

Background: Nucleolar disruption is involved in cellular stress response and is sufficient for p53 activation. p53 tetramerization is crucial to exert its activity.
Results: The nucleolar protein MYBBP1A enhanced p53 tetramerization by directly interacting with p53.
Conclusion: p53 tetramerization by MYBBP1A is indispensable for activating p53 under nucleolar stress.
Significance: This is the first report to describe the possible mechanism underlying p53 tetramerization in cells under nucleolar stress.
Keywords: Cell Death, Nucleolus, p300, p53, Post-translational Modification, Stress Response
Abstract
Tetramerization of p53 is crucial to exert its biological activity, and nucleolar disruption is sufficient to activate p53. We previously demonstrated that nucleolar stress induces translocation of the nucleolar protein MYBBP1A from the nucleolus to the nucleoplasm and enhances p53 activity. However, whether and how MYBBP1A regulates p53 tetramerization in response to nucleolar stress remain unclear. In this study, we demonstrated that MYBBP1A enhances p53 tetramerization, followed by acetylation under nucleolar stress. We found that MYBBP1A has two regions that directly bind to lysine residues of the p53 C-terminal regulatory domain. MYBBP1A formed a self-assembled complex that provided a molecular platform for p53 tetramerization and enhanced p300-mediated acetylation of the p53 tetramer. Moreover, our results show that MYBBP1A functions to enhance p53 tetramerization that is necessary for p53 activation, followed by cell death with actinomycin D treatment. Thus, we suggest that MYBBP1A plays a pivotal role in the cellular stress response.
Introduction
The tumor suppressor p53 is a critical mediator of the cellular stress response because it maintains genomic integrity and prevents oncogenic transformation (1). The p53 protein regulates many target genes that induce cell cycle arrest or apoptosis (2, 3). The p53 protein level is tightly regulated and maintained at low levels in unstressed cells by its negative regulator HDM2. HDM2 is a ubiquitin ligase that ubiquitinates p53, thereby targeting p53 for proteasome-mediated degradation (4–6). HDM2 is inactivated in response to various stressors such as DNA damage; thus, p53 is rapidly stabilized and activated in cells that sustain various types of stressors (3).
DNA damage activates p53 through post-translational modifications (7, 8) such as phosphorylation, ubiquitination, and acetylation, which play critical roles regulating p53 function (9–15). Phosphorylation of p53 inhibits its binding to HDM2 and represses p53 ubiquitination (16). The p300/CBP protein possesses histone acetyltransferase activity, acetylates p53, acts as a coactivator of p53, and augments p53 transcriptional activity (17–19). Acetylation of p53 occurs at multiple lysine residues in the C-terminal regulatory domain (CRD)4 of p53 (residues 370, 372, 373, 381, 382, and 386) in response to DNA-damaging agents (20–22). Acetylation of these lysine residues stabilizes the p53 protein by direct competition with ubiquitination of the same lysine residues (23). Acetylation of p53-CRD also inhibits p53 sumoylation (24), not necessarily correlated with its binding to DNA. In contrast, acetylation of p53-CRD reportedly plays a role in recruitment of transcriptional coactivators to p53 (25). Based on these observations, acetylation of p53 is considered to play a vital role in p53 activation (26, 27).
It has been recently demonstrated that p53 tetramerization is essential for acetylation of its C-terminal lysine (28). Under unstressed conditions, p53 exists in a monomeric state (29–31) or in a dimeric state (or both) (32). The protein functions most efficiently as a tetramer because tetramers have a higher binding affinity for DNA (33–36). Thus, tetramerization-deficient p53 mutants exhibit much lower affinities for DNA than the wild-type protein (p53-WT) (37). Tetramerization of p53 occurs by direct interaction with the 325–356 spanning residues (tetramerization domain (TET)) (32, 38). Moreover, the TET mutant (p53-mTET), which has a dimer-dimer interface disrupted by replacement of Leu-344 with alanine, only forms the dimer (39).
In addition, tetramerization regulates p53 post-translational modifications (40). p300 interacts and promotes acetylation of p53-WT but does not interact and promote p53 mutant proteins, which are unable to form tetramers. Activation of p53 in response to DNA damage begins with tetramerization of p53, which provides appropriate binding sites for p300 and leads to binding of p300 and subsequent acetylation of p53 C-terminal lysine residues. The acetylation, in turn, further tightens the p300-p53 complex, stabilizes the p53 protein, and binds to the promoter sequence to facilitate recruitment of coactivators and transactivation of target genes (28). Therefore, tetramerization of p53 is vital to its function and plays a pivotal role in regulating its activity.
DNA damage also induces repression of ribosomal RNA transcription by RNA polymerase I, resulting in disruption of the nucleolar structure (41, 42). Low concentrations of actinomycin D (ActD) specifically inhibit RNA polymerase I-driven transcription but do not affect RNA polymerase II-driven transcription (43, 44). Therefore, ActD treatment causes nucleolar disruption (45). Recent studies on the cellular response to nucleolar stress have demonstrated that several nucleolar proteins are involved in activating p53. Ribosomal proteins such as RPS7, RPL5, RPL11, and RPL23 directly bind to HDM2 and inhibit HDM2-mediated p53 ubiquitination (45–51). Similarly, the nucleolar proteins NPM, NCL, NS, and ARF directly bind to HDM2 and inhibit degradation of p53 (52–56).
We reported previously that nucleolar disruption induces acetylation and accumulation of p53 without phosphorylation. The nucleolar protein Myb-binding protein 1A (MYBBP1A) binds to p53-CRD lysine residues (57) and facilitates p53 acetylation to enhance p53-mediated transcription by enhancing p53-p300 interactions in a manner that differs from that of other nucleolar proteins (58). However, the mechanism by which MYBBP1A enhances p53 acetylation and induces p53 activity remains unclear.
In this study, we demonstrated that MYBBP1A has two p53-binding regions and that it enhances p53 tetramerization by dimerizing itself, thereby promoting the interaction between p53 and p300 and acetylation of p53. Thus, we suggest that MYBBP1A plays a pivotal role in the cellular stress response.
EXPERIMENTAL PROCEDURES
Cell Culture and Treatments
MCF-7 human breast cancer cells and H1299 p53-deficient human lung cancer cells were maintained in DMEM (Sigma). LNCaP human prostate cancer cells were maintained in RPMI 1640 (Nacalai Tesque, Kyoto, Japan). All media were supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin solution (Nacalai Tesque). Cells were maintained at 37 °C in an atmosphere containing 5% CO2 and 100% humidity. To induce nucleolar stress, cells were exposed to ActD (5 nm), UV irradiation (25 J/m2), and adriamycin (0.5 mg/ml). Cell viability after ActD treatment was determined by the trypan blue dye exclusion test.
siRNA and Plasmid DNA Transfection
Cells at 30–50% confluency were siRNA-transfected using Lipofectamine RNAi MAX (Invitrogen), according to the manufacturer's protocol. All siRNAs were purchased from Invitrogen. The siRNA duplex was siMYBBP1A, 5′-UCUUUCAGUCAGGUCGGCUGGUGAA-3′. Stealth RNAi negative control medium or high GC was used as the negative control. Protein and RNA were extracted 48 h after siRNA transfection. Cells at 70–80% confluency were transfected with plasmid DNA using Lipofectamine LTX (Invitrogen) according to the manufacturer's protocol. Protein was extracted 24 h after transfection of plasmid DNA.
Expression Vectors and Antibodies
cDNAs encoding full-length and the indicated mutants of p53 and MYBBP1A were amplified by PCR and subcloned into the pcDNA3 plasmid (Invitrogen) containing sequences encoding FLAG, FLAG-HA, HA, or Myc sequences. β-Actin (Sigma) and anti-human-p53 (DO-1; Santa Cruz Biotechnology, Santa Cruz, CA) monoclonal antibodies and rabbit anti-p53-K382Ac (Cell Signaling Technology, Danvers, MA) polyclonal antibodies were used according to the manufacturer's instructions. Rabbit anti-human MYBBP1A antibody was raised against a synthetic peptide corresponding to amino acids 1265–1328 of human MYBBP1A.
Electrophoretic Mobility Shift Assay
EMSA was performed using the LightShiftTM EMSA optimization and control kit and chemoluminescent nucleic acid detection modules (Pierce). Binding reactions containing 1 μl of p53 and/or MYBBP1A-overexpressing cell lysates, 10 mm Tris, 50 mm KCl, 1 mm DTT, 2.5% glycerol, 5 mm MgCl2, 0.05% Nonidet P-40, and the double-stranded oligonucleotide probe were incubated in a final volume of 10 μl for 20 min at room temperature. After the incubation, the samples were separated on 5% SDS-PAGE gel, transferred to nylon membranes by electroblotting, and UV cross-linked. DNA-protein complexes were visualized with streptavidin-horseradish peroxidase, followed by chemiluminescence detection. According to a published procedure (59), EMSA was performed using p21 dsDNA (5′-CGCGAACATGTTCGAACATGTTCGCG-3′), which is similar to the p21-5′ p53-binding site, as described under “Experimental Procedures” for the ChIP assay.
Real Time Quantitative PCR (RT-qPCR)
RT-qPCR was performed as described previously (60). Cells were homogenized in 1 ml of Isogen (Nippon Gene, Toyama, Japan), and total RNA was extracted according to the manufacturer's instructions. cDNA was synthesized from total RNA using ReverTra Ace reverse transcriptase (Toyobo, Osaka, Japan) and oligo(dT) primers. Real time PCR was used to amplify fragments representing the indicated mRNAs. The primer sequences used were as follows: GAPDH forward primer, 5′-GTATGACTCCACTCACGGCAAA-3′; GAPDH reverse primer, 5′-GGTCTCGCTCCTGGAAGATG-3′; p21 forward primer, 5′-GGAGACTCTCAGGGTCGAAA-3′; p21 reverse primer, 5′-TTAGGGCTTCCTCTTGGAGA-3′; HDM2 forward prime,: 5′-AGCAAACTGGTGCTCAAGG-3′; HDM2 reverse primer: 5′-CTGTTGCAATGTGATGGAAGG-3′; PUMA forward primer, 5′-GGGCCCAGACTGTGAATCCT-3′; and PUMA reverse primer, 5′-ACGTGCTCTCTCTAAACCTATGCA-3′.
GST Pulldown Assay
cDNAs encoding full-length human p53 or MYBBP1A and its deletion mutant derivatives were cloned into pGEX-4T-1 (Amersham Biosciences). GST fusion proteins were expressed in BL-21 cells following induction with isopropyl β-d-thiogalactopyranoside and purified with glutathione-Sepharose 4B beads (Amersham Biosciences). In vitro translated MYBBP1A was synthesized using an in vitro transcription/translation-coupled reticulocyte lysate system (Promega, Madison, WI). Binding was performed in TNE buffer (150 mm NaCl, 0.5% Nonidet P-40, 50 mm Tris-HCl, pH 8.0, 5 mm EDTA) for 30 min under rotation at 4 °C, and the beads were washed five times with TNE buffer. Beads were boiled in loading buffer for 5 min, and the supernatants were loaded onto SDS-polyacrylamide gels, followed by immunoblotting.
Coimmunoprecipitation (Co-IP) and Immunoblotting
Cells were lysed in TNE buffer supplemented with 1 m phenylmethylsulfonyl fluoride and 1 g/ml aprotinin. Extracted proteins were immunoprecipitated with antibody-coated protein G-Sepharose (Amersham Biosciences) beads. Bound proteins were separated by SDS-PAGE, transferred to polyvinylidene difluoride membranes (Millipore, Milford, MA), and detected with appropriate primary antibodies and horseradish peroxidase-conjugated secondary antibodies. Specific proteins were visualized using an enhanced chemiluminescence immunoblot detection system (Amersham Biosciences).
Protein Cross-linking Assay
Cells were transfected with the indicated siRNA or plasmids and lysed in TNE buffer after 6 h of ActD treatment. Glutaraldehyde (GA) was added to the lysates at the indicated concentrations. After incubating the lysates on ice for 20 min, the GA reactions were stopped by adding 2× loading buffer, and the samples were heated at 100 °C for 5 min and resolved by SDS-PAGE. Western blot analysis was performed with anti-p53 antibody (DO-1).
Gel Filtration Chromatography
Cell lysates were fractionated with a fast protein liquid chromatography protein purification system on a HiPrep 16/60 Sephacry S-300 HR (GE Healthcare). The column was equilibrated with Tris buffer (20 mm Tris, pH 8.0, 150 mm NaCl, 0.1% (v/v) Nonidet P-40, 2 mm EDTA), and lysates (10 mg) were applied to and eluted from the column with the same buffer. The flow rate was 0.5 ml/min, and 1.5-ml fractions were collected. The column was calibrated with Sigma gel filtration standards, including thyroglobulin (669 kDa), apoferritin (443 kDa), γ-amylase (200 kDa), alcohol dehydrogenase (150 kDa), albumin (66 kDa), and carbonic anhydrase (29 kDa).
ChIP and RT-qPCR Detection
The ChIP assay was performed according to a published procedure (61). The distal p21 site (p21-5′) between the two p53-binding sites within the p21 promoter region (62) was detected by RT-qPCR using the following primers: p21–5′ forward primer, 5′-GTGGCTCTGATTGGCTTTCTG-3′; and p21–5′ reverse primer, 5′-CTGAAAACAGGCAGCCCAAG-3′.
Duolink in Situ Proximity Ligation Assay (PLA)
The Duolink in situ PLA was performed according to the manufacturer's protocol (Olink Bioscience, San Francisco, CA). In brief, H1299 cells transfected with the indicated plasmids and grown on chamber slides were rinsed three times with PBS (140 mm NaCl, 2.7 mm KCl, 1.5 mm KH2PO4, and 8.1 mm Na2HPO4) and fixed in 4% formaldehyde in PBS for 10 min. After rinsing three times with PBS, the cells were permeabilized in 0.5% Triton X-100 in 20 mm HEPES (pH 7.5) with 150 mm KCl and blocked with TBS-T buffer containing 3% BSA for 1 h at 37 °C. After blocking, the cells were incubated for 2 h at 37 °C with mouse anti-p53 and rabbit anti-FLAG antibodies in PBS containing 1% BSA. Cells were washed three times with PBS and incubated with secondary rabbit PLUS and mouse MINUS antibodies for 1 h at 37 °C in the dark. Cells were washed three times in PBS before detecting the probe using the in situ PLA detection kit (Olink Bioscience). Duolink and DAPI signals were detected using an LSM-700 microscope (Carl Zeiss, Oberkochen, Germany).
RESULTS
MYBBP1A Contains Two p53-binding Sites
To determine the region responsible for p53 binding to MYBBP1A, we performed the GST pulldown assay using full-length wild-type MYBBP1A (MYBBP1A-WT) or the indicated MYBBP1A deletion mutants (Fig. 1A). The GST pulldown assays indicated that both regions from amino acids (a.a.) 643 to 1150 and from a.a. 1151 to 1328 of MYBBP1A interacted with p53. To limit the MYBBP1A-responsive region for p53 binding, we generated four additional MYBBP1A deletion mutants (a.a. 1–1150, 643–1150, 775–1150, and 1151–1271). The GST pulldown assay revealed that MYBBP1A had bound to p53 via the middle region (a.a. 643–1150) and the basic repeat-rich region (a.a. 1151–1271) within the C-terminal region of MYBBP1A (Fig. 1A). MYBBP1A 1–1150 mutant was referred hereafter as dominant-negative mutant of MYBBP1A (MYBBP1A-DN) (the details are provided in the text for Fig. 8, C and D).
FIGURE 1.

MYBBP1A contains two p53-binding domains. A, domain structures of MYBBP1A-WT and deletion mutants of MYBBP1A (left panel). White squares, dark gray regions, and black regions show the leucine zipper-like motif, the acidic region, and the basic repeat, respectively. a.a. 1–1150, 643–1150, and 1151–1271 MYBBP1A regions directly bound to p53 (right). In vitro translated FLAG and HA (F/H)-tagged MYBBP1A-WT or its deletion mutant were incubated with GST-fused p53-WT (GST-p53) or GST proteins (GST). Bound proteins were analyzed by immunoblotting. Input, 25% of the in vitro translated MYBBP1A used in the GST pulldown assay. The deletion mutant (a.a. 1–1150) is referred to as MYBBP1A-DN (the details are provided in the text for Fig. 8, C and D). B, p53 interacted with MYBBP1A via a.a. 643–1150 or 1151–1328 regions of MYBBP1A in cells. H1299 cells were transfected with a combination of expression plasmids encoding F/H-MYBBP1A-WT, its various deletion mutants, and p53-WT, as indicated. The cells were treated with ActD for 6 h at 24 h after transfection. MYBBP1A was immunoprecipitated from the cell lysates using anti-FLAG antibodies, and the p53 association was detected by immunoblotting using anti-p53 antibody. C, MYBBP1A interacted with p53 via two independent regions in intact cells. H1299 cells transfected with the indicated plasmids were treated with ActD for 6 h. The cells were fixed and subjected to the Duolink in situ PLA using anti-FLAG and anti-p53 antibodies. The Duolink signal is shown in red. The nuclei were stained using DAPI and are shown in blue.
FIGURE 8.

Dimerization of MYBBP1A is necessary for p53 tetramerization. A, MYBBP1A interacted with MYBBP1A in cells. H1299 cells were transfected with a combination of expression plasmids encoding F/H-MYBBP1A-WT and Myc-MYBBP1A-WT, as indicated. The cells were treated with ActD for 6 h at 24 h post-transfection. MYBBP1A was immunoprecipitated from the cell lysates using anti-FLAG antibodies and subjected to immunoblotting using anti-Myc or anti-FLAG antibodies. B, MYBBP1A directly bound to MYBBP1A via a.a. 1151–1328 MYBBP1A region. In panel a, domain structures of full-length WT and deletion mutants of MYBBP1A are shown. White squares, dark gray regions, and black regions show the leucine zipper-like motif, the acidic region, and the basic repeat, respectively. In panel b, in vitro translated F/H-MYBBP1A was incubated with the GST-fused truncated MYBBP1A proteins, as shown in panel a. Bound proteins were then analyzed by immunoblotting. The deletion mutant (a.a. 1–1150) was referred to as MYBBP1A-DN. C, MYBBP1A-DN did not form a dimer with MYBBP1A in the cells. H1299 cells were transfected with a combination of expression plasmids encoding F/H-MYBBP1A-WT, F/H-MYBBP1A-DN, and Myc-MYBBP1A-WT, as indicated. The cells were treated with ActD for 6 h at 24 h post-transfection. MYBBP1A was immunoprecipitated from the cell lysates using anti-FLAG antibody and subjected to immunoblotting using anti-Myc or FLAG antibodies. D, MYBBP1A-DN inhibited the formation of p53 tetramers. In panel a, MCF-7 cells were transfected with the expression plasmid encoding EGFP (OE Control) or Myc-MYBBP1A (OE-MYBBP1A-DN or OE-MYBBP1A-WT). To validate the efficiency of MYBBP1A overexpression, MYBBP1A protein abundance was determined by immunoblotting with anti-Myc and anti-MYBBP1A antibodies. Note: Anti-MYBBP1A antibody does not recognize MYBBP1A-DN because MYBBP1A-DN is lacking the anti-MYBBP1A antibody epitope. In panel b, MCF-7 cells were transfected with the expression plasmid encoding EGFP (OE-Control), MYBBP1A-DN (OE-MYBBP1A-DN), or MYBBP1A-WT (OE-MYBBP1A-WT). The cells were treated with ActD for 6 h at 24 h post-transfection and were subjected to the cross-linking assay in GA at final concentrations of 0, 0.005, 0.01, or 0.02%. In panel c, MCF-7 cells were transfected with a combination of expression plasmids encoding MYBBP1A-WT and MYBBP1A-DN, as indicated. MCF-7 cells were treated with ActD for 6 h at 24 h post-transfection and were subjected to the cross-linking assay in GA at a final concentration of 0.04%.
We subsequently tested the interaction of these domains with p53 in cells. We introduced p53 expression plasmids into H1299 cells with or without expression plasmids for the full-length or deletion mutants of MYBBP1A. Co-IP assays showed the association between p53 and MYBBP1A-WT in H1299 cells under ActD treatment (Fig. 1B, lane 2). Interactions of p53 with MYBBP1A deletion mutants were also detectable (Fig. 1B, lanes 3 and 4). To directly show the interaction between MYBBP1A and p53 in intact cells, we performed immunofluorescent staining using the Duolink in situ PLA, which enables visualization of the MYBBP1A-p53 interaction. Similar to MYBBP1A-WT, the N-terminal (a.a. 1–1150) and C-terminal (a.a. 1151–1328) regions of MYBBP1A, each of which contains a single p53-bindig site, bound to p53-mTET (Fig. 1C).
Taken together, these data indicate that MYBBP1A has two p53-binding sites. However, it remains to be determined whether these two regions of MYBBP1A simultaneously and cooperatively interact with the p53 dimer.
MYBBP1A Binds the p53 Dimer
We performed Co-IP assays using expression plasmids for FLAG and HA-tagged p53 (F/H-p53) and Myc-tagged p53 (Myc-p53) and found that Myc-p53 bound to F/H-p53 (Fig. 2A, lane 2). Following this, we performed Co-IP assays in the presence of Myc-tagged MYBBP1A-WT (Myc-MYBBP1A-WT) and found that MYBBP1A significantly enhanced the binding between F/H-p53 and Myc-p53 (Fig. 2A, lane 2 versus lane 4, top panel, and p < 0.01, Student's t test, n = 3, bottom panel).
FIGURE 2.

MYBBP1A binds to the p53 dimer. A, H1299 cells transfected with the indicated plasmids were treated with ActD for 6 h. p53 was immunoprecipitated from cell lysates using anti-FLAG antibodies and detected by immunoblotting using anti-p53 antibodies (top panel). Immunoprecipitated signal intensity of Myc-p53-WT in the absence or presence of Myc-MYBBP1A-WT was independently quantified three times, and the intensity of each was normalized by that of total Myc-p53-WT. The quantitative data were compared using the Student's t test (**, p < 0.01, bottom panel). B, MYBBP1A was immunoprecipitated from cell lysates using anti-FLAG (first) antibody and were eluted with FLAG peptide and immunoprecipitated again with anti-Myc (second) antibody. MYBBP1A and p53 were detected in immunoblotting precipitates.
To more clearly address whether the MYBBP1A complex included two p53 molecules, we further performed sequential Co-IP assays. We performed transfection using expression plasmids for F/H-MYBBP1A, Myc-p53, and HA-tagged p53 (HA-p53) and then sequentially performed immunoprecipitation with anti-FLAG (first) and anti-Myc (second) antibodies. Immunoblotting demonstrated the presence of HA-p53 in the final immunoprecipitant by coexpression with MYBBP1A-WT (Fig. 2B, lane 4). In contrast, neither the MYBBP1A-DN (a.a. 1–1150) nor the MYBBP1A 1–642, 1151–1328 mutant (truncated from a.a. 643 to 1150), each of which possessed one p53-binding domain, coexpressing samples contained HA-p53 in the final immunoprecipitant (Fig. 2B, lanes 8 and 12). Taken together, these results suggest that MYBBP1A binds to the p53 dimer via two distinct MYBBP1A sites.
MYBBP1A Dissociates from p53 in a p53-CRD Acetylation-dependent Manner
We previously reported that MYBBP1A-WT did not bind to the p53–6KA mutant, which bears simultaneous lysine substitutions to alanines at positions 370, 372, 373, 381, 382, and 386 within the p53-CDR region (57). Thus, we investigated whether these lysine residues within p53-CRD regions were critical in the p53-MYBBP1A interaction via the middle (a.a. 643–1150) and C-terminal (a.a. 1151–1328) regions of MYBBP1A. We performed the GST pulldown assay and found that similar to MYBBP1A-WT, neither the MYBBP1A 643–1150 nor 1151–1328 mutant was bound to the p53–6KA mutant (Fig. 3A). Furthermore, we performed Co-IP assay using the p53–6KA mutant and confirmed that MYBBP1A-WT and MYBBP1A deletion mutants did not interact with the p53–6KA mutant in H1299 cells (Fig. 3B). These data indicate that MYBBP1A directly interacts with lysine residues within the p53-CRD region via the middle (a.a. 643–1150) and C-terminal (a.a. 1151–1328) regions of MYBBP1A.
FIGURE 3.
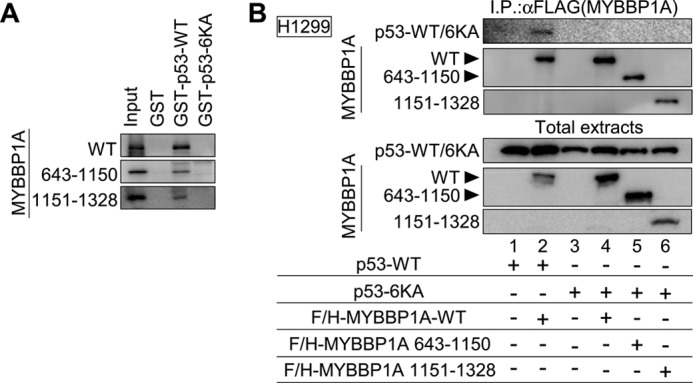
MYBBP1A dissociates from p53 in a p53-CRD acetylation-dependent manner. A, a.a. 643–1150 and 1151–1328 regions of MYBBP1A did not directly bind to the p53–6KA mutant. In vitro translated F/H-MYBBP1A-WT or its deletion mutant was incubated with GST-fused p53-WT (GST-p53-WT), p53–6KA (GST-p53–6KA), or with GST. Bound proteins were analyzed as in Fig. 1A. B, p53 interacted with MYBBP1A via its lysine residues in the CRD region in cells. H1299 cells were transfected with a combination of expression plasmids encoding F/H-MYBBP1A-WT, its various deletion mutants, and p53-WT, as indicated. The cells were treated with ActD for 6 h at 24 h post-transfection. MYBBP1A was immunoprecipitated from the cell lysates using anti-FLAG antibody and analyzed as in Fig. 1B.
Our previous report also revealed that the p53–6KQ mutant (K370Q/K372Q/K373Q/K381Q/K382Q/K386Q), known as an acetylation mimicking mutation, does not bind to MYBBP1A (57). In contrast, the p53–6KR mutant (K370R/K372R/K373R/K381R/K382R/K386R), which is generally used as an acetylation-deficient missense mutant, binds to MYBBP1A more strongly than p53-WT (57). Taken together, we conclude that MYBBP1A dissociates from p53 in an acetylation-dependent manner with the lysine residues within p53-CRD.
MYBBP1A Promotes Tetramerization of p5
p53 exerts its activity by oligomerization (33–37). MYBBP1A enhances p53 activity (58), and we found that MYBBP1A has two p53-binding domains. These findings suggest that MYBBP1A may regulate p53 oligomerization. We next performed gel filtration assay to examine p53 oligomerization. The gel filtration assay revealed that p53 was eluted in higher molecular weight fractions (fractions 1–9, 200–669 kDa) in the presence of ectopically expressed MYBBP1A compared with that eluted in the absence of MYBBP1A (Fig. 4, (OE-Control) versus (OE-MYBBP1A-WT)). In contrast, we observed that the amount of p53 eluted in these fractions decreased in the presence of MYBBP1A-DN (a.a. 1–1150) compared with that eluted in the presence of MYBBP1A-WT (Fig. 4, (OE-MYBBP1A-WT) versus (OE-MYBBP1A-DN)). These results suggest that MYBBP1A affects p53 oligomerization. However, we could not exclude the effects of p53 interactants other than MYBBP1A because these assays were performed using cell lysates in which a large number of p53 interactants existed.
FIGURE 4.
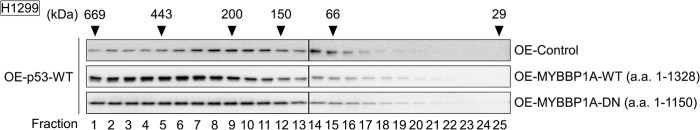
MYBBP1A enhances p53 oligomerization. Expression plasmids encoding wild-type p53 (OE-p53-WT) were transfected with that encoding EGFP (OE-Control), MYBBP1A-WT (a.a. 1–1138) (OE-MYBBP1A-WT), or MYBBP1A-DN (a.a. 1–1150) (OE-MYBBP1A-DN) into H1299 cells. The cells were treated with ActD for 6 h at 24 h post-transfection. The lysates were subjected to size exclusion chromatography. Fractions were analyzed by immunoblotting for the presence of p53. The elution positions of the molecular size markers are shown.
Next, we performed a protein cross-linking assay. Although siRNA knockdown of MYBBP1A hardly affected p53 dimerization under ActD treatment and other types of DNA damage, the p53 tetramer substantially decreased by knocking down MYBBP1A in MCF-7 (Fig. 5A) and LNCaP cells (data not shown). Similar results were also obtained by using the distinct siMYBBP1A (5′-GGUCCGAGAUGAAAUAUGCCCUGAA-3′) (data not shown).
FIGURE 5.

MYBBP1A promotes p53 tetramerization. A, knockdown of MYBBP1A reduced the formation of p53 tetramers. MCF-7 cells were transfected with either control siRNA (siControl) or MYBBP1A siRNA (siMYBBP1A). To validate the efficiency of MYBBP1A knockdown, protein abundance was determined by immunoblotting with anti-MYBBP1A antibody in panel a. At 48 h post-transfection, the cells were treated with ActD for 6 h in panel b, with UV irradiation in panel c, and with adriamycin for 6 h in panel d. The lysates were subjected to a protein cross-linking assay in GA at final concentrations of 0, 0.005, 0.01, or 0.02%, followed by immunoblotting. B, overexpression of MYBBP1A enhanced the formation of p53 tetramers in wild-type p53-expressing H1299 cells. Expression plasmids encoding p53-WT (OE-p53-WT), p53–6KA (OE-p53–6KA), or p53-mTET (OE-p53-mTET) were transfected into H1299 cells with that encoding EGFP (OE-Control) or F/H-MYBBP1A-WT (OE-MYBBP1A). The cells were treated with ActD for 6 h at 24 h post-transfection. To validate the efficiency of overexpression of p53-WT or p53–6KA with or without MYBBP1A, the protein abundance of p53 and MYBBP1A was determined by immunoblotting with anti-p53 or anti-MYBBP1A antibodies in panels a. The lysates were subjected to a cross-linking assay in GA at final concentrations of 0, 0.005, 0.01, or 0.02% in panels b–d. C, dose effect of MYBBP1A on p53 tetramerization. The H1299 cell lysate of OE-MYBBP1A was mixed with that of siMYBBP1A#1 at the indicated ratio and was incubated for 1 h on ice. Following this, the mixed lysates were cross-linked by GA at a final concentration of 0.04%.
On the other hand, we transfected expression plasmids encoding p53 and/or MYBBP1A into H1299 cells (Fig. 5B, panel a). Overexpression of MYBBP1A enhanced p53 tetramerization in WT p53-expressing H1299 cells under ActD treatment (Fig. 5B, panel b). In contrast, overexpression of MYBBP1A did not influence p53 tetramerization in p53 6KA mutant-expressing H1299 cells (Fig. 5B, panel c). Moreover, p53-mTET did not form tetramers even when MYBBP1A was overexpressed (Fig. 5B, panel d), although it interacted with MYBBP1A (Fig. 6).
FIGURE 6.
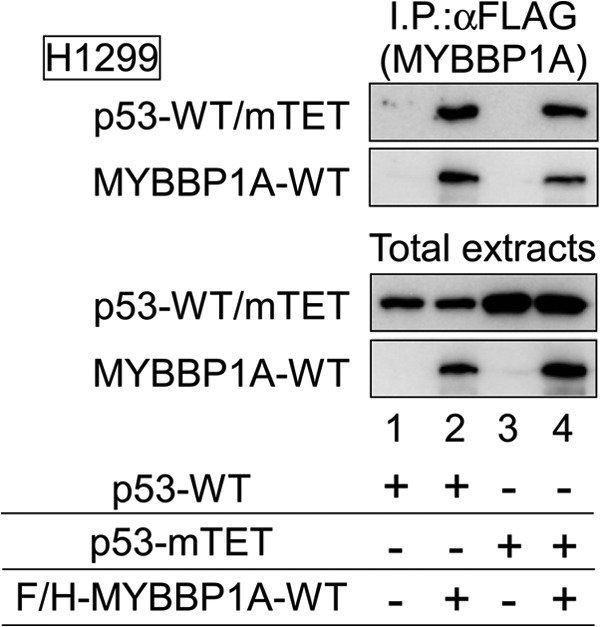
p53-mTET interacts with MYBBP1A. H1299 cells were transfected with a combination of expression plasmids encoding F/H-MYBBP1A-WT, p53-WT, and p53-mTET, as indicated. The cells were treated with ActD for 6 h at 24 h post-transfection. MYBBP1A was immunoprecipitated from the cell lysates using anti-FLAG antibody and subjected to immunoblotting using anti-FLAG or anti-p53 antibodies.
To perform a more convincing protein cross-linking assay, we titrated MYBBP1A to investigate the dose effect of MYBBP1A on p53 tetramerization. MYBBP1A was titrated by mixing two types of lysates: one prepared from cells in which MYBBP1A was overexpressed and the other prepared from cells in which MYBBP1A was knocked down. As expected, we found that p53 tetramerization was enhanced in a MYBBP1A-dependent manner (Fig. 5C). Collectively, these findings indicate that p53 tetramerization is enhanced by MYBBP1A under nucleolar stress. These findings also indicate that p53 forms a dimer independent of MYBBP1A, in good agreement with a previous study suggesting that major p53 itself can autonomously form a dimer (32).
To further confirm the effects of MYBBP1A on p53 tetramerization because p53 exerts its DNA binding affinity under conditions in which it forms a tetramer (32, 38), we next performed EMSA using the lysates extracted from p53-WT-overexpressing H1299 cells. p53-WT interacted with the p21 promoter DNA probe (Fig. 7, lane 3), which was further potentiated by MYBBP1A co-overexpression (Fig. 7, lane 4). Moreover, the mobility of these bands was further retarded by anti-p53 antibody (Fig. 7, lanes 7 and 8). In contrast, p53-mTET did not associate with the DNA probe (Fig. 7, lanes 5 and 6). Therefore, these data indicate that MYBBP1A promotes not only p53 tetramerization but also binding to promoter DNA.
FIGURE 7.
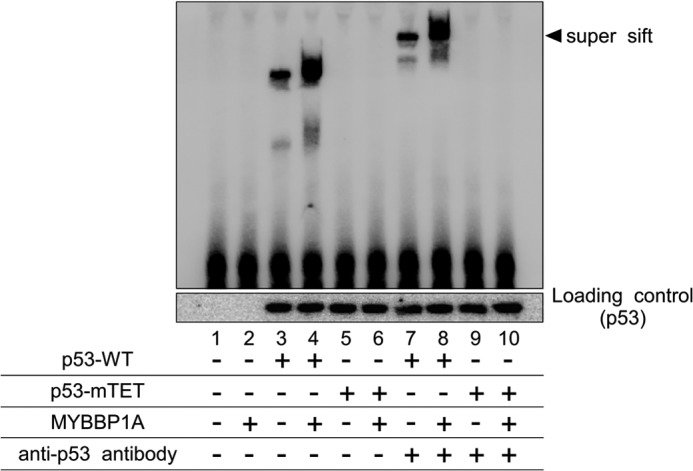
MYBBP1A enhances tetramerization-dependent p53 promoter binding. H1299 cells were transfected with the indicated plasmids and treated with ActD for 6 h. Nuclear extracts were subjected to an EMSA.
MYBBP1A Forms a Dimer via the C-terminal Region, Which Is Necessary for p53 Tetramerization
It has been previously reported that MYBBP1A forms a dimer (63). Therefore, we investigated whether MYBBP1A could bind to two p53 molecules and whether MYBBP1A dimerization enhances p53 tetramerization. First, we examined the MYBBP1A-MYBBP1A interaction by Co-IP assays and observed that Myc-MYBBP1A coimmunoprecipitated with F/H-MYBBP1A (Fig. 8A). This result indicates that MYBBP1A interacts with MYBBP1A. To further investigate the mode of the MYBBP1A-MYBBP1A interaction, we generated MYBBP1A deletion mutants and tested the interaction in the GST pulldown assay (Fig. 8B, panel a). The GST pulldown assay determined that the region from a.a. 1151 to 1328 of MYBBP1A interacted with MYBBP1A-WT. However, the regions from a.a. 1 to 642 and from a.a. 643 to 1150 of MYBBP1A did not interact with MYBBP1A-WT (Fig. 8B, panel b, upper panel). Moreover, the region from a.a. 1151 to 1328 of MYBBP1A associated with the region itself (Fig. 8B, panel b, lower panel). Taken together, these results show that MYBBP1A interacts with itself via the region from a.a. 1151 to 1328.
To test whether MYBBP1A forms a self-assembled complex via a.a. 1151–1328 in intact cells, we transfected expression plasmids for MYBBP1A-WT or MYBBP1A-DN (a.a. 1–1150) into H1299 cells. Co-IP assays demonstrated that MYBBP1A-DN did not interact with MYBBP1A-WT (Fig. 8C). These data indicate that the region from a.a. 1151 to 1328 is indispensable for MYBBP1A dimerization in cells.
Although MYBBP1A-DN (a.a. 1–1150), which contains a single p53-binding site (Fig. 1), can only interact with the p53 monomer (Fig. 2), the mutant did not form a dimer in MYBBP1A-WT cells (Fig. 8C). Thus, we expected that MYBBP1A-DN would act as a dominant-negative MYBBP1A mutant by inhibiting p53 tetramerization. To examine this possibility, we performed a protein cross-linking assay using MYBBP1A-DN-overexpressing cells (Fig. 8D, panel a). Overexpression of MYBBP1A-DN significantly reduced tetramerized p53 in MCF-7 (Fig. 8D, panel b) and LNCaP cells (data not shown). We also found that MYBBP1A-DN inhibited the effect of exogenous MYBBP1A-WT on p53 tetramerization (Fig. 8D, panel c). These data indicate that MYBBP1A dimerization and/or interactions with two p53 molecules are necessary for p53 tetramerization.
MYBBP1A Enhances the p53-p300 Interaction and p53 Acetylation by Regulating p53 Tetramerization
It has been previously demonstrated that tetramerization of p53 is necessary for p300-mediated p53 acetylation (28). Consistent with these findings, our results indicate that acetylation of p53-mTET was not enhanced by overexpressing p300 (Fig. 9A). Therefore, we next examined whether overexpression of MYBBP1A-DN, which inhibited p53 tetramerization (Fig. 8), affected p53 acetylation. Overexpression of MYBBP1A-DN reduced protein and Lys-382 acetylation (K382Ac) levels of p53 compared with those of the control (Fig. 9B, lane 3 versus lane 4). We further increased the amount of total extract (Fig. 9B, lanes 4–7) and found that MYBBP1A-DN significantly reduced K382Ac levels of p53 rather than the protein levels of p53 (Fig. 9B, lane 3 versus lane 7). These results indicate that p53 is not acetylated without tetramerization promoted by MYBBP1A. Next, a combination of p53 and p300 expression plasmids was introduced into H1299 cells in the presence or absence of the MYBBP1A-DN expression plasmid to test the effects of MYBBP1A on the interaction between p53 and p300. Co-IP assays showed that the association between p53 and p300 in the presence of ActD significantly decreased by overexpressing MYBBP1A-DN (Fig. 9C).
FIGURE 9.

MYBBP1A enhances the p53-p300 interaction and p53 acetylation by regulating p53 tetramerization. A, p53-mTET was not acetylated by overexpressing p300. The expression plasmids encoding p53-WT or p53-mTET were transfected into H1299 cells with or without p300, 24 h before treatment with ActD for 6 h. Cell lysates were analyzed by immunoblotting using anti-p53 or site-specifically acetylated p53 antibodies. B, overexpression of MYBBP1A-DN decreased Lys-382 acetylation (K382Ac) levels of p53. MCF-7 cells were transfected with the expression plasmid encoding EGFP (OE-Control) or MYBBP1A-DN (OE-MYBBP1A-DN), 24 h before treatment with ActD for 6 h. Cell lysates were analyzed by immunoblotting using anti-p53 or site-specifically acetylated p53 antibodies. ×1, ×2, ×3, and ×4 indicate the ratios of total extract volumes applied to SDS-PAGE. C, overexpression of MYBBP1A-DN decreased binding of p300 to p53. H1299 cells were transfected with a combination of expression plasmids encoding FLAG-p300, Myc-MYBBP1A-DN, and p53-WT, as indicated. The cells were treated with ActD for 6 h at 24 h after transfection. FLAG-p300 was immunoprecipitated using anti-FLAG antibody, and the immunoprecipitates were eluted using FLAG peptides and analyzed by immunoblotting using the indicated antibodies. D, overexpression of MYBBP1A-DN reduced the recruitment of p53 and p300 to the p21 promoter in ActD-treated cells. MCF-7 cells were transfected with the expression plasmid encoding EGFP (OE-Control) or FH-MYBBP1A-DN (OE-MYBBP1A-DN) for 24 h before treatment with ActD for 6 h. The CHIP assay was performed using normal rabbit IgG, anti-p53, and anti-p300 antibodies. The p53-binding region of the p21 promoter was amplified and analyzed by RT-qPCR. The values are given as the means ± standard deviation for triplicate assays.
Next, we tested the effects of MYBBP1A-DN overexpression on recruitment of p53 and p300 to the promoter region of p21 using the ChIP assay (Fig. 9D). Recruitment of p53 and p300 were abrogated in the presence of MYBBP1A-DN as compared with those in the presence of MYBBP1A-WT (lanes 2 versus 6 for p53 and lanes 3 versus 7 for p300). By comparison, MYBBP1A itself did not bind to the promoter region (lanes 4 and 8). Consistent with these results, we previously revealed that the recruitment of p53 was abrogated by knocking down MYBBP1A or by overexpressing MYBBP1A-DN. In contrast, overexpression of MYBBP1A-WT increased the recruitment of p53 to the p21 promoter, whereas MYBBP1A was not recruited to the promoter (57).
Taken together, these data indicate that MYBBP1A enhances the tetramerization, p300-mediated acetylation, and recruitment of p53 to the target gene promoters. We conclude that MYBBP1A itself is dissociated from p53 by p300-mediated acetylation of p53 and that MYBBP1A is absent from DNA-bound p53 on the promoter.
MYBBP1A Regulates p53 Activation by Enhancing Tetramerization
Our results indicate that MYBBP1A promotes p53 tetramerization (Fig. 5). A previous report has suggested that p53 tetramerization is indispensable for the transactivating capabilities of p53 (64). Therefore, we examined whether promotion of tetramerization by MYBBP1A contributes to p53 function under cell stress.
The effects of MYBBP1A on the induction levels of p53 target gene products were examined. mRNA and protein levels of p21, HDM2, and PUMA in MCF-7 (Fig. 10A) and LNCaP (data not shown) cells substantially decreased in cells in which MYBBP1A was knocked down. Similarly, overexpression of MYBBP1A-DN reduced mRNA and protein levels.
FIGURE 10.

MYBBP1A regulates p53 activation by enhancing tetramerization. A, overexpression of MYBBP1A-DN and knockdown of MYBBP1A reduced mRNA and protein levels of p53 target genes, p21, HDM2, and PUMA. MCF-7 cells were transfected with indicated siRNAs or plasmids for 48 or 24 h, respectively, followed by ActD treatment. In panel a, total RNAs were prepared and expression of the indicated genes was analyzed by RT-qPCR. In panel b, total extracts were prepared, and expression of the indicated genes was analyzed by immunoblotting. B, overexpression of MYBBP1A-DN and knockdown of MYBBP1A decreased the percentage of apoptosis induced by ActD treatment. p53-positive MCF-7 (panel a) and p53-null H1299 (panel b) cells were treated with the indicated siRNAs or plasmids and cultured for 24 h. Following this, the cells were treated with or without ActD at a final concentration of 5 nm. The percentage of dead cells was measured by a trypan blue exclusion assay at the indicated times after ActD treatment. The values are means ± standard deviation for triplicate assays.
We then investigated the effects of MYBBP1A-dependent p53 tetramerization on apoptosis induced under cell stress (Fig. 10B). In agreement with the previous results, ActD treatment increased the number of dead MCF-7 cells (Fig. 10B, panel a, OE-control or siControl). ActD-induced cell death was enhanced by overexpressing MYBBP1A-WT (Fig. 10B, panel a, OE-control versus OE-MYBBP1A-WT). However, cell death induced by ActD was significantly suppressed by knockdown of MYBBP1A or by overexpression of MYBBP1A-DN in MCF-7 (Fig. 10B, panel a, siMYBBP1A or OE-MYBBP1A-DN) and LNCaP cells (data not shown). Furthermore, we investigated this effect on p53-null H1299 cells by overexpressing MYBBP1A-WT in H1299 cells. In contrast to the p53-positive MCF-7 cells (Fig. 10B, panel a), ActD-induced cell death did not increase by overexpressing MYBBP1A-WT in p53-null H1299 cells (Fig. 10B, panel b, OE-control versus OE-MYBBP1A-WT). These results clearly indicate that promotion of p53 tetramerization by MYBBP1A is a fundamental mechanism for activating p53 under cell stress.
DISCUSSION
MYBBP1A Enhances p53 Tetramerization and Acetylation in Response to Nucleolar Disruption
Based on the findings of our studies and those of previous reports, we propose the following model in which MYBBP1A enhances tetramerization and activation of p53 (Fig. 11). Under nucleolar stress, rRNA transcription is inhibited and rRNA content in the nucleolus decreases. Loss of nucleolar rRNA, which serves as a scaffold for nucleolar proteins, results in nucleolar disruption and translocation of MYBBP1A to the nucleoplasm (58). At the same time, p53 is stabilized by stress-induced inactivation of HDM2 (3) and forms a dimer independent of MYBBP1A. The nucleoplasm-located MYBBP1A may bind to the nonacetylated p53 dimer via the middle (a.a. 643–1150) and C-terminal (a.a. 1151–1328) regions of MYBBP1A. Following this, tetramerization of nonacetylated p53 is enhanced by dimerizing MYBBP1A to which the p53 dimer is bound. The transcriptional coactivator p300 binds to the nonacetylated p53 tetramer in a complex with the MYBBP1A dimer. After p300 acetylates p53 within the complex, MYBBP1A dissociates from the acetylated p53 tetramer in a complex with p300 (57). Finally, the acetylated p53 tetramer in a complex with p300 binds to the promoters and induces expression of target genes related to apoptosis or cell cycle arrest. However, whether p53 acetylation by p300 precedes DNA binding of the p300-p53 complex (dashed arrow) remains a significant future issue.
FIGURE 11.
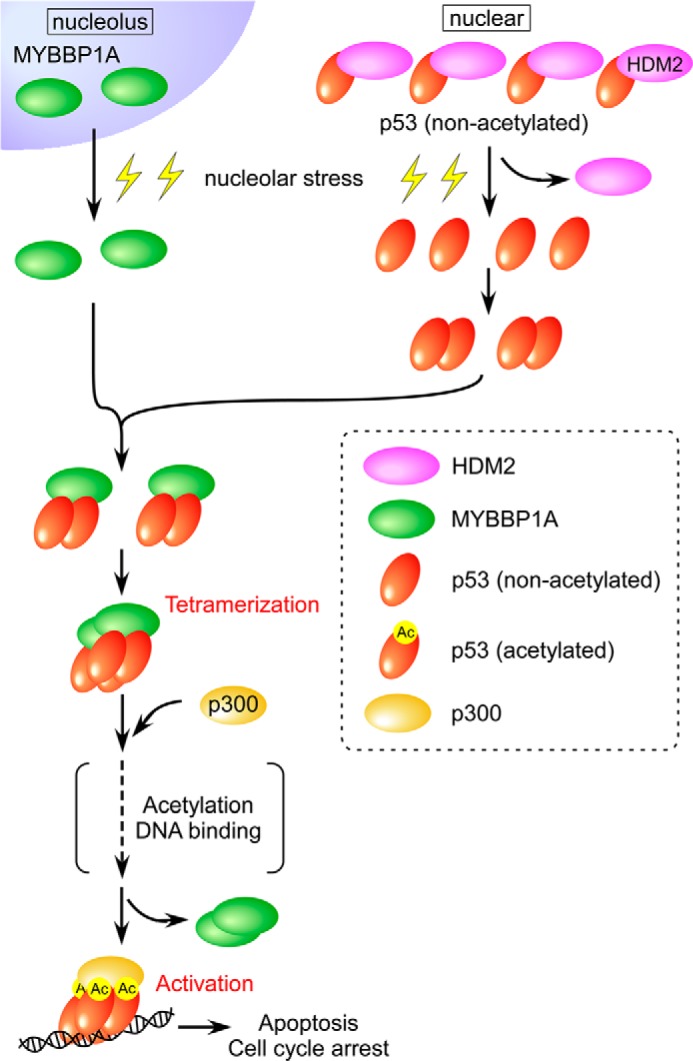
Proposed model for the role of MYBBP1A in p53 tetramerization following nucleolar stress. The details are provided in the text.
Nucleolar Stress and p53 Activation
A number of external and internal insults induce nucleolar stress by disrupting the nucleolar structure (41, 42). Nucleolar stress inhibits rRNA transcription and impairs ribosomal biogenesis, thereby leading to nucleolar disruption. Grummt et al. demonstrated that loss of rRNA transcription resulting in nucleolar disruption is sufficient for inducing p53-dependent apoptosis (65).
Over the last decade, the connection between p53, the nucleolus, and the nucleolar protein has become well established. Prives and Hall (51) demonstrated that the ribosomal protein S7 activates p53 in response to nucleolar stress. Moreover, several nucleolar proteins, including RPL5, RPL11, RPL23, nucleophosmin, nucleolin, nucleostemin, and ARF, activate p53 under nucleolar disrupted conditions (45–50, 52–56). In contrast to these nucleolar proteins that interact with and inhibit HDM2 to stabilize p53 proteins, MYBBP1A directly binds to p53 and enhances its tetramerization and acetylation under cellular stress. Thus, the mode of MYBBP1A action is distinct from those of other nucleolar proteins in terms of regulating p53 activation.
Tetramerization of p53 and Its Regulatory Proteins
Tetramerization of p53 is regulated by the TET domain in the C terminus, spanning residues 325–356, which can by itself form tetramers in vitro (38). p53 forms a homodimer or a tetramer by directly interacting with TET domains (32). TET domain deletion mutants can neither form oligomers nor be activated under DNA damage conditions because they cannot associate with DNA (66).
Thus, tetramerization of p53 plays an important role in its activation. It has been reported that a few proteins enhance tetramerization of p53 (67–69). Among these proteins, 14-3-3σ-dependent tetramerization of p53 has been described relatively well. 14-3-3σ binds to the p53 C terminus and stabilizes p53 tetramerization through DNA damage-dependent phosphorylation of p53 at Ser-378 (59, 70).
Nucleolar disruption reportedly induces p53 activation without phosphorylation (58). Therefore, nucleolar stress-mediated p53 activation may not be accounted for by 14-3-3σ. In this study, we observed enhanced p53 tetramerization through the p53 CRD region by interaction with MYBBP1A, which acts as a platform for tetramerization. This regulatory cascade begins with nucleolar disruption, followed by translocation of MYBBP1A. This is the first report to describe a possible mechanism underlying p53 tetramerization under nucleolar stress.
Relationship between Tetramerization and Acetylation of p53
Gu and co-workers (26) showed that acetylation, among many other p53 post-translational modifications, plays a crucial role in regulating p53 activity. It has also been shown that p53 tetramerization plays an essential role in regulating p53 activity (28). Recent evidence indicates that post-translational modifications, including p53 acetylation, are regulated by p53 tetramerization (40). Tetramerization provides appropriate binding sites for p300 and leads to p300 binding and subsequent acetylation of p53 C-terminal lysine residues (28). Our findings highlight the relationship between tetramerization and acetylation in the mechanisms of p53 activation. We demonstrated that knockdown of MYBBP1A-WT or overexpression of MYBBP1A-DN reduced p53 tetramerization and acetylation. In contrast, immunoprecipitation assays have revealed that MYBBP1A does not associate with acetylated p53 (57). These results suggest that MYBBP1A interacts with p53 and facilitates p53 tetramerization, subsequently enhancing acetylation of p53. Thus, consistent with previous reports, our findings also suggest that tetramerization and subsequent acetylation are essential for p53 activation.
Physiological Significance of p53 Activation by MYBBP1A
p53 plays a critical role in cancer prevention, given that p53 suppresses tumorigenesis by inducing cell cycle arrest and apoptosis through its transcriptional activity (71). We recently demonstrated that MYBBP1A plays a role in tumor prevention in the context of p53 activation (72). We conducted extensive analysis and demonstrated that MYBBP1A expression is associated with breast cancer tumorigenesis in clinical samples. In vivo and in vitro assays demonstrated that MYBBP1A regulates p53-dependent tumorigenesis. These studies showed that promotion of p53 tetramerization by MYBBP1A is essential for p53 activation.
Approximately half of human tumors carry inactivating mutations in TP53 (73, 74). Numerous reports have shown that many mutations occur in the p53 DNA-binding domain (75, 76). Correspondingly, TET domain mutations occur in patients with Li-Fraumeni and Li-Fraumeni-like syndromes, suggesting the importance of p53 tetramerization in cancer progression (77–79). In contrast, a mutation in the p53-CRD region, which interacts with MYBBP1A, rarely occurs in cancer (74). In view of these observations and our results, inactivation of the p53-CRD region during tumorigenesis may be caused by down-regulation of a protein such as MYBBP1A rather than by the emergence of a mutation in the CRD region.
This work was supported by a Grant-in-Aid for the Japan Society for the Promotion of Science.
- CRD
- C-terminal regulatory domain
- TET
- tetramerization domain
- ActD
- actinomycin D
- RT-qPCR
- real time quantitative PCR
- Co-IP
- coimmunoprecipitation
- GA
- glutaraldehyde
- PLA
- proximity ligation assay
- a.a.
- amino acids
- MYBBP1A
- Myb-binding protein 1A.
REFERENCES
- 1. Vogelstein B., Lane D., Levine A. J. (2000) Surfing the p53 network. Nature 408, 307–310 [DOI] [PubMed] [Google Scholar]
- 2. Levine A. J. (1997) p53, the cellular gatekeeper for growth and division. Cell 88, 323–331 [DOI] [PubMed] [Google Scholar]
- 3. Prives C., Hall P. A. (1999) The p53 pathway. J. Pathol. 187, 112–126 [DOI] [PubMed] [Google Scholar]
- 4. Michael D., Oren M. (2003) The p53-Mdm2 module and the ubiquitin system. Semin. Cancer Biol. 13, 49–58 [DOI] [PubMed] [Google Scholar]
- 5. Brooks C. L., Gu W. (2006) p53 ubiquitination. Mdm2 and beyond. Mol. Cell 21, 307–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shi D., Gu W. (2012) Dual roles of MDM2 in the regulation of p53. Ubiquitination dependent and ubiquitination independent mechanisms of MDM2 repression of p53 activity. Genes Cancer 3, 240–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Feng L., Lin T., Uranishi H., Gu W., Xu Y. (2005) Functional analysis of the roles of posttranslational modifications at the p53 C terminus in regulating p53 stability and activity. Mol. Cell Biol. 25, 5389–5395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meek D. W., Anderson C. W. (2009) Posttranslational modification of p53. Cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 1, a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Appella E., Anderson C. W. (2001) Post-translational modifications and activation of p53 by genotoxic stresses. Eur. J. Biochem. 268, 2764–2772 [DOI] [PubMed] [Google Scholar]
- 10. Brooks C. L., Gu W. (2003) Ubiquitination, phosphorylation and acetylation. The molecular basis for p53 regulation. Curr. Opin. Cell Biol. 15, 164–171 [DOI] [PubMed] [Google Scholar]
- 11. Bode A. M., Dong Z. (2004) Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 4, 793–805 [DOI] [PubMed] [Google Scholar]
- 12. Olsson A., Manzl C., Strasser A., Villunger A. (2007) How important are post-translational modifications in p53 for selectivity in target-gene transcription and tumour suppression? Cell Death Differ. 14, 1561–1575 [DOI] [PubMed] [Google Scholar]
- 13. Vousden K. H., Lane D. P. (2007) p53 in health and disease. Nat. Rev. Mol. Cell Biol. 8, 275–283 [DOI] [PubMed] [Google Scholar]
- 14. Carter S., Vousden K. H. (2009) Modifications of p53. Competing for the lysines. Curr. Opin. Genet. Dev. 19, 18–24 [DOI] [PubMed] [Google Scholar]
- 15. Kruse J. P., Gu W. (2009) Modes of p53 regulation. Cell 137, 609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shieh S. Y., Ikeda M., Taya Y., Prives C. (1997) DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91, 325–334 [DOI] [PubMed] [Google Scholar]
- 17. Avantaggiati M. L., Ogryzko V., Gardner K., Giordano A., Levine A. S., Kelly K. (1997) Recruitment of p300/CBP in p53-dependent signal pathways. Cell 89, 1175–1184 [DOI] [PubMed] [Google Scholar]
- 18. Gu W., Shi X. L., Roeder R. G. (1997) Synergistic activation of transcription by CBP and p53. Nature 387, 819–823 [DOI] [PubMed] [Google Scholar]
- 19. Lill N. L., Grossman S. R., Ginsberg D., DeCaprio J., Livingston D. M. (1997) Binding and modulation of p53 by p300/CBP coactivators. Nature 387, 823–827 [DOI] [PubMed] [Google Scholar]
- 20. Gu W., Roeder R. G. (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90, 595–606 [DOI] [PubMed] [Google Scholar]
- 21. Sakaguchi K., Herrera J. E., Saito S., Miki T., Bustin M., Vassilev A., Anderson C. W., Appella E. (1998) DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev. 12, 2831–2841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu L., Scolnick D. M., Trievel R. C., Zhang H. B., Marmorstein R., Halazonetis T. D., Berger S. L. (1999) p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell Biol. 19, 1202–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ito A., Lai C. H., Zhao X., Saito S., Hamilton M. H., Appella E., Yao T. P. (2001) p300/CBP-mediated p53 acetylation is commonly induced by p53-activating agents and inhibited by MDM2. EMBO J. 20, 1331–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu S. Y., Chiang C. M. (2009) Crosstalk between sumoylation and acetylation regulates p53-dependent chromatin transcription and DNA binding. EMBO J. 28, 1246–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Barlev N. A., Liu L., Chehab N. H., Mansfield K., Harris K. G., Halazonetis T. D., Berger S. L. (2001) Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell 8, 1243–1254 [DOI] [PubMed] [Google Scholar]
- 26. Tang Y., Zhao W., Chen Y., Zhao Y., Gu W. (2008) Acetylation is indispensable for p53 activation. Cell 133, 612–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Muñoz-Fontela C., González D., Marcos-Villar L., Campagna M., Gallego P., González-Santamaría J., Herranz D., Gu W., Serrano M., Aaronson S. A., Rivas C. (2011) Acetylation is indispensable for p53 antiviral activity. Cell Cycle 10, 3701–3705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Itahana Y., Ke H., Zhang Y. (2009) p53 Oligomerization is essential for its C-terminal lysine acetylation. J. Biol. Chem. 284, 5158–5164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sakaguchi K., Sakamoto H., Lewis M. S., Anderson C. W., Erickson J. W., Appella E., Xie D. (1997) Phosphorylation of serine 392 stabilizes the tetramer formation of tumor suppressor protein p53. Biochemistry 36, 10117–10124 [DOI] [PubMed] [Google Scholar]
- 30. Mateu M. G., Sánchez Del Pino M. M., Fersht A. R. (1999) Mechanism of folding and assembly of a small tetrameric protein domain from tumor suppressor p53. Nat. Struct. Biol. 6, 191–198 [DOI] [PubMed] [Google Scholar]
- 31. Poon G. M., Brokx R. D., Sung M., Gariépy J. (2007) Tandem dimerization of the human p53 tetramerization domain stabilizes a primary dimer intermediate and dramatically enhances its oligomeric stability. J. Mol. Biol. 365, 1217–1231 [DOI] [PubMed] [Google Scholar]
- 32. Friedman P. N., Chen X., Bargonetti J., Prives C. (1993) The p53 protein is an unusually shaped tetramer that binds directly to DNA. Proc. Natl. Acad. Sci. U.S.A. 90, 3319–3323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kern S. E., Kinzler K. W., Bruskin A., Jarosz D., Friedman P., Prives C., Vogelstein B. (1991) Identification of p53 as a sequence-specific DNA-binding protein. Science 252, 1708–1711 [DOI] [PubMed] [Google Scholar]
- 34. Halazonetis T. D., Kandil A. N. (1993) Conformational shifts propagate from the oligomerization domain of p53 to its tetrameric DNA binding domain and restore DNA binding to select p53 mutants. EMBO J. 12, 5057–5064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Balagurumoorthy P., Sakamoto H., Lewis M. S., Zambrano N., Clore G. M., Gronenborn A. M., Appella E., Harrington R. E. (1995) Four p53 DNA-binding domain peptides bind natural p53-response elements and bend the DNA. Proc. Natl. Acad. Sci. U.S.A. 92, 8591–8595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nagaich A. K., Zhurkin V. B., Durell S. R., Jernigan R. L., Appella E., Harrington R. E. (1999) p53-induced DNA bending and twisting. p53 tetramer binds on the outer side of a DNA loop and increases DNA twisting. Proc. Natl. Acad. Sci. U.S.A. 96, 1875–1880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Weinberg R. L., Veprintsev D. B., Fersht A. R. (2004) Cooperative binding of tetrameric p53 to DNA. J. Mol. Biol. 341, 1145–1159 [DOI] [PubMed] [Google Scholar]
- 38. Jeffrey P. D., Gorina S., Pavletich N. P. (1995) Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science 267, 1498–1502 [DOI] [PubMed] [Google Scholar]
- 39. Waterman J. L., Shenk J. L., Halazonetis T. D. (1995) The dihedral symmetry of the p53 tetramerization domain mandates a conformational switch upon DNA binding. EMBO J. 14, 512–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Warnock L. J., Knox A., Mee T. R., Raines S. A., Milner J. (2008) Influence of tetramerisation on site-specific post-translational modifications of p53. Comparison of human and murine p53 tumor suppressor protein. Cancer Biol. Ther. 7, 1481–1489 [DOI] [PubMed] [Google Scholar]
- 41. Rubbi C. P., Milner J. (2003) Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 22, 6068–6077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mayer C., Grummt I. (2005) Cellular stress and nucleolar function. Cell Cycle 4, 1036–1038 [DOI] [PubMed] [Google Scholar]
- 43. Perry R. P., Kelley D. E. (1970) Inhibition of RNA synthesis by actinomycin D. Characteristic dose-response of different RNA species. J. Cell Physiol. 76, 127–139 [DOI] [PubMed] [Google Scholar]
- 44. Iapalucci-Espinoza S., Franze-Fernández M. T. (1979) Effect of protein synthesis inhibitors and low concentrations of actinomycin D on ribosomal RNA synthesis. FEBS Lett. 107, 281–284 [DOI] [PubMed] [Google Scholar]
- 45. Lohrum M. A., Ludwig R. L., Kubbutat M. H., Hanlon M., Vousden K. H. (2003) Regulation of HDM2 activity by the ribosomal protein L11. Cancer Cell 3, 577–587 [DOI] [PubMed] [Google Scholar]
- 46. Marechal V., Elenbaas B., Piette J., Nicolas J. C., Levine A. J. (1994) The ribosomal L5 protein is associated with mdm-2 and mdm-2-p53 complexes. Mol. Cell Biol. 14, 7414–7420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang Y., Wolf G. W., Bhat K., Jin A., Allio T., Burkhart W. A., Xiong Y. (2003) Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol. Cell Biol. 23, 8902–8912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dai M. S., Lu H. (2004) Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J. Biol. Chem. 279, 44475–44482 [DOI] [PubMed] [Google Scholar]
- 49. Dai M. S., Zeng S. X., Jin Y., Sun X. X., David L., Lu H. (2004) Ribosomal protein L23 activates p53 by inhibiting MDM2 function in response to ribosomal perturbation but not to translation inhibition. Mol. Cell Biol. 24, 7654–7668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jin A., Itahana K., O'Keefe K., Zhang Y. (2004) Inhibition of HDM2 and activation of p53 by ribosomal protein L23. Mol. Cell Biol. 24, 7669–7680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhu Y., Poyurovsky M. V., Li Y., Biderman L., Stahl J., Jacq X., Prives C. (2009) Ribosomal protein S7 is both a regulator and a substrate of MDM2. Mol. Cell 35, 316–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kamijo T., Weber J. D., Zambetti G., Zindy F., Roussel M. F., Sherr C. J. (1998) Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc. Natl. Acad. Sci. U.S.A. 95, 8292–8297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pomerantz J., Schreiber-Agus N., Liégeois N. J., Silverman A., Alland L., Chin L., Potes J., Chen K., Orlow I., Lee H. W., Cordon-Cardo C., DePinho R. A. (1998) The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2's inhibition of p53. Cell 92, 713–723 [DOI] [PubMed] [Google Scholar]
- 54. Kurki S., Peltonen K., Latonen L., Kiviharju T. M., Ojala P. M., Meek D., Laiho M. (2004) Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell 5, 465–475 [DOI] [PubMed] [Google Scholar]
- 55. Saxena A., Rorie C. J., Dimitrova D., Daniely Y., Borowiec J. A. (2006) Nucleolin inhibits Hdm2 by multiple pathways leading to p53 stabilization. Oncogene 25, 7274–7288 [DOI] [PubMed] [Google Scholar]
- 56. Dai M. S., Sun X. X., Lu H. (2008) Aberrant expression of nucleostemin activates p53 and induces cell cycle arrest via inhibition of MDM2. Mol. Cell Biol. 28, 4365–4376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ono W., Akaogi K., Waku T., Kuroda T., Yokoyama W., Hayashi Y., Kimura K., Kishimoto H., Yanagisawa J. (2013) Nucleolar protein, Myb-binding protein 1A, specifically binds to nonacetylated p53 and efficiently promotes transcriptional activation. Biochem. Biophys. Res. Commun. 434, 659–663 [DOI] [PubMed] [Google Scholar]
- 58. Kuroda T., Murayama A., Katagiri N., Ohta Y. M., Fujita E., Masumoto H., Ema M., Takahashi S., Kimura K., Yanagisawa J. (2011) RNA content in the nucleolus alters p53 acetylation via MYBBP1A. EMBO J. 30, 1054–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rajagopalan S., Jaulent A. M., Wells M., Veprintsev D. B., Fersht A. R. (2008) 14-3-3 activation of DNA binding of p53 by enhancing its association into tetramers. Nucleic Acids Res. 36, 5983–5991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kajiro M., Hirota R., Nakajima Y., Kawanowa K., So-ma K., Ito I., Yamaguchi Y., Ohie S. H., Kobayashi Y., Seino Y., Kawano M., Kawabe Y., Takei H., Hayashi S., Kurosumi M., Murayama A., Kimura K., Yanagisawa J. (2009) The ubiquitin ligase CHIP acts as an upstream regulator of oncogenic pathways. Nat. Cell Biol. 11, 312–319 [DOI] [PubMed] [Google Scholar]
- 61. Murayama A., Ohmori K., Fujimura A., Minami H., Yasuzawa-Tanaka K., Kuroda T., Oie S., Daitoku H., Okuwaki M., Nagata K., Fukamizu A., Kimura K., Shimizu T., Yanagisawa J. (2008) Epigenetic control of rDNA loci in response to intracellular energy status. Cell 133, 627–639 [DOI] [PubMed] [Google Scholar]
- 62. Weinberg R. L., Veprintsev D. B., Bycroft M., Fersht A. R. (2005) Comparative binding of p53 to its promoter and DNA recognition elements. J. Mol. Biol. 348, 589–596 [DOI] [PubMed] [Google Scholar]
- 63. Yamauchi T., Keough R. A., Gonda T. J., Ishii S. (2008) Ribosomal stress induces processing of Mybbp1a and its translocation from the nucleolus to the nucleoplasm. Genes Cells 13, 27–39 [DOI] [PubMed] [Google Scholar]
- 64. Hupp T. R., Lane D. P. (1994) Allosteric activation of latent p53 tetramers. Curr. Biol. 4, 865–875 [DOI] [PubMed] [Google Scholar]
- 65. Yuan X., Zhou Y., Casanova E., Chai M., Kiss E., Gröne H. J., Schütz G., Grummt I. (2005) Genetic inactivation of the transcription factor TIF-IA leads to nucleolar disruption, cell cycle arrest, and p53-mediated apoptosis. Mol. Cell 19, 77–87 [DOI] [PubMed] [Google Scholar]
- 66. Li M., Luo J., Brooks C. L., Gu W. (2002) Acetylation of p53 inhibits its ubiquitination by Mdm2. J. Biol. Chem. 277, 50607–50611 [DOI] [PubMed] [Google Scholar]
- 67. Waterman M. J., Stavridi E. S., Waterman J. L., Halazonetis T. D. (1998) ATM-dependent activation of p53 involves dephosphorylation and association with 14-3-3 proteins. Nat. Genet. 19, 175–178 [DOI] [PubMed] [Google Scholar]
- 68. Hanson S., Kim E., Deppert W. (2005) Redox factor 1 (Ref-1) enhances specific DNA binding of p53 by promoting p53 tetramerization. Oncogene 24, 1641–1647 [DOI] [PubMed] [Google Scholar]
- 69. Meng X., Yue J., Liu Z., Shen Z. (2007) Abrogation of the transactivation activity of p53 by BCCIP down-regulation. J. Biol. Chem. 282, 1570–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Schumacher B., Mondry J., Thiel P., Weyand M., Ottmann C. (2010) Structure of the p53 C-terminus bound to 14-3-3. Implications for stabilization of the p53 tetramer. FEBS Lett. 584, 1443–1448 [DOI] [PubMed] [Google Scholar]
- 71. Lacroix M., Toillon R. A., Leclercq G. (2006) p53 and breast cancer, an update. Endocr. Relat. Cancer 13, 293–325 [DOI] [PubMed] [Google Scholar]
- 72. Akaogi K., Ono W., Hayashi Y., Kishimoto H., Yanagisawa J. (2013) MYBBP1A suppresses breast cancer tumorigenesis by enhancing the p53 dependent anoikis. BMC Cancer 13, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hainaut P., Hollstein M. (2000) p53 and human cancer. The first ten thousand mutations. Adv. Cancer Res. 77, 81–137 [DOI] [PubMed] [Google Scholar]
- 74. Petitjean A., Mathe E., Kato S., Ishioka C., Tavtigian S. V., Hainaut P., Olivier M. (2007) Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype. Lessons from recent developments in the IARC TP53 database. Hum. Mutat. 28, 622–629 [DOI] [PubMed] [Google Scholar]
- 75. Hollstein M., Sidransky D., Vogelstein B., Harris C. C. (1991) p53 mutations in human cancers. Science 253, 49–53 [DOI] [PubMed] [Google Scholar]
- 76. Pavletich N. P., Chambers K. A., Pabo C. O. (1993) The DNA-binding domain of p53 contains the four conserved regions and the major mutation hot spots. Genes Dev. 7, 2556–2564 [DOI] [PubMed] [Google Scholar]
- 77. Lomax M. E., Barnes D. M., Hupp T. R., Picksley S. M., Camplejohn R. S. (1998) Characterization of p53 oligomerization domain mutations isolated from Li-Fraumeni and Li-Fraumeni like family members. Oncogene 17, 643–649 [DOI] [PubMed] [Google Scholar]
- 78. DiGiammarino E. L., Lee A. S., Cadwell C., Zhang W., Bothner B., Ribeiro R. C., Zambetti G., Kriwacki R. W. (2002) A novel mechanism of tumorigenesis involving pH-dependent destabilization of a mutant p53 tetramer. Nat. Struct. Biol. 9, 12–16 [DOI] [PubMed] [Google Scholar]
- 79. Zambetti G. P. (2007) The p53 mutation “gradient effect” and its clinical implications. J. Cell Physiol. 213, 370–373 [DOI] [PubMed] [Google Scholar]