (Santa Cruz Biotechnology), TLR4 (Santa Cruz Biotechnology) or β-actin (Sigma Aldrich, St Louis, MO) were diluted 1 : 1000 in 5% BSA–0·1% Tween-20–TBS and added to the membrane. After overnight incubation at 4°, the membrane was washed four times with TTBS. Next, the horseradish peroxidase-conjugated secondary antibody was added and incubated for 1 hr at room temperature. After TTBS washing, signals were detected using the enhanced chemiluminescence detection kit and Hyperfilm-ECL reagents (Amersham Pharmacia).
Immunohistochemistry of the RA synovium
Immunohistochemical staining for TLR2, TLR3, TLR4 and IL-17 was performed on sections of synovial tissue. Briefly, the synovium samples were obtained from RA and OA patients, fixed in 4% paraformaldehyde solution overnight at 4°, dehydrated with alcohol, washed, embedded in paraffin, and sectioned into thick slices. Tissue sections (7 μm) were depleted of endogenous peroxidase activity with methanolic H2O2, blocked with normal serum for 30 min, and incubated overnight at 4° with anti-TLR2, TLR3, TLR4 (Santa Cruz Biotechnology), IL-17 (R&D Systems, Minneapolis MN), STAT3, or p-STAT3-Ser727 (Cell Signaling, Beverly, MA). The samples were incubated with the secondary antibody (biotinylated IgG) for 20 min and then with streptavidin–peroxidase complex (Vector, Peterborough, UK) for 1 hr followed by 3,3′-diaminobenzidine (Dako, Glostrup, Denmark). A negative isotype-matched control was included in each run. The sections were counterstained with haematoxylin, and the samples were photographed with an Olympus photomicroscope (Tokyo, Japan).
Enzyme-linked immunosorbent assay for IL-17
The concentration of IL-17 was measured by sandwich ELISA as follows. Anti-human IL-17 monoclonal antibody (R&D Systems) was added to a 96-well plate (Nunc, Roskilde, Denmark) and incubated overnight at 4°. The wells were blocked with blocking solution (PBS containing 1% BSA and 0·05% Tween-20) for 2 hr at room temperature; the test samples and the standard recombinant IL-17 (R&D Systems) were added to separate wells of the 96-well plate, and the plate was incubated at room temperature for 2 hr. The plate was washed, biotinylated IL-17 polyclonal antibody (R&D Systems) was added, and the reaction was allowed to proceed for 2 hr at room temperature. The plate was washed, 2000-fold diluted ExtrAvidin–alkaline phosphate (Sigma Aldrich) was added, and the reaction was allowed to proceed for a further 2 hr. The plate was washed, and 50 μl of p-nitrophenyl phosphate disodium salt (Pierce Chemical Company, Rockford, IL) diluted to 1 mg/ml in diethanolamine buffer was applied.
Statistical analysis
All data are expressed as the mean ± SD. Statistical analysis was performed using spss 10.0 for Windows (SPSS Inc., Chicago, IL). The differences between groups were analysed using an unpaired Student's t-test, assuming equal variances. P < 0·05 was considered significant.
Results
Increased level of IL-17 protein and mRNA in RA and OA
We first examined the expression level of IL-17 in the serum and synovial fluid from RA and OA patients. The concentration of IL-17 was significantly higher in synovial fluid than in serum. The IL-17 concentration was higher in RA synovial fluid than in OA synovial fluid (Fig. 1a). Next, we examined the IL-17 mRNA level in the PBMC and SFMC from patients. The IL-17 mRNA level was also higher in PBMC and SFMC from RA patients than from OA patients. The IL-17 mRNA level was significantly higher in RA SFMC than in OA SFMC (Fig. 1b).
Figure 1.
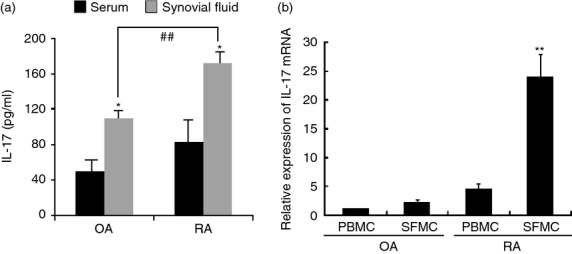
Increased level of interleukin-17 (IL-17) protein and mRNA in rheumatoid arthritis (RA) and osteoarthritis (OA). (a) Expression level of IL-17 protein was measured in the serum and synovial fluid of RA (n = 5) and OA (n = 5) patients by ELISA. (b) Level of IL-17 mRNA in peripheral blood mononuclear cells (PBMC) and synovial fluid mononuclear cells (SFMC) was measured by RT-PCR. Data are expressed as mean ± SD of five different patients. *P < 0·05 compared with serum, **P < 0·01 compared with OA SFMC serum, ##P < 0·01 compared with OA synovial fluid.
Immunohistochemistry staining for IL-17, TLR2, TLR3 and TLR4 in synovial tissue of patients
We examined whether the expression of IL-17, TLR2, TLR3 and TLR4 was higher in RA synovial tissue than in OA synovial tissue using specific monoclonal antibodies to these molecules. Expression levels of IL-17, TLR2, TLR3 and TLR4 were higher in RA synovial tissue than in OA synovial tissue (Fig. 2). Expression of IL-17 was higher in SFMC and synovial tissue from RA patients than in those from OA patients. These results suggest that the synovium of RA patients produced an inflammatory environment, in which a variety of inflammatory molecules, such as IL-17 and TLR that interact with each other, can lead to excessive inflammation.
Figure 2.
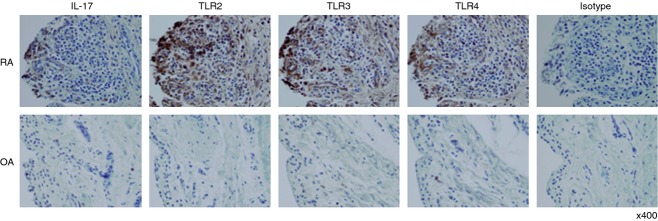
Immunohistochemical staining of interleukin-17 (IL-17), Toll-like receptor 2 (TLR2), TLR3 and TLR4 in synovium from patients. Immunohistochemical staining for TLR2, TLR3, TLR4 and IL-17 of the synovium from patients with rheumatoid arthritis (RA) or osteoarthritis (OA). Expression of IL-17, TLR2, TLR3 and TLR4 was detected in the synovium of patients with RA and OA using immunohistochemical staining, and isotype as a positive control. The synovial tissues were stained with antibodies to anti-human IL-17, -TLR2, -TLR3, -TLR4 or an isotype-control antibody. The brown colour shows the target. All tissues were counterstained with haematoxylin (original magnification × 400).
Expression of TLR2, TLR3 and TLR4 was increased by IL-17 in RA FLS
We investigated whether an increase in IL-17 concentration up-regulates TLR expression in RA FLS. The RA and OA FLS were incubated with IL-17 (1 or 10 ng/ml) for 24 hr, and the mRNA levels for TLR2, TLR3 and TLR4 were measured by RT-PCR. TLR2, TLR3 and TLR4 were increased by IL-17 stimulation. In contrast, the IL-17 had no effect in OA FLS (Fig. 3a,b). Protein levels of TLR2, TLR3 and TLR4 were increased by IL-17, also (Fig. 3c). These results suggest that IL-17-mediated cell signalling increased the expression of TLR2, TLR3 and TLR4 mRNA in RA FLS.
Figure 3.

Interleukin-17 (IL-17) -increased expression of Toll-like receptor 2 (TLR2), TLR3 and TLR4 in rheumatoid arthritis (RA) fibroblast-like synoviocytes (FLS). RA (n = 4) and osteoarthritis (OA) (n = 4) FLS (1 × 105 cells) were incubated with of recombinant human IL-17 (rhIL-17, 1 ng/ml or 10 ng/ml) for 24 hr. (a) and (b) The mRNA levels of TLR2, TLR3 and TLR4 were measured by RT-PCR. The expressions of TLR2, TLR3 and TLR4, relative to β-actin were measured. The relative gene expressions are shown on the right. (c) RA (n = 4) and OA (n = 4) FLS (3 × 105 cells) were incubated with rhIL-17 (1 ng/ml or 10 ng/ml) for 24 hr. The protein level of TLR2, TLR3, TLR4 and β-actin were detected by Western blot. Data are expressed as mean ± SD of four different patients. *P < 0·05 compared with nil (control condition), **P < 0·01 compared with nil (control condition).
IL-23 augmented IL-17-induced expression of TLR2, TLR3 and TLR4 in RA FLS
Interleukin-23, a cytokine with pro-inflammatory properties, stimulates the production of IL-17 and the IL-23–IL-17 axis plays a central role in autoimmune inflammation of joints. Levels of IL-23 correlate with IL-17 levels in the synovial fluid and the serum from RA patients. We next investigated whether IL-23 has a synergistic effect with IL-17 on TLR expression. Both RA and OA FLS were incubated with IL-17 (1 ng/ml) in the presence or absence of IL-23 (10 ng/ml) for 24 hr. The IL-23 had a synergistic effect to that of IL-17 in increasing TLR expression (Fig. 4). The mRNA expression of TLR2, TLR3 and TLR4 was increased by the combination of IL-17 and IL-23 more than by the single stimulation. This result suggests an important role for the IL-23–IL-17 axis in joint inflammation and TLR expression.
Figure 4.

Interleukin-23 (IL-23) and IL-17 increased expression of Toll-like receptor 2 (TLR2), TLR3 and TLR4 in rhemaotoid arthritis fibroblast-like synoviocytes (RA FLS). (a) and (b) RA (n = 4) and OA (n = 4) FLS (1 × 105 cells) were incubated with recombinant human (rh) IL-17 (1 ng/ml), rhIL-23 (10 ng/ml), or rhIL-17 plus rhIL-23 for 24 hr. TLR2, TLR3 and TLR4 expression was measured by RT-PCR. The expressions of TLR2, TLR3 and TLR4, relative to β-actin were measured. The relative gene expressions are shown on the right. Data are expressed as mean ± SD of four different patients. **P < 0·01 compared with nil (control condition).
IL-17-induced increase in TLR expression via the STAT3 pathway in RA FLS
We investigated whether the STAT3 pathway is involved in the effects of IL-17 and IL-23 on TLR expression. The STAT3 pathway was blocked by S3I-201 as a STAT3 inhibitor,16 and RA and OA FLS were stimulated with 1 ng/ml of IL-17. The mRNA expression of TLR3 was decreased by STAT3 inhibitor, S3I-201, but the TLR2 and TLR4 were not changed by STAT3 inhibitor (Fig. 5a). The pSTAT3 and TLR3 staining was greater in RA synovial tissue than in OA synovial tissue (Fig. 5b).
Figure 5.
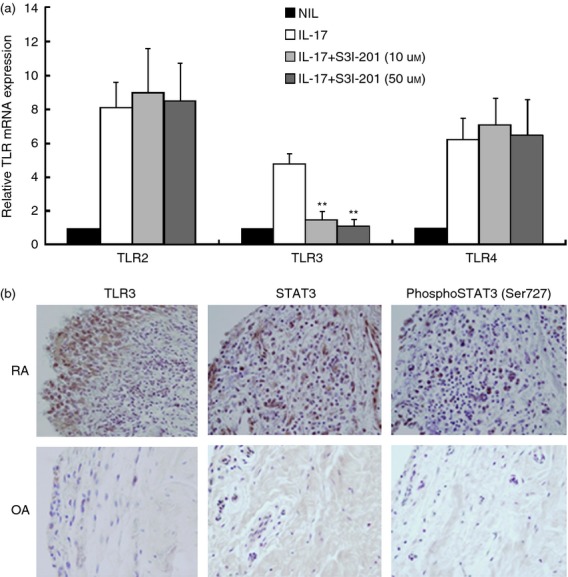
Interleukin-17 (IL-17) increased expression of Toll-like receptor 3 (TLR3) via the phospho-signal transducer and activator of transcription 3 (STAT3) pathway in rheumatoid arthritis fibroblast-like synoviocytes (RA FLS). (a) The STAT3 pathway was blocked with S3I-201 (10 μm or 50 μm), and RA (n = 4) and osteoarthritis (OA) (n = 4) FLS were stimulated with recombinant human (rh) IL-17 (1 ng/ml). The FLS were pre-treated for 1 hr with S3I-201 and then were cultured with rhIL-17 for 24 hr, and the mRNA levels of TLR2, TLR3 and TLR4 were measured by RT-PCR. The expressions of TLR2, TLR3 and TLR4, relative to β-actin were measured. (b) Immunohistochemical staining for TLR3, STAT3 and phospho-STAT3-Ser727 were performed on sections of synovium from RA and OA patients. The sections were stained with specific antibodies to anti-human-TLR3, -STAT3, and -phospho-STAT3. The brown colour shows the positive cells. All tissues were counterstained with haematoxylin (original magnification × 400). Data are expressed as mean ± SD of four different patients. **P < 0·01 compared with IL-17 stimulation.
Discussion
We found higher IL-17 protein concentration and mRNA expression in serum and synovial fluid from RA patients. Immunohistochemical staining showed greater IL-17, TLR2, TLR3 and TLR4 expression in synovial tissues from RA compared with OA patients. Incubation of FLS with IL-23 augmented the effect of IL-17 on the expression of TLR2, TLR3 and TLR4. Immunohistochemical staining also showed that TLR3 and activated STAT3 were highly expressed in RA FLS. We confirmed that the STAT3 blocker S3I-201 reduces TLR3 mRNA expression in RA FLS.
The Th17 cells are the most characterized source of IL-17 during the adaptive immune response and chronic inflammatory response.17 Th17 cells are associated with the induction of autoimmunity and, under the influence of IL-23, these cells produce IL-17, IL-6, IL-22, TNF-α and other factors.17,18 Interleukin-17, a cytokine with pro-inflammatory properties, stimulates the production of the IL-23 p19 subunit by the activating mitogen-activated protein kinase nuclear factor-κB and phosphatidylinositol-3-kinase/Akt signalling pathways.19 The IL-23–IL-17 axis, rather than IL-12 or interferon-γ, plays a central role in autoimmune inflammation in joints. The IL-23–IL-17 loop is essential for both the onset phase and destruction phases of RA.19 We found that IL-17 protein levels are high in the serum and synovial fluid of RA patients and that IL-17 is expressed in the RA synovium. Our data suggest that IL-17 is an important cytokine associated with RA pathogenesis. Interleukin-23 had a synergistic effect to that of IL-17 in increasing TLR expression. Taken together, our data suggest that the IL-23–IL-17 axis plays an important role in joint inflammation and TLR expression.
The FLS from RA are part of the innate immune system. These cells express TLRs and produce cytokines, chemokines and other factors. They are an essential cellular component of RA pathogenesis, and are influenced by each other. In the CIA model, TLR2, TLR4 and TLR9 expression increased in the synovium. In our current study, IL-17 increased TLR expression in RA FLS. Most studies of RA FLS show high expression of TLR2, TLR3 and TLR4.1,8 The role of TLR3 in RA pathogenesis seems to involve TLR3 activation of RANKL through IL-1β.9 Our previous studies have shown expression of TLR2, TLR410 and TLR39,19 in RA FLS. Our current data show the simultaneous TLR profiles in RA FLS. Our results coincide with those of other investigators.1,8
Toll-like receptor stimulation induces the production of inflammatory cytokines and induction of their signalling. TLR2 and TLR4 ligands induce RANKL expression in RA FLS. TLR stimulation of RA FLS also induces the production of IL-1β and TNF-α to a lesser extent, but has no effect on IL-17 production.10 The TLR3 activation also induces the production of IL-1β but has no effect on IL-17 or TNF-α production in RA FLS.9 Brentano et al.1 found strong up-regulation of IL-6, MMP-1 and MMP-3 by all three TLR ligands; the greatest up-regulation was achieved by the TLR3 ligand poly(I-C) in RA FLS.
The results of TLR stimulation seem to vary between cells and in different diseases. Both TLR4 and TLR9 are generally thought to induce a Th1 response by driving IL-12 production by dendritic cells. The TLR2 activation might induce a Th2-biased immune response through the production of IL-10 and IL-13; TLR2 may also trigger the proliferation of regulatory T cells, which play a crucial role in the induction of tolerance to self antigens and protection against autoimmunity.20,21 TLR2 controls the function of regulatory T cells in a MyD88-dependent fashion and regulation of IFN-γ-producing Th1 cells.11,22 In contrast, TLR4 contributes to more severe disease by modulating the Th17 cell population and IL-17 production in IL-1 receptor antagonist knockout mice.11 In macrophages, a major type of synovial lining cell, TLRs activate opposing negative and positive feedback loops. They induce the production of both inflammatory cytokines such as TNF-α and anti-inflammatory molecules such as IL-10. Interleukin-10 inhibits the production of TNF via a STAT3-dependent feedback loop. Interferon-γ primes cells to increase their TNF production by disrupting the IL-10–STAT3 feedback loop.14 In RA FLS, TLR stimulation induces the production of pro-inflammatory cytokines such as IL-1β and TNF-α, and activates osteoclastogenesis by up-regulating RANKL expression. Inhibiting the expression of TLRs in RA FLS may help to control inflammation and osteoclastogenesis.
In our recent study of a mouse CIA model, we found that IL-17 increased TLR2, TLR4 and TLR9 expression in the synovium. That a pro-inflammatory cytokine, IL-17, can up-regulate TLRs is an important observation. We confirmed that IL-17 induces greater expression of TLR2, TLR3 and TLR4 in RA FLS than in OA FLS. Higher IL-17 inducibility of TLR expression in RA indicated that inflammation and pathogenesis of RA is different from those of OA. In RA, IL-17 is produced by Th17 cells and IL-17 induces TLR2, TLR3 and TLR4 expression. The TLR stimulation produces inflammatory cytokines and activates osteoclastogenesis. These results suggest that another mechanism is responsible for the amplifications of synovial inflammation; i.e. that IL-17 helps to control innate immunity by regulating TLR expression in RA FLS. It may be a respectable hypothesis that adaptive immunity and innate immunity are each amplifying inflammation through TLR induction via IL-17.
What can reduce or block the IL-17 stimulation of TLR expression? STAT3 is required for the development of Th17 cells. STAT3, which is activated by IL-6 and IL-23, may serve as a selective STAT protein in cytokine-mediated Th17 cell differentiation;5,6 IL-17 induction of IL-23 production by FLS leads to a positive feedback mechanism that perpetuates synovial inflammation in RA.15,19 Our result showing that IL-23 and IL-17 have a synergistic effect in increasing TLR expression in RA FLS suggests that the IL-23–IL-17 axis is involved in stimulation of TLR expression. In addition, there is a complex cross-regulation between the major macrophage-activating pathways mediated by TLRs, immunoreceptor tyrosine-based activation motif-coupled immunoreceptors and cytokines/STATs at the level of signal transduction.15 This led us to investigate whether the STAT3 pathway is involved in IL-17-induced changes in TLR expression. Blocking STAT3 with S3I-20116 reduced the IL-17-stimulated expression of only TLR3; TLR2 and TLR4 expression levels did not change.
Interleukin-17 increases TLR2, TLR3 and TLR4 expression in RA FLS. Interestingly, in our study IL-17 increased only TLR3 expression via the STAT3 pathway in RA FLS. In a CIA mouse model, IL-17 increases the expression of TLR2 and TLR4 by stimulating the production of IL-1β and IL-6.4 Our data suggest that different pathways or cytokines are involved in the IL-17-stimulated increase in TLR expression in RA FLS.
In summary, our results show that IL-17 increases TLR expression in RA FLS. The TLRs usually induce production of inflammatory cytokines by diverse signalling pathways. However, the inflammatory cytokine IL-17 continues to stimulate TLR expression, suggesting that another mechanism of inflammatory amplification is involved in autoimmune diseases. Interleukin-17 acts via the STAT3 pathway only for TLR3 expression. Blocking the STAT3 pathway may be a new means of controlling IL-17-induced TLR3 expression and may be an interesting therapeutic target for limiting the role of IL-17 in causing synovial inflammation in RA.
Acknowledgments
This work was supported by the Basic Science Research Programme through the National Research Foundation of Korea (NRF), funded by the Ministry of Education, Science and Technology (grant number 2009-0081791) and by a grant (A092258) from the Korea Healthcare Technology R&D Project, Ministry for Health, Welfare, and Family Affairs, Republic of Korea.
Disclosures
The authors have no conflict of interest of any kind to declare in the materials or services referred to in this article.
References
- 1.Brentano F, Kyburz D, Gay S. Toll-like receptors and rheumatoid arthritis. Methods Mol Biol. 2009;517:329–43. doi: 10.1007/978-1-59745-541-1_20. [DOI] [PubMed] [Google Scholar]
- 2.Ospelt C, Neidhart M, Gay RE, Gay S. Synovial activation in rheumatoid arthritis. Front Biosci. 2004;9:2323–34. doi: 10.2741/1399. [DOI] [PubMed] [Google Scholar]
- 3.Shahrara S, Huang Q, Mandelin AM, 2nd, Pope RM. TH-17 cells in rheumatoid arthritis. Arthritis Res Ther. 2008;10:R93. doi: 10.1186/ar2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee JH, Cho ML, Kim JI, et al. Interleukin 17 (IL-17) increases the expression of Toll-like receptor-2, 4, and 9 by increasing IL-1β and IL-6 production in autoimmune arthritis. J Rheumatol. 2009;36:684–92. doi: 10.3899/jrheum.080169. [DOI] [PubMed] [Google Scholar]
- 5.Paradowska-Gorycka A, Grzybowska-Kowalczyk A, Wojtecka-Lukasik E, Maslinski S. IL-23 in the pathogenesis of rheumatoid arthritis. Scand J Immunol. 2010;71:134–45. doi: 10.1111/j.1365-3083.2009.02361.x. [DOI] [PubMed] [Google Scholar]
- 6.Cho ML, Kang JW, Moon YM, et al. STAT3 and NF-κB signal pathway is required for IL-23-mediated IL-17 production in spontaneous arthritis animal model IL-1 receptor antagonist-deficient mice. J Immunol. 2006;176:5652–61. doi: 10.4049/jimmunol.176.9.5652. [DOI] [PubMed] [Google Scholar]
- 7.Kim HR, Kim HS, Park MK, Cho ML, Lee SH, Kim HY. The clinical role of IL-23p19 in patients with rheumatoid arthritis. Scand J Rheumatol. 2007;36:259–64. doi: 10.1080/03009740701286813. [DOI] [PubMed] [Google Scholar]
- 8.Ospelt C, Brentano F, Rengel Y, et al. Overexpression of toll-like receptors 3 and 4 in synovial tissue from patients with early rheumatoid arthritis: toll-like receptor expression in early and longstanding arthritis. Arthritis Rheum. 2008;58:3684–92. doi: 10.1002/art.24140. [DOI] [PubMed] [Google Scholar]
- 9.Kim KW, Cho ML, Oh HJ, Kim HR, Kang CM, Heo YM, Lee SH, Kim HY. TLR-3 enhances osteoclastogenesis through upregulation of RANKL expression from fibroblast-like synoviocytes in patients with rheumatoid arthritis. Immunol Lett. 2009;124:9–17. doi: 10.1016/j.imlet.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 10.Kim KW, Cho ML, Lee SH, et al. Human rheumatoid synovial fibroblasts promote osteoclastogenic activity by activating RANKL via TLR-2 and TLR-4 activation. Immunol Lett. 2007;110:54–64. doi: 10.1016/j.imlet.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 11.Abdollahi-Roodsaz S, Joosten LA, Koenders MI, et al. Stimulation of TLR2 and TLR4 differentially skews the balance of T cells in a mouse model of arthritis. J Clin Invest. 2008;118:205–16. doi: 10.1172/JCI32639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loures FV, Pina A, Felonato M, Calich VL. TLR2 is a negative regulator of Th17 cells and tissue pathology in a pulmonary model of fungal infection. J Immunol. 2009;183:1279–90. doi: 10.4049/jimmunol.0801599. [DOI] [PubMed] [Google Scholar]
- 13.Nichols JR, Aldrich AL, Mariani MM, Vidlak D, Esen N, Kielian T. TLR2 deficiency leads to increased Th17 infiltrates in experimental brain abscesses. J Immunol. 2009;182:7119–30. doi: 10.4049/jimmunol.0802656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu X, Chen J, Wang L, Ivashkiv LB. Crosstalk among Jak-STAT, Toll-like receptor, and ITAM-dependent pathways in macrophage activation. J Leukoc Biol. 2007;82:237–43. doi: 10.1189/jlb.1206763. [DOI] [PubMed] [Google Scholar]
- 15.Hu X, Chakravarty SD, Ivashkiv LB. Regulation of interferon and Toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol Rev. 2008;226:41–56. doi: 10.1111/j.1600-065X.2008.00707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siddiquee K, Zhang S, Guida WC, et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci USA. 2007;104:7391–6. doi: 10.1073/pnas.0609757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8:337–48. doi: 10.1038/nri2295. [DOI] [PubMed] [Google Scholar]
- 18.Brennan FM, McInnes IB. Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest. 2008;118:3537–45. doi: 10.1172/JCI36389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim HR, Cho ML, Kim KW, et al. Up-regulation of IL-23p19 expression in rheumatoid arthritis synovial fibroblasts by IL-17 through PI3-kinase-, NF-κB- and p38 MAPK-dependent signalling pathways. Rheumatology (Oxford) 2007;46:57–64. doi: 10.1093/rheumatology/kel159. [DOI] [PubMed] [Google Scholar]
- 20.Verreck FA, de Boer T, Langenberg DM, et al. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc Natl Acad Sci USA. 2004;101:4560–5. doi: 10.1073/pnas.0400983101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miossec P. An update on the cytokine network in rheumatoid arthritis. Curr Opin Rheumatol. 2004;16:218–22. doi: 10.1097/00002281-200405000-00009. [DOI] [PubMed] [Google Scholar]
- 22.Sutmuller RP, den Brok MH, Kramer M, et al. Toll-like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest. 2006;116:485–94. doi: 10.1172/JCI25439. [DOI] [PMC free article] [PubMed] [Google Scholar]