

Abstract
Loss-of-function mutations in the Caenorhabditis elegans gene sup-18 suppress the defects in muscle contraction conferred by a gain-of-function mutation in SUP-10, a presumptive regulatory subunit of the SUP-9 two-pore domain K+ channel associated with muscle membranes. We cloned sup-18 and found that it encodes the C. elegans ortholog of mammalian iodotyrosine deiodinase (IYD), an NADH oxidase/flavin reductase that functions in iodine recycling and is important for the biosynthesis of thyroid hormones that regulate metabolism. The FMN-binding site of mammalian IYD is conserved in SUP-18, which appears to require catalytic activity to function. Genetic analyses suggest that SUP-10 can function with SUP-18 to activate SUP-9 through a pathway that is independent of the presumptive SUP-9 regulatory subunit UNC-93. We identified a novel evolutionarily conserved serine-cysteine-rich region in the C-terminal cytoplasmic domain of SUP-9 required for its specific activation by SUP-10 and SUP-18 but not by UNC-93. Since two-pore domain K+ channels regulate the resting membrane potentials of numerous cell types, we suggest that the SUP-18 IYD regulates the activity of the SUP-9 channel using NADH as a coenzyme and thus couples the metabolic state of muscle cells to muscle membrane excitability.
Author Summary
Iodotyrosine deiodinase (IYD) controls the recycling of iodide in the biogenesis of thyroid hormones that regulate metabolism. Defects in IYD result in congenital hypothyroidism, a multisystem disorder that can lead to growth failure and severe mental retardation. We identified the gene sup-18 of the nematode Caenorhabditis elegans as a regulator of the SUP-9/UNC-93/SUP-10 two-pore domain potassium channel complex and showed that SUP-18 is an ortholog of IYD, a member of the NADH oxidase/flavin reductase family. SUP-18 IYD is required for the activation of the channel complex by a gain-of-function mutation of the SUP-10 protein. SUP-9 channel activation by SUP-18 requires a conserved serine-cysteine-rich region in the C-terminus of SUP-9 and is independent of the function of the conserved multi-transmembrane protein UNC-93. We propose that SUP-18 uses NADH as a coenzyme to activate the SUP-9 channel in response to the activity of SUP-10 and the metabolic state of muscle cells.
Introduction
Hypothyroidism, one of the most common endocrine disorders, can cause many different symptoms and can lead to defects in brain development and maturation and retarded postnatal development [1]. For thyroid hormone biosynthesis, iodide is recycled by iodotyrosine deiodinase through the deiodination of monoiodotyrosine and diiodotyrosine, two byproducts in the generation of thyroid hormones [2]–[6]. In humans, this deiodination is catalyzed by human iodotyrosine dehalogenase (DEHAL1)/iodotyrosine deiodinase (IYD), an NADH oxidase/flavin reductase [7]–[10]. Mutations in IYD cause congenital hypothyroidism [11]–[13]. How the activity of IYD is regulated in vivo and whether IYD has other functions remain to be elucidated.
Four transmembrane/two-pore domain K+ channels play a key role in establishing the resting membrane potentials of many cell types and in modulating their responses to neurotransmitters and second messengers [14]–[16]. To date, 15 human two-pore domain K+ channels have been identified [14], [16], [17]. The activities of two-pore domain K+ channels can be regulated by multiple chemical and physical factors, including temperature [18], membrane stretch [19], [20], arachidonic acid [21], pH [22], [23], volatile anesthetics [24], [25] and neurotransmitters [26], [27].
The gene sup-9 of the nematode Caenorhabditis elegans encodes a two-pore domain K+ channel [28]. sup-9(n1550) gain-of-function (gf) mutants are egg-laying defective and display a flaccid paralysis and a rubberband uncoordinated (Unc) behavior: when prodded on the head, a sup-9(n1550gf) worm contracts and relaxes along its entire body without moving backwards, while a wild-type worm contracts its anterior end and moves away [29]. Loss-of-function (lf) mutations in sup-9 or two other genes, sup-10 and unc-93, completely suppress these sup-9(n1550gf) defects [29]–[31]. In addition, gf mutations in sup-10 and unc-93 themselves induce a rubberband Unc paralysis, which in turn are suppressed by lf mutations in sup-9, sup-10 and unc-93 [30]–[32]. lf mutants of unc-93, sup-9 and sup-10 do not have obviously abnormal phenotypes [29]–[31], [33]. The SUP-9 two-pore domain K+ channel is most closely related to human TASK-3 [28], [34], [35]. unc-93 encodes a conserved multi-pass transmembrane protein [33]. An UNC-93 homolog, UNC93b1, is involved in innate immune responses in mammals [36], [37]. sup-10 encodes a novel type-I transmembrane protein [35]. Genetic analyses and the molecular identities of these genes suggest that in vivo SUP-10 and UNC-93 form a protein complex with the SUP-9 two-pore domain K+ channel and modulate its activity as regulatory subunits [28], [33].
Mutations in the gene sup-18 suppress the muscle defects caused by gf mutations in these three genes, strongly suppressing the locomotory defects of sup-10(n983gf) mutants, partially suppressing the locomotory defects of the strong unc-93(e1500gf) mutants, the weak unc-93(n200gf) mutants and the strong sup-9(n1550gf)/+ heterozygous mutants, and suppressing only the lethality of sup-9(n1550gf) mutants [29], [30] (also see Table 1 below). In this study we report that sup-18 encodes the C. elegans ortholog of mammalian iodotyrosine deiodinase/dehalogenase (IYD) [7], [8], [10]. Our findings suggest that SUP-18 is a functional regulator of the SUP-9/SUP-10/UNC-93 two-pore domain K+ channel complex in vivo and that IYD might function with two-pore domain K+ channel complexes in mammals.
Table 1. sup-18(lf) mutations specifically suppress sup-10(n983gf) locomotory defects.
| Genotype | LR ± SEM | n |
| Wild-type | 27.3±0.5 | 36 |
| sup-9(n1550gf) * | Inviable | |
| sup-9(n1550gf)/+ | 1.3±0.4 | 16 |
| sup-10(n983gf) | 5.1±0.5 | 16 |
| unc-93(e1500gf) | 0.2±0.1 | 32 |
| unc-93(n200gf) | 16.1±0.5 | 24 |
| sup-18(n1030) | 25.3±0.7 | 16 |
| sup-9(n1550gf); sup-18(n1030) | 0.0±0.0 | 12 |
| sup-9(n1550gf)/+; sup-18(n1030) | 4.6±0.5 | 16 |
| unc-93(e1500gf) sup-18(n1030) | 1.0±0.3 | 35 |
| unc-93(n200gf) sup-18(n1030) | 21.4±0.5 | 36 |
| sup-18(n1030); sup-10(n983gf) | 26.4±0.6 | 16 |
| sup-9(n1913); unc-93(e1500gf) | 27.1±0.5 | 11 |
| unc-93(e1500gf); sup-10(n4025) | 13.9±0.6 | 30 |
| unc-93(e1500gf) sup-18(n1030); sup-10(n4025) | 19.1±0.4 | 30 |
| unc-93(e1500gf); sup-10(n4026) | 14.9±0.5 | 30 |
| unc-93(e1500gf) sup-18(n1030); sup-10(n4026) | 19.6±0.4 | 30 |
Young adult hermaphrodites were assayed for the number of body bends made on a bacterial lawn in 1 min interval.
: sup-9(n1550gf) homozygous animals are inviable [35].
LR: locomotory rate (bodybends/min).
Results
sup-18 has gene-specific effects on the rubberband Unc phenotype
sup-10(n983gf) mutants have a reduced locomotory rate (Table 1). A loss-of-function mutation in sup-18, n1030, restores wild-type locomotion to sup-10(n983gf) mutants (Table 1) [30]. unc-93(n200gf) causes a less severe rubberband Unc phenotype than sup-10(n983gf), yet the unc-93(n200gf) phenotype is still only partially suppressed by sup-18(n1030) (Table 1). unc-93(e1500gf) mutants, which have a more severe rubberband Unc phenotype than sup-10(n983gf) mutants, similarly are only weakly suppressed by sup-18(n1030). These results suggest that the differential suppression of the rubberband Unc mutants by sup-18(n1030) is caused by gene-specific effects rather than by differential severity of paralysis in these mutants.
We further tested this notion using weakly paralyzed double mutants carrying the unc-93(e1500gf) mutation and a partial lf allele of sup-10. Introduction of the sup-18(n1030) mutation into partially suppressed unc-93(e1500gf); sup-10(n4025) or unc-93(e1500gf); sup-10(n4026) mutants only weakly improved their locomotory rates from approximately 14 to 19 body-bends/minute (Table 1). These results confirm that sup-18(n1030) only weakly suppresses gf mutations in unc-93.
The suppression of sup-10(n983gf) depends on the dosage of the sup-18 allele [29]. We found that sup-18(n1030)/+; sup-10(n983gf) males exhibit an intermediate phenotype (15.2 bends/min) between those of the more severely paralyzed sup-10(n983gf) males (4.7 bends/min) and the strongly suppressed sup-18(n1030); sup-10(n983gf) males (31.7 bends/min) (Table 2). This dose-dependent effect was observed for all lf alleles of sup-18 tested (Table 2). By contrast, the suppression of sup-10(n983gf) by sup-9(n1913), a channel null allele, was recessive.
Table 2. sup-18(lf) mutations exhibit dosage-dependent suppression of the locomotory defects of sup-10(n983gf) mutant.
| Genotype | LR ± SEM | n |
| Wild-type | 33.0±1.2 | 36 |
| sup-10(n983gf) | 4.7±0.9 | 25 |
| sup-18(n1030); sup-10(n983gf) | 31.7±0.7 | 15 |
| sup-18(n1030)/+; sup-10(n983gf) | 15.2±0.7 | 25 |
| sup-18(n1033)/+; sup-10(n983gf) | 13.8±0.6 | 25 |
| sup-18(n1014)/+; sup-10(n983gf) | 14.6±0.6 | 25 |
| sup-18(n1036)/+; sup-10(n983gf) | 12.3±0.7 | 25 |
| sup-18(n1471)/+; sup-10(n983gf) | 14.0±0.7 | 25 |
| sup-9(n1913); sup-10(n983gf) | 33.2±1.1 | 15 |
| sup-9(n1913)/+; sup-10(n983gf) | 5.6±0.6 | 20 |
| unc-93(e1500gf); sup-10(n4025) | 10.0±0.5 | 30 |
| unc-93(e1500gf) sup-18(n1030)/+; sup-10(n4025) | 9.8±0.7 | 20 |
| unc-93(e1500gf) sup-18(n1030); sup-10(n4025) | 17.5±0.9 | 30 |
| sup-9(n1550gf)/+ | 3.8±0.5 | 24 |
| sup-9(n1550gf)/+; sup-18(n1030)/+ | 5.0±0.5 | 39 |
| sup-9(n1550gf)/+; sup-18(n1030) | 9.8±0.5 | 22 |
Young adult males were assayed for the number of bends made on a bacterial lawn during 1 min interval. LR: locomotory rate (bodybends/min).
Because the weak suppression of the locomotory defect of unc-93(e1500gf) mutants by sup-18(lf) mutations (Table 1) [30] makes a dosage analysis of sup-18(lf) suppression of unc-93(e1500gf) difficult, we examined weakly paralyzed unc-93(e1500gf); sup-10(n4025) males, which are more visibly suppressed by sup-18(n1030) (Table 2). We found that the locomotory rate of unc-93(e1500gf); sup-10(n4025) males heterozygous for sup-18(n1030) was similar to that of males wild-type for sup-18 (10.0 vs. 9.8, respectively) (Table 2). Similarly, sup-9(n1550gf)/+; sup-18(n1030)/+ males had only slightly improved locomotion compared to sup-9(n1550gf)/+ males (5.0 vs. 3.8, respectively) (Table 2). We conclude that the dose-dependent suppression of rubberband Unc mutants by sup-18 alleles is also gene-specific: the sup-10(n983gf) phenotype is much more sensitive to sup-18 levels than is that of the other rubberband mutants.
sup-18 encodes the C. elegans ortholog of mammalian iodotyrosine deiodinase
sup-18 had previously been mapped to the interval between daf-4 and unc-32 on LGIII [30]. Using three-point mapping we further localized sup-18 to the interval between ncl-1 and unc-36 (see Materials and Methods) (Figure 1A). Transgene rescue experiments with cosmids spanning the ncl-1-to-unc-36 interval and with smaller cosmid subclones identified a 4.5 kb minimal rescuing fragment from cosmid C02C2: as a transgene, this fragment restored the rubberband Unc phenotype to sup-18(n1010); sup-10(n983gf) mutants (Figure 1A). This rescuing fragment contained a single predicted gene, C02C2.5 [www.wormbase.org]. We screened a mixed-stage cDNA library [38] using the smallest cosmid subclone with sup-18 rescuing activity and obtained a single partial cDNA of this predicted gene. We defined the structure of this gene from RT-PCR and RACE experiments (see Materials and Methods) (Figure 1B).
Figure 1. sup-18 encodes the C. elegans ortholog of mammalian iodotyrosine deiodinase.

(A) Top, genetic map of the sup-18 region of linkage group III (LG III). Horizontal lines below sup-18 represent the cosmids tested for rescue of sup-18(n1010); sup-10(n983gf) mutants. Bottom, physical map of cosmid C02C2. Open boxes, coding regions; horizontal brackets, cosmid subclones. Rescued lines, number of independently derived transgenic lines/total number of lines scored. Rescue was scored as the appearance of animals with the phenotype of rubberband Unc paralysis. (B) Intron-exon structure of sup-18 as inferred by comparison of the cDNA and genomic sequences. Dark boxes, coding regions; open boxes, untranslated regions; arrow, direction of transcription. The sup-18 open reading frame is 978 bp, the 5′ UTR is 57 bp and the 3′UTR is 44 bp. (C) Sequence alignment of SUP-18 and iodotyrosine deiodinases from other species. Amino acids conserved in at least three species are darkly shaded, while amino acids with similar physical properties in at least three species are lightly colored. *: sup-18 missense mutations (see Table 3). Residues mutated in human hypothyroidism patients are indicated by red boxes. The FMN cofactor binding residues in mouse IYD are indicated by yellow boxes. The transmembrane domain and NADH oxidase/flavin reductase domain are underlined and labeled. Genebank accession numbers are as follows: SUP-18, JX978835; Drosophila, AAM11009; M. musculus, AAH23358; H. sapiens, NP_981932.
sup-18 encodes a predicted protein of 325 amino acids. This protein is the only C. elegans ortholog of mammalian iodotyrosine deiodinase (IYD), which belongs to the NADH oxidase/flavin reductase superfamily (Fig. 1C) [7]–[10]. IYD catalyzes the recycling of iodide by deiodinating 3′-monoiodotyrosine and 3′, 5′-diiodotyrosine, the main byproducts in the process of thyroid hormone biogenesis [2]–[5], [7], [8]. The identity between SUP-18 and human IYD protein variant 2 (also named DEHAL1) [8] is 31% overall and 45% over the NADH oxidase/flavin reductase domain (Figure 1C). Like IYDs of Drosophila, mouse and human, SUP-18 has a hydrophobic region that precedes the NADH oxidase/flavin reductase domain and might serve as a transmembrane domain.
We identified molecular lesions in the sup-18 coding sequence of all 18 mutant strains analyzed (Table 3, Fig. 1C). The sup-18(n1033) mutation leads to the substitution of an isoleucine for the initiator methionine, which should cause any translational products to be nonfunctional. (The next three ATG sequences in the sup-18 cDNA are out of frame.) The sup-18(n1030) and sup-18(n1548) mutations cause premature stop codons that likely generate truncated protein products. Four mutations (n1038, n527, n463, n1539) cause a frameshift. Another four mutations (n1036, n1035, n1015, n1558) affect splice donor or acceptor sites. The remaining seven missense mutations (n1010, n1554, n1471, n1556, n1014, n1022, n528) disrupt residues within the NADH oxidase/flavin reductase domain.
Table 3. sup-18 loss-of-function mutations.
| Allele | Mutation | Effect | Mutagen | Background |
| n1033 | ATG to ATT | M1I | EMS | sup-10(n983gf) |
| n1030 | CGA to TGA | R65stop | EMS | sup-10(n983gf) |
| n1038 | 916 bp deletion | 97+frameshift | EMS | unc-93(e1500gf) |
| n527 | 13 bp deletion | 154+frameshift | Spont | unc-93(e1500gf) |
| n1548 | TGG to TAG | W170stop | EMS | sup-10(n983gf) |
| n463 | 4 bp deletion | 175+frameshift | Spont | unc-93(e1500gf) |
| n1539 | Tc3 Insertion | 320+frameshift | Spont | sup-10(n983gf) |
| n1036 | agGT to aaGT | 3rd splice acceptor | EMS | sup-10(n983gf) |
| n1035 | agGC to aaGC | 5th splice acceptor | EMS | sup-10(n983gf) |
| n1015 | GTgt to GTat | 8th splice donor | EMS | sup-10(n983gf) |
| n1558 | agAT to aaAT | 8th splice acceptor | EMS | sup-10(n983gf) |
| n1010 | AGT to AAT | S137N | EMS | sup-10(n983gf) |
| n1554 | GGC to AGC | G258D | EMS | sup-10(n983gf) |
| n1471 | GGC to GAC | G258S | Gamma | sup-10(n983gf) |
| n1556 | ACT to ATT | T271I | EMS | sup-10(n983gf) |
| n1014 | GGA to AGA | G280R | EMS | sup-10(n983gf) |
| n1022 | AGG to AAG | R289K | EMS | sup-10(n983gf) |
| n528 | ACC to CCC | T322P | Spont | unc-93(e1500gf) |
DNA sequences were determined for both strands of sup-18 exons and intron/exon boundaries of each mutant. For splice-junction mutants, the intron sequence is indicated by lowercase and the exon sequence by uppercase letters. EMS, ethyl methanesulfonate; Spont, spontaneous. Frameshift, mutations causing frameshift after the indicated codons.
SUP-18 and SUP-10 are similarly localized within muscles
To examine the expression pattern of sup-18, we introduced the coding sequence of gfp between codons 88 and 89 of a genomic clone of sup-18, generating a sup-18 translation fusion transgene (see Materials and Methods). Similar to transgenic animals expressing a Psup-10::gfp translational fusion transgene, Psup-18::gfp transgenic animals displayed GFP fluorescence in body-wall (Fig. 2A, D), defecation (Fig. 2B, E) and vulval muscles (Fig. 2C, F). In body-wall muscle cells (Fig. 2A, D), the SUP-10::GFP and SUP-18::GFP fusion proteins both localized to cell-surface regions aligned with dense bodies, the functional analogs to vertebrate Z-lines that connect the myofibril lattice to the cell membrane [39]. In addition to muscles, three neurons in the head of Psup-18::gfp transgenic animals also displayed GFP staining (I. de la Cruz and H. R. Horvitz, unpublished observations). We previously reported expression of a Psup-9::gfp reporter in the four SIA interneurons [28]. We stained the Psup-18::gfp transgenic animals with an anti-CEH-17 antibody, which labels the four SIA neurons and the ALA neuron [40], and found that the neurons expressing the SUP-18::GFP fusion protein were not the SIAs (I. de la Cruz and H. R. Horvitz, unpublished observations).
Figure 2. SUP-18 is expressed predominantly in muscles and co-localizes subcellularly with SUP-10.
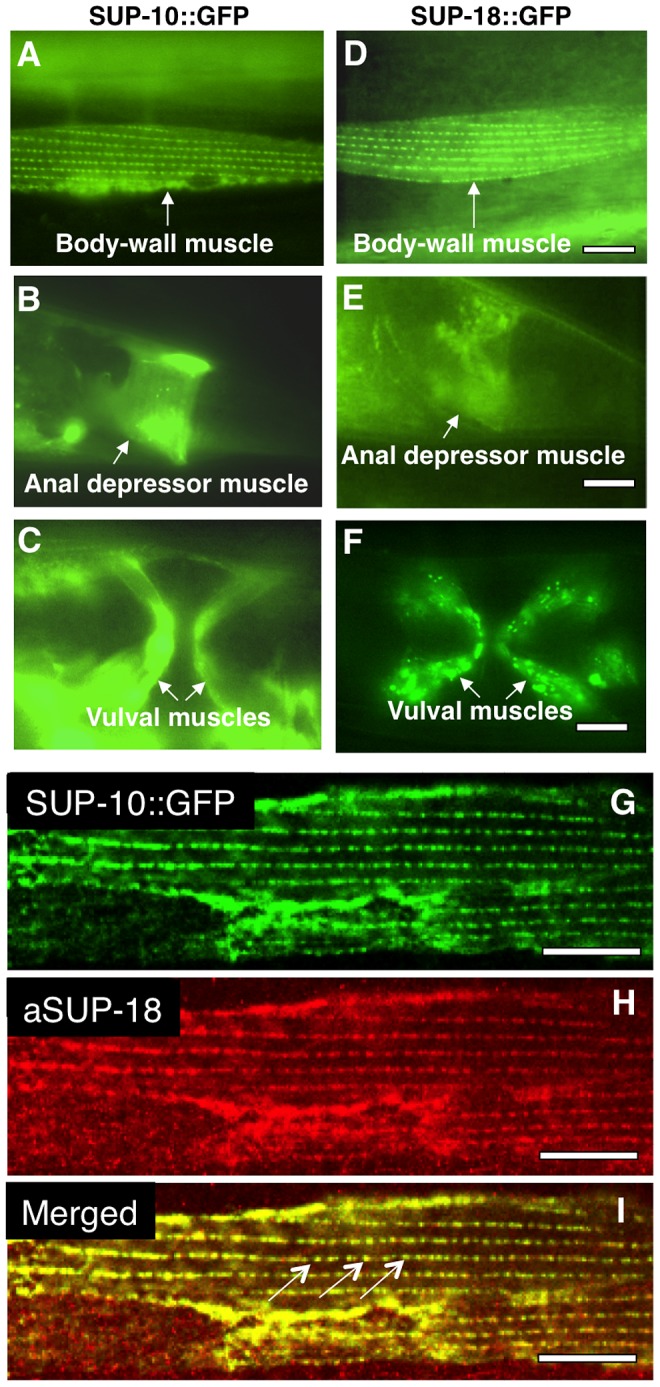
Epifluorescence images of worms carrying (A–C) a Psup-10::gfp translational fusion transgene or (D–F) a Psup-18::gfp translational fusion transgene. (A, D) Body-wall muscle cells displaying GFP fluorescence in dense body-like structures. (B, E) Tail regions of transgenic animals showing fluorescence in the anal depressor muscles (arrows). (C, F) Ventral views of transgenic animals showing fluorescence in vulval muscles (arrows). (G, H, I) Confocal microscopic images of an animal expressing a Psup-10::gfp translational fusion transgene and a Pmyo-3sup-18 transgene. SUP-10::GFP fusion protein (G) was visualized by GFP signals and SUP-18 (H) was detected by immunostaining with a rabbit anti-SUP-18 polyclonal antibody (see Materials and Methods). (I) The merged picture indicates colocalization of SUP-10::GFP and SUP-18 in the dense bodies (arrows). Scale bars, 10 µm.
We generated a rabbit anti-SUP-18 antibody (see Materials and Methods). In immunostained animals, this antibody could detect overexpressed SUP-18 but failed to detect endogenous SUP-18, probably because of the low level of SUP-18 expression. We next generated transgenic animals co-expressing a Psup-10::gfp fusion transgene and sup-18 under control of a myo-3 promoter [41] and examined the subcellular expression of SUP-18 using the antibody and of SUP-10::GFP using GFP fluorescence. We found that SUP-10 and SUP-18 colocalize in subcellular structures, including the dense bodies in the body-wall muscles (Fig. 2G, H, I). Since GFP fusions to SUP-9 and UNC-93 localize similarly [28], this result suggests that SUP-18 colocalizes with a SUP-9/UNC-93/SUP-10 complex.
SUP-18 is a type-I transmembrane protein that can function independently of membrane anchoring
Mammalian IYD is a transmembrane protein [7], [8]. The presence of a possible transmembrane domain in the predicted SUP-18 protein sequence (Fig. 1C) suggests that SUP-18 is also a transmembrane protein. To distinguish whether the NADH oxidase/flavin reductase domain of SUP-18 resides intracellularly or extracellularly, we generated transgenic animals expressing different SUP-18::β-galactosidase fusion proteins and assayed β-galactosidase activity in vivo in fixed animals (Fig. 3A). When β-galactosidase is localized intracellularly it is enzymatically active, whereas extracellular localization results in loss of β-galactosidase activity [42], [43]. The use of β-galactosidase activity to elucidate the membrane topology of C. elegans proteins in vivo has been reported previously for the presenilin SEL-12 protein [44] and for the MEC-4 sodium channel subunit [45].
Figure 3. SUP-18 is a type-I transmembrane protein with an NADH oxidase/flavin reductase domain that resides intracellularly and can function without plasma membrane localization.

(A) Transgenes expressing a SUP-18::β-galactosidase fusion protein were transformed into wild-type animals using the dominant rol-6 coinjection marker. β-galactosidase assays were performed as described [77]. At least 100 animals per transgenic line were scored for lacZ staining using Nomarski optics. Quantification was as following: staining of adult vulval muscles and larvae easily observed at 10× magnification in >30% of animals (++); staining of 3–30% of animals (+); staining of fewer than 3% of animals (−); no transgenic lines, not labeled. Schematic: open boxes, SUP-18 transmembrane domain; black boxes, a synthetic transmembrane domain; EC, SUP-18 extracellular domain; SS, signal sequence; NR, NADH oxidase/flavin reductase domain. (B) Lines of sup-18(n1033); sup-10(n983gf) animals carrying transgenes (Tgs) driven by the myo-3 promoter, as indicated, were scored for locomotory rates. Pmyo-3 gfp (pPD93.97) was used as a coinjection marker to allow the identification of transgenic animals by their GFP fluorescence. Non-transgenic sup-10(n983gf) and sup-10(n1033); sup-10(n983gf) mutants were scored as controls. Error bars, mean ± SEM; n = 12 for each transgenic line or control genotype. sup-18(intra): transgenes expressing the SUP-18 intracellular domain (amino acid 66–325) fused with GFP.
Fixed transgenic animals expressing β-galactosidase fused to either the C-terminal region of SUP-18 or immediately C-terminal to the putative transmembrane domain showed robust β-galactosidase activity (Fig. 3A). Introduction of a synthetic transmembrane domain [45] between SUP-18 and β-galactosidase in these chimeras eliminated β-galactosidase enzymatic activity, presumably because the membrane orientation of β-galactosidase had been reversed (Fig. 3A).
These results strongly suggest that SUP-18 is a transmembrane protein and that the NADH oxidase/flavin reductase domain of SUP-18 resides intracellularly. But they do not distinguish between a type-I transmembrane protein (single-pass transmembrane protein with the N-terminal domain located extracellularly) and a cytoplasmic protein that simply localizes at the cell surface, e.g., by interacting with another membrane protein or by linking to a GPI anchor [46]. To test if the putative transmembrane domain of SUP-18 can indeed behave as a transmembrane domain, we inserted a signal sequence at the N-terminus of SUP-18 (see Materials and Methods). While a fusion containing the presumptive extracellular domain of SUP-18 but lacking the putative transmembrane domain resided intracellularly as expected, the introduction of a signal sequence led to its secretion and loss of β-galactosidase enzymatic activity (Fig. 3A). By contrast, when either the SUP-18 putative transmembrane domain or the synthetic transmembrane domain [45] was added to this SUP-18::β-galactosidase fusion, the enzymatic activity was restored. These results indicate that the putative transmembrane domain of SUP-18 can indeed function as a transmembrane domain and suggest that SUP-18 is likely a type-I integral membrane protein, like IYD.
To establish an assay for in vivo SUP-18 activity, we expressed the sup-18 coding sequence under the control of the myo-3 promoter [41] in sup-18(n1033); sup-10(n983) mutant animals. While sup-10(n983gf) mutant animals are defective in locomotion, double mutants carrying the sup-18(n1033) null mutation had improved locomotory rates (Fig. 3B). Expression of Pmyo-3 gfp in sup-18(n1033); sup-10(n983gf) animals had little effect on their locomotory rate, whereas expression of a Pmyo-3 sup-18(+) transgene restored sup-10(n983gf) paralysis (Fig. 3B). By contrast, expression of two Pmyo-3 sup-18 mutant constructs containing either the n1554 missense mutation or the n1010 mutation (which affects a conserved amino acid in the NADH oxidase/flavin reductase domain; Fig. 1C and Table 3) did not restore the rubberband Unc phenotype to sup-18(n1033); sup-10(n983gf) mutants (Fig. 3B).
We found that the mouse IYD gene could not substitute for sup-18 in vivo in restoring the rubberband Unc phenotype of sup-18(n1033); sup-10(n983gf) animals (Figure 3B). We tagged mouse IYD with GFP at its C-terminus and found that C. elegans expressing the fusion protein displayed GFP fluorescence in body-wall muscle structures similar to that observed for the SUP-18::GFP fusion (I. de la Cruz and H. R. Horvitz, unpublished observations). These results suggest that mouse IYD had been expressed properly and that mouse IYD might be inactive or otherwise incapable of substituting for SUP-18 in C. elegans.
Interestingly, transgenic expression of the SUP-18 intracellular domain alone (amino acids 66–325) was sufficient to restore rubberband Unc paralysis to sup-18(n1033); sup-10(n983gf) animals, although the rescue was less robust than that conferred by full-length SUP-18 (Fig. 3B). This finding suggests that the extracellular and transmembrane domains of SUP-18 are not essential for its in vivo function and is consistent with the conclusion that the NADH oxidase/flavin reductase domain is intracellular.
Increased sup-18(+) expression in body-wall muscles specifically enhances the behavioral defects of sup-10(n983gf) mutants
The overexpression of sup-18(+) from a Pmyo-3 sup-18(+) transgene in sup-18(n1033): sup-10(n983gf) mutants not only restored the rubberband Unc phenotype but also apparently enhanced that phenotype beyond that of sup-10(n983gf) single mutants (Fig. 3B). This finding indicates a dose-dependent effect of sup-18(+) and is consistent with our gene-dosage observation that sup-18(lf)/+ can partially improve the locomotory rate of sup-10(n983gf) mutants (Table 2). Overexpression of sup-18(+) with the coinjection marker lin-15(+) in lin-15 mutant animals did not cause obvious differences in locomotion compared to animals injected with lin-15(+) alone (Table 4), indicating that overexpression of sup-18(+) itself did not slow locomotion.
Table 4. Overexpression of sup-18 in body-wall muscles enhances the defects of sup-10(n983gf) but not unc-93(e1500gf) mutants.
| Genotype | LR ± SEM | Brood size |
| Wild-type | 26.8±0.4 | ND |
| lin-15; nEx[lin-15(+)#1] | 26.5±0.9 | ND |
| lin-15; nEx[lin-15(+)#2] | 27.3±0.7 | ND |
| lin-15; nEx[lin-15(+); sup-18(+)#1] | 27.1±0.8 | ND |
| lin-15; nEx[lin-15(+); sup-18(+)#2] | 26.9±0.8 | ND |
| lin-15; nEx[lin-15(+); sup-10(+); sup-18(+)#1] | 24.5±0.8 | ND |
| lin-15; nEx[lin-15(+); sup-10(+); sup-18(+)#2] | 25.2±0.8 | ND |
| sup-10(n983gf) lin-15; nEx[lin-15(+)#1] | 5.7±0.4 | 74±5 |
| sup-10(n983gf) lin-15; nEx[lin-15(+)#2] | 5.4±0.4 | 75±4 |
| sup-10(n983gf) lin-15; nEx[lin-15(+); sup-18(+)#1] | 0.1±0.1 | 27±3 |
| sup-10(n983gf) lin-15; nEx[lin-15(+); sup-18(+)#2] | 0.0±0.0 | 17±3 |
| sup-10(n983gf) lin-15; nEx[lin-15(+); sup-10(n983gf)#1] | 5.9±1.2 | ND |
| sup-10(n983gf) lin-15; nEx[lin-15(+); sup-10(n983gf)#2] | 6.6±0.8 | ND |
| sup-10(n983gf) lin-15; nEx[lin-15(+); sup-9(+)#1] | 6.0±0.6 | ND |
| sup-10(n983gf) lin-15; nEx[lin-15(+); sup-9(+)#2] | 5.1±0.4 | ND |
| sup-10(n983gf) lin-15; nEx[lin-15(+); unc-93(+)#1] | 5.1±0.3 | ND |
| sup-10(n983gf) lin-15; nEx[lin-15(+); unc-93(+)#2] | 5.8±0.5 | ND |
| unc-93(e1500gf); lin-15; nEx[lin-15(+)#1] | 0.0±0.0 | 35±2 |
| unc-93(e1500gf); lin-15; nEx[lin-15(+)#2] | 0.0±0.0 | 43±2 |
| unc-93(e1500gf); lin-15; nEx[lin-15(+); sup-18(+)#1] | 0.0±0.0 | 37±2 |
| unc-93(e1500gf); lin-15; nEx[lin-15(+); sup-18(+)#2] | 0.0±0.0 | 40±3 |
| sup-9(n1550gf); sup-18(n1030) | 0.0±0.0 | 24±1 |
Locomotion rate, mean for 12 animals. Brood size, mean for 10 animals. Four extrachromosomal arrays (nEx) were generated, two containing lin-15(+) alone and two containing both lin-15(+) and sup-18(+), and were introduced into the different genetic backgrounds by mating to ensure consistent gene dosage among experiments. ND, not determined. LR: locomotory rate (bodybends/min).
We introduced the extrachromosomal arrays containing the transgenes from two independently-derived strains carrying sup-18(+) and the lin-15(+) coinjection marker into sup-10(n983gf) lin-15 double mutants by mating, so that each resulting strain would contain the same transgenes as the parental strain and therefore would overexpress sup-18(+) at equivalent levels. sup-18(+) overexpression caused a severe paralysis of sup-10(n983gf) lin-15 animals relative to control transgenic animals expressing lin-15 alone (0.1 and 0.0 vs. 5.7 and 5.4, bends/minute, respectively) (Table 4). sup-10(n983gf) mutants overexpressing sup-18(+) were smaller in size (Fig. 4A–D) and resembled severely paralyzed mutants carrying a sup-9(n1550gf) mutation (compare Figs. 4B and 4D).
Figure 4. Overexpression of a sup-18 transgene enhances the rubberband Unc phenotype of sup-10(n983gf) but not unc-93(e1500gf) mutants.
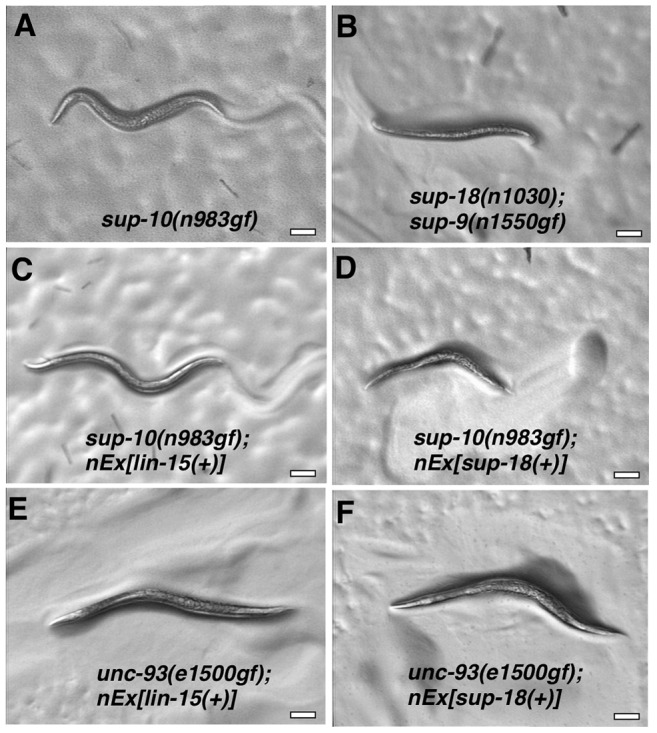
Hermaphrodites were photographed at least 2 µm. Genotypes were as follows: (A) sup-10(n983gf). (B) sup-18(n1030); sup-9(n1550gf). (C) sup-10(n983gf); lin-15(n765); nEx[lin-15(+)]. (D) sup-10(n983gf); lin-15(n765); nEx[lin-15(+), Pmyo-3 sup-18(+)]. (E) unc-93(e1500gf); lin-15(n765); nEx[lin-15(+)]. (F) unc-93(e1500gf); lin-15(n765); nEx[lin-15(+), Pmyo-3 sup-18(+)].
We next tested if overexpression of sup-9(+), unc-93(+) or sup-10(n983gf) itself could enhance the sup-10(n983gf) defect as did overexpression of sup-18(+). Overexpression of these other genes under the control of the myo-3 promoter did not affect the locomotory rate of transgenic sup-10(n983gf) mutant animals compared to animals transgenic for lin-15 alone (Table 4). These results suggest that the activity of SUP-18 might be enhanced by increased expression, while increased expression of SUP-9, UNC-93 and SUP-10 does not increase the biological effects of these proteins.
We tested if overexpression of sup-18(+) could enhance the defects of unc-93(e1500gf) mutants and found no obvious difference in appearance compared to control animals overexpressing lin-15 alone (Fig. 4E, F). Because the locomotory rate of unc-93(e1500gf) mutants transgenic for either sup-18(+) or lin-15(+) transgenes was zero (Table 4) and an enhancement of locomotory defects could not be scored, we turned to a different aspect of the phenotype of rubberband mutants, a reduced brood size [29]. Consistent with the enhancement of locomotory defects, overexpression of sup-18(+) reduced the brood size of sup-10(n983gf) mutants by three-fold, from an average of 74 and 75 progeny for the two transgenic lines, to 17 and 27, respectively (Table 4). These low brood sizes are comparable to those of severely paralyzed sup-9(n1550gf); sup-18(n1030) mutants (Table 4). By contrast, the brood sizes of unc-93(e1500gf) mutants did not change in response to sup-18(+) overexpression (35 and 43 vs. 37 and 40, respectively). Thus, the effects of sup-18(+) overexpression on the locomotion and brood size of rubberband Unc mutants are gene-specific: the sup-10(n983gf) phenotype is more sensitive to increased sup-18 levels than is that of unc-93(e1500gf) mutants.
The sup-9(n1435) mutation specifically suppresses the behavioral defects of sup-10(n983gf) mutants
Like sup-18 mutations, the sup-9 allele n1435 strongly suppresses the locomotory defects of sup-10(n983gf) but not those of unc-93(e1500gf) mutants (Table 5) [29]. By contrast, null mutations in sup-9, such as sup-9(n1913), completely suppress the defects caused by gf mutations in both sup-10 and unc-93 (Table 5) [30], [31]. To determine if other sup-9 alleles exhibit similar gene-specific effects, we assayed 13 previously isolated sup-9 missense mutations [34], [35], [36], [39]. Four sup-9 mutations that had been isolated as sup-10(n983gf) suppressors and nine that had been isolated as unc-93(e1500gf) suppressors all strongly suppressed unc-93(e1500gf) and sup-10(n983gf) defects equally well (Table 5), confirming that sup-9(n1435) represents a rare class of sup-9 mutations.
Table 5. Suppression of sup-10(n983gf) and unc-93(e1500gf) locomotory defects (bodybends) by sup-9 mutations.
| sup-9 alleles | sup-10(n983gf) | unc-93(e1500gf) |
| Wild-type | 5.1±0.5 | 0.1±0.1 |
| n1913 (null) | 25.8±0.7 | 26.8±0.8 |
| n1435 | 26.4±0.8 | 0.3±0.1 |
| n1016 | 25.9±0.9 | 25.6±0.8 |
| n1025 | 23.6±0.8 | 26.9±0.4 |
| n1472 | 24.5±0.5 | 22.3±0.9 |
| n1557 | 24.8±0.9 | 28.4±0.8 |
| lr35 | 24.9±0.5 | 26.4±0.7 |
| lr45 | 26.5±0.6 | 23.4±0.8 |
| lr100 | 24.3±0.6 | 24.6±0.7 |
| lr129 | 26.0±0.7 | 23.3±0.5 |
| lr142 | 24.1±0.5 | 25.6±0.4 |
| n213 | 27.2±0.7 | 24.2±0.5 |
| n264 | 22.4±0.6 | 22.3±0.6 |
| n233 | 26.1±0.6 | 26.7±0.7 |
| lr38 | 26.7±0.5 | 26.4±0.6 |
Each assay represents the mean ± SEM of hermaphrodite bodybends assayed in 1 min intervals. n = 12 for all strains.
The similarity of sup-18(lf) mutations and sup-9(n1435) in preferentially suppressing sup-10(n983gf) defects compared to those of unc-93(e1500gf) mutants suggests that sup-18(lf) mutations and the sup-9(n1435) mutation might act via the same mechanism. If so, n1435 might have no suppressive activity in the absence of sup-18. Indeed, the locomotory rate of the sup-9(n1435); unc-93(e1500gf) sup-18(n1030) triple mutant was similar to that of either the sup-9(n1435); unc-93(e1500gf) or the unc-93(e1500gf) sup-18(n1030) double mutant (Fig. 5A). This effect appears to be specific for sup-9(n1435), as a different weak sup-9 allele, n264, was enhanced by sup-18(n1030) (Fig. 5A). We also assayed the brood size of unc-93(e1500gf) mutants in the presence of either or both sup-18(n1030) and sup-9(n1435). For example, although the low brood size of unc-93(e1500gf) mutants was restored to wild-type levels by the null mutation sup-9(n1913) (Fig. 5B), sup-9(n1435) and sup-18(n1030) single mutations or sup-9(n1435); sup-18(n1030) double mutations only partially rescued the brood size of unc-93(e1500gf) mutants and the double mutations acted similarly to the sup-18(n1030) single mutation (Fig. 5B). As was the case for locomotion, for brood size sup-18(n1030) enhanced the effect of the weak loss-of-function allele, sup-9(n264) on unc-93(e1500gf) mutants (Fig. 5B). The lack of an additive effect of sup-18(n1030) and sup-9(n1435) in suppressing the locomotion and brood size defects of unc-93(e1500gf) mutants suggests that sup-9(n1435) and sup-18(n1030) mutations likely act through the same pathway.
Figure 5. The sup-9(n1435) and sup-18(lf) mutations act similarly in suppressing rubberband Unc mutant phenotypes.
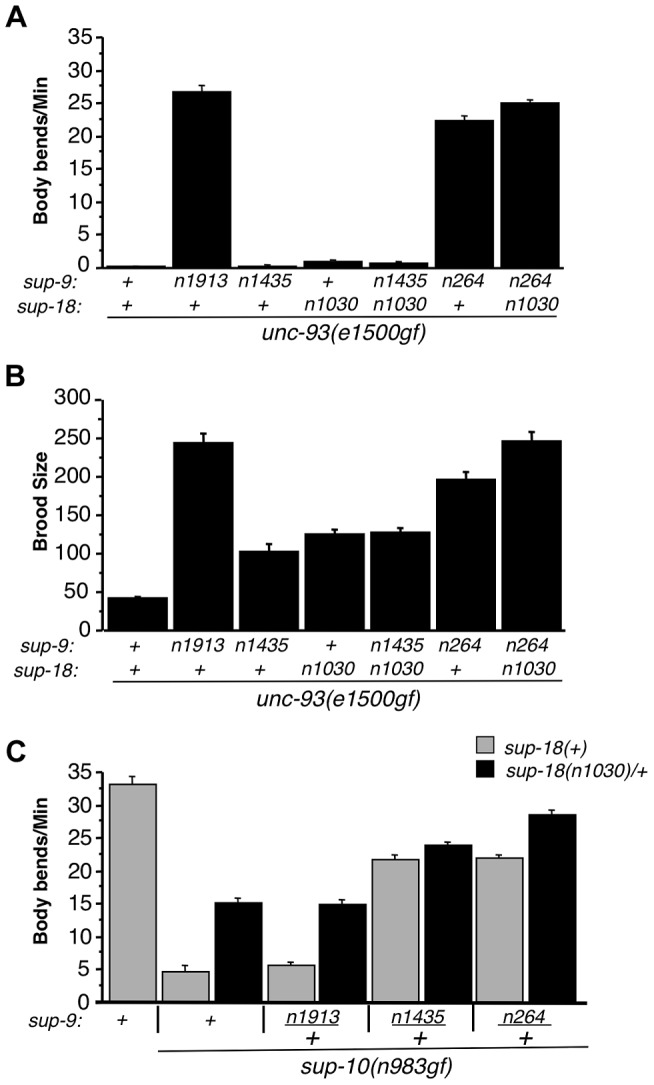
(A) The effects of sup-9(n1435) and sup-18(n1030) mutations on the locomotion (body bends) of unc-93(gf) mutants are not additive. At least 12 young hermaphrodites were assayed for each genotype. Error bars, means ± SEMs. (B) The suppressive effects of sup-9(n1435) and sup-18(n1030) on the brood size defects of unc-93(e1500gf) mutants are not additive. Brood sizes of hermaphrodites were determined by picking L4 hermaphrodites to single plates, passaging them each day to a new plate and scoring the number of progeny on each plate. At least 11 animals were assayed for each genotype. Error bars, means ± SEMs. (C) Body bend assays of young males. At least 15 animals were assayed for each genotype. Error bars, means ± SEMs.
To further examine this hypothesis, we tested for an additive effect between sup-18(n1030)/+ and sup-9(n1435)/+ in their suppression of the locomotory defects of sup-10(n983gf) mutants. (A test for an additive effect of sup-18(n1030) and sup-9(n1435) homozygous mutations would not be informative, as both mutations fully suppress the locomotory defect of sup-10(n983gf) mutants.) We found that sup-10(n983gf) males heterozygous for either sup-9(n1435)/+ or sup-18(n1030)/+ are partially suppressed for the locomotory defects (Fig. 5C). The sup-9(n1435)/+; sup-18(n1030)/+; sup-10(n983gf) male triple mutant moved only slightly better than sup-9(n1435)/+; sup-10(n983gf) mutants (23.7±0.6 vs. 21.6±0.9, mean ± SEM, respectively) (Fig. 5C), suggesting a very weak additive effect of sup-18(n1030)/+ and sup-9(n1435)/+. [This small effect might be caused by the presence in these animals of wild-type SUP-9 dimers at a fourth the wild-type level; this SUP-9 would respond to sup-18(n1030)/+ effects.] To verify the specificity of the interaction between sup-18(n1030) and sup-9(n1435), we tested sup-9(n264). sup-9(n264)/+ is as strong as sup-9(n1435)/+ in suppressing the locomotory defects of sup-10(n983gf) mutants. However, unlike sup-9(n1435)/+; sup-18(n1030)/+; sup-10(n983gf) mutants, sup-9(n264)/+; sup-18(n1030)/+; sup-10(n983gf) mutants moved better than sup-9(n264)/+; sup-10(n983gf) mutants (28.5±0.5 vs. 21.3±0.6 bends/minute, mean ± SEM, respectively) (Fig. 5C). This result is consistent with the finding that sup-18(n1030) and sup-9(n1435) lack an obviously additive effect in suppressing the locomotion and egg-laying defects of unc-93(e1500gf) mutants (Fig. 5A and B) and supports our conclusion that sup-9(n1435) and sup-18(lf) alleles act in the same pathway in affecting rubberband Unc mutants.
The sup-9(n1435) mutation affects a conserved region in the C-terminal domain of SUP-9
We determined the sup-9 coding sequences in sup-9(n1435) mutants and identified a C-to-T transition within codon 292, leading to a serine-to-phenylalanine substitution within the predicted intracellular C-terminal domain of SUP-9 (Fig. 6A). Although SUP-9 is 41%–47% identical in amino acid sequence over its entire region to several TASK-family two-pore domain K+ channels [28], the C-terminal cytoplasmic domain of SUP-9 is poorly conserved among these channels (Fig. 6A). However, the serine affected by the n1435 mutation is located in a small conserved stretch of amino acids with the sequence SxxSCxCY (Fig. 6A). We named this region the SC (Serine-Cysteine-rich)-box. The residues in the SC-box do not correspond to any reported motifs, including phosphorylation sites, as defined by the protein motif database PROSITE [47]. Variations of the SC-box are found in the human TASK-1 and TASK-3 channels and in two Drosophila two-pore domain K+ channels (Fig. 6A). We have not found an SC Box in other human two-pore domain K+ channels (I. de la Cruz and H. R. Horvitz, unpublished observations) or in TWK-4 (C40C9.1), a C. elegans two-pore domain K+ channel that is 41% identical to and the most closely related C. elegans channel to SUP-9 (Fig. 6A).
Figure 6. sup-9(n1435) is mutated in a conserved SC box at the C-terminus of two-pore domain K+ channels.

(A) Top, schematic diagram of the proposed structure of the SUP-9 channel. Cylinders represent the four presumptive transmembrane domains. Middle, alignment of the C-terminal cytoplasmic tail of SUP-9 (amino acids 247–329) and related channels: human TASK-1 (aa 248–394) Genebank Acc#NP_002237; human TASK-3 (aa 248–374) Genebank Acc#Q9NPC2; Drosophila predicted protein CG9637 (aa 238–398) Genebank Acc# AAF54970; Drosophila predicted protein CG9361 (aa 249–340) Genebank Acc# AAF54374; C. elegans TWK-4 (aa 255–334) Genebank Acc#AAC32857. Residues conserved among at least four of the channels are shaded. Box, SC box; black triangle, site of the n1435 mutation; white triangle, site of SUP-9::TWK-4 C-terminal fusion. Bottom, alignment of SC boxes from SUP-9 and related channels. Residues conserved among SUP-9 and at least two other channels are shaded. (B, C) Mutant animals of the indicated genotypes were assayed for egg-laying rate. Young adult hermaphrodites were allowed to lay eggs for 3 hrs on a bacterial lawn, and eggs or larvae on the plate were counted. Independently derived transgenic lines of (B) sup-10(n983gf) lin-15 or (C) unc-93(e1500gf) sup-18(n1030); lin-15 animals containing both a lin-15-rescuing transgene and the Pmyo-3 sup-9 derivatives are indicated. Error bars, means ± SEMs of at least 14 animals.
To determine if other residues in the SC-box of SUP-9 might function like the S292F substitution, we performed an in vivo mutagenesis study of the SC-box. We mutated residues S289, C293, C295 and Y296 to alanine individually and compared their effects in suppressing the egg-laying defects of the sup-10(n983gf) and unc-93(e1500gf) sup-18(n1030) double mutants. When assayed over a 3 hr period, both mutant strains laid fewer than three eggs, and a sup-9(n1913) null mutation drastically increased egg laying by both strains (Fig. 6B, C). As a control, overexpression of a sup-9(+) cDNA driven by the myo-3 promoter (Pmyo-3 sup-9(+)) in either sup-10(n983gf) or unc-93(e1500gf) sup-18(n1030) mutants did not increase egg-laying in each of three independent transgenic lines. By contrast, overexpression of a sup-9 cDNA containing the n1435 mutation (Pmyo-3 sup-9(n1435)) dominantly suppressed the egg-laying defects of sup-10(n983gf) mutants (Fig. 6B) but not those of unc-93(e1500gf) sup-18(n1030) animals (Fig. 6C). These results establish an in vivo assay for identifying mutations in sup-9 that preferentially suppress sup-10(n983gf) over unc-93(e1500gf) mutations.
A Pmyo-3 sup-9(S289A) and a Pmyo-3 sup-9(Y296A) transgene suppressed the defects of sup-10(n983gf) mutants but not of unc-93(e1500gf) sup-18(n1030) mutants, suggesting that the S289A and Y296A mutations act similarly to n1435 to mediate the gene-specific effects of sup-18(lf) mutations. By contrast, the cysteine-to-alanine mutations at residues 293 and 295 of SUP-9 suppressed both sup-10(n983gf) and unc-93(e1500gf) sup-18(n1030) mutants (Fig. 6B, C). We suggest that these mutations when overexpressed have a dominant-negative effect on the wild-type sup-9 allele.
To further understand how its C-terminal domain affects SUP-9 activity, we deleted in the sup-9 cDNA the region encoding the SUP-9 C-terminal cytoplasmic domain. We also replaced this region with the corresponding region of twk-4, which encodes a two-pore domain K+ channel without an SC-box, or of TASK-3, a mammalian homolog that contains an SC-box (Fig. 6). Deletion of the SUP-9 C-terminal domain caused suppression of both the sup-10(n983gf) and unc-93(e1500gf) sup-18(n1030) mutant phenotypes, suggesting that the truncated form of SUP-9 acts as a dominant-negative protein. Interestingly, both the sup-9::twk-4 and sup-9::TASK-3 fusion transgenes suppressed the sup-10(n983gf) egg-laying defect (Fig. 6B) but failed to suppress that of the unc-93(e1500gf) sup-18(n1030) mutants (Fig. 6C), suggesting that these fusion transgenes act similarly to sup-9(n1435) and affect rubberband Unc mutants in a gene-specific manner.
To identify more sup-9 mutations that act similarly to sup-9(n1435), we performed a genetic screen for mutations that semidominantly suppressed the sup-10(n983gf) rubberband phenotype (see Materials and Methods). We isolated eight mutations of sup-9 that define seven novel alleles (n3975 (n4265), n3976, n3977, n3935, n4259, n4262 and n4269) (Fig. 7A) and three additional mutations (n3942, n4253, n4254) that contained the same C-to-T transition and therefore caused the same S292F substitution as sup-9(n1435). As heterozygotes, five of the seven novel alleles (n3977, n3935, n4259, n4262, n4269) were stronger suppressors of sup-10(n983gf) mutants like sup-9(n1435)/+ (∼23 bends/minute), while the other two (n3975, n3976) were weaker (Fig. 7B). These mutations affect six different regions of SUP-9 (Fig. 7A), including the first (n3975) and second (n3977) transmembrane domains, the first pore domain (n3976), the beginning of the C-terminal cytoplasmic domain (n3935), the SC-box (n4259 and n4262), and a region C-terminal to the SC-box (n4269)
Figure 7. Characterization of novel sup-9 alleles.

(A) Schematic of proposed SUP-9 functional domains and identified missense mutations. (B) Effects of various combinations of sup-9 and sup-18 alleles on the locomotion of sup-10(n983gf) animals. (C) Effects of sup-9 alleles on the locomotion of unc-93(e1500gf) animals.
To determine if these novel sup-9 mutations conferred resistance to sup-18 activation or if they were simply dominant-negative lf mutations, we tested their responsiveness to changes in sup-18 levels in a similar manner to that used for testing sup-9(n1435) (Table 2 and Fig. 5). By comparing the locomotion of sup-9(mut)/+; sup-18(n1030)/+; sup-10(n983gf) mutants to that of sup-18(n1030)/+; sup-10(n983gf) mutants, we found that sup-9(n3935)/+, sup-9(n4259)/+, sup-9(n4262)/+ and sup-9(n4269)/+ caused a weak effect similar to that by sup-9(n1435)/+, while n3975/+, n3976/+ and n3977/+ caused a significant improvement in locomotory rate in response to a change in sup-18 levels (Fig. 7B). This result suggests that the channels generated by the three mutations n3975, n3976 and n3977 have impaired ability to generate K+ currents but retain regulation by SUP-18.
In addition to its sup-18 insensitivity, sup-9(n1435) was also a weak suppressor of the unc-93(e1500gf) locomotory defect, while the null mutation sup-9(n1913) completely suppressed the unc-93(gf) defect (Tables 1 and 5). Similarly, sup-9(n4259), sup-9(n4262) and sup-9(n4269) only weakly suppressed the locomotory defects of unc-93(e1500gf) animals (Fig. 7C), suggesting that these mutations belong to the class of sup-9 alleles defined by sup-9(n1435). However, sup-9(n3935) completely suppressed the locomotory defects of unc-93(e1500gf) animals (Fig. 7C), indicating that sup-9(n3935) was not only insensitive to sup-18 but also resistant to the activating effects of unc-93(e1500gf). Thus, mutations affecting different residues of SUP-9 confer differential channel sensitivity to its regulatory subunits.
Discussion
sup-18 encodes a transmembrane protein orthologous to mammalian iodotyrosine deiodinase
Two-pore domain K+ channels are widely expressed and play important roles in regulating resting membrane potentials of cells [15], [17]. However, very little is known about protein factors with which these channels interact. We previously identified UNC-93 and SUP-10 as presumptive regulatory subunits of the SUP-9 two-pore domain K+ channel. We now suggest that SUP-18 also regulates the SUP-9/UNC-93/SUP-10 channel complex.
sup-18 encodes the C. elegans ortholog of mammalian iodotyrosine deiodinase (IYD), which belongs to the NADH oxidase/flavin reductase superfamily [7], [8]. By oxidizing NADH using flavin mononucleotide (FMN) as a cofactor, IYD catalyzes the recycling of iodide from monoiodotyrosine and diiodotyrosine, two major byproducts in the synthesis of thyroid hormones [7], [8]. Lack of IYD function can lead to congenital hypothyroidism [12], [13]. In C. elegans, no SUP-18 function besides regulating the SUP-9 channel has been identified. The enzymatic activity of SUP-18 remains to be defined.
Little is known about the metabolism and function of iodide in nematodes. The C. elegans genome contains two genes, ZK822.5 and F52H2.4, that encode homologs of the mammalian sodium/iodide symporter, which enriches iodide in the thyroid cells by active membrane transport [48]. The presence of both SUP-18 IYD and sodium/iodide symporter-like proteins suggests that iodide functions biologically in C. elegans. Although iodide appears not to be an essential trace element in the culture medium of C. elegans [49], it is possible that residual iodide in components of that medium can provide sufficient nutritional support for survival. C. elegans lacks homologs of mammalian iodothyronine deiodinase (I. de la Cruz, L. Ma and H. R. Horvitz, unpublished observations), enzymes that remove the iodine moieties from the precursor thyroxine (T4) and generate the more potent thyroid hormone 3, 5, 3′-triiodothyronine [50], which suggests that thyroid hormones might not be synthesized in C. elegans.
IYDs across metazoan species share a similar enzymatic activity in reductive deiodination of diiodotyrosine [51], and it seems likely that SUP-18 acts similarly in C. elegans. Like mammalian IYDs, SUP-18 contains a presumptive N-terminal transmembrane domain that is required for full activity. Interestingly, the SUP-18 intracellular region lacking the transmembrane domain could still partially activate the SUP-9 channel, suggesting that membrane association is not absolutely required for SUP-9 activation by SUP-18. Membrane association is important for mammalian IYD enzymatic activities [5], [52], [53].
The presence of a transmembrane domain suggests that SUP-18 IYD might interact with other transmembrane proteins. The genetic interactions we observe between sup-18 and the genes that encode the SUP-9/UNC-93/SUP-10 two-pore domain K+ channel complex support this hypothesis. Based on expression studies, we conclude that SUP-18 and SUP-10 localize to similar subcellular structures within muscle cells, further supporting the idea that SUP-18 and the channel complex interact physically. We found that transgenic expression of the SUP-18 intracellular domain could enhance the expression of the rubberband phenotype, suggesting that plasma membrane localization is not essential for SUP-18 function. Nonetheless, the expression of the full-length SUP-18 was more potent than the expression of the SUP-18 intracellular domain in rescuing the rubberband Unc phenotypes of sup-18(lf); sup-10(n983gf) mutants, suggesting that the presence of a transmembrane domain in SUP-18 IYD could enhance the activity of SUP-18 by targeting SUP-18 to the plasma membrane.
The crystal structure of mouse IYD reveals that eight residues contact the FMN cofactor: R96, R97, S98, R100, P123, S124, T235 and R275 [54]. Except T235, which is replaced by a serine in SUP-18, these residues are completely conserved (Figure 1C, yellow boxes). Furthermore, the sup-18(n1010) missense mutation leads to an S137N substitution at the position equivalent to the mouse S98 residue, likely disrupting the binding of FMN. This high degree of conservation at the cofactor binding site suggests that SUP-18 likely retains the ability to bind FMN and likely has a catalytic activity.
Three IYD missense mutations that cause hypothyroidism (R101W, I116T, and A220T) affect residues that are conserved in SUP-18 [12], [55] (Fig. 1C, red boxes). A fourth human mutation replaces F105 and I106 with a leucine [8]. The phenylalanine at position 105 is conserved in SUP-18 (Fig. 1C). The conservation of residues associated with IYD function supports the hypothesis that SUP-18 regulates the SUP-9 two-pore domain K+ channel complex via an enzymatic activity. The SUP-18 substrate remains to be elucidated.
That SUP-18 might function as a NADH oxidase/flavin reductase raises the intriguing possibility that SUP-18 might couple the metabolic state of muscle cells with membrane excitability. Mammalian Kvβ voltage-gated K+ channel regulatory subunits [56], which belong to the aldo-keto reductase superfamily [57], [58], have similarly been proposed to couple metabolic state with cell excitability based on indirect evidence. Kvβ2 has an NADP+ cofactor bound in its active site and a catalytic triad spaced appropriately to engage in enzymatic activity [58]. Although suggestive of an enzymatic activity, no substrate has been reported for Kvβ subunits. While Kvβ2 knockout mice have seizures and reduced lifespans, mice carrying a catalytic null mutation in Kvβ2 have a wild-type phenotype, suggesting that if an enzymatic activity for Kvβ2 exists, it is functionally dispensable in vivo [59]. By contrast, the predicted catalytic mutation sup-18(n1010) behaves like a null mutation in its inability to activate the SUP-9 channel, even though the SUP-18(n1010) protein is synthesized and localized normally to the cell surface of muscle cells (I. de la Cruz and H. R. Horvitz, unpublished observations). Five other sup-18 mutations affecting highly conserved residues in the NADH oxidase/flavin reductase domain also behave like null mutations, consistent with the hypothesis that SUP-18 enzymatic activity is essential for its function.
sup-18(lf) mutations define a new class of gene-specific suppressors of the rubberband Unc mutants
sup-18(lf) mutations strongly suppress sup-10(n983gf) mutants and weakly suppress unc-93(e1500gf) mutants. Certain specific mutations of sup-9, including n1435, n4259, n4262, and n4269, act similarly to sup-18(lf) and are strong suppressors of sup-10(n983gf) mutants and weak suppressors of unc-93(e1500gf) mutants. Together these sup-9 mutations and sup-18(lf) mutations represent a novel class of mutations that exhibit gene-specific suppression of the rubberband Unc mutants and are distinct from another class of gene-specific suppressors we identified previously, mutations in three splicing factor genes that strongly suppress unc-93(e1500gf) and sup-10(n983gf) but do not obviously suppress unc-93(n200gf) or sup-9(n1550gf) [60]–[62]. The difference between sup-18(lf) and sup-9(n1435, n4259, n4262, n4269) mutations and the splicing factor mutations in their patterns of suppressing the rubberband Unc mutants suggests that these two classes of suppressors function by distinct mechanisms.
SUP-18 is an activator of the SUP-9 two-pore domain K+ channel
SUP-9 is closely related to the subfamily of two-pore domain K+ channels that include human TASK-1 and TASK-3 [28]. TASK-1 is activated by multiple factors, including extracellular pH [22], [23], [63], inhalational anesthetics such as halothane [24] and oxygen [64]. TASK-1 is directly inhibited by sub-micromolar levels of the cannabinoid neurotransmitter anandamide [65] and by neuromodulators such as thyrotropin releasing hormone (TRH) [27]. A histidine residue in the first P-domain of TASK-1 modulates its sensitivity to pH [66], while a six amino acid stretch following its fourth transmembrane domain is required for both halothane activation and TRH suppression [24], [67]. Deletion of the TASK intracellular C-terminal domain, which contains the SC-box, does not change its basal activity or activation by halothane [24], [67], suggesting that the TASK-1 C-terminal domain and probably the SC-box represent an activation region that is required by some types of channel activator (e.g., human IYD) but not by others (e.g., halothane and pH). It remains to be determined whether IYD is involved in the inhibition of TASK-1 channel activity by TRH.
From our genetic analysis of the sup-9(n1435) mutation and site-directed mutagenesis of sup-9, we have defined the SC-box, a domain of SUP-9 required for SUP-10(n983gf)-specific activation. The importance of the SC-box in mediating this activation is supported by the results of a genetic screen in which we isolated additional sup-9 mutations (Fig. 7) that act like sup-9(n1435) and cause distinct amino acid changes in (n4259 (S292A), n4262 (S294A)) or near (n4269 (L303P)) the SC-box. Although conserved in the human TASK-1 and TASK-3 channels (Fig. 6A), no function has yet been assigned to the SC-box. Our analyses suggest that the SC-box and the C-terminal domain of SUP-9 likely mediate the functional interaction between SUP-9 and SUP-10/SUP-18 but are dispensable for interaction with UNC-93. We found that replacing the C-terminal domain of SUP-9 with the corresponding region of TWK-4 (which lacks an SC-box) or of TASK-3 (with an SC-box) makes the fusion channels behave like SUP-9(n1435), consistent with the model that the SC-box is required for SUP-9 activation by SUP-10(n983gf) and SUP-18 (based on the TWK-4 data) and suggests that SC-box-dependent activation requires one or more nearby residues in the C-terminal domain (based on the TASK-3 data). The unc-93(e1500gf) mutation results in a glycine-to-arginine substitution at amino acid 388 in one of the putative transmembrane domains [33], suggesting that the UNC-93(gf) protein activates SUP-9 through an interaction involving transmembrane domains, without a need for the SC-box or the rest of the cytoplasmic domain.
We describe three important properties of the unusual sup-9(n1435) mutation. First, SUP-9(n1435) channels cannot be activated by SUP-10(n983gf). Second, SUP-9(n1435) channels are insensitive to SUP-18 activity. Third, SUP-9(n1435) channels can be activated by UNC-93(e1500gf). The existence of a channel mutation that is insensitive to both SUP-18 and SUP-10(n983gf) suggests that these two inputs act through a common pathway. A mutant channel that can be activated by UNC-93(e1500gf) but not by SUP-10(n983gf) suggests that there is an independent pathway for SUP-9 activation by UNC-93.
We propose a model to explain the functional interactions between SUP-18 and SUP-9/UNC-93/SUP-10 (Fig. 8). In this model, SUP-10 and UNC-93 have an essential role in and are both required for activating SUP-9 channel, since the n1550 gf mutation in sup-9 is completely suppressed by sup-10(lf) and unc-93(lf) mutations [38]. SUP-18 activates SUP-9 only weakly and relies on SUP-10 for this activation (Fig. 8). SUP-10(n983gf) enhances the activity of SUP-18 and results in over-activation of SUP-9 by SUP-18. Our model is consistent with the genetic and molecular evidence described in this and previous studies [28]–[31], [33] and should provide a framework for understanding the interactions of SUP-18 and the SUP-9/UNC-93/SUP-10 channel complex. Our results do not distinguish whether SUP-18 regulates the SUP-9/UNC-93/SUP-10 complex via a direct physical interaction or indirectly through an unknown factor or factors.
Figure 8. A model for activation of the SUP-9 channel by multiple subunits.
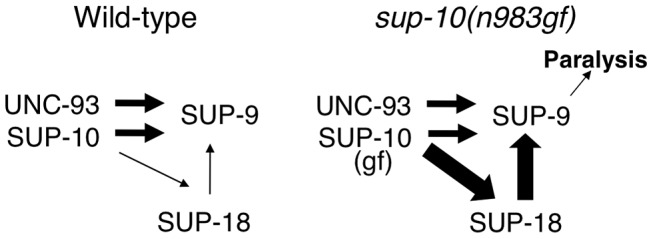
In this model, SUP-10 and UNC-93 act independently of SUP-18 to activate SUP-9. In addition, SUP-10 enhances SUP-18, which activates SUP-9 through a distinct pathway. SUP-10(n983gf) over-enhances SUP-18, which over-activates SUP-9 and leads to paralysis. The widths of the arrows pointing at SUP-9 are representative of their relative strengths in sustaining gf activity, with thicker arrows representing a larger contribution.
In short, we identified SUP-18 IYD as a functional regulator of the SUP-9/UNC-93/SUP-10 two-pore domain K+ channel complex. We also defined an evolutionarily conserved serine-cysteine-rich domain, the SC-box, in the C-terminal region of SUP-9 and showed that this region is required for activation of the channel by SUP-18. Since IYD is likely to be an NADH oxidase/flavin reductase that uses the ubiquitous energy carrier molecule NADH as a coenzyme, our study suggests that IYD might couple cellular metabolic state to two-pore domain K+ channel activities. Future molecular analyses should reveal the mechanism underlying the interaction between the SUP-9 two-pore domain K+ channels and SUP-18 IYD.
Materials and Methods
Strains and genetics
C. elegans strains were cultured as described [49], except that E. coli strain HB101 was used instead of OP50 as a food source. Strains were grown at 20°C unless otherwise noted. The following mutations were used in this study:
LGII sup-9(n213, n233, n264 [31], n1016, n1025 [30], n1435, n1550gf [29], lr35, lr38, lr45, lr100, lr129, lr142, n1472, n1557, n1913 [28], n3935, n3942, n3975, n3976, n3977, n4253, n4254, n4259, n4262, n4265, n4269 (this study)).
LGIII unc-93(e1500gf, n200gf) [31], sma-3(e491) [49], mec-14(u55) [68], ncl-1(e1865) [69], unc-36(e251) [49]. sup-18(n463, n527, n528, n1010, n1014, n1015, n1022, n1030, n1033, n1035, n1036 [30], n1038, n1471, n1539, n1548, n1554, n1556, n1558 (this study)).
LGX sup-10(n183 [31], n1008, n983gf [30], n4025, n4026 (this study)), lin-15(n765ts) [70].
Isolation of partially suppressed unc-93(e1500gf) mutants
Since lf mutations in sup-10 completely suppress the paralysis of unc-93(e1500gf) mutants [31], we reasoned that partial lf mutations of sup-10 would partially suppress the unc-93(e1500gf) locomotory phenotype. To isolate such partial lf sup-10 mutations, we performed an EMS F2 genetic screen for partial suppressors of the locomotory defects of unc-93(e1500gf) mutants. From 17,500 haploid genomes screened, we isolated over 30 strong suppressors and seven weak suppressors. We assigned two of the seven weak suppressors, n4025 and n4026, to the sup-10 locus by complementation tests and three others to the unc-93 locus. All seven were saved for future analyses.
Mapping and cloning of sup-18
34 Sma non-Unc and 23 Unc non-Sma progeny were isolated from a sma-3 mec-14 ncl-1 unc-36/sup-18 parent. Scoring of the ncl-1 and sup-18 phenotypes identified the 57 recombination events to be distributed in the three relevant intervals as follows: sma-3 (30/57) ncl-1 (3/57) sup-18 (24/57) unc-36. A pool of cosmids C33C3, C08C3, C27D11, C02C2, C39F10 and C44C9 at 1 ng/µL each and a rol-6 marker [71] at 80 ng/µL were injected into sup-18(n1010); sup-10(n983gf) animals. Two Rol transgenic lines were obtained, one of which generated rubberband Unc animals. The four middle cosmids were injected separately, and C02C2 yielded 5/5 rescued lines, while transgenes containing cosmids C08C3 (0/7), C27D11 (0/5) or C39F10 (0/9) showed no rescue.
RT-PCR was performed on cDNA from the wild-type N2 strain using the primers 5′-TTGAAAACCCCTGTTAAATAC-3′ and 5′-CGAGTTTCTAATAAAAATAAACC-3′. PCR products were cloned into pBSKII (Stratagene), and their sequences determined. 5′ and 3′ RACE were performed using the corresponding kits (Gibco).
Molecular biology
Genomic subclones of cosmid C02C2 were generated in pBSKII (Stratagene). The subclones, in the order shown in Figure 1, spanned the following sequences (Genebank Acc#L23649): EcoRV (9,790) - EcoRV (21,098); PstI (23,699) - PstI (32,833); PstI (23,699) - SacI (28,185); BstBI (24,448)-SacI (28,185); and HindIII (24,671) - HindIII (27,169).
All PCR amplifications used in plasmid constructions were performed using Pfu polymerase, and the sequences of their products were determined. The Pmyo-3 sup-18 vectors for ectopic expression of wild-type or mutant sup-18 alleles were generated by PCR amplification of the respective coding regions from sup-18 cDNAs using primers that introduced NheI and SacI sites at the 5′ and 3′ ends, respectively, and cloned into vector pPD95.86 (from A. Fire). Pmyo-3 sup-18(intra) was similarly constructed, except that the 5′ primer began at codon 66 of sup-18. The gfp-tagged version of this vector was created by PCR amplification of the gfp coding sequence from vector pPD95.77 (from A. Fire) and subcloned into Pmyo-3 sup-18(intra) just prior to the start codon of the sup-18 sequence.
Pmyo-3 mIYD (mouse IYD) was generated by PCR amplification of the coding region of the mouse cDNA (Gene Bank AK002363) with 5′ and 3′ primers containing NheI and EcoRV sites, respectively, and subcloning the PCR products into pPD95.86 at the NheI and SacI (blunted) sites. Pmyo-3 mIYD::gfp was generated by a similar strategy using a 5′ primer containing an NheI site and a 3′ primer that did not include the stop codon of mIYD but instead contained a BamHI site. The myo-3 promoter from pPD95.86 was subcloned into pPD95.77, such that upon subcloning of the mIYD PCR fragment into the NheI and BamHI sites of the vector the myo-3 promoter drove expression of the mIYD gene fused in-frame at its 3′ end to gfp.
The sup-18::gfp genomic fusion was constructed by introducing SphI sites at the ends of a gfp cassette by PCR amplification of plasmid pPD95.77 (from A. Fire) and subsequent subcloning into the single SphI site contained within a 9.1 kb PstI genomic sup-18 rescuing fragment. The resulting fusion contained 6.5 kb of promoter sequence, the entire sup-18 coding region with gfp inserted between the transmembrane and NADH oxidase/flavin reductase domains and 1.1 kb of 3′ UTR and downstream sequence.
The sup-10::gfp fusion used in colocalization studies was constructed by subcloning a 7.3 kb MfeI genomic fragment from cosmid C27G6 containing sup-10 into the EcoRI site of pBSKII. A 6.4 kb Pst I fragment was subcloned from this vector into pPD95.77, which contained 3.5 kb of promoter sequence and the sup-10 coding region. Using PCR, we introduced a SalI site immediately preceding the stop codon of sup-10 to create an in-frame fusion with the gfp coding sequence.
sup-18::β-galactosidase fusions were created by PCR amplification of 1869 bp of 5′ sup-18 promoter sequence and subcloning the product into the SphI and PstI sites of pPD34.110 (from A. Fire) to generate Psup-18 TM-β-Gal, which contains a synthetic transmembrane sequence [45] followed by the β-galactosidase coding sequence [72]. sup-18 genomic coding sequence spanning codons 1–42, 1–70 and 1–301 were PCR-amplified from the minimal rescuing fragment with 5′ and 3′ primers that contained PstI and BamHI sites, respectively, and subcloned into these sites in Psup-18 TM-β-Gal. The synthetic transmembrane domain was deleted from these plasmids by excising the KpnI fragment containing this domain. A signal sequence [73] was inserted into these vectors using standard PCR techniques.
The GST::sup-18(N) and MBP::sup-18(N) fusion genes used to generate and purify anti-SUP-18 antibodies were generated by PCR amplification of codons 1–258 of the sup-18 cDNA and subcloning the products into pGEX-2T (Pharmacia) and pMal-2c (NEB) vectors.
The full-length twk-4 cDNA was cloned by RT-PCR with primers 5′-CTCTGCTAGCAATGCATCAAATTGACGGAAAATCTGC-3′ and 5′-AGAGGATCCATATAGTTCAAGATCCACCAGATG-3′ from wild-type mixed-stage RNA. The sequence of the twk-4 cDNA obtained was in agreement with its predicted sequence (GenBank Acc#AF083646). The C-terminal cytoplasmic domain of sup-9 from the Pmyo-3 sup-9 vector (codons 257–329 of sup-9) was replaced by twk-4 codons (265–365) using standard PCR ligation techniques to generate Pmyo-3 sup-9::twk-4. Site-directed mutagenesis of the SC-box in the Pmyo-3 sup-9 vector was likewise performed.
Body-bend assay
Young adults were individually picked to plates with HB101 bacteria, and body-bends were counted for one minute using a dissecting microscope as described [74].
Antibody and immunostaining
A GST::SUP-18(N) fusion protein was expressed in E. coli and the insoluble protein was purified by SDS-PAGE and used to immunize rabbits. Antisera were purified by binding to the MBP::SUP-18 protein immobilized on nitrocellulose strips and elution with 100 mM glycine-HCl (pH 2.5). This antibody could detect SUP-18 overexpressed in the body-wall muscles (Fig. 2H) but failed to detect endogenous SUP-18.
For immunofluorescence experiments, worms at mixed stages were fixed in 1% paraformaldehyde for 2 hrs at 4°C and permeabilized as described [75]. For colocalization studies, transgenic worms were stained with primary antibodies at 1∶200 dilution and a secondary goat-anti-rabbit antibody conjugated with Texas Red (Jackson Labs). Worms were viewed using confocal microscopy.
Transgenic animals
Germline transformation experiments were performed using standard methods [71]. Transgenic strains carrying the lin-15(n765ts) mutation contained the coinjection marker pL15EK(lin-15(+)) at 50 ng/µL [70], and transgenic animals were identified by their non-Muv phenotype at 22.5°C. The dominant rol-6 plasmid [71] was used at 100 ng/µl during cosmid rescue experiments, and transgenic animals were identified by their Rol phenotype. The dominant myo-3::gfp fusion vector pPD93.97 (from A. Fire) was used where indicated at 80 ng/µl, and transgenic animals were identified by GFP fluorescence. Experimental DNA was injected at 30–50 ng/µl.
Isolation of novel sup-9 alleles
One plausible genetic strategy for isolating sup-9 alleles similar to sup-9(n1435) would be to perform an F2 screen for suppressors of the sup-10(n983gf) locomotory defect and then test these suppressors for their effects on the locomotory defect of unc-93(e1500gf) mutants. Most sup-9 alleles isolated from such a screen would be typical lf alleles rather than rare alleles that would result in a SUP-9 protein specifically impaired in activation by SUP-10(gf) and SUP-18(+). We therefore opted for an alternative strategy based on the semidominance of the sup-9(n1435) mutation. While sup-9 null mutations, such as n1913, recessively suppress the locomotory defects of sup-10(n983gf) mutants, sup-9(n1435) caused a strong semidominant suppression (Fig. 5C). As two-pore domain K+ channels are homodimers [66], [76], this semidominance likely reflects the formation of nonfunctional heterodimers composed of n1435 and wild-type SUP-9 proteins. The strength of this semidominance (∼23 vs. ∼5 bends/minute for sup-9(n1435)/+; sup-10(n983gf) vs. sup-10(n983gf) mutants, respectively) formed the basis of an F1 screen for suppressors of the sup-10(n983gf) locomotory defect.
sup-10(n983gf) L4 hermaphrodites were mutagenized with EMS, and approximately 550,000 F1 progeny (1.1×106 genomes) were screened for improved locomotion on agar plates. From 89 candidate suppressors, 35 mutants retested in the next generation, representing at least 31 independent isolates. To quantify the semidominant character of these mutants (sup(new)), wild-type males were crossed with homozygous mutant hermaphrodites to generate sup(new)/+; sup-10(n983gf)/0 males, and their locomotory rate was scored. Because sup-10 is on the X chromosome, this strategy generates males hemizygous for sup-10(n983gf) while heterozygous for autosomal mutations, providing a convenient assay of semidominance. Four mutations completely suppressed the rubberband Unc phenotype of males, with locomotory rates very similar to that of wild-type animals (∼33 bends/minute). We reasoned that these four mutants were likely lf alleles of sup-10, as such animals would be hemizygous for sup-10. We confirmed this assignment by determining the sequences of the sup-10 locus and found mutations in all four strains (I. de la Cruz and H. R. Horvitz, unpublished observations). For the remaining strong mutants, we performed complementation tests with sup-9, sup-18 and unc-93 strains and identified 11 semidominant alleles of sup-9 (see Results).
Acknowledgments
We thank Dan Denning, Dengke Ma, Nicolas Paquin and Craig Ceol for advice concerning this manuscript and Craig Ceol for assistance with some experiments; Andy Fire for C. elegans gfp and LacZ expression vectors; Beth Castor for assistance with DNA sequencing; Beth DeStasio for her enthusiastic support and discussions; and all members of the Horvitz laboratory for their support.
Funding Statement
This work was funded by NIH Grant GM24663 to HRH. IPdlC was funded by an NIH predoctoral training grant and an NSF Graduate Fellowship. LM is supported by NSFC Grant 31371253. HRH is an Investigator of the Howard Hughes Medical Institute. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Park SM, Chatterjee VK (2005) Genetics of congenital hypothyroidism. J Med Genet 42: 379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Roche J, Michel R, Michel O, Lissitzky S (1952) Enzymatic dehalogenation of iodotyrosine by thyroid tissue; on its physiological role. Biochim Biophys Acta 9: 161–169. [DOI] [PubMed] [Google Scholar]
- 3. Querido A, Stanbury JB, Kassenaar AA, Meijer JW (1956) The metabolism of iodotyrosines. III. Di-iodotyrosine deshalogenating activity of human thyroid tissue. J Clin Endocrinol Metab 16: 1096–1101. [DOI] [PubMed] [Google Scholar]
- 4. Stanbury JB, Kassenaar AA, Meijer JW (1956) The metabolism of iodotyrosines. I. The fate of mono- and di-iodotyrosine in normal subjects and in patients with various diseases. J Clin Endocrinol Metab 16: 735–746. [DOI] [PubMed] [Google Scholar]
- 5. Goswami A, Rosenberg IN (1979) Characterization of a flavoprotein iodotyrosine deiodinase from bovine thyroid. Flavin nucleotide binding and oxidation-reduction properties. J Biol Chem 254: 12326–12330. [PubMed] [Google Scholar]
- 6. Rosenberg IN, Goswami A (1979) Purification and characterization of a flavoprotein from bovine thyroid with iodotyrosine deiodinase activity. J Biol Chem 254: 12318–12325. [PubMed] [Google Scholar]
- 7. Friedman JE, Watson JA Jr, Lam DW, Rokita SE (2006) Iodotyrosine deiodinase is the first mammalian member of the NADH oxidase/flavin reductase superfamily. J Biol Chem 281: 2812–2819. [DOI] [PubMed] [Google Scholar]
- 8. Gnidehou S, Caillou B, Talbot M, Ohayon R, Kaniewski J, et al. (2004) Iodotyrosine dehalogenase 1 (DEHAL1) is a transmembrane protein involved in the recycling of iodide close to the thyroglobulin iodination site. FASEB J 18: 1574–1576. [DOI] [PubMed] [Google Scholar]
- 9. Moreno JC (2003) Identification of novel genes involved in congenital hypothyroidism using serial analysis of gene expression. Horm Res 60: 96–102. [DOI] [PubMed] [Google Scholar]
- 10. Moreno JC, Keijser R, Aarraas S, De Vijlder JJM, Ris-Stalpers C (2002) Cloning and characterization of a novel thyroidal gene encoding proteins with a conserved nitroreductase domain. J Endocrinol Invest 25: 23. [Google Scholar]
- 11. Choufoer JC, Kassenaar AA, Querido A (1960) The syndrome of congenital hypothyroidism with defective dehalogenation of iodotyrosines. Further observations and a discussion of the pathophysiology. J Clin Endocrinol Metab 20: 983–1003. [DOI] [PubMed] [Google Scholar]
- 12. Moreno JC, Klootwijk W, van Toor H, Pinto G, D'Alessandro M, et al. (2008) Mutations in the iodotyrosine deiodinase gene and hypothyroidism. N Engl J Med 358: 1811–1818. [DOI] [PubMed] [Google Scholar]
- 13. Moreno JC, Visser TJ (2010) Genetics and phenomics of hypothyroidism and goiter due to iodotyrosine deiodinase (DEHAL1) gene mutations. Mol Cell Endocrinol 322: 91–98. [DOI] [PubMed] [Google Scholar]
- 14. Enyedi P, Czirjak G (2010) Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev 90: 559–605. [DOI] [PubMed] [Google Scholar]
- 15. Goldstein SA, Bockenhauer D, O'Kelly I, Zilberberg N (2001) Potassium leak channels and the KCNK family of two-P-domain subunits. Nat Rev Neurosci 2: 175–184. [DOI] [PubMed] [Google Scholar]
- 16. Mathie A, Al-Moubarak E, Veale EL (2010) Gating of two pore domain potassium channels. J Physiol 588: 3149–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goldstein SA, Bayliss DA, Kim D, Lesage F, Plant LD, et al. (2005) International Union of Pharmacology. LV. Nomenclature and molecular relationships of two-P potassium channels. Pharmacol Rev 57: 527–540. [DOI] [PubMed] [Google Scholar]
- 18. Maingret F, Lauritzen I, Patel AJ, Heurteaux C, Reyes R, et al. (2000) TREK-1 is a heat-activated background K(+) channel. EMBO J 19: 2483–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maingret F, Fosset M, Lesage F, Lazdunski M, Honore E (1999) TRAAK is a mammalian neuronal mechano-gated K+ channel. J Biol Chem 274: 1381–1387. [DOI] [PubMed] [Google Scholar]
- 20. Patel AJ, Honore E, Maingret F, Lesage F, Fink M, et al. (1998) A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J 17: 4283–4290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fink M, Lesage F, Duprat F, Heurteaux C, Reyes R, et al. (1998) A neuronal two P domain K+ channel stimulated by arachidonic acid and polyunsaturated fatty acids. EMBO J 17: 3297–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Duprat F, Lesage F, Fink M, Reyes R, Heurteaux C, et al. (1997) TASK, a human background K+ channel to sense external pH variations near physiological pH. EMBO J 16: 5464–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Leonoudakis D, Gray AT, Winegar BD, Kindler CH, Harada M, et al. (1998) An open rectifier potassium channel with two pore domains in tandem cloned from rat cerebellum. J Neurosci 18: 868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Patel AJ, Honore E, Lesage F, Fink M, Romey G, et al. (1999) Inhalational anesthetics activate two-pore-domain background K+ channels. Nat Neurosci 2: 422–426. [DOI] [PubMed] [Google Scholar]
- 25. Sirois JE, Lei Q, Talley EM, Lynch C 3rd, Bayliss DA (2000) The TASK-1 two-pore domain K+ channel is a molecular substrate for neuronal effects of inhalation anesthetics. J Neurosci 20: 6347–6354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Millar JA, Barratt L, Southan AP, Page KM, Fyffe RE, et al. (2000) A functional role for the two-pore domain potassium channel TASK-1 in cerebellar granule neurons. Proc Natl Acad Sci U S A 97: 3614–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Talley EM, Lei Q, Sirois JE, Bayliss DA (2000) TASK-1, a two-pore domain K+ channel, is modulated by multiple neurotransmitters in motoneurons. Neuron 25: 399–410. [DOI] [PubMed] [Google Scholar]
- 28. de la Cruz IP, Levin JZ, Cummins C, Anderson P, Horvitz HR (2003) sup-9, sup-10, and unc-93 may encode components of a two-pore K+ channel that coordinates muscle contraction in Caenorhabditis elegans . J Neurosci 23: 9133–9145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Levin JZ, Horvitz HR (1993) Three new classes of mutations in the Caenorhabditis elegans muscle gene sup-9 . Genetics 135: 53–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Greenwald I, Horvitz HR (1986) A visible allele of the muscle gene sup-10X of C. elegans . Genetics 113: 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Greenwald IS, Horvitz HR (1980) unc-93(e1500): A behavioral mutant of Caenorhabditis elegans that defines a gene with a wild-type null phenotype. Genetics 96: 147–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. De Stasio E, Lephoto C, Azuma L, Holst C, Stanislaus D, et al. (1997) Characterization of revertants of unc-93(e1500) in Caenorhabditis elegans induced by N-ethyl-N-nitrosourea. Genetics 147: 597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Levin JZ, Horvitz HR (1992) The Caenorhabditis elegans unc-93 gene encodes a putative transmembrane protein that regulates muscle contraction. J Cell Biol 117: 143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim Y, Bang H, Kim D (2000) TASK-3, a new member of the tandem pore K(+) channel family. J Biol Chem 275: 9340–9347. [DOI] [PubMed] [Google Scholar]
- 35. Rajan S, Wischmeyer E, Xin Liu G, Preisig-Muller R, Daut J, et al. (2000) TASK-3, a novel tandem pore domain acid-sensitive K+ channel. An extracellular histiding as pH sensor. J Biol Chem 275: 16650–16657. [DOI] [PubMed] [Google Scholar]
- 36. Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, et al. (2006) Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 314: 308–312. [DOI] [PubMed] [Google Scholar]
- 37. Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, et al. (2006) The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol 7: 156–164. [DOI] [PubMed] [Google Scholar]
- 38. Barstead RJ, Waterston RH (1989) The basal component of the nematode dense-body is vinculin. J Biol Chem 264: 10177–10185. [PubMed] [Google Scholar]
- 39. Waterston RH, Thomson JN, Brenner S (1980) Mutants with altered muscle structure of Caenorhabditis elegans . Dev Biol 77: 271–302. [DOI] [PubMed] [Google Scholar]
- 40. Pujol N, Torregrossa P, Ewbank JJ, Brunet JF (2000) The homeodomain protein CePHOX2/CEH-17 controls antero-posterior axonal growth in C. elegans . Development 127: 3361–3371. [DOI] [PubMed] [Google Scholar]
- 41. Okkema PG, Harrison SW, Plunger V, Aryana A, Fire A (1993) Sequence requirements for myosin gene expression and regulation in Caenorhabditis elegans . Genetics 135: 385–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Froshauer S, Green GN, Boyd D, McGovern K, Beckwith J (1988) Genetic analysis of the membrane insertion and topology of MalF, a cytoplasmic membrane protein of Escherichia coli . J Mol Biol 200: 501–511. [DOI] [PubMed] [Google Scholar]
- 43. Silhavy TJ, Beckwith JR (1985) Uses of lac fusions for the study of biological problems. Microbiol Rev 49: 398–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li X, Greenwald I (1996) Membrane topology of the C. elegans SEL-12 presenilin. Neuron 17: 1015–1021. [DOI] [PubMed] [Google Scholar]
- 45. Lai CC, Hong K, Kinnell M, Chalfie M, Driscoll M (1996) Sequence and transmembrane topology of MEC-4, an ion channel subunit required for mechanotransduction in Caenorhabditis elegans . J Cell Biol 133: 1071–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fujita M, Kinoshita T (2012) GPI-anchor remodeling: potential functions of GPI-anchors in intracellular trafficking and membrane dynamics. Biochim Biophys Acta 1821: 1050–1058. [DOI] [PubMed] [Google Scholar]
- 47. Bairoch A, Bucher P (1994) PROSITE: recent developments. Nucleic Acids Res 22: 3583–3589. [PMC free article] [PubMed] [Google Scholar]
- 48. De La Vieja A, Dohan O, Levy O, Carrasco N (2000) Molecular analysis of the sodium/iodide symporter: impact on thyroid and extrathyroid pathophysiology. Physiol Rev 80: 1083–1105. [DOI] [PubMed] [Google Scholar]
- 49. Brenner S (1974) The genetics of Caenorhabditis elegans . Genetics 77: 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bianco AC, Kim BW (2006) Deiodinases: implications of the local control of thyroid hormone action. J Clin Invest 116: 2571–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Phatarphekar A, Buss JM, Rokita SE (2014) Iodotyrosine deiodinase: a unique flavoprotein present in organisms of diverse phyla. Mol Biosyst 10: 86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Goswami A, Rosenberg IN (1977) Studies on a soluble thyroid iodotyrosine deiodinase: activation by NADPH and electron carriers. Endocrinology 101: 331–341. [DOI] [PubMed] [Google Scholar]
- 53. Watson JA Jr, McTamney PM, Adler JM, Rokita SE (2008) Flavoprotein iodotyrosine deiodinase functions without cysteine residues. Chembiochem 9: 504–506. [DOI] [PubMed] [Google Scholar]
- 54. Thomas SR, McTamney PM, Adler JM, Laronde-Leblanc N, Rokita SE (2009) Crystal structure of iodotyrosine deiodinase, a novel flavoprotein responsible for iodide salvage in thyroid glands. J Biol Chem 284: 19659–19667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Afink G, Kulik W, Overmars H, de Randamie J, Veenboer T, et al. (2008) Molecular characterization of iodotyrosine dehalogenase deficiency in patients with hypothyroidism. J Clin Endocrinol Metab 93: 4894–4901. [DOI] [PubMed] [Google Scholar]
- 56. Pongs O, Leicher T, Berger M, Roeper J, Bahring R, et al. (1999) Functional and molecular aspects of voltage-gated K+ channel beta subunits. Ann N Y Acad Sci 868: 344–355. [DOI] [PubMed] [Google Scholar]
- 57. McCormack T, McCormack K (1994) Shaker K+ channel beta subunits belong to an NAD(P)H-dependent oxidoreductase superfamily. Cell 79: 1133–1135. [DOI] [PubMed] [Google Scholar]
- 58. Gulbis JM, Mann S, MacKinnon R (1999) Structure of a voltage-dependent K+ channel beta subunit. Cell 97: 943–952. [DOI] [PubMed] [Google Scholar]
- 59. McCormack K, Connor JX, Zhou L, Ho LL, Ganetzky B, et al. (2002) Genetic analysis of the mammalian K+ channel beta subunit Kvbeta 2 (Kcnab2). J Biol Chem 277: 13219–13228. [DOI] [PubMed] [Google Scholar]
- 60. Ma L, Gao X, Luo J, Huang L, Teng Y, et al. (2012) The Caenorhabditis elegans gene mfap-1 encodes a nuclear protein that affects alternative splicing. PLoS Genet 8: e1002827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ma L, Horvitz HR (2009) Mutations in the Caenorhabditis elegans U2AF large subunit UAF-1 alter the choice of a 3′ splice site in vivo . PLoS Genet 5: e1000708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ma L, Tan Z, Teng Y, Hoersch S, Horvitz HR (2011) In vivo effects on intron retention and exon skipping by the U2AF large subunit and SF1/BBP in the nematode Caenorhabditis elegans . RNA 17: 2201–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lopes CM, Gallagher PG, Buck ME, Butler MH, Goldstein SA (2000) Proton block and voltage gating are potassium-dependent in the cardiac leak channel Kcnk3. J Biol Chem 275: 16969–16978. [DOI] [PubMed] [Google Scholar]
- 64. Lewis A, Hartness ME, Chapman CG, Fearon IM, Meadows HJ, et al. (2001) Recombinant hTASK1 is an O(2)-sensitive K(+) channel. Biochem Biophys Res Commun 285: 1290–1294. [DOI] [PubMed] [Google Scholar]
- 65. Maingret F, Patel AJ, Lazdunski M, Honore E (2001) The endocannabinoid anandamide is a direct and selective blocker of the background K(+) channel TASK-1. EMBO J 20: 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lopes CM, Zilberberg N, Goldstein SA (2001) Block of Kcnk3 by protons. Evidence that 2-P-domain potassium channel subunits function as homodimers. J Biol Chem 276: 24449–24452. [DOI] [PubMed] [Google Scholar]
- 67. Talley EM, Bayliss DA (2002) Modulation of TASK-1 (Kcnk3) and TASK-3 (Kcnk9) potassium channels: volatile anesthetics and neurotransmitters share a molecular site of action. J Biol Chem 277: 17733–17742. [DOI] [PubMed] [Google Scholar]
- 68. Huang M, Chalfie M (1994) Gene interactions affecting mechanosensory transduction in Caenorhabditis elegans . Nature 367: 467–470. [DOI] [PubMed] [Google Scholar]
- 69. Hedgecock EM, Herman RK (1995) The ncl-1 gene and genetic mosaics of Caenorhabditis elegans . Genetics 141: 989–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Clark SG, Lu X, Horvitz HR (1994) The Caenorhabditis elegans locus lin-15, a negative regulator of a tyrosine kinase signaling pathway, encodes two different proteins. Genetics 137: 987–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mello CC, Kramer JM, Stinchcomb D, Ambros V (1991) Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequences. EMBO J 10: 3959–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Fire A, Harrison SW, Dixon D (1990) A modular set of lacZ fusion vectors for studying gene expression in Caenorhabditis elegans . Gene 93: 189–198. [DOI] [PubMed] [Google Scholar]
- 73. Perry MD, Li W, Trent C, Robertson B, Fire A, et al. (1993) Molecular characterization of the her-1 gene suggests a direct role in cell signaling during Caenorhabditis elegans sex determination. Genes Dev 7: 216–228. [DOI] [PubMed] [Google Scholar]
- 74. Sawin ER, Ranganathan R, Horvitz HR (2000) C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron 26: 619–631. [DOI] [PubMed] [Google Scholar]
- 75. Finney M, Ruvkun G (1990) The unc-86 gene product couples cell lineage and cell identity in C. elegans . Cell 63: 895–905. [DOI] [PubMed] [Google Scholar]
- 76. Czirjak G, Enyedi P (2002) Formation of functional heterodimers between the TASK-1 and TASK-3 two-pore domain potassium channel subunits. J Biol Chem 277: 5426–5432. [DOI] [PubMed] [Google Scholar]
- 77. Fire A (1992) Histochemical techniques for locating Escherichia coli beta-galactosidase activity in transgenic organisms. Genet Anal Tech Appl 9: 151–158. [DOI] [PubMed] [Google Scholar]