

Abstract
Alzheimer’s disease is characterized by pathological aggregation of protein tau and amyloid-β peptides, both of which are considered to be toxic to neurons. Naturally occurring dietary flavonoids have received considerable attention as alternative candidates for Alzheimer’s therapy taking into account their antiamyloidogenic, antioxidative, and anti-inflammatory properties. Experimental evidence supports the hypothesis that certain flavonoids may protect against Alzheimer’s disease in part by interfering with the generation and assembly of amyloid-β peptides into neurotoxic oligomeric aggregates and also by reducing tau aggregation. Several mechanisms have been proposed for the ability of flavonoids to prevent the onset or to slow the progression of the disease. Some mechanisms include their interaction with important signaling pathways in the brain like the phosphatidylinositol 3-kinase/Akt and mitogen-activated protein kinase pathways that regulate prosurvival transcription factors and gene expression. Other processes include the disruption of amyloid-β aggregation and alterations in amyloid precursor protein processing through the inhibition of β-secretase and/or activation of α-secretase, and inhibiting cyclin-dependent kinase-5 and glycogen synthase kinase-3β activation, preventing abnormal tau phosphorylation. The interaction of flavonoids with different signaling pathways put forward their therapeutic potential to prevent the onset and progression of Alzheimer’s disease and to promote cognitive performance. Nevertheless, further studies are needed to give additional insight into the specific mechanisms by which flavonoids exert their potential neuroprotective actions in the brain of Alzheimer’s disease patients.
Keywords: Flavonoids, Alzheimer’s disease, amyloid precursor protein, amyloid beta, BACE-1, tau, signaling
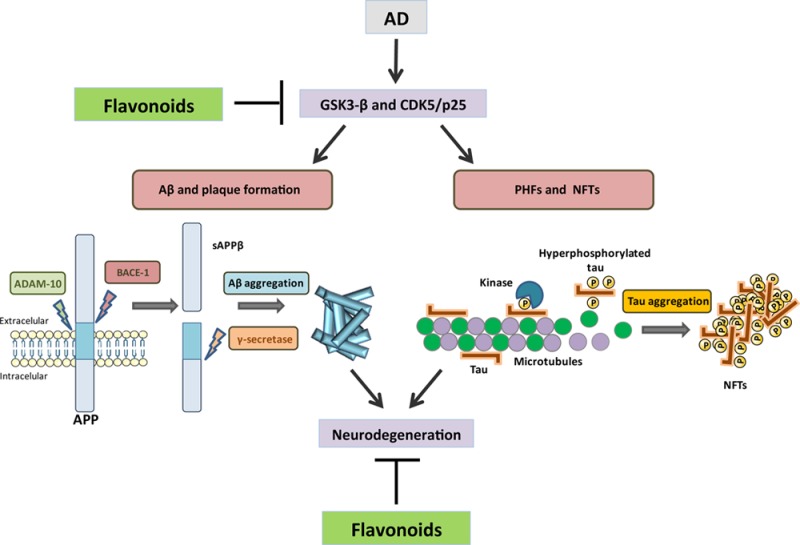
Alzheimer’s disease (AD) is a neurodegenerative disorder and the most common form of dementia worldwide. The major histopathological hallmarks of AD include proteinous aggregates in the form of neurofibrillary tangles (NFTs), consisting of hyperphosphorylated tau1,2 and extracellular senile plaques, which are deposits of heterogeneously sized small peptides of amyloid-β (Aβ) that are formed via sequential proteolytic cleavages of the amyloid precursor protein (APP) (3) (Figure 1). Dominant mutations in APP, presenilin-1 (PS1) or PS2 are responsible for the early onset or familial form of AD. These mutations have been shown to profoundly alter APP metabolism, favoring the production of aggregation-prone Aβ species, these findings formed the basis for the “amyloid cascade hypothesis” of AD pathogenesis. This broadly accepted hypothesis states that the generation of neurotoxic Aβ peptides by β-secretase and γ-secretase are at the basis of AD pathophysiology. Other hallmarks of this disease, like neurotransmitter changes4,5 and neuronal and synapse loss in the neocortex and the hippocampus6,7 develop as a consequence of this event.
Figure 1.

β-Amyloid plaques and neurofibrillary tangles are hallmark deposits of Alzheimer’s disease. The major protein component of the plaques is Aβ that results from APP by proteolytic cleavage. β-Secretase (BACE-1) generates the amino terminus of Aβ, and γ-secretase defines its length. Aβ forms toxic oligomeric aggregates that then deposit as plaques. In AD, tau is hyperphosphorylated and dissociates from MTs, causing them to depolymerize. Tau is then deposited in aggregates such as NFTs.
Amyloid Precursor Protein
APP belongs to a protein family that includes APP-like protein 1 and 2 in mammals.8,9 All are single-pass transmembrane proteins with large extracellular domains, and all are similarly processed. Though the family shares several other conserved domains such as the E1 and E2 domains in the extracellular sequence, the Aβ domain is unique to the APP protein. Alternative splicing of the APP transcript generates eight isoforms, of which three are most common: the 695 amino acid form, which is expressed predominantly in the central nervous system, and the 751 and 770 amino acid forms, which are more ubiquitously expressed.10 APP is synthesized in the endoplasmic reticulum and then transported through the Golgi apparatus to the trans-Golgi-network (TGN) where the highest concentration of APP is found in neurons at steady state.11,12 From the TGN, APP can be transported in TGN-derived secretory vesicles to the cell surface where it can be proteolytically cleaved directly by α-secretase and then γ-secretase, producing a soluble molecule (sAPPα),13 a process that does not generate Aβ. Alternatively, it can be reinternalized in clathrin-coated pits into another endosomal compartment containing the proteases β-secretase 1 (BACE-1) and γ-secretase,14,15 resulting in the production of Aβ, which is then released into the extracellular space following vesicle recycling or otherwise degraded in lysosomes.
BACE-1 Secretase and APP Cleavage
The first step in Aβ generation is APP cleavage by the β-secretase; BACE-1 is the major β-secretase.16,17 BACE-1 is a membrane-bound aspartyl protease with a characteristic type I transmembrane domain near the C-terminus.16 Overexpression or downregulation of BACE-1 induces or inhibits cleavage of APP, confirming that BACE-1 is the β-secretase involved in APP metabolism and its activity is the rate-limiting factor in Aβ generation.16,18
Several studies have investigated the potential of BACE-1 as a therapeutic target. BACE-1 knockout mice do not produce detectable levels of Aβ and have no severe phenotypic abnormalities.19,20 Suppression of this secretase by RNA interference reduced APP processing and Aβ production in primary cortical neurons derived from both wild-type and Swedish APP mutant transgenic mice.21 Moreover, disruption of the BACE-1 gene rescued memory deficits and cholinergic dysfunction in Swedish APP mice.22 Studies have also found that BACE-1 protein and activity levels are elevated in brain regions affected by AD23,24 and that oral administration of a potent and selective BACE-1 inhibitor decreased β-cleavage and Aβ production in APP transgenic mice in vivo.25 Nevertheless, some studies also show that BACE-1 as a drug target may not be as safe as first assumed. For example, BACE-1 KO mice were reported to present hypomyelination of peripheral nerves and aberrant axonal segregation,26 suggesting that inhibition of β-secretase may have unwanted serious collateral effects.
Tau Protein and Alzheimer’s Disease
Tau is one of the microtubule associated proteins that is thought to have a role in the stabilization of neuronal microtubules, providing the tracks for intracellular transport. In AD, tau protein is not able to keep the cytoskeleton well organized in the axonal process, since this protein loses its capacity to bind to microtubules due to conformational changes and misfoldings,27,28 leading to its aberrant aggregation as fibrillary structures inside neurons.29 Other tau modifications related to this dementia include phosphorylation, proteolysis, and ubiquitination, where abnormal phosphorylation is considered the most critical modification. Tau aggregation into paired helical filaments (PHFs) results from its hyperphosphorylated state, and culminates in the formation of NFTs which constitute one of the earliest AD markers (Figure 1). Moreover, abnormal hyperphosphorylated tau detached from microtubules also leads to increased intraneuronal soluble tau concentration, due to sequestration of normal tau from microtubules, further facilitating tau aggregation into PHFs.30
Changes in tau protein, affecting stabilization of microtubules, are likely to impair axonal transport,31,32 leading to changes in synaptic proteins and mitochondria axonal transport and ultimately culminating in “dying back” axons.
Kinases and Phosphatases: Involvement in Alzheimer’s Disease
In AD pathogenesis, the proteins that bind and interact with APP are different according to the phosphorylation state of APP.33,34 Several studies have been performed to clarify the role of APP phosphorylation at specific residues. Within the APP molecule, Thr668 is considered the major phosphorylation site, although other amino acids in the APP cytoplasmic domain are also phosphorylated.35−37 CDK5 (cyclin-dependent kinase-5) and GSK-3β (glycogen synthase kinase-3β) are thought to phosphorylate APP at Thr668 in neurons38−40 and when cells are subjected to a stress stimulus, c-Jun N-terminal kinase (JNK) may also phosphorylate APP at Thr668.41,42 Hyperphosphorylation of APP in AD patients’ brain may also be explained by the Aβ inhibition of phosphoprotein phosphatase 1 (PPP1)35 or PPP2, thus contributing to the phosphorylation of APP at Thr668.43
As already referred, another hallmark in AD is the hyperphosphorylation of tau protein (Figure 1), and phosphorylation of tau regulates its binding activity to microtubules stimulating their assembly. Basal phosphorylation levels are required for optimal tau function, whereas, as mentioned previously, in an hyperphosphorylated state tau loses its biological activity. Around 85 tau phosphorylation sites, of which 28 are exclusively phosphorylated in AD brains, have been described.44 In AD, abnormal tau phosphorylation may be the result of upregulation of tau kinase(s) or downregulation of tau phosphatase(s), although they may not be mutually exclusive.45 The kinases that are assumed to play the most significant role in brain tau phosphorylation are GSK-3β, CDK5, cAMP-dependent protein kinase (PKA), and calcium/calmodulin-dependent kinase II (CaMK-II).46 Among these kinases, GSK-3β may play a major role in regulating tau phosphorylation in both physiological and pathological conditions. GSK-3β can phosphorylate tau at several residues and a complementary and PPP1, PPP2A, PPP2B, and PPP2C are all possible candidates that can dephosphorylate tau protein.47 In general tau phosphoprotein is at least three- to 4-fold more hyperphosphorylated in the brain of AD patients when compared to that of aged nondemented individuals.48
Flavonoids
An increasing body of evidence demonstrates the neuroprotective potential of flavonoids either by preventing the onset or by slowing the progression of age-related neurodegenerative diseases. Dietary supplementation studies using flavonoid-rich plant or food extracts have shown their ability to influence cognition and learning in humans and also in animal models of diseases.49−55 Presently, there is no direct association between flavonoid consumption and improvement in neurological health. Nevertheless, the potential beneficial effect of flavonoids in the brain seems to be related to their ability to interact with intracellular neuronal and glial signaling pathways, thus influencing the peripheral and cerebral vascular system, protecting vulnerable neurons, enhancing existing neuronal function, or stimulating neuronal regeneration.54
Flavonoids are naturally occurring polyphenolic compounds widely spread in plants. They are present in foods and beverages of plant origin such as a variety of fruits, vegetables, cocoa, cereals, tea and wine.56 The six main subclasses of flavonoids include the (1) flavonols (e.g., kaempferol, quercetin), which are present in onions, leeks, and broccoli; (2) flavones (e.g., apigenin, luteolin), present in parsley and celery; (3) isoflavones (e.g., daidzein, genistein), which are mainly found in soy and soy products; (4) flavanones (e.g., hesperetin, naringenin), mainly found in citrus fruit and tomatoes; (5) flavanols (e.g., catechin, epicatechin, epigallocatechin, epigallocatechin gallate (EGCG)), which are abundant in green tea, red wine, and chocolate; and finally (6) anthocyanidins (e.g., pelargonidin, cyanidin, malvidin), whose sources include berry fruits and also red wine57 (Figure 2).
Figure 2.

Major classes of natural flavonoids and their dietary sources.
It was thought that the ability of flavonoids to promote memory, learning, and cognitive function was mediated by their antioxidant capacity.58 Nevertheless, due to their limited absorption and their low bioavailability in the brain, increasing evidence demonstrates that they are able to interact with the cellular and molecular components of the brain responsible for memory, having the potential to protect vulnerable neurons, enhance existing neuronal function, stimulate neuronal regeneration, and induce neurogenesis.58,59
Effect of Flavonoids on APP Processing
Several flavonoids have been shown to inhibit the development of AD and to reverse cognitive deficits in rodent models, indicating their potential therapeutic utility. Since altered APP processing leading to increased Aβ production is a key pathogenic feature of AD, several studies have been directed toward the antiamyloidogenic properties of flavonoids. In this regard, it was recently demonstrated that anthocyanin-enriched bilberry and black currant extracts have the ability to modulate APP processing and alleviate behavioral abnormalities in the APP/PS1 mouse model of AD.60 In the transgenic PSAPP mouse model of cerebral amyloidosis, oral administration of tannic acid for 6 months prevented transgene-associated behavioral impairment and defective spatial reference memory. Several other studies further support the efficacy of flavonoids on memory and learning, as for example, nobiletin, a citrus flavonoid, which proved to ameliorate Aβ-induced memory impairment and decrease the Aβ burden and plaques in the hippocampus of a transgenic mouse model of AD.61 Moreover, grape derived polyphenols (GSPE) administered orally for 5 months to Tg2576 mice, attenuates cognitive deterioration coincidently with reduced levels of high-molecular-weight soluble Aβ oligomers in the brain.62
Another citrus flavonoid, luteolin, was shown to reduce Aβ peptide generation in both human “Swedish” mutant APP transgene-bearing neuron-like cells and primary neurons, also decreasing the amyloidogenic gamma-secretase APP processing.63 Additionally, consumption of polyphenol-rich grape seed extract or curcumin for 9 months prevented amyloid-beta deposition in the brain of an AD mouse model.64
At the APP processing level, it was demonstrated that long-term treatment (16 months) with Ginkgo biloba extract (EGb761) significantly lowered APP protein levels in a transgenic mouse model of AD, suggesting that its potential neuroprotective properties may be, at least partly, related to its APP lowering effects.65 Also, brain parenchymal and cerebral vascular β-amyloid deposits were diminished in tannic acid treated PSAPP mice, suggesting that it acts as a natural β-secretase inhibitor.66 Natural flavonoids were shown to potently inhibit BACE-1 activity and reduce the level of secreted Aβ in primary cortical neurons,67 whereas epigallocatechin-3-gallate and curcumin suppress amyloid beta-induced BACE-1 upregulation in neuronal cultures.68
Several studies have focused on studying the beneficial properties of regular intake of green tea. Green tea polyphenol (−)-epigallocatechin-3-gallate (EGCG) has proved to have a beneficial role in reducing brain Aβ levels, by modulating amyloid precursor protein (APP) processing.69,70 ADAM10 activation is necessary for EGCG promotion of nonamyloidogenic (α-secretase cleavage) APP processing.71 Furthermore, EGCG-mediated enhancement of nonamyloidogenic processing of APP was found to be mediated by the maturation of ADAM10, via an estrogen receptor-α/phosphoinositide 3-kinase/Ak-transforming dependent mechanism. Since estrogen depletion following menopause has been correlated with an increased risk of developing AD, selective estrogen receptor modulation could be a therapeutic target and the use of EGCG could be considered as an alternative to estrogen therapy in the prophylaxis and treatment of this disease.72
EGCG may also have a neuroprotective action by possessing the ability to inhibit the formation of β-sheet-rich amyloid fibrils. It was demonstrated that it inhibits the fibrillogenesis of Aβ by directly binding to the natively unfolded polypeptides and preventing their conversion into on-pathway aggregation, intermediates which are toxic to neurons.73 Moreover, EGCG has the ability to convert large amyloid-beta fibrils into smaller, amorphous protein aggregates that are nontoxic and therefore suggesting that EGCG is a potent remodeling agent of mature amyloid fibrils.74 Besides EGCG, other flavonoids have also shown antiamyloidogenic properties especially myricetin, that exerts an antiamyloidogenic effect in vitro by preferentially and reversibly binding to the amyloid fibril structure of Aβ, rather than to Aβ monomers.75,76
Overall, these studies suggest that certain flavonoids are able to disrupt fibrillization by leading to the production of off-target Aβ oligomers, function as BACE-1 inhibitors or act by enhancing ADAM10 activity, consequently reducing Aβ production (Figure 3). However, more studies are needed to discover which flavonoid structures have the greatest beneficial potential and their underlying mechanisms of action.
Figure 3.

APP processing and flavonoid activity. Flavonoids can reduce Aβ production either by enhancing α-secretase (ADAM10) activity or by inhibiting β-secretase (BACE-1). Additionally, flavonoids may lead to the production of off-target Aβ oligomers, thereby disrupting fibrillization.
Tau and Flavonoids
Potential beneficial effects of flavonoids in AD may play a role in downstream targets such as tau phosphorylation. Concerning this, some studies have elucidated aspects on the effect of flavonoids in the tau protein, which may impact AD. Myrecetin and epicatechin-5-gallate have been shown to inhibit heparin-induced tau formation77 and EGCG administration in Alzheimer transgenic mice leads to modulated tau profiles, with suppression of sarkosyl-soluble phosphorylated tau isoforms.70
Other studies using GSPE have also shown its capability to inhibit tau neuropathology in a mouse model of AD, inhibiting tau peptide aggregations, as well as dissociating preformed tau peptide aggregates and disrupting PHFs.78−81 Interaction of flavonoids with signaling pathways important for tau phosphorylation will be further discussed in the next section.
Flavonoids and Their Interaction with Signaling Pathways
There is extensive evidence indicating that certain flavonoids82 and some of their metabolites83−85 are capable of exerting beneficial effects on neurological processes through their interaction with neuronal signaling pathways.
Potential flavonoid-binding sites on neurons include the adenosine,86 GABAA,87−89 δ-opioid,90,91 nicotinic,92,93 TrkB,94 estrogen,72 and testosterone receptors,95 as well as a specific brain plasma membrane binding site for polyphenols.96 Flavonoids and their metabolites have been shown to exert neuronal effects through their interactions with a number of protein kinase and lipid kinase signaling cascades, such as the PI3K/Akt, tyrosine kinase, protein kinase C, and MAPK signaling pathways and the nuclear factor-κB pathway.58,59,83,97−103 Inhibitory or stimulatory actions at these pathways are likely to greatly affect neuronal function, by changing the phosphorylation state of target molecules and/or by modulating gene expression. Consequently, this can lead to changes in plasticity, synaptic protein synthesis, and morphological changes involved in neurodegenerative processes and memory. Mitogen-activated kinases (MAPKs) belong to the superfamily of serine/threonine kinases and regulate several cellular mechanisms by transducing extracellular signals into intracellular responses.104,105 It has been suggested that flavonoids and their metabolites may interact selectively with the MAPK signaling pathways.102,106 The action of flavonoids on the ERK pathway83,99,107−110 appears to be mediated by interactions with MAPK kinases MEK1 and MEK2 and potentially membrane receptors.102,111,112 In fact, flavonoids have close structural homology to specific pharmacological modulators of ERK signaling such as PD98059 (2′-amino-3′-methoxyflavone). Moreover, activation of ERK can result in downstream activation of cAMP response element binding protein (CREB), which may lead to changes in synaptic plasticity and memory113,114 and to upregulation of neuroprotective pathways. It was demonstrated that memory performance in rats supplemented with blueberry, which contains high amounts of flavanols and anthocyanins, correlates with the activation of CREB and with increases in both pro- and mature levels of BDNF in the hippocampus,115 both of which are linked to the control of synaptic plasticity and long-term memory. Moreover, administration of green tea catechins for 6 months prevented spatial learning and memory impairments in senescence-accelerated mouse prone-8 mice by decreasing Aβ (1–42) oligomers, increasing the activity of the protein kinase A/cAMP-response element binding protein (PKA/CREB) pathway, and by upregulating synaptic plasticity-related proteins in the hippocampus.116
Additionally flavonoids modulate PI3-kinase, via direct interactions with its ATP binding site.98,117 One of the most selective PI3-kinase inhibitors available, LY294002, was modeled on the structure of quercetin.100,118 LY294002 and quercetin fit into the binding pocket of the enzyme although with different orientations.119 Substitution of hydroxyl groups on the flavonoid B ring and the degree of unsaturation of the C2–C3 bond in the C ring are important determinants of this particular bioactivity. Regarding this, it appears that different flavonoids are likely to express different cellular outcomes depending on their degree of interaction with either receptors or downstream kinases, meaning that the interactions with signaling pathways may be structure-dependent. One example of this is the flavonol quercetin and some of its in vivo metabolites which were shown to inhibit prosurvival Akt/PKB signaling pathways by inhibiting PI3-kinase activity,83 whereas flavanones, such as hesperetin, have been shown to activate Akt/PKB signaling to confer prosurvival properties in cortical neurons.108 Furthermore, it has been shown that EGCG stimulates extracellular signal-regulated kinase (ERK)- and PI3K-dependent increase in CREB phosphorylation and upregulates GluR2 levels in cortical neurons and can therefore act as a modulator in neurotransmission, plasticity, and synaptogenesis.107
Supplementing the diet of aged animals with blueberry for 12 weeks, has been shown to induce the phosphorylation of hippocampal Akt, the activation downstream of mTOR, and the increased expression of activity-regulated cytoskeletal-associated protein (Arc/Arg3.1).115 Since Arc is known to be important in LTP and has been proposed to be under regulatory control of both BDNF,120 such changes may underlie events related to spatial memory through the facilitation of alterations in synaptic strength, and the induction of morphological changes.121 These possibly include alterations in neuronal spine density and morphology, which are considered vital for learning and memory.122 Studies indicating that changes in neuronal morphology can occur in response to flavonoid supplementation support this hypothesis,50,123 and certain flavonoids can influence neuronal dendrite outgrowth in vitro.124
Tau hyperphosphorylation and accumulation in neurofibrillary tangles is strongly correlated with cognitive deficits, and among the kinases that phosphorylate tau, GSK-3β is strongly implicated in AD pathogenesis. Flavonoids have proved to have beneficial effects by inhibiting the activity of certain kinases that contribute to this pathology. It was demonstrated that indirubins inhibit GSK-3β and CDK5/p25; both of these protein kinases are involved in abnormal tau phosphorylation in AD (125) (Figure 4). Moreover, the flavonoid morin is capable of inhibiting GSK-3β activity and blocking GSK-3β-induced tau phosphorylation in vitro. Morin also attenuated Aβ-induced tau phosphorylation and protected human neuroblastoma cells against Aβ cytotoxicity. Treatment of 3xTg-AD mice with morin resulted in a decrease in tau hyperphosphorylation in hippocampal neurons.126 In the Tg2576 mouse model of AD, luteolin is able to decrease soluble Aβ levels, reduce GSK-3 activity, and disrupt PS1-APP association,63 and recently it was demonstrated that cyanidin 3-O-glucoside (Cy3G) can rescue the cognitive impairments that are induced by Aβ via the modulation of GSK-3β/tau in rats, suggesting a potential therapeutic role of Cy3G in AD.127 It seems reasonable to deduce that imbalances in the phosphorylation system are therefore one of the causes for hyperphosphorylation of cytoskeletal proteins in AD.
Figure 4.
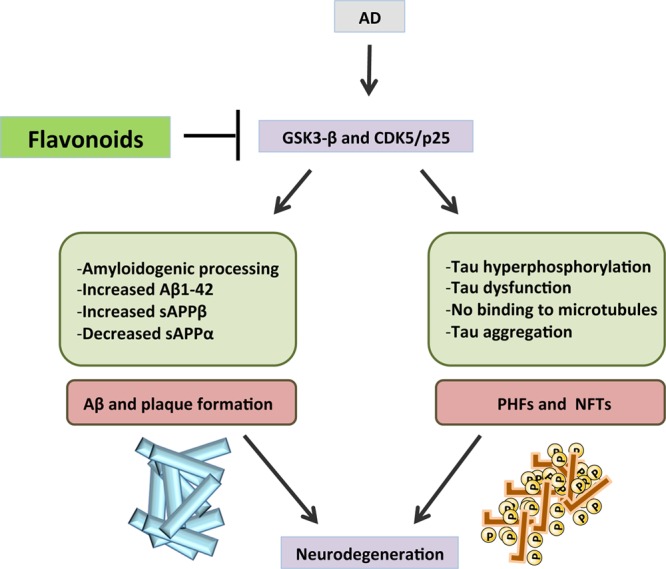
Flavonoids beneficial effects are exerted by inhibiting the activity GSK-3β and CDK5/p25, therefore preventing the activation of signaling cascades that contribute to neurodegeneration in AD.
Although there has been intense interest in the ability of flavonoids to modulate kinases, there is no indication that they may affect signaling pathways via a modulation of phosphatase activity. Considering that phosphatases reverse the activity of kinases and since phosphatases are integral to many signaling pathways, it is conceivable that changes in ERK activation and related transcription factors may result from flavonoid-induced modulation of phosphatase activity.82 Nevertheless, future studies are needed to evaluate the potential of flavonoids to inhibit, or activate phosphatases and their mechanisms of action.
Conclusions
Flavonoids are widely available in natural foods, and as a result, treatments for AD with such natural compounds through diet or dietary supplements are considered an attractive alternative. Flavonoids have demonstrated to have beneficial properties against the general mechanisms of AD in a variety of cell culture and animal models. Nevertheless, more studies addressing the specific mechanisms by which flavonoids exert their potential neuroprotective actions are required, before novel flavonoid-based dietary applications are applied in practice to reduce AD risk. Advances in the understanding of the mechanisms underlying flavonoid–protein interactions in AD, may represent a promising goal for developing novel neuroprotective strategies for neurodegenerative diseases.
Glossary
Abbreviations
- AD
Alzheimer’s disease
- NFTs
neurofibrillary tangles
- Aβ
amyloid-β peptide
- APP
amyloid precursor protein
- PS1
presenilin-1
- TGN
trans-Golgi-network
- BACE-1
beta-secretase 1
- PHFs
Paired helical filaments
- CDK5
cyclin-dependent kinase-5
- GSK-3β
glycogen synthase kinase-3β
- PPP1
phosphoprotein phosphatase 1
- EGCG
(-)-epigallocatechin-3-gallate
- ADAM10
A disintegrin and metalloproteinase domain-containing protein 10
This work was supported by CENTRO-07-ST24-FEDER-002034 – MI-PI-43-ARH/2012 (Project: New Strategies Applied to Neuropathological Disorders), QREN (Mais Centro-Programa Operacional Regional do Centro e União Europeia/Fundo Europeu de Desenvolvimento Regional), PEST-OE/SAU/UI0482/2013, PTDC/QUI-BIQ/101317/2008 and PTDC/BEX-BCM/0493/2012.
The authors declare no competing financial interest.
References
- Holtzman D. M.; Morris J. C.; Goate A. M. (2011) Alzheimer’s disease: the challenge of the second century. Sci. Transl. Med. 3, 77sr71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron T. F.; Petrucelli L. (2009) The role of tau in neurodegeneration. Mol. Neurodegener. 4, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D. J. (1998) The cell biology of beta-amyloid precursor protein and presenilin in Alzheimer’s disease. Trends Cell Biol. 8, 447–453. [DOI] [PubMed] [Google Scholar]
- Francis P. T. (2005) The interplay of neurotransmitters in Alzheimer’s disease. CNS Spectrums 10, 6–9. [DOI] [PubMed] [Google Scholar]
- Reinikainen K. J.; Soininen H.; Riekkinen P. J. (1990) Neurotransmitter changes in Alzheimer’s disease: implications to diagnostics and therapy. J. Neurosci. Res. 27, 576–586. [DOI] [PubMed] [Google Scholar]
- Scheff S. W.; Price D. A.; Schmitt F. A.; Mufson E. J. (2006) Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 27, 1372–1384. [DOI] [PubMed] [Google Scholar]
- Desikan R. S.; Sabuncu M. R.; Schmansky N. J.; Reuter M.; Cabral H. J.; Hess C. P.; Weiner M. W.; Biffi A.; Anderson C. D.; Rosand J.; Salat D. H.; Kemper T. L.; Dale A. M.; Sperling R. A.; Fischl B. (2010) Selective disruption of the cerebral neocortex in Alzheimer’s disease. PloS One 5, e12853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasco W.; Gurubhagavatula S.; Paradis M. D.; Romano D. M.; Sisodia S. S.; Hyman B. T.; Neve R. L.; Tanzi R. E. (1993) Isolation and characterization of APLP2 encoding a homologue of the Alzheimer’s associated amyloid beta protein precursor. Nat. Genet. 5, 95–100. [DOI] [PubMed] [Google Scholar]
- Coulson E. J.; Paliga K.; Beyreuther K.; Masters C. L. (2000) What the evolution of the amyloid protein precursor supergene family tells us about its function. Neurochem. Int. 36, 175–184. [DOI] [PubMed] [Google Scholar]
- Bayer T. A.; Cappai R.; Masters C. L.; Beyreuther K.; Multhaup G. (1999) It all sticks together--the APP-related family of proteins and Alzheimer’s disease. Mol. Psychiatry 4, 524–528. [DOI] [PubMed] [Google Scholar]
- Hartmann T.; Bieger S. C.; Bruhl B.; Tienari P. J.; Ida N.; Allsop D.; Roberts G. W.; Masters C. L.; Dotti C. G.; Unsicker K.; Beyreuther K. (1997) Distinct sites of intracellular production for Alzheimer’s disease A beta40/42 amyloid peptides. Nat. Med. 3, 1016–1020. [DOI] [PubMed] [Google Scholar]
- Greenfield J. P.; Tsai J.; Gouras G. K.; Hai B.; Thinakaran G.; Checler F.; Sisodia S. S.; Greengard P.; Xu H. (1999) Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer beta-amyloid peptides. Proc. Natl. Acad. Sci. U.S.A. 96, 742–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisodia S. S. (1992) Beta-amyloid precursor protein cleavage by a membrane-bound protease. Proc. Natl. Acad. Sci. U.S.A. 89, 6075–6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordstedt C.; Caporaso G. L.; Thyberg J.; Gandy S. E.; Greengard P. (1993) Identification of the Alzheimer beta/A4 amyloid precursor protein in clathrin-coated vesicles purified from PC12 cells. J. Biol. Chem. 268, 608–612. [PubMed] [Google Scholar]
- Caporaso G. L.; Takei K.; Gandy S. E.; Matteoli M.; Mundigl O.; Greengard P.; De Camilli P. (1994) Morphologic and biochemical analysis of the intracellular trafficking of the Alzheimer beta/A4 amyloid precursor protein. J. Neurosci. 14, 3122–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R.; Bennett B. D.; Babu-Khan S.; Kahn S.; Mendiaz E. A.; Denis P.; Teplow D. B.; Ross S.; Amarante P.; Loeloff R.; Luo Y.; Fisher S.; Fuller J.; Edenson S.; Lile J.; Jarosinski M. A.; Biere A. L.; Curran E.; Burgess T.; Louis J. C.; Collins F.; Treanor J.; Rogers G.; Citron M. (1999) Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741. [DOI] [PubMed] [Google Scholar]
- Yan R.; Bienkowski M. J.; Shuck M. E.; Miao H.; Tory M. C.; Pauley A. M.; Brashier J. R.; Stratman N. C.; Mathews W. R.; Buhl A. E.; Carter D. B.; Tomasselli A. G.; Parodi L. A.; Heinrikson R. L.; Gurney M. E. (1999) Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature 402, 533–537. [DOI] [PubMed] [Google Scholar]
- Cai H.; Wang Y.; McCarthy D.; Wen H.; Borchelt D. R.; Price D. L.; Wong P. C. (2001) BACE1 is the major beta-secretase for generation of Abeta peptides by neurons. Nat. Neurosci. 4, 233–234. [DOI] [PubMed] [Google Scholar]
- Luo Y.; Bolon B.; Kahn S.; Bennett B. D.; Babu-Khan S.; Denis P.; Fan W.; Kha H.; Zhang J.; Gong Y.; Martin L.; Louis J. C.; Yan Q.; Richards W. G.; Citron M.; Vassar R. (2001) Mice deficient in BACE1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat. Neurosci. 4, 231–232. [DOI] [PubMed] [Google Scholar]
- Roberds S. L.; Anderson J.; Basi G.; Bienkowski M. J.; Branstetter D. G.; Chen K. S.; Freedman S. B.; Frigon N. L.; Games D.; Hu K.; Johnson-Wood K.; Kappenman K. E.; Kawabe T. T.; Kola I.; Kuehn R.; Lee M.; Liu W.; Motter R.; Nichols N. F.; Power M.; Robertson D. W.; Schenk D.; Schoor M.; Shopp G. M.; Shuck M. E.; Sinha S.; Svensson K. A.; Tatsuno G.; Tintrup H.; Wijsman J.; Wright S.; McConlogue L. (2001) BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer’s disease therapeutics. Hum. Mol. Genet. 10, 1317–1324. [DOI] [PubMed] [Google Scholar]
- Kao S. C.; Krichevsky A. M.; Kosik K. S.; Tsai L. H. (2004) BACE1 suppression by RNA interference in primary cortical neurons. J. Biol. Chem. 279, 1942–1949. [DOI] [PubMed] [Google Scholar]
- Ohno M.; Sametsky E. A.; Younkin L. H.; Oakley H.; Younkin S. G.; Citron M.; Vassar R.; Disterhoft J. F. (2004) BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer’s disease. Neuron 41, 27–33. [DOI] [PubMed] [Google Scholar]
- Yang L. B.; Lindholm K.; Yan R.; Citron M.; Xia W.; Yang X. L.; Beach T.; Sue L.; Wong P.; Price D.; Li R.; Shen Y. (2003) Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 9, 3–4. [DOI] [PubMed] [Google Scholar]
- Johnston J. A.; Liu W. W.; Todd S. A.; Coulson D. T.; Murphy S.; Irvine G. B.; Passmore A. P. (2005) Expression and activity of beta-site amyloid precursor protein cleaving enzyme in Alzheimer’s disease. Biochem. Soc. Trans. 33, 1096–1100. [DOI] [PubMed] [Google Scholar]
- Hussain I.; Hawkins J.; Harrison D.; Hille C.; Wayne G.; Cutler L.; Buck T.; Walter D.; Demont E.; Howes C.; Naylor A.; Jeffrey P.; Gonzalez M. I.; Dingwall C.; Michel A.; Redshaw S.; Davis J. B. (2007) Oral administration of a potent and selective non-peptidic BACE-1 inhibitor decreases beta-cleavage of amyloid precursor protein and amyloid-beta production in vivo. J. Neurochem. 100, 802–809. [DOI] [PubMed] [Google Scholar]
- Willem M.; Garratt A. N.; Novak B.; Citron M.; Kaufmann S.; Rittger A.; DeStrooper B.; Saftig P.; Birchmeier C.; Haass C. (2006) Control of peripheral nerve myelination by the beta-secretase BACE1. Science 314, 664–666. [DOI] [PubMed] [Google Scholar]
- Fox N.; Harvey R. J.; Rossor M. N. (1996) Protein folding, nucleation phenomena and delayed neurodegeneration in Alzheimer’s disease. Rev. Neurosci. 7, 21–28. [DOI] [PubMed] [Google Scholar]
- Hyman B. T.; Augustinack J. C.; Ingelsson M. (2005) Transcriptional and conformational changes of the tau molecule in Alzheimer’s disease. Biochim. Biophys. Acta 1739, 150–157. [DOI] [PubMed] [Google Scholar]
- Garcia-Sierra F.; Ghoshal N.; Quinn B.; Berry R. W.; Binder L. I. (2003) Conformational changes and truncation of tau protein during tangle evolution in Alzheimer’s disease. J. Alzheimer’s Dis. 5, 65–77. [DOI] [PubMed] [Google Scholar]
- Alonso A. D.; Grundke-Iqbal I.; Barra H. S.; Iqbal K. (1997) Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc. Natl. Acad. Sci. U.S.A. 94, 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebneth A.; Godemann R.; Stamer K.; Illenberger S.; Trinczek B.; Mandelkow E. (1998) Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer’s disease. J. Cell Biol. 143, 777–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Martin T.; Cuchillo-Ibanez I.; Noble W.; Nyenya F.; Anderton B. H.; Hanger D. P. (2013) Tau phosphorylation affects its axonal transport and degradation. Neurobiol. Aging 34, 2146–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Cruz e Silva O. A.; Fardilha M.; Henriques A. G.; Rebelo S.; Vieira S.; da Cruz e Silva E. F. (2004) Signal transduction therapeutics: relevance for Alzheimer’s disease. J. Mol. Neurosci. 23, 123–142. [DOI] [PubMed] [Google Scholar]
- Nakaya T.; Suzuki T. (2006) Role of APP phosphorylation in FE65-dependent gene transactivation mediated by AICD. Genes Cells 11, 633–645. [DOI] [PubMed] [Google Scholar]
- Lee M. S.; Kao S. C.; Lemere C. A.; Xia W.; Tseng H. C.; Zhou Y.; Neve R.; Ahlijanian M. K.; Tsai L. H. (2003) APP processing is regulated by cytoplasmic phosphorylation. J. Cell Biol. 163, 83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Cruz e Silva E. F.; da Cruz e Silva O. A. (2003) Protein phosphorylation and APP metabolism. Neurochem. Res. 28, 1553–1561. [DOI] [PubMed] [Google Scholar]
- Rebelo S.; Vieira S. I.; Esselmann H.; Wiltfang J.; da Cruz e Silva E. F.; da Cruz e Silva O. A. (2007) Tyrosine 687 phosphorylated Alzheimer’s amyloid precursor protein is retained intracellularly and exhibits a decreased turnover rate. Neurodegener. Dis. 4, 78–87. [DOI] [PubMed] [Google Scholar]
- Iijima K.; Ando K.; Takeda S.; Satoh Y.; Seki T.; Itohara S.; Greengard P.; Kirino Y.; Nairn A. C.; Suzuki T. (2000) Neuron-specific phosphorylation of Alzheimer’s beta-amyloid precursor protein by cyclin-dependent kinase 5. J. Neurochem. 75, 1085–1091. [DOI] [PubMed] [Google Scholar]
- Aplin A. E.; Gibb G. M.; Jacobsen J. S.; Gallo J. M.; Anderton B. H. (1996) In vitro phosphorylation of the cytoplasmic domain of the amyloid precursor protein by glycogen synthase kinase-3beta. J. Neurochem. 67, 699–707. [DOI] [PubMed] [Google Scholar]
- Liu F.; Su Y.; Li B.; Zhou Y.; Ryder J.; Gonzalez-DeWhitt P.; May P. C.; Ni B. (2003) Regulation of amyloid precursor protein (APP) phosphorylation and processing by p35/Cdk5 and p25/Cdk5. FEBS Lett. 547, 193–196. [DOI] [PubMed] [Google Scholar]
- Standen C. L.; Brownlees J.; Grierson A. J.; Kesavapany S.; Lau K. F.; McLoughlin D. M.; Miller C. C. (2001) Phosphorylation of thr(668) in the cytoplasmic domain of the Alzheimer’s disease amyloid precursor protein by stress-activated protein kinase 1b (Jun N-terminal kinase-3). J. Neurochem. 76, 316–320. [DOI] [PubMed] [Google Scholar]
- Taru H.; Iijima K.; Hase M.; Kirino Y.; Yagi Y.; Suzuki T. (2002) Interaction of Alzheimer’s beta -amyloid precursor family proteins with scaffold proteins of the JNK signaling cascade. J. Biol. Chem. 277, 20070–20078. [DOI] [PubMed] [Google Scholar]
- Sontag E.; Nunbhakdi-Craig V.; Sontag J. M.; Diaz-Arrastia R.; Ogris E.; Dayal S.; Lentz S. R.; Arning E.; Bottiglieri T. (2007) Protein phosphatase 2A methyltransferase links homocysteine metabolism with tau and amyloid precursor protein regulation. J. Neurosci. 27, 2751–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L.; Latypova X.; Wilson C. M.; Magnaudeix A.; Perrin M. L.; Yardin C.; Terro F. (2013) Tau protein kinases: involvement in Alzheimer’s disease. Ageing Res. reviews 12, 289–309. [DOI] [PubMed] [Google Scholar]
- Buee L.; Bussiere T.; Buee-Scherrer V.; Delacourte A.; Hof P. R. (2000) Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Rev. 33, 95–130. [DOI] [PubMed] [Google Scholar]
- Gong C. X.; Iqbal K. (2008) Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Curr. Med. Chem. 15, 2321–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila J.; Lucas J. J.; Perez M.; Hernandez F. (2004) Role of tau protein in both physiological and pathological conditions. Physiol. Rev. 84, 361–384. [DOI] [PubMed] [Google Scholar]
- Kopke E.; Tung Y. C.; Shaikh S.; Alonso A. C.; Iqbal K.; Grundke-Iqbal I. (1993) Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 268, 24374–24384. [PubMed] [Google Scholar]
- Galli R. L.; Shukitt-Hale B.; Youdim K. A.; Joseph J. A. (2002) Fruit polyphenolics and brain aging: nutritional interventions targeting age-related neuronal and behavioral deficits. Ann. N.Y. Acad. Sci. 959, 128–132. [DOI] [PubMed] [Google Scholar]
- Haque A. M.; Hashimoto M.; Katakura M.; Tanabe Y.; Hara Y.; Shido O. (2006) Long-term administration of green tea catechins improves spatial cognition learning ability in rats. J. Nutr. 136, 1043–1047. [DOI] [PubMed] [Google Scholar]
- Kuriyama S.; Hozawa A.; Ohmori K.; Shimazu T.; Matsui T.; Ebihara S.; Awata S.; Nagatomi R.; Arai H.; Tsuji I. (2006) Green tea consumption and cognitive function: a cross-sectional study from the Tsurugaya Project 1. Am. J. Clin. Nutr. 83, 355–361. [DOI] [PubMed] [Google Scholar]
- Unno K.; Takabayashi F.; Kishido T.; Oku N. (2004) Suppressive effect of green tea catechins on morphologic and functional regression of the brain in aged mice with accelerated senescence (SAMP10). Exp. Gerontol. 39, 1027–1034. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Wang L.; Wu J.; Cai J. (2006) The in vivo synaptic plasticity mechanism of EGb 761-induced enhancement of spatial learning and memory in aged rats. Br. J. Pharmacol. 148, 147–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youdim K. A.; Joseph J. A. (2001) A possible emerging role of phytochemicals in improving age-related neurological dysfunctions: a multiplicity of effects. Free Radical Biol. Med. 30, 583–594. [DOI] [PubMed] [Google Scholar]
- Sokolov A. N.; Pavlova M. A.; Klosterhalfen S.; Enck P. (2013) Chocolate and the brain: Neurobiological impact of cocoa flavanols on cognition and behavior. Neurosci. Biobehav. Rev. 37(10 Pt 2), 2445–2453 10.1016/j.neubiorev.2013.06.013. [DOI] [PubMed] [Google Scholar]
- Beecher G. R. (2003) Overview of dietary flavonoids: nomenclature, occurrence and intake. J. Nutr. 133, 3248S–3254S. [DOI] [PubMed] [Google Scholar]
- Pietta P. G. (2000) Flavonoids as antioxidants. J. Nat. Prod. 63, 1035–1042. [DOI] [PubMed] [Google Scholar]
- Spencer J. P. (2010) Beyond antioxidants: the cellular and molecular interactions of flavonoids and how these underpin their actions on the brain. Proc. Nutr. Soc. 69, 244–260. [DOI] [PubMed] [Google Scholar]
- Spencer J. P. (2009) The impact of flavonoids on memory: physiological and molecular considerations. Chem. Soc. Rev. 38, 1152–1161. [DOI] [PubMed] [Google Scholar]
- Vepsalainen S.; Koivisto H.; Pekkarinen E.; Makinen P.; Dobson G.; McDougall G. J.; Stewart D.; Haapasalo A.; Karjalainen R. O.; Tanila H.; Hiltunen M. (2013) Anthocyanin-enriched bilberry and blackcurrant extracts modulate amyloid precursor protein processing and alleviate behavioral abnormalities in the APP/PS1 mouse model of Alzheimer’s disease. J. Nutr. Biochem. 24, 360–370. [DOI] [PubMed] [Google Scholar]
- Onozuka H.; Nakajima A.; Matsuzaki K.; Shin R. W.; Ogino K.; Saigusa D.; Tetsu N.; Yokosuka A.; Sashida Y.; Mimaki Y.; Yamakuni T.; Ohizumi Y. (2008) Nobiletin, a citrus flavonoid, improves memory impairment and Abeta pathology in a transgenic mouse model of Alzheimer’s disease. J. Pharmacol. Exp. Ther. 326, 739–744. [DOI] [PubMed] [Google Scholar]
- Wang J.; Ho L.; Zhao W.; Ono K.; Rosensweig C.; Chen L.; Humala N.; Teplow D. B.; Pasinetti G. M. (2008) Grape-derived polyphenolics prevent Abeta oligomerization and attenuate cognitive deterioration in a mouse model of Alzheimer’s disease. J. Neurosci. 28, 6388–6392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezai-Zadeh K.; Douglas Shytle R.; Bai Y.; Tian J.; Hou H.; Mori T.; Zeng J.; Obregon D.; Town T.; Tan J. (2009) Flavonoid-mediated presenilin-1 phosphorylation reduces Alzheimer’s disease beta-amyloid production. J. Cell. Mol. Med. 13, 574–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. J.; Thomas P.; Zhong J. H.; Bi F. F.; Kosaraju S.; Pollard A.; Fenech M.; Zhou X. F. (2009) Consumption of grape seed extract prevents amyloid-beta deposition and attenuates inflammation in brain of an Alzheimer’s disease mouse. Neurotoxic. Res. 15, 3–14. [DOI] [PubMed] [Google Scholar]
- Augustin S.; Rimbach G.; Augustin K.; Schliebs R.; Wolffram S.; Cermak R. (2009) Effect of a short- and long-term treatment with Ginkgo biloba extract on amyloid precursor protein levels in a transgenic mouse model relevant to Alzheimer’s disease. Arch. Biochem. Biophys. 481, 177–182. [DOI] [PubMed] [Google Scholar]
- Mori T.; Rezai-Zadeh K.; Koyama N.; Arendash G. W.; Yamaguchi H.; Kakuda N.; Horikoshi-Sakuraba Y.; Tan J.; Town T. (2012) Tannic acid is a natural beta-secretase inhibitor that prevents cognitive impairment and mitigates Alzheimer-like pathology in transgenic mice. J. Biol. Chem. 287, 6912–6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimmyo Y.; Kihara T.; Akaike A.; Niidome T.; Sugimoto H. (2008) Flavonols and flavones as BACE-1 inhibitors: structure-activity relationship in cell-free, cell-based and in silico studies reveal novel pharmacophore features. Biochim. Biophys. Acta 1780, 819–825. [DOI] [PubMed] [Google Scholar]
- Shimmyo Y.; Kihara T.; Akaike A.; Niidome T.; Sugimoto H. (2008) Epigallocatechin-3-gallate and curcumin suppress amyloid beta-induced beta-site APP cleaving enzyme-1 upregulation. NeuroReport 19, 1329–1333. [DOI] [PubMed] [Google Scholar]
- Rezai-Zadeh K.; Shytle D.; Sun N.; Mori T.; Hou H.; Jeanniton D.; Ehrhart J.; Townsend K.; Zeng J.; Morgan D.; Hardy J.; Town T.; Tan J. (2005) Green tea epigallocatechin-3-gallate (EGCG) modulates amyloid precursor protein cleavage and reduces cerebral amyloidosis in Alzheimer transgenic mice. J. Neurosci. 25, 8807–8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezai-Zadeh K.; Arendash G. W.; Hou H.; Fernandez F.; Jensen M.; Runfeldt M.; Shytle R. D.; Tan J. (2008) Green tea epigallocatechin-3-gallate (EGCG) reduces beta-amyloid mediated cognitive impairment and modulates tau pathology in Alzheimer transgenic mice. Brain Res. 1214, 177–187. [DOI] [PubMed] [Google Scholar]
- Obregon D. F.; Rezai-Zadeh K.; Bai Y.; Sun N.; Hou H.; Ehrhart J.; Zeng J.; Mori T.; Arendash G. W.; Shytle D.; Town T.; Tan J. (2006) ADAM10 activation is required for green tea (-)-epigallocatechin-3-gallate-induced alpha-secretase cleavage of amyloid precursor protein. J. Biol. Chem. 281, 16419–16427. [DOI] [PubMed] [Google Scholar]
- Fernandez J. W.; Rezai-Zadeh K.; Obregon D.; Tan J. (2010) EGCG functions through estrogen receptor-mediated activation of ADAM10 in the promotion of non-amyloidogenic processing of APP. FEBS Lett. 584, 4259–4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrnhoefer D. E.; Bieschke J.; Boeddrich A.; Herbst M.; Masino L.; Lurz R.; Engemann S.; Pastore A.; Wanker E. E. (2008) EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 15, 558–566. [DOI] [PubMed] [Google Scholar]
- Bieschke J.; Russ J.; Friedrich R. P.; Ehrnhoefer D. E.; Wobst H.; Neugebauer K.; Wanker E. E. (2010) EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. U.S.A. 107, 7710–7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirohata M.; Hasegawa K.; Tsutsumi-Yasuhara S.; Ohhashi Y.; Ookoshi T.; Ono K.; Yamada M.; Naiki H. (2007) The anti-amyloidogenic effect is exerted against Alzheimer’s beta-amyloid fibrils in vitro by preferential and reversible binding of flavonoids to the amyloid fibril structure. Biochemistry 46, 1888–1899. [DOI] [PubMed] [Google Scholar]
- Ono K.; Yoshiike Y.; Takashima A.; Hasegawa K.; Naiki H.; Yamada M. (2003) Potent anti-amyloidogenic and fibril-destabilizing effects of polyphenols in vitro: implications for the prevention and therapeutics of Alzheimer’s disease. J. Neurochem. 87, 172–181. [DOI] [PubMed] [Google Scholar]
- Taniguchi S.; Suzuki N.; Masuda M.; Hisanaga S.; Iwatsubo T.; Goedert M.; Hasegawa M. (2005) Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J. Biol. Chem. 280, 7614–7623. [DOI] [PubMed] [Google Scholar]
- Wang J.; Santa-Maria I.; Ho L.; Ksiezak-Reding H.; Ono K.; Teplow D. B.; Pasinetti G. M. (2010) Grape derived polyphenols attenuate tau neuropathology in a mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 22, 653–661. [DOI] [PubMed] [Google Scholar]
- Pasinetti G. M.; Ksiezak-Reding H.; Santa-Maria I.; Wang J.; Ho L. (2010) Development of a grape seed polyphenolic extract with anti-oligomeric activity as a novel treatment in progressive supranuclear palsy and other tauopathies. J. Neurochem. 114, 1557–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L.; Yemul S.; Wang J.; Pasinetti G. M. (2009) Grape seed polyphenolic extract as a potential novel therapeutic agent in tauopathies. J. Alzheimer’s Dis. 16, 433–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ksiezak-Reding H.; Ho L.; Santa-Maria I.; Diaz-Ruiz C.; Wang J.; Pasinetti G. M. (2012) Ultrastructural alterations of Alzheimer’s disease paired helical filaments by grape seed-derived polyphenols. Neurobiol. Aging 33, 1427–1439. [DOI] [PubMed] [Google Scholar]
- Spencer J. P. (2007) The interactions of flavonoids within neuronal signalling pathways. Genes Nutr. 2, 257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer J. P.; Rice-Evans C.; Williams R. J. (2003) Modulation of pro-survival Akt/protein kinase B and ERK1/2 signaling cascades by quercetin and its in vivo metabolites underlie their action on neuronal viability. J. Biol. Chem. 278, 34783–34793. [DOI] [PubMed] [Google Scholar]
- Incani A.; Deiana M.; Corona G.; Vafeiadou K.; Vauzour D.; Dessi M. A.; Spencer J. P. (2010) Involvement of ERK, Akt and JNK signalling in H2O2-induced cell injury and protection by hydroxytyrosol and its metabolite homovanillic alcohol. Mol. Nutr. Food Res. 54, 788–796. [DOI] [PubMed] [Google Scholar]
- Vauzour D.; Vafeiadou K.; Spencer J. P. (2007) Inhibition of the formation of the neurotoxin 5-S-cysteinyl-dopamine by polyphenols. Biochem. Biophys. Res. Commun. 362, 340–346. [DOI] [PubMed] [Google Scholar]
- Ji X. D.; Melman N.; Jacobson K. A. (1996) Interactions of flavonoids and other phytochemicals with adenosine receptors. J. Med. Chem. 39, 781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez S. P.; Mewett K. N.; Hanrahan J. R.; Chebib M.; Johnston G. A. (2008) Flavan-3-ol derivatives are positive modulators of GABA(A) receptors with higher efficacy for the alpha(2) subtype and anxiolytic action in mice. Neuropharmacology 55, 900–907. [DOI] [PubMed] [Google Scholar]
- Adachi N.; Tomonaga S.; Tachibana T.; Denbow D. M.; Furuse M. (2006) (−)-Epigallocatechin gallate attenuates acute stress responses through GABAergic system in the brain. Eur. J. Pharmacol. 531, 171–175. [DOI] [PubMed] [Google Scholar]
- Hanrahan J. R.; Chebib M.; Johnston G. A. (2011) Flavonoid modulation of GABA(A) receptors. Br. J. Pharmacol. 163, 234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katavic P. L.; Lamb K.; Navarro H.; Prisinzano T. E. (2007) Flavonoids as opioid receptor ligands: identification and preliminary structure-activity relationships. J. Nat. Prod. 70, 1278–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panneerselvam M.; Tsutsumi Y. M.; Bonds J. A.; Horikawa Y. T.; Saldana M.; Dalton N. D.; Head B. P.; Patel P. M.; Roth D. M.; Patel H. H. (2010) Dark chocolate receptors: epicatechin-induced cardiac protection is dependent on delta-opioid receptor stimulation. A. J. Physiol.: Heart Circ. Physiol. 299, H1604–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B. H.; Choi S. H.; Shin T. J.; Pyo M. K.; Hwang S. H.; Kim B. R.; Lee S. M.; Lee J. H.; Kim H. C.; Park H. Y.; Rhim H.; Nah S. Y. (2010) Quercetin enhances human alpha7 nicotinic acetylcholine receptor-mediated ion current through interactions with Ca(2+) binding sites. Mol. Cells 30, 245–253. [DOI] [PubMed] [Google Scholar]
- Lee B. H.; Choi S. H.; Shin T. J.; Pyo M. K.; Hwang S. H.; Lee S. M.; Paik H. D.; Kim H. C.; Nah S. Y. (2011) Effects of quercetin on alpha9alpha10 nicotinic acetylcholine receptor-mediated ion currents. Eur. J. Pharmacol. 650, 79–85. [DOI] [PubMed] [Google Scholar]
- Jang S. W.; Liu X.; Yepes M.; Shepherd K. R.; Miller G. W.; Liu Y.; Wilson W. D.; Xiao G.; Blanchi B.; Sun Y. E.; Ye K. Q. (2010) A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc. Natl. Acad. Sci. U.S.A. 107, 2687–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nifli A. P.; Bosson-Kouame A.; Papadopoulou N.; Kogia C.; Kampa M.; Castagnino C.; Stournaras C.; Vercauteren J.; Castanas E. (2005) Monomeric and oligomeric flavanols are agonists of membrane androgen receptors. Exp. Cell Res. 309, 329–339. [DOI] [PubMed] [Google Scholar]
- Han Y. S.; Bastianetto S.; Dumont Y.; Quirion R. (2006) Specific plasma membrane binding sites for polyphenols, including resveratrol, in the rat brain. J. Pharmacol. Exp. Ther. 318, 238–245. [DOI] [PubMed] [Google Scholar]
- Goyarzu F.; Malin D. H.; Lau F. C.; Taglialatela G.; Moon W. D.; Jennings R.; Moy E.; Moy D.; Lippold S.; Shukitt-Hale B.; Joseph J. A. (2004) Blueberry supplemented diet: Effects on object recognition memory and nuclear factor-kappa B levels in aged rats. Nutr. Neurosci. 7, 75–83. [DOI] [PubMed] [Google Scholar]
- Gamet-Payrastre L.; Manenti S.; Gratacap M. P.; Tulliez J.; Chap H.; Payrastre B. (1999) Flavonoids and the inhibition of PKC and PI 3-kinase. Gen. Pharmacol. 32, 279–286. [DOI] [PubMed] [Google Scholar]
- Schroeter H.; Spencer J. P.; Rice-Evans C.; Williams R. J. (2001) Flavonoids protect neurons from oxidized low-density-lipoprotein-induced apoptosis involving c-Jun N-terminal kinase (JNK), c-Jun and caspase-3. Biochem. J. 358, 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matter W. F.; Brown R. F.; Vlahos C. J. (1992) The inhibition of phosphatidylinositol 3-kinase by quercetin and analogs. Biochem. Biophys. Res. Commun. 186, 624–631. [DOI] [PubMed] [Google Scholar]
- Agullo G.; Gamet-Payrastre L.; Manenti S.; Viala C.; Remesy C.; Chap H.; Payrastre B. (1997) Relationship between flavonoid structure and inhibition of phosphatidylinositol 3-kinase: a comparison with tyrosine kinase and protein kinase C inhibition. Biochem. Pharmacol. 53, 1649–1657. [DOI] [PubMed] [Google Scholar]
- Kong A. N. T.; Yu R.; Chen C.; Mandlekar S.; Primiano T. (2000) Signal transduction events elicited by natural products: Role of MAPK and caspase pathways in homeostatic response and induction of apoptosis. Arch. Pharm. Res. 23, 1–16. [DOI] [PubMed] [Google Scholar]
- Williams R. J.; Spencer J. P.; Rice-Evans C. (2004) Flavonoids: antioxidants or signalling molecules?. Free. Radical Biol. Med. 36, 838–849. [DOI] [PubMed] [Google Scholar]
- Cobb M. H.; Goldsmith E. J. (1995) How Map Kinases Are Regulated. J. Biol. Chem. 270, 14843–14846. [DOI] [PubMed] [Google Scholar]
- Cargnello M.; Roux P. P. (2011) Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75, 50–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobuchi H.; Roy S.; Sen C. K.; Nguyen H. G.; Packer L. (1999) Quercetin inhibits inducible ICAM-1 expression in human endothelial cells through the JNK pathway. Am. J. Physiol.: Cell Physiol. 277, C403–C411. [DOI] [PubMed] [Google Scholar]
- Schroeter H.; Bahia P.; Spencer J. P.; Sheppard O.; Rattray M.; Cadenas E.; Rice-Evans C.; Williams R. J. (2007) (-)Epicatechin stimulates ERK-dependent cyclic AMP response element activity and up-regulates GluR2 in cortical neurons. J. Neurochem. 101, 1596–1606. [DOI] [PubMed] [Google Scholar]
- Vauzour D.; Vafeiadou K.; Rice-Evans C.; Williams R. J.; Spencer J. P. (2007) Activation of pro-survival Akt and ERK1/2 signalling pathways underlie the anti-apoptotic effects of flavanones in cortical neurons. J. Neurochem. 103, 1355–1367. [DOI] [PubMed] [Google Scholar]
- Maher P.; Akaishi T.; Abe K. (2006) Flavonoid fisetin promotes ERK-dependent long-term potentiation and enhances memory. Proc. Natl. Acad. Sci. U.S.A. 103, 16568–16573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan A.; Xu C. J.; Nair S. S.; Chen C.; Hebbar V.; Kong A. N. (2006) Modulation of activator protein-1 (AP-1) and MAPK pathway by flavonoids in human prostate cancer PC3 cells. Arch. Pharm. Res. 29, 633–644. [DOI] [PubMed] [Google Scholar]
- Llorens F.; Garcia L.; Itarte E.; Gomez N. (2002) Apigenin and LY294002 prolong EGF-stimulated ERK1/2 activation in PC12 cells but are unable to induce full differentiation. FEBS Lett. 510, 149–153. [DOI] [PubMed] [Google Scholar]
- Schroeter H.; Boyd C.; Spencer J. P.; Williams R. J.; Cadenas E.; Rice-Evans C. (2002) MAPK signaling in neurodegeneration: influences of flavonoids and of nitric oxide. Neurobiol. Aging 23, 861–880. [DOI] [PubMed] [Google Scholar]
- Impey S.; Smith D. M.; Obrietan K.; Donahue R.; Wade C.; Storm D. R. (1998) Stimulation of cAMP response element (CRE)-mediated transcription during contextual learning. Nat. Neurosci. 1, 595–601. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S.; Tavazoie S. F.; Maloratsky A.; Jacobs K. M.; Harris K. M.; Greenberg M. E. (1997) CREB: a major mediator of neuronal neurotrophin responses. Neuron 19, 1031–1047. [DOI] [PubMed] [Google Scholar]
- Williams C. M.; El Mohsen M. A.; Vauzour D.; Rendeiro C.; Butler L. T.; Ellis J. A.; Whiteman M.; Spencer J. P. (2008) Blueberry-induced changes in spatial working memory correlate with changes in hippocampal CREB phosphorylation and brain-derived neurotrophic factor (BDNF) levels. Free Radical Biol. Med. 45, 295–305. [DOI] [PubMed] [Google Scholar]
- Li Q.; Zhao H. F.; Zhang Z. F.; Liu Z. G.; Pei X. R.; Wang J. B.; Li Y. (2009) Long-term green tea catechin administration prevents spatial learning and memory impairment in senescence-accelerated mouse prone-8 mice by decreasing Abeta1–42 oligomers and upregulating synaptic plasticity-related proteins in the hippocampus. Neuroscience 163, 741–749. [DOI] [PubMed] [Google Scholar]
- Vlahos C. J.; Matter W. F.; Hui K. Y.; Brown R. F. (1994) A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 269, 5241–5248. [PubMed] [Google Scholar]
- Ferriola P. C.; Cody V.; Middleton E. Jr. (1989) Protein kinase C inhibition by plant flavonoids. Kinetic mechanisms and structure-activity relationships. Biochem. Pharmacol. 38, 1617–1624. [DOI] [PubMed] [Google Scholar]
- Walker E. H.; Pacold M. E.; Perisic O.; Stephens L.; Hawkins P. T.; Wymann M. P.; Williams R. L. (2000) Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol. Cell 6, 909–919. [DOI] [PubMed] [Google Scholar]
- Yin Y.; Edelman G. M.; Vanderklish P. W. (2002) The brain-derived neurotrophic factor enhances synthesis of Arc in synaptoneurosomes. Proc. Natl. Acad. Sci. U.S.A. 99, 2368–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltereit R.; Dammermann B.; Wulff P.; Scafidi J.; Staubli U.; Kauselmann G.; Bundman M.; Kuhl D. (2001) Arg3.1/Arc mRNA induction by Ca2+ and cAMP requires protein kinase A and mitogen-activated protein kinase/extracellular regulated kinase activation. J. Neurosci. 21, 5484–5493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris K. M.; Kater S. B. (1994) Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic function. Annu. Rev. Neurosci. 17, 341–371. [DOI] [PubMed] [Google Scholar]
- van Praag H.; Lucero M. J.; Yeo G. W.; Stecker K.; Heivand N.; Zhao C.; Yip E.; Afanador M.; Schroeter H.; Hammerstone J.; Gage F. H. (2007) Plant-derived flavanol (-)epicatechin enhances angiogenesis and retention of spatial memory in mice. J. Neurosci. 27, 5869–5878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reznichenko L.; Amit T.; Youdim M. B.; Mandel S. (2005) Green tea polyphenol (−)-epigallocatechin-3-gallate induces neurorescue of long-term serum-deprived PC12 cells and promotes neurite outgrowth. J. Neurochem. 93, 1157–1167. [DOI] [PubMed] [Google Scholar]
- Leclerc S.; Garnier M.; Hoessel R.; Marko D.; Bibb J. A.; Snyder G. L.; Greengard P.; Biernat J.; Wu Y. Z.; Mandelkow E. M.; Eisenbrand G.; Meijer L. (2001) Indirubins inhibit glycogen synthase kinase-3 beta and CDK5/p25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer’s disease. A property common to most cyclin-dependent kinase inhibitors?. J. Biol. Chem. 276, 251–260. [DOI] [PubMed] [Google Scholar]
- Gong E. J.; Park H. R.; Kim M. E.; Piao S.; Lee E.; Jo D. G.; Chung H. Y.; Ha N. C.; Mattson M. P.; Lee J. (2011) Morin attenuates tau hyperphosphorylation by inhibiting GSK3beta. Neurobiol. Dis. 44, 223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L.; Zhang J.; Qin M. (2013) Protective effect of cyanidin 3-O-glucoside on beta-amyloid peptide-induced cognitive impairment in rats. Neurosci. Lett. 534, 285–288. [DOI] [PubMed] [Google Scholar]