

Background: Flavin-based electron bifurcation explains the energy metabolism of anaerobic microorganisms.
Results: Kinetic, structural, and spectral data revealed a detailed picture of the bifurcation process.
Conclusion: NADH reduces β-FAD of Etf, which bifurcates one electron to Bcd via α-FAD and the other to ferredoxin. Repetition leads to reduction of crotonyl-CoA.
Significance: The mechanism can be extended to other bifurcating systems.
Keywords: Bioenergetics/Electron Transfer Complex, Coenzyme A, Crystal Structure, Electron Transfer, Energy Metabolism, Enzyme Mechanisms
Abstract
Electron bifurcation is a fundamental strategy of energy coupling originally discovered in the Q-cycle of many organisms. Recently a flavin-based electron bifurcation has been detected in anaerobes, first in clostridia and later in acetogens and methanogens. It enables anaerobic bacteria and archaea to reduce the low-potential [4Fe-4S] clusters of ferredoxin, which increases the efficiency of the substrate level and electron transport phosphorylations. Here we characterize the bifurcating electron transferring flavoprotein (EtfAf) and butyryl-CoA dehydrogenase (BcdAf) of Acidaminococcus fermentans, which couple the exergonic reduction of crotonyl-CoA to butyryl-CoA to the endergonic reduction of ferredoxin both with NADH. EtfAf contains one FAD (α-FAD) in subunit α and a second FAD (β-FAD) in subunit β. The distance between the two isoalloxazine rings is 18 Å. The EtfAf-NAD+ complex structure revealed β-FAD as acceptor of the hydride of NADH. The formed β-FADH− is considered as the bifurcating electron donor. As a result of a domain movement, α-FAD is able to approach β-FADH− by about 4 Å and to take up one electron yielding a stable anionic semiquinone, α-FAD⨪, which donates this electron further to Dh-FAD of BcdAf after a second domain movement. The remaining non-stabilized neutral semiquinone, β-FADH•, immediately reduces ferredoxin. Repetition of this process affords a second reduced ferredoxin and Dh-FADH− that converts crotonyl-CoA to butyryl-CoA.
Introduction
Electron bifurcation proposed by Peter Mitchell in 1976 (1) is the key event of the Q (ubiquinone)-cycle used by the bc1 complex to double the efficiency of the proton-motive process. In 2008, a second type of electron bifurcation was discovered that is based on flavin cofactors bound to a soluble cytoplasmic protein complex and not on ubiquinone used by the transmembrane bc1 complex (2, 3). This so-called flavin-based electron bifurcation, considered as a new mode of energy coupling, plays a crucial role in diverse energy-limited anaerobic organisms and is proposed to operate since the early evolution of life (4). So far, five such systems are experimentally established; for a recent review, see Ref. 5. The latest additions are caffeyl-CoA reductase of Acetobacterium woodii (6) and formate dehydrogenase of Clostridium acidiurici (7). All enzyme complexes possess at least one flavin, either FAD or riboflavin 5′-phosphate, and catalyze the reduction of a low-potential ferredoxin. The functional understanding of these enzyme complexes allows an explanation of the bioenergetics of many fermenting bacteria, methanogens, and acetogens.
The best studied flavin-based electron bifurcating system is the electron transferring flavoprotein-butyryl-CoA dehydrogenase (Etf-Bcd)5 complex, which is a key enzyme in butyrate producing clostridia. This Etf-Bcd complex couples the exergonic reduction of crotonyl-CoA to butyryl-CoA (E0′ = −10 mV) by NADH with the endergonic reduction of ferredoxin (Fd) also by oxidation of NADH to NAD+ (E0′ = −320 mV). According to current knowledge, the electron pair of NADH is split at the two-electron acceptor and one-electron donor FAD; one electron reduces the Dh-FAD (dehydrogenase FAD) of Bcd and the other goes to ferredoxin (Fd−) characterized by a low redox potential (E0′ = −405 mV) (8). Repetition of this process leads to a second reduced ferredoxin and butyryl-CoA upon hydride transfer from FADH− of Bcd to crotonyl-CoA. The “energy-rich” reduced ferredoxin contributes to the energy conservation of the organism either by regeneration of NADH via the H+/Na+-pumping ferredoxin-NAD+ reductase also called Rnf (9–11) or by reduction of protons to H2, which increases the substrate-level phosphorylation via the oxidative branch of the fermentation.
To gain detailed mechanistic insights into electron bifurcation, we chose as a model EtfAf and BcdAf of the Gram-negative glutamate fermenting Acidaminococcus fermentans, in which Etf and Bcd are separate proteins (12), in contrast to the non-dissociable Etf-Bcd complexes of several clostridia. We solved the crystal structures of EtfAf and BcdAf, and characterized the proteins further by biochemical, kinetic, and spectroscopic methods.
EXPERIMENTAL PROCEDURES
Crotonyl-CoA and Butyryl-CoA
Both were synthesized by acylation of CoASH in aqueous 1 m KHCO3 using 1 m crotonic anhydride or butyric anhydride in acetonitrile with a slight molar excess. After acidification the CoA-thioesters were purified on C18 columns and stored as lyophilized powders at −80 °C (13). The concentration of crotonyl-CoA was determined by the NAD+-dependent β-oxidation to acetyl-CoA and acetyl phosphate as described for the assay of glutaconyl-CoA decarboxylase (14). HPLC of crotonyl-CoA and butyryl-CoA was performed on a C18 Kinetex column (5-μm particle size, 100 Å pore size, 250 × 4.6 mm, Phenomenex, Aschaffenburg, Germany) at a flow rate of 1 ml/min in 50 mm KH2PO4, pH 5.3, and 5% acetonitrile. During 20 min a linear gradient up to 60% acetonitrile was applied. The ferricenium (Fc+) solution for the Bcd assay was prepared in 10 mm HCl to a final concentration of 2 mm set at 617 nm with ϵ617 = 0.41 mm−1 cm−1 (15).
Protein concentrations were estimated with the Bradford assay (16) (Bio-Rad-Microassay reagent, Bio-Rad-Laboratories). Bovine serum albumin (Sigma) served as standard. SDS-PAGE was performed as described by the method of Laemmli (17).
Purification of EtfAf
A. fermentans VR4 (DSM 20731) was grown in a 100-liter glutamate-yeast extract medium under anaerobic conditions at 37 °C (14). Wet packed cells (10 g) were suspended in 20 ml of 50 mm potassium phosphate, pH 7.0 (buffer A), and opened by three passages through a French press. The supernatant, obtained by centrifugation at 150,000 × g for 1 h at 4 °C was loaded on a DEAE column and fractionated with 10 column volumes of 0–100% 1 m NaCl in buffer A. Etf eluted from the column at around 19% NaCl. The fractions with the highest activity (iodonitrosotetrazolium chloride assay) were combined and dialyzed overnight against buffer A. The dialysate was concentrated by ultrafiltration and mixed with an equal volume of 3 m (NH4)2SO4 to make a final concentration of 1.5 m. This mixture was loaded on a phenyl-Sepharose column and fractionated with 10 column volumes of 0–100% 1.5 m (NH4)2SO4 in buffer A. The fractions eluting around 58% were run on SDS-PAGE, which showed both the subunits of EtfAf.
Heterologous Production of EtfAf
The genes Acfer_0555 (β-subunit) and Acfer_0556 (α-subunit) were cloned in this order as one fragment by applying the StargateTM system according to the instructions of IBA, Göttingen, Germany. Genomic DNA of A. fermentans was used for PCR as template with the forward primer 5′-AGCTCTTCAATGAACATCGTTGTATGTGT-3′ and the reverse primer 5′-AAGCTCTTCACCCGGATTCTTTGAAGCCTTGA-3′. The PCR product was cloned into the p-ENTRY vector and introduced into Escherichia coli DH5α by chemical transformation. After confirmation of the desired sequence, the genes were transferred from the donor vector to expression vector pASG IBA33 and further into E. coli BL21(GroEL). The recombinant cells were cultivated in 4 liters of Standard I nutrient broth (Merck, Darmstadt, Germany) containing ampicillin (100 μg/ml) and chloramphenicol (34 μg/ml). When the A600 nm was 0.5, both anhydrotetracycline (0.2 μg/ml) and 0.1 mm isopropylthiogalactoside were added to the culture medium to induce the overproduction of Etf and GroEL. The cultures were incubated at room temperature overnight. After harvesting, the cells (17 g wet mass) were suspended in 34 ml of 10 mm imidazole in 10 mm sodium phosphate, pH 7.4, and disrupted by passing the suspension three times through a French press cell at 140 megapascals. Cell debris was removed by centrifugation at 30,000 × g for 1 h and 4 °C to obtain 34 ml of cell extract containing 34 mg of protein/ml. The purification process was performed under oxic conditions at 4 °C. The supernatant was applied on a 20 ml of nickel-nitrilotriacetic acid column, which was equilibrated with buffer A. The recombinant protein was eluted with 500 mm NaCl and 150 mm imidazole in 10 mm sodium phosphate, pH 7.4. The purified protein was concentrated by ultrafiltration, washed with 50 mm potassium phosphate, pH 7.0, and stored at −80 °C. The prosthetic group was quantitatively extracted with both trichloroacetic acid and heat denaturation of the protein and identified by thin layer chromatography on TLC plates (Silica Gel 60 F254) from Merck KGaA, Germany. The solvent system was n-butyl alcohol:acetic acid:water (4:3:3) (18). The titration of EtfAf with NADH was performed under anoxic conditions in 50 mm potassium phosphate, pH 7.0.
Purification of Butyryl-CoA Dehydrogenase (BcdAf)
The supernatant obtained by centrifugation at 150,000 × g (see purification of Etf) was saturated with ammonium sulfate to 60%. After centrifugation at 50,000 × g for 1 h at 4 °C, the clear supernatant was dialyzed against buffer A. The supernatant was then loaded on a DEAE-Sepharose column equilibrated with buffer A. After washing with this buffer, proteins were eluted by linear gradient from 0 to 1 m NaCl in buffer A. The fractions containing green BcdAf were analyzed by SDS-PAGE. The purest fractions were collected, washed with buffer A by ultrafiltration, and stored at −80 °C.
The yellow form of BcdAf was prepared by adding a molar excess of solid dithionite to the solution in buffer A under anaerobic conditions. After standing on ice for 1 h the enzyme was desalted by passing through a PD10 column in anaerobic 0.1 m potassium phosphate, pH 6.8. Then the enzyme was washed three times under air with the same but aerobic buffer by ultrafiltration using Centricon 30 (19).
Partial Purification of Hydrogenase
Clostridium pasteurianum (DSM 525) was grown on a medium containing 100 mm glucose, 70 mm NaHCO3, and yeast extract (2 g/liter). Wet packed cells (10 g) were suspended in 50 mm potassium phosphate, pH 7.4 (buffer B), and broken by three passages through a French press at 140 megapascal (20,000 p.s.i.). The broken cells were centrifuged at 7,000 × g for 20 min. The supernatant was heated at 55–60 °C for 10 min under a hydrogen atmosphere. After keeping on ice for 10 min, precipitated protein was removed by centrifugation at 20,000 × g for 20 min. The supernatant containing the hydrogenase (6 mg of protein/ml) was stored at −20 °C until use (20).
Purification of Ferredoxin
Clostridium tetanomorphum (DSM 526) was grown on the same medium as used for A. fermentans from which biotin was omitted. The purification was carried out under strictly anoxic conditions in an atmosphere of 95% N2 and 5% H2 (Coy Anaerobic Chamber). Wet packed cells (10 g) were suspended in buffer B. After sonication, the supernatant, obtained by centrifugation at 150,000 × g for 1 h at 4 °C, was loaded on a DEAE column that was equilibrated by buffer B. After washing the unbound proteins, ferredoxin was eluted with a linear gradient from 0 to 100% 2 m NaCl in buffer B. The active fractions found by the bifurcation assay were collected and concentrated with a 3-kDa ultrafiltration membrane. The concentrated sample was loaded to a Superdex 75 column that was equilibrated with 150 mm NaCl in buffer B. The fractions containing ferredoxin were concentrated and stored under anoxic conditions at −80 °C. To calculate the concentration of ferredoxin, a molecular mass of 6 kDa was used (8).
Enzyme Assays
EtfAf activity was measured in a 1-ml cuvette (d = 1 cm) containing 50 mm potassium phosphate, pH 7.0, 250 μm NADH, and 100 μm iodonitrosotetrazolium chloride. The formation of the red formazane was followed at 492 nm, ϵ = 19.2 mm−1 cm−1 (12). BcdAf activity was measured in 50 mm potassium phosphate, pH 7.0, with 0.2 mm ferricenium hexafluorophosphate (Fc+) and 0.1 mm butyryl-CoA. The decrease of absorbance was followed at 310 nm, ϵ = 2 × 4.3 mm−1 cm−1, because 2 mol of Fc+ are required to oxidize 1 mol of butyryl-CoA (21). Unless otherwise indicated the assay for EtfAf/BcdAf activity (bifurcation assay) was done under anoxic conditions containing 250 μm NADH, 100 μm crotonyl-CoA, 0.5 μm EtfAf, 1 μm BcdAf, 1 μm ferredoxin, crude hydrogenase (30 μg/ml), and 50 mm potassium phosphate, pH 7.0 (3). The decrease in NADH concentration was monitored at 340 nm, ϵ = 6.3 mm−1 cm−1 (22). When appropriate the data were fitted to the Michealis-Menten equation using GraphPad Prism 5 software.
X-ray Structure Analysis of EtfAf and BcdAf
EtfAf was concentrated to 20 mg/ml in 10 mm MOPS and 1 mm FAD, pH 7.0, BcdAf to 15 mg/ml in 10 mm MOPS, pH 6.8, and 1 mm FAD. Crystallization experiments were performed with the sitting drop method at 4 and 18 °C using a CrystalMationTM system from Rigaku and commercially available screens. Initial conditions for both enzymes were found in the PACT++ screen, which were further optimized (see Table 1). Data were collected at the PXII beamline at the Swiss-Light-Source in Villigen and processed with XDS (23). Phase determination is described in Table 1. Crystallization of the EtfAf-NAD+ complex was performed under related conditions (Table 1). The structure of BcdAf was solved by molecular replacement using the coordinates of Bcd of Megasphaera elsdenii (1buc) as model (24). Model errors were corrected within COOT (25). The refinement was performed with REFMAC5 (26). Data quality and refinement statistics were listed in Table 1. Figs. 3–7 were generated with PyMOL (Schrödinger, LLC).
TABLE 1.
Crystallization conditions and x-ray analysis statistics
| Data set | EtfAf | EtfAf (Hg edge)a | EtfAf + NAD+ | BcdAf |
|---|---|---|---|---|
| Crystallization | 18% PEG 3350, | 18% PEG 3350, | 17% PEG 3350, | 225% PEG1500, |
| Conditionsb | 0.1 m Bistris | 0.1 m Bistris | 0.1 m Bistris | 10% SPG buffer, |
| propane, pH 7.5 | propane, pH 7.5 | propane, pH 7.5 | pH 9.0 | |
| 0.1 m Na formate | 0.1 m Na formate | 0.1 m Na formate | ||
| 1 mm FAD | 1 mm FAD | 1 mm FAD | ||
| Protein concentration | 20 mg/ml | 20 mg/ml | 20 mg/ml | 15 mg/ml |
| Temperature | 4 °C | 4 °C | 4 °C | 4 °C |
| Soaking conditions | 0.1 mm CH3HgAc, | |||
| 5 h | ||||
| Freezing conditions | +25% | +25% | +25% | 25% |
| (w/v) glycerol | (w/v) glycerol | (w/v) glycerol | (v/v) 1,2-propandiol | |
| Data collection | ||||
| Wavelength (Å) | 1.0 | 1.006 | 1.0 | 0.9999 |
| Space group | P212121 | P212121 | P212121 | P21212 |
| Unit cell parameter | ||||
| a, b, c (Å) | 80.2, 84.9, 106.7 | 78.2, 85.7, 106.6 | 79.5, 84.8, 106.4 | 109.1, 141.4, 64.1 |
| Number of molecules per asymmetric unit | 1 | 1 | 1 | 1 |
| Resolution range (Å) | 30.0-1.6 | 30.0-3.0 | 30.0-1.45 | 30.0-1.8 |
| (highest shell) | (1.7-1.6) | (3.1-3.0) | (1.55-1.45) | (1.9-1.8) |
| Redundancy | 2.4 (2.4) | 4.6 (4.8) | 3.9 (4.0) | 7.9 (8.1) |
| Completeness (%) | 93.2 (91.8) | 99.6 (99.8) | 97.6 (96.4) | 99.9 (100) |
| Rmerge (%) | 6.9 (48.8) | 9.6 (150.9) | 4.5 (63.5) | 14.4 (78.8) |
| I/σ(I) | 11.8 (2.5) | 11.8 (1.5) | 15.6 (3.0) | 13.2 (3.9) |
| Refinement | ||||
| Resolution limit (Å) | 1.6-50.0 | 1.45-50.0 | 1.8-50.0 | |
| (highest shell) | 1.6-1.64 | 1.45-1.49 | 1.8-1.84 | |
| Rwork/Rfree (%) | 16.1/19.1 | 15.1/20.8 | 15.3/18.3 | |
| (highest shell) | 26.4/28.1 | 24.5/37.5 | 22.0/25.2 | |
| Root mean square deviation bond lengths (Å) | 0.024 | 0.023 | 0.022 | |
| Root mean square deviation bond angles (°) | 2.3 | 2.36 | 2.13 | |
| Average B (Å2) | 18.2 | 23.3 | 18.7 | |
a Phase determination. Initial SIRAS phases were obtained from native Etf data and from measurements of a crystal soaked with CH3HgAc using SHELXD (50), SHARP (51), and SOLOMON (52). After phase determination the model was built automatically at 2.5-Å resolution with PHENIX (53) supported by a model of human Etf that could be correctly placed into the unit cell and further improved at 1.6-Å resolution with ARP/WARP (54).
b Applied crystallization screens: JBScreen HTS I+II, pentaerythritol, PACT++ (JBS), JCSG Core Suite I–IV (Qiagen), and PGA (MDL).
FIGURE 3.
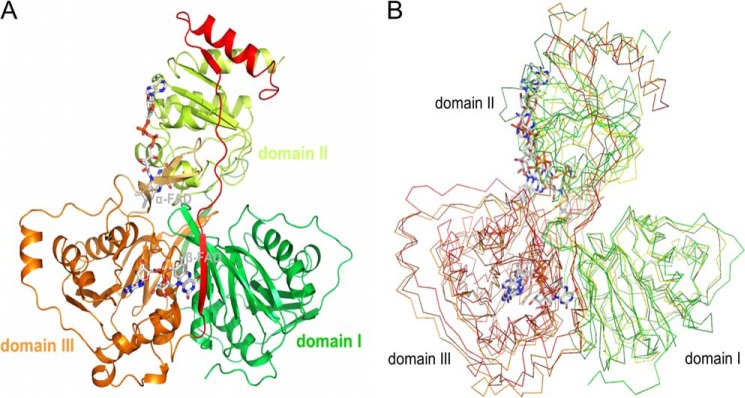
Structures of Etf. A, Etf of A. fermentans. Subunit α is composed of domains I (green) and II (yellow green) and subunit β of domain III (orange), the β-hairpin (light orange), and the C-terminal arm (red). α-FAD and β-FAD are drawn as stick models with the carbons in gray. B, structures of EtfAf, human Etf, and Methylophilus methylotrophus Etf superimposed. Domains I (green, yellow, forest; 5–199. EtfAf numbering) and II (green, yellow, forest; 215–340) form subunit α and domain III (orange, brown, red; 1–219) forms subunit β.
FIGURE 4.
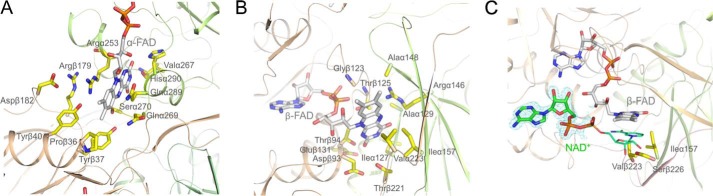
A, binding mode of α-FAD. The isoalloxazine ring is located between domains II and III. One side of α-FAD is attached along the C-terminal side of the parallel β-sheet of domain II and the opposite side is largely exposed to bulk solvent. N5 is hydrogen-bonded to Ser-270-OG. B, binding mode of β-FAD. The isoalloxazine ring of compressed β-FAD primarily binds to domains III and I. Arg-α146-NH2 is projected toward the bottom of the isoalloxazine ring and interacts with N5 of the isoalloxazine ring. C, binding of NAD+. The ADP part (in stick mode) of NAD+ (carbons in green) is predominantly fixed by its contacts to β-FAD. The nicotinamide-ribose part (in line mode) is disordered but can be modeled to the re-face of the isoalloxazine. Ile-α157, Val-β223, and Ser-β226 interfere with the nicotinamide-ribose in the conformation capable for hydride transfer and have to be displaced upon NADH binding. The electron density (in blue) is contoured at 1.5.
FIGURE 5.
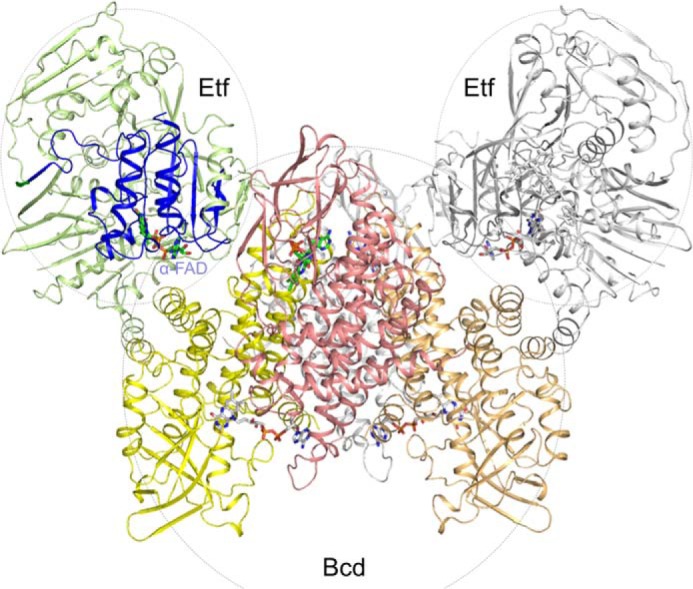
Modeling of the EtfAf and BcdAf complex. The EtfAf and EtfAf and BcdAf structures were separately determined. The first Etf is drawn in green (domains I +III) and in blue (domain II); the second Etf in gray. The subunits of tetrameric Bcd are shown in yellow, orange, red, and gray. Docking is based on the structure of the Etf-MCAD structure (PDB code 2A1T). The isoalloxazine rings of α-FAD of EtfAf and Dh-FAD of BcdAf are more than 30 Å apart from each other. Therefore, a productive electron transfer requires a large-scale conformational change of domain II or a different EtfAf-BcdAf interface.
FIGURE 6.
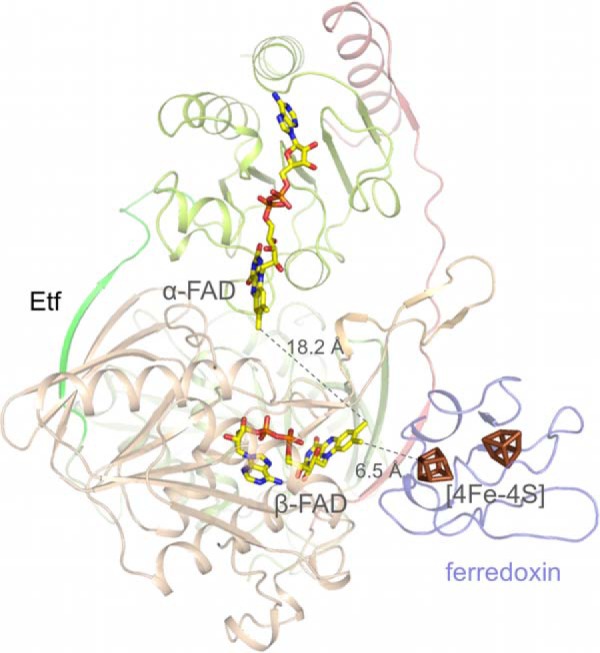
Docking of ferredoxin of C. acidici-urici to EtfAf. Ferredoxin was modeled to domain III in a manner that the distance between its [4Fe-4S] cluster and the isoalloxazine ring of the bifurcating β-FAD is minimal. The resulting edge-to-edge distance at about 6.5 Å allows a rapid electron transfer. It is unclear whether NAD+ is released to bulk solvent prior to bifurcation. This short distance is adjustable because the isoalloxazine of β-FAD directly sits at the protein surface. Ferredoxin could also be attached to domain II at the EtfAf-BcdAf interface (not shown) to provide a suitable electron transfer distance between the redox centers. However, the distance between the [4Fe-4S] cluster and α-FAD is about 14 Å significantly longer than that between the [4Fe-4S] cluster and β-FAD and thus the electron transfer is less likely.
FIGURE 7.
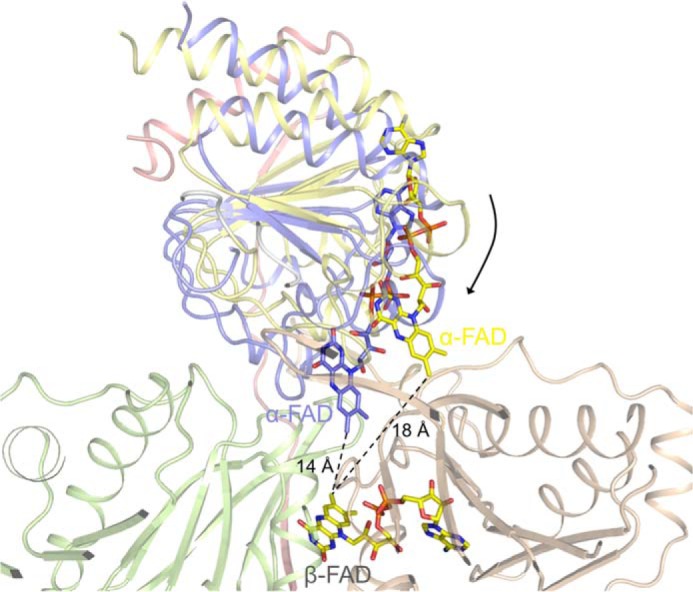
Modeling of domain II of EtfAf in a conformation in that the distance between the isoalloxazine rings of α-FAD and β-FAD are about 14 Å sufficient for an effective electron transfer. Because of the soft interface between the base of domain II and the domains I + III, a rotation of domain II is not severely restricted. It can also not be excluded that, alternatively, selective electron transfer between α-FAD and β-FAD is provided by fixing them in a distance that the rate is determined by the polypeptide tuned one-electron redox potentials of the isoalloxazine rings.
RESULTS AND DISCUSSION
Biochemical Characterization
To find out which of the two gene clusters encoding EtfAf are expressed under the conditions of glutamate fermentation by A. fermentans VR4 (DSM 20731) (27), the protein was purified from a cell-free extract, kinetically characterized by a formazane producing NADH-dependent assay (specific activity 2.5 units/mg of protein) (12) and analyzed by N-terminal sequencing and peptide mapping with MALDI-TOF mass spectrometry for gene identification. Accordingly, EtfAf is encoded by the genes Acfer_0556 and Acfer_0555, composed of 340 and 263 amino acids for subunits α and β, respectively.
To obtain EtfAf in higher amounts, we overproduced the enzyme with a C-terminal His tag at subunit α in E. coli. According to gel filtration, the pure protein (Fig. 1) is a heterodimer with a molecular mass of around 66 kDa (theoretically 37.6 + 28.4 kDa). Non-covalently bound FAD (and not AMP and other flavins such as riboflavin, riboflavin-5′-phosphate or hydroxylated flavins (28)) was identified by HPLC and MALDI-TOF analysis. Quantitative analyses using UV-visible spectroscopy (ϵ450 = 11.3 mm−1 cm−1) (29) revealed about 0.7 FAD/heterodimer, which could be raised to 2 upon incubation with 1 mm FAD, which agrees well with the structural data (see below). Thereby the specific activity of the EtfAf in the formazane assay increased 4-fold. Bcd of A. fermentans (BcdAf) was purified (Fig. 1) as homotetrameric flavoprotein (4 × 42 kDa) with a specific activity of 20 units/mg of protein (21). BcdAf is encoded by the Acfer_1477 gene (acyl-CoA dehydrogenase, 383 amino acids).
FIGURE 1.

SDS-PAGE of native BcdAf (lanes 1–3) and recombinant His-tagged EtfAf (lanes 4 and 5) at different purification stages. Each lane contained 10 μg of protein. Lane 1, cell-free extract of A. fermentans. Lane 2, supernatant after ammonium sulfate precipitation at 60% saturation. Lane 3, DEAE-Sepharose eluate. Lane 4, cell-free extract of E. coli producing EtfAf. Lane 5, eluate from the nickel-nitrilotriacetic acid column. EtfAf has an apparent molecular mass of 34 kDa for subunit α and 27 kDa for subunit β.
Kinetic Characterization
Although EtfAf and BcdAf are stable under air, all the following experiments were performed in an anaerobic chamber under an atmosphere of 95% N2 and 5% H2, since ferredoxin and the reduced forms of flavin are oxygen sensitive. Incubation of catalytic amounts of EtfAf and BcdAf with NADH and crotonyl-CoA caused oxidation of NADH at the low rate of 0.01 units mg−1 of Etf (units = μmol min−1). However, upon addition of catalytic amounts of ferredoxin (1 μm) and hydrogenase the rate increased to 1.5 units mg−1. The products of the reaction were besides NAD+, butyryl-CoA (determined by MALDI-TOF mass spectrometry and HPLC) and molecular hydrogen (determined by gas chromatography (3)). Hence, EtfAf + BcdAf catalyzed the reduction of crotonyl-CoA to butyryl-CoA coupled to the reduction of ferredoxin (Fd; Equation 1), whereby hydrogenase recycled the oxidized ferredoxin (Equation 2) resulting in Equation 3 (3).
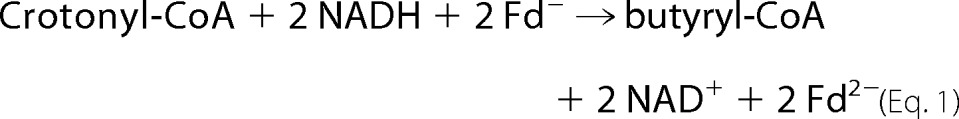 |
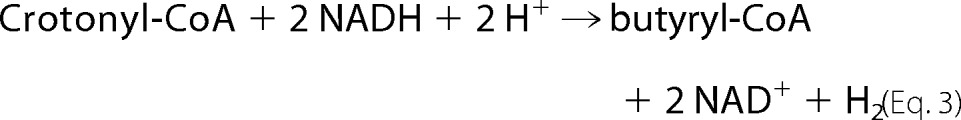 |
Omission of EtfAf, BcdAf, ferredoxin, or crotonyl-CoA gave no activity. No reaction was observed when NADH was replaced by NADPH or crotonyl-CoA by butyryl-CoA. The required stoichiometry of NADH:crotonyl-CoA = 2.1 ± 0.1 was measured (Fig. 2A) with a solution of the CoA thioester, whose concentration was calibrated by a coupled NAD-dependent assay using β-oxidation enzymes from A. fermentans (14). Because EtfAf and BcdAf are found as separated proteins in solution, a transient Bcd-Etf complex is sufficient to perform a bifurcation process.
FIGURE 2.

Bifurcation assays. See “Experimental Procedures” for details. A, stoichiometry between NADH and crotonyl-CoA of 2.1:1. The assays were performed with limiting amounts of crotonyl-CoA. B, dependence of the specific activity of NADH oxidation (μmol of NADH/mg of EtfAf) on the concentration of the BcdAf monomer. Each assay contained 0.5 μm EtfAf, 1 μm ferredoxin and hydrogenase (30 μg/ml). C, dependence of the specific activity of NADH oxidation (μmol of NADH/mg of EtfAf) on the concentration of ferredoxin used in the assay. Each assay contained 0.5 μm EtfAf, 1 μm BcdAf monomer and hydrogenase; Km (ferredoxin) = 0.2 μm. D, dependence of the specific activity of NADH oxidation (μmol of NADH/mg of EtfAf) on the concentration of NADH used in the assay. Each bifurcation assay contained 0.5 μm EtfAf, 1 μm BcdAf monomer, 1 μm ferredoxin and hydrogenase. E, complete reduction of 15 μm ferredoxin in the bifurcation assay without hydrogenase. (a) Spectrum of oxidized ferredoxin before starting the bifurcation; (b) excess dithionite was added; (c) after bifurcation has terminated due to limiting amounts of ferredoxin.
Titration of EtfAf with BcdAf increased the rate of NADH oxidation until a molar ratio of 2 Etf:1 Bcd (tetramer) was reached (Fig. 2B). This optimal ratio agrees well with the molecular masses of the stable clostridial Bcd-Etf complexes of Clostridium kluyveri (3), Clostridium difficile (30), and C. tetanomorphum (31, 32) with compositions of Etf1-Bcd(dimer) or Etf2-Bcd(tetramer) (see also Fig. 5) (5).
The optimal ratio of ferredoxin:Etf: tetrameric Bcd was determined to be 2:1:0.5 based on the titration of the EtfAf-BcdAf system with ferredoxin in the presence of hydrogenase (Fig. 2C). These data suggest that during steady state ferredoxin takes up only one electron and hands it over to the hydrogenase. Most likely the electron acceptor is the semi-reduced form (Fd−), which is completely reduced to Fd2− (see Equations 1 and 2). This behavior agrees well with the different redox potentials of the A. fermentans ferredoxin (E0′ = −340 for Fd/Fd− and −405 mV for Fd−/Fd2−) (8).
The dependence of the NADH oxidation rate on the concentration of crotonyl-CoA followed Michaelis-Menten kinetics, Km = 12 μm. By increasing the NADH concentrations the rate passed through a maximum already at about 70 μm NADH (Fig. 2D). This inhibition suggests equilibrium between the free proteins and their complex (EtfAf + BcdAf ⇌ EtfAf-BcdAf) whereby some of the free EtfAf gets fully reduced by NADH (see spectroscopic characterization) and cannot participate in electron bifurcation. In contrast, the tight non-dissociating bifurcating Etf-Bcd complex of C. difficile exhibits normal Michaelis-Menten kinetics for NADH, Km = 145 μm (30).
Using stoichiometric amounts of ferredoxin (15 μm) in the absence of hydrogenase, identical spectra in the range of 400–500 nm were observed, regardless of whether the reduction was performed by dithionite or by NADH and crotonyl-CoA mediated by EtfAf-BcdAf (Fig. 2E). As demonstrated for other flavin-based electron bifurcating systems (5, 6, 30), ferredoxin reduction is almost 100% indicating a tight energetic coupling between ferredoxin and crotonyl-CoA reduction. Limiting amounts of crotonyl-CoA under these conditions resulted in the reduction of 1 mol of ferredoxin/mol of crotonyl-CoA; hence at equilibrium the completely oxidized Fd accepts two electrons, one by each [4Fe-4S] cluster. In contrast to flavodoxin (33) the fully oxidized and half-reduced ferredoxins absorb at the same wavelength and therefore cannot be distinguished spectroscopically.
Structural Characterization
The structural basis of FAD-based electron bifurcation requires information about (i) the proteins Etf containing α-FAD and β-FAD, and Bcd containing Dh-FAD, (ii) the complexes of Etf-NAD+, Etf-ferredoxin, and Etf-Bcd, as well as (iii) the electron transfer routes between the redox centers.
Overall Structure of EtfAf
Structural analysis of recombinantly produced EtfAf resulted in an R/Rfree factor of 16.1/19.1% at 1.6-Å resolution (see Table 1). As crystallization was performed under aerobic conditions without reducing agents, both FADs are oxidized. Architecturally, heterodimeric Etf is composed of three domains: domains I (5–199) and II (215–340) from subunit α and domain III (1–219) from subunit β (Fig. 3A). A comparison with the structurally related electron-accepting Etfs (34–36) is presented in Fig. 3B. Domains I and III, essentially sharing the same fold (root mean square deviation of 4.6 Å; 208 Cα atoms used), consist of an α/β structure and a tightly associated β-meander. The polypeptides of subunits α and β proceed by strands 200:213 and 220:228 that enlarges the β-meander of the respective other subunit to a four-stranded antiparallel β-sheet. The major difference between domains I and III is an exposed β-hairpin-like segment following strand 2:7 of domain III (Fig. 3A). Domain II of subunit α is composed of a flavodoxin-like fold enlarged by a sixth strand and one helix from subunit β termed as the C-terminal arm (229–262) (Fig. 3A). Domains I and III are tightly associated (contact area of 4200 Å2). Domain II sits above this base in a shallow bowl and is hung up on opposite sides by the flexible linker connecting domains I and II (B-factor 210:213: >50 Å2) and the mentioned C-terminal arm of subunit β (Fig. 3A). Domains I + III and domain II are only loosely attached via a layer of solvent molecules mostly visible in the electron density at 1.6 Å but only by a few direct non-covalent polypeptide interactions.
α-FAD Binding
The completely occupied, non-covalently bound α-FAD is present in a stretched conformation (Fig. 4A) similar to that found in other Etf family members (34). The planar isoalloxazine ring embedded into a pocket at the domain II-III interface is flanked from the re-face by Arg-α253 and Tyr-β40 and from the si-face by Gln-α269, Gln-α289, and His-α290 (Fig. 4A). In contrast to the other structurally characterized Etfs, the guanidinium group of Arg-α253 is oriented coplanar to the isoalloxazine ring and interacts with subunit β via Tyr-β40, Asp-β182 (solvent-mediated) and via Arg-β173. Both guanidinium groups are stacked to each other (Fig. 4A). The N5 atom of the dihydropyrazine ring is completely shielded from bulk solvent and hydrogen bonded to Ser-α270-OG. The N1-C2 = O group of the pyrimidine ring is hydrogen bonded to the 4′-OH of ribityl, Val-α267-O, His-α290-ND1, and Arg-α253-NH, respectively, the latter being positioned at the N-terminal end of helix 253:258. These interactions, well conserved in Etfs (34–36) are considered a major structural feature to neutralize the negatively charged FAD⨪ and FADH− states (Fig. 4A) reflected in high redox potentials of the α-FAD/α-FAD⨪ couple (E0′ = +81 mV for EtfMe) and in a weakened form also of the α-FAD⨪/α-FADH− couple (E0′ = −136 mV for EtfMe) (37, 38). The higher value of the latter couple is attributable to the unfavorable Ser-α270-OG-N5(protonated) interactions. EtfMe is the Etf of M. elsdenii, a relative of A. fermentans, which also contains two FAD per heterodimer and shares 49% sequence identity. Altogether, α-FAD is not attractive as bifurcating flavin due to the high redox potential and the accompanied weak reduction power.
β-FAD Binding
The 1.6-Å electron density of EtfAf revealed that the non-covalently bound β-FAD is also completely occupied but arranged in a compressed S-shaped conformation at the C-terminal side of the central β-sheet of domain III (Figs. 3A and 4B). The AMP binding mode of EtfAf corresponds to that of the other structurally characterized Etfs (34) (Fig. 4B). The isoalloxazine ring protrudes beyond domain III toward domain I (Fig. 3A) with its nonpolar xylene ring being largely exposed to bulk solvent. The planar tricyclic ring is flanked from the si-face by Thr-β25, Gly-β123, Arg-α146, and Ala-α148 and from the re-face by Ile-α157, Thr-β221, and Val-β223 (Fig. 4B). The pyrimidine ring of β-FAD and the polypeptide are hydrogen-bonded via N3 and Leu-α127-O, O4 and Ala-α129-NH as well as O2 and Thr-β94-NH and -OG1, the latter being located at the positively charged N-terminal side of helix 94:108. Interestingly, O2 is further contacted by the carboxylate groups of Asp-β93 and Glu-β131 (exchanged in human Etf by Gly and Lys), which destabilizes a negatively charged state of the isoalloxazine ring (Fig. 4B). This finding is in agreement with the rather low redox potential (E0′ = −280 mV) of the β-FAD/β-FADH− couple and the observation that in EtfMe β-FAD binds stronger than α-FAD, and α-FADH− stronger than β-FADH− (38). An important function is also attributed to Arg-α146-NH2 in adjusting an even lower β-FAD/β-FADH• potential as it forms a stronger hydrogen bond with N5 in β-FAD than in β-FADH• and β-FADH−. As a result, β-FAD appears to be suitable as bifurcating flavin.
Structure of the EtfAf-NAD+ Complex
Binding of NAD+ was characterized on the basis of an EtfAf-NAD+ structure at 1.5-Å resolution after co-crystallizing EtfAf and NAD+ (Table 1). Its electron density only exhibits the ADP part, whereas the ribose-nicotinamide part is completely disordered (Fig. 4C). NAD+ appears to be primarily fixed to its binding site by interactions with β-FAD. A hydrogen bond is formed between the 2′-OH of the AMP-ribose of NAD+ and the ribitol hydroxyl groups of β-FAD. The diphosphate and ribose fraction of ADP is largely exposed to bulk solvent, whereas the adenine ring is sandwiched between the loops following strands 56:61 and 80:84. In agreement with the kinetic data, NADPH cannot replace NADH due to a collision between the 2′-phospho group and the protein scaffold.
Considering the ADP moiety as anchor point, the ribose-nicotinamide moiety can be reasonably modeled to the re-side of β-FAD in a stacking arrangement thereby underlining the biological relevance of the detected ADP as part of the NAD+ binding site. C4 of the nicotinamide ring and N5 of the isoalloxazine ring can be approached to around 3.5 Å (Fig. 4C) but the ribose-nicotinamide part clashes with the side chains of Ile-β157, Val-β223, and Ser-β226. As demonstrated by spectroscopic characterizations (see next paragraph) NADH reduces β-FAD in the absence of ferredoxin or Bcd and consequently can displace them. This induced displacement by NADH of about 3 Å might significantly affect the conformation of the C-terminal arm of subunit β, the β-meander of subunit α (domain I), and the extended β-hairpin following strand 2:7 of domain III (Fig. 4C).
Structure of BcdAf
The BcdAf structure was established at 1.9-Å resolution (R/Rfree = 15.3/18.3%) based on the M. elsdenii enzyme (24) as a model for molecular replacement calculations. The architecture of BcdAf and Bcd of M. elsdenii corresponds to each other with a root mean square deviation of 0.6 Å (95% of the Cα atoms) and a sequence identity of 66%. BcdAf is a homotetramer built up of a dimer of two tightly associated dimers (Fig. 5). Each subunit is composed of an amino-terminal α-helical bundle domain, a medial seven-stranded β-sheet domain, and a second α-helical domain at the carboxyl terminus. Dh-FAD is embedded between the two latter domains and its conformation and polypeptide surrounding is virtually identical in the two enzymes. Although no substrate analog was supplemented to the BcdAf solution, CoA persulfide (39) was bound to the re-side of the isoalloxazine ring.
Modeled Structures of the EtfAf-Ferredoxin and EtfAf-BcdAf Complexes
The ferredoxin of C. acidi-urici (40) (the structure of the closely related ferredoxin of C. tetanomorphum used in this work is not known) can be docked to domain III of Etf in a manner that the proximal [4Fe-4S] cluster and the isoalloxazine ring of β-FAD are below 8 Å apart from each other in the presence of NADH and nearly in van der Waals contact in its absence (Fig. 6).
The EtfAf-BcdAf complex can be modeled (Fig. 5) on the basis of the MCAD-Etf (medium chain acyl-CoA dehydrogenase-Etf from Homo sapiens) complex structures (36, 41). After superposition of EtfAf and BcdAf onto the MCAD-Etf complex, the distance between α-FAD and Dh-FAD of BcdAf is more than 30 Å. Different EtfAf-BcdAf contact areas were tested, but no orientation was found that meets the 14 Å distance criteria for an appropriate electron transfer rate between α-FAD and Dh-FAD (42). Therefore, we suggest a reorientation of domain II analogous to that observed between human Etf in the isolated and in complex with MCAD (43). The distance between the two FAD can thus be shortened to about 10 Å.
Electron Transfer between α-FAD and β-FAD
Based on the described findings, the one-electron transfer steps between β-FAD and α-FAD are a necessary prerequisite to explain the experimentally observed coupling between NADH oxidation at β-FAD and crotonyl-CoA reduction at Bcd mainly attached to domain II carrying α-FAD. However, the isoalloxazine rings of β-FAD and α-FAD have an edge-to-edge distance of 18 Å in the present EtfAf structure (Fig. 3A), which appears to be too long for an efficient electron transfer at biologically relevant rates (42). The EtfAf structure offers as solution a rigid-body movement of domain II relative to domains I + III, which reduces the distances between the isoalloxazine rings to about 14 Å (Fig. 7). These conformational changes are realistic because of the soft solvent-mediated domain II-(I + III) interface and their realization in the related Etf-MCAD and Etf-TMADH systems (36, 41). It might be triggered by productive NADH binding conveyed to domain II and/or by Bcd binding (43).
Spectroscopic Characterizations
The UV-visible absorption spectrum of recombinant EtfAf as isolated from E. coli containing 0.7 FAD/heterodimer is shown in Fig. 8. Upon overnight incubation at 4 °C with 1 mm FAD and removal of the excess FAD by gel filtration the spectrum changes to a much higher absorption of the two peaks at 375 and 450 nm and a less deep trough between the two peaks (Fig. 8). This reconstituted EtfAf contained 2 FAD/heterodimer. Stepwise addition of NADH in 2.0 μm amounts to 10 μm reconstituted EtfAf (occupied with 20 μm FAD) under anoxic conditions and in the absence of Bcd and ferredoxin (Fig. 9A) revealed a shift of the peak at 375 to 370 nm and a decrease of the 450 nm peak resulting in an equal intensity to the 370 nm peak (at 4–6 μm NADH). We attribute this 370 nm peak to a stable red anionic semiquinone (44). After addition of about 10 μm NADH the spectrum corresponds to a mixture of an oxidized and a reduced FAD. The unusual difference spectrum-oxidized half-reduced EtfAf with maxima at 400 and 450 nm is shown in Fig. 9B. Complete reduction of the FAD content was achieved at around 20 μm NADH (NADH/FAD = 1.0). Very similar spectra were recently reported from EtfMe (37, 38).
FIGURE 8.

UV-visible absorption spectrum of the electron transferring flavoprotein of A. fermentans. EtfAf as isolated from E. coli (green), reconstituted EtfAf (red), and KBr-treated EtfAf (blue).
FIGURE 9.

Formation of stable red anionic FAD semiquinones. A, to 10 μm Etf NADH was added stepwise: 0 (black line), 4 (khaki), 6 (red), 10 (blue), 14 (green), 18 (pink), and 20 μm (magenta). B, difference spectrum between unreduced EtfAf − EtfAf reduced with 10 μm NADH. It shows the UV-visible spectrum of α-FAD bound to EtfAf. C, to 10 μm Bcd-subunit (blue) and 10 μm Etf, NADH was added stepwise: 0 (black line), 2.5 (brown), 5 (pink), 7.5 (green), 10 (red), 12.5 (black), 15 (dark brown), 17.5 (light blue), 20 (magenta), and 22.5 μm (gray). D, same as C but instead of NADH, 100 μm butyryl-CoA was added. E, to 20 μm Bcd-subunit (black), 100 μm butyryl-CoA (red) or 200 μm butyryl-CoA (blue) were added.
To remove the weakly bound FAD, the protein was mixed with an equal volume of 2 m KBr and washed in a Centricon (30 kDa cut off) two times with 1 m KBr, followed by 50 mm potassium phosphate, pH 6.8 (38). The KBr-treated Etf exhibited a spectrum with absorbance at 375 and 450 nm of only 50% of that of the reconstituted Etf. Hence one of the two FADs was completely removed. Furthermore, there was a deep trough between the two peaks, like that of free FAD. A very similar behavior has been described for Etf from M. elsdenii (EtfMe) (37). Notably, titration of the KBr-treated EtfAf (10 μm) with NADH indicated a progressive reduction of both peaks, without the transient formation of the anionic semiquinone. Complete reduction was achieved with 10 μm NADH.
In context of the structural results we concluded that EtfAf contained a strongly bound β-FAD and a weekly bound α-FAD. Removal of α-FAD with KBr gave an Etf, whose β-FAD was smoothly reduced by NADH to β-FADH− without semiquinone intermediate. Upon reduction of the reconstituted Etf (2 FAD/Etf) by NADH, one electron of β-FADH− is further transferred to α-FAD yielding the stable anionic red semiquinone α-FAD⨪ (44). The remaining electron on the presumably blue neutral semiquinone β-FADH⨪ is not observable at 590 nm (44). We postulate an immediate intermolecular oxidation of the unstable β-FADH⨪ by reducing α-FAD of another oxidized EtfAf molecule to α-FAD⨪. Hence, the intermolecular electron transfer from β-FADH• to α-FAD should be faster than the intramolecular transfer of the second electron to α-FAD⨪. The observations that the maximum of the peak at 370 nm was already reached at 0.5 NADH/2 FAD and that both FAD are located at the surface of the EtfAf structure were compatible with an intermolecular electron transfer. Half-reduction of EtfAf and EtfMe (1 NADH/2 FAD) was interpreted as reduced α-FADH− and oxidized β-FAD (38), whereby the spectrum of α-FAD with a maximum at 400 nm turned out to be untypical for flavoproteins (Fig. 9B).
In the presence of 5 μm tetrameric BcdAf (20 μm Bcd subunit) and 10 μm EtfAf the peak at 370 nm reached its maximum at a ratio of 1 NADH/Etf (Fig. 9C) in contrast to 0.5 NADH/Etf in the absence of Bcd. Moreover, the peak of α-FAD⨪ at 370 nm rose much higher with than without BcdAf. Apparently, BcdAf binding (± crotonyl-CoA) to EtfAf increases the concentration of α-FAD⨪ by suppressing its further reduction to α-FADH− to some extent and/or changes its surrounding. The rotation of domain II predicted on the basis of the presented structural data would increase the distance between β-FAD and α-FAD and turn α-FAD from an accessible to a more shielded position. This reorientation might also implicate a decrease of the extremely high redox potential E0′ = +81 mV of α-FAD/α-FAD⨪ (for EtfMe) to ensure an electron transfer from α-FAD⨪ to Dh-FAD and eventually to crotonyl-CoA (E0′ = −10 mV). An increased peak at 370 nm was also achieved by adding butyryl-CoA to EtfAf + BcdAf instead of NADH (Fig. 9D). Obviously electrons can also flow in the reverse direction generating α-FAD⨪. Thus, EtfAf can also act as an electron carrier like the AMP containing Etfs. In a control experiment with Bcd alone no anionic semquinone was observed (Fig. 9E).
The additional absorbance increase between 500 and 700 nm (Fig. 9, C–E) could be due to an extremely broad band of a neutral semiquinone in Bcd, although the absorption maximum at 590 nm (44) is missing. A similar featureless band was observed with the FAD-containing 4-hydroxybutyryl-CoA dehydratase after addition of the substrate crotonyl-CoA (45). Concomitantly, EPR spectroscopy detected the formation of a neutral semiquinone (46, 47). Reduction of the dehydratase by light or with dithionite, however, gave the “normal” spectrum of a semiquinone with a maximum at 590 nm.
Proposed Mechanism
The reaction starts with a hydride transfer from NADH to the re-side of β-FAD of EtfAf forming β-FADH− (Fig. 10, A and B). Taking the two-electron redox potential of −280 mV for β-FAD/β-FADH− (38), bifurcation could lead to estimated one-electron potentials downhill to α-FAD of about −60 mV and uphill to ferredoxin of about −500 mV. The one-electron transfer from β-FADH− to α-FAD is thermodynamically feasible and presumably requires a reorientation of domain II to reduce the distance between the two isoalloxazine rings from 18 to 14 Å (Fig. 7). Simultaneously or subsequently the remaining β-FADH•, whose redox potential has dropped to <−500 mV, donates the other electron to the nearby ferredoxin (Fig. 10, C and D). See Fig. 6 for the predicted proximity between its [4Fe-4S] cluster and the isoalloxazine of β-FAD.
FIGURE 10.

Proposed mechanism of the flavin-based electron bifurcation process of the Etf/Bcd system on a structural ground. The figure shows the Bcd dimers (green and gray and light green) interacting with Etf (light blue, domain I; medium blue, domain II; dark blue, domain III). We propose that domain II changes its position relative to domains I and III. The small rectangles depict FAD in the quinone state (yellow), anionic semiquinone state (red), neutral semiquinone state (light blue in Etf and dark blue in Bcd), and hydroquinone state (white). For the sake of simplicity addition of one electron to oxidized ferredoxin (brown) yields reduced ferredoxin (white). The cycle starts with A containing three oxidized FAD. Butyryl-CoA and reduced ferredoxin, the products of the last turnover, are exchanged by the substrates crotonyl-CoA and NADH. As soon as bound, NADH immediately reduces β-FAD to β-FADH− (B). The formed NAD+ is released and ferredoxin binds. Bifurcation concomitant with a conformational change of domain II yields β-FADH• and α-FAD⨪, which swings to Dh-FAD (D-FAD) (C). Two one-electron transfers to ferredoxin and to Bcd end the first half of the cycle (D). Release of the first reduced ferredoxin and binding of the second NADH starts the second half of the cycle (E-F-A), the only difference being the action of Dh-FADH• rather than Dh-FAD as electron acceptor in Bcd. After the second bifurcation and electron transfers to ferredoxin and Dh-FADH•, the second reduced ferredoxin and butyryl-CoA are formed. It should be noted that only the α-FAD⨪ has been shown experimentally. The existence of the two neutral semiquinones, β-FADH• and Dh-FADH•, remains to be established. Also the binding order of NADH, crotonyl-CoA, and ferredoxin is without experimental evidence.
This electron flow from β-FADH• to ferredoxin is only accomplished if the thermodynamically more favorable electron transfer to α-FAD⨪ is prevented. Therefore, after the first electron transfer to α-FAD, we postulate a rotation of domain II toward the FAD binding site of BcdAf based on spectroscopic and structural data. This conformational change, concomitantly, also reduces the distance between α-FAD⨪ and Dh-FAD from about 30 to about 10 Å according to modeling studies with the determined EtfAf and BcdAf structures (Fig. 5) on the basis of the reported Etf-MCAD structure (41). Thus, α-FAD embedded into the weakly associated domain II serves as a shuttle between the electron-donating β-FADH− and the electron-accepting Dh-FAD. How the shuttle process is coordinated remains open. According to the presented data and the well explored Etf/acyl-CoA dehydrogenase systems (48), Bcd binding but also productive NADH, ferredoxin, and crotonyl-CoA binding might trigger domain II rearrangement. Interestingly, the electron transfer between the semiquinone in the bifurcating bc1 complex to the low potential heme bL is also ensured by an interruption of the second electron transfer between ubiquinone and the high potential [2Fe-2S] cluster because of a conformational change of the Rieske protein (49).
After reorientation of domain II, α-FAD⨪ donates an electron to Dh-FAD forming presumably Dh-FADH• (Fig. 10, C and D) (19). Repetition of this process affords a second reduced ferredoxin (Fd−) or Fd2− and the generated FADH− of Bcd transfers a hydride to crotonyl-CoA forming butyryl-CoA (Fig. 10, E-F-A).
The mechanism presented for flavin-based electron bifurcation in the EtfAf/BcdAf system can be extended to the other systems. Most likely, their flavin functions as β-FAD and the low-potential ferredoxin can be closely attached to it. Interestingly, in most cases α-FAD has been replaced by iron-sulfur clusters that should have potentials higher than that of ferredoxin and in the same range than that of the terminal high-potential electron acceptors. It might be difficult to tune a flavin in a way that it works in the other systems. The same arguments may explain the presence of the Rieske iron-sulfur cluster in the bifurcating bc1 complex of the respiratory chain.
Acknowledgments
We thank Professor Rolf K. Thauer (MPI of terrestrial Microbiology) for helpful discussions, Professor Hartmut Michel (MPI of Biophysics) for continuous support, the Biochemisches Institut, Universität Giessen, for the determination of the N-terminal sequences of Etf, Yvonne Thielmann and Barbara Rathmann of the Core Center (MPI of Biophysics) for performing initial crystallization screenings and the staff of the Swiss-Light-Source, Villigen, for help during data collection, Dr. Seigo Shima (MPI of terrestrial Microbiology) for help in flavin identification. N. P. C. acknowledges Dr. Anutthaman Parthasarathy (Stanford University) and Dr. Ankan Banerjee (MPI of terrestrial Microbiology) for constant encouragement.
This work was supported by grants from the Max-Planck Society, the German Research Foundation (DFG), and SYNMIKRO of the Philipps-Universität Marburg.
The atomic coordinates and structure factors (codes 4KPU, 4L21, and 4L1F) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- Etf
- electron transferring flavoprotein
- EtfAf
- Etf of A. fermentans
- EtfMe
- Etf of M. elsdenii
- Bcd
- butyryl-CoA dehydrogenase
- BcdAf
- Bcd of A. fermentans
- Fd/Fd−/Fd2−
- oxidized/semi-reduced/reduced ferredoxin
- BisTris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- Dh-FAD
- dehydrogenase FAD.
REFERENCES
- 1. Mitchell P. (1975) The protonmotive Q cycle. A general formulation. FEBS Lett. 59, 137–139 [DOI] [PubMed] [Google Scholar]
- 2. Herrmann G., Jayamani E., Mai G., Buckel W. (2008) Energy conservation via electron-transferring flavoprotein in anaerobic bacteria. J. Bacteriol. 190, 784–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Li F., Hinderberger J., Seedorf H., Zhang J., Buckel W., Thauer R. K. (2008) Coupled ferredoxin and crotonyl coenzyme A (CoA) reduction with NADH catalyzed by the butyryl-CoA dehydrogenase/Etf complex from Clostridium kluyveri. J. Bacteriol. 190, 843–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sousa F. L., Thiergart T., Landan G., Nelson-Sathi S., Pereira I. A., Allen J. F., Lane N., Martin W. F. (2013) Early bioenergetic evolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368, 20130088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Buckel W., Thauer R. K. (2013) Energy conservation via electron bifurcating ferredoxin reduction and proton/Na+ translocating ferredoxin oxidation. Biochim. Biophys. Acta 1827, 94–113 [DOI] [PubMed] [Google Scholar]
- 6. Bertsch J., Parthasarathy A., Buckel W., Müller V. (2013) An electron-bifurcating caffeyl-CoA reductase. J. Biol. Chem. 288, 11304–11311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang S., Huang H., Kahnt J., Thauer R. K. (2013) Clostridium acidurici electron-bifurcating formate dehydrogenase. Appl. Environ. Microbiol. 79, 6176–6179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thamer W., Cirpus I., Hans M., Pierik A. J., Selmer T., Bill E., Linder D., Buckel W. (2003) A two [4Fe-4S]-cluster-containing ferredoxin as an alternative electron donor for 2-hydroxyglutaryl-CoA dehydratase from Acidaminococcus fermentans. Arch. Microbiol. 179, 197–204 [DOI] [PubMed] [Google Scholar]
- 9. Boiangiu C. D., Jayamani E., Brügel D., Herrmann G., Kim J., Forzi L., Hedderich R., Vgenopoulou I., Pierik A. J., Steuber J., Buckel W. (2005) Sodium ion pumps and hydrogen production in glutamate fermenting anaerobic bacteria. J. Mol. Microbiol. Biotechnol. 10, 105–119 [DOI] [PubMed] [Google Scholar]
- 10. Biegel E., Müller V. (2010) Bacterial Na+-translocating ferredoxin:NAD+ oxidoreductase. Proc. Natl. Acad. Sci. U.S.A. 107, 18138–18142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Imkamp F., Biegel E., Jayamani E., Buckel W., Müller V. (2007) Dissection of the caffeate respiratory chain in the acetogen Acetobacterium woodii. Identification of an Rnf-type NADH dehydrogenase as a potential coupling site. J. Bacteriol. 189, 8145–8153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wohlfarth G., Buckel W. (1985) A sodium ion gradient as energy source for Peptostreptococcus asaccharolyticus. Arch. Microbiol. 142, 128–135 [DOI] [PubMed] [Google Scholar]
- 13. Parthasarathy A., Pierik A. J., Kahnt J., Zelder O., Buckel W. (2011) Substrate specificity of 2-hydroxyglutaryl-CoA dehydratase from Clostridium symbiosum. Toward a Bio-based production of adipic acid. Biochemistry 50, 3540–3550 [DOI] [PubMed] [Google Scholar]
- 14. Buckel W. (1986) Biotin-dependent decarboxylases as bacterial sodium pumps. Purification and reconstitution of glutaconyl-CoA decarboxylase from Acidaminococcus fermentans. In Methods of Enzymology (Fleischer S., Fleischer B., eds.), Vol. 125, pp. 547–558, Academic Press, New York [Google Scholar]
- 15. Hetzel M., Brock M., Selmer T., Pierik A. J., Golding B. T., Buckel W. (2003) Acryloyl-CoA reductase from Clostridium propionicum. An enzyme complex of propionyl-CoA dehydrogenase and electron-transferring flavoprotein. Eur. J. Biochem. 270, 902–910 [DOI] [PubMed] [Google Scholar]
- 16. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 17. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 18. Mayhew S. G., Massey V. (1969) Purification and Characterization of flavodoxin from Peptostreptococcus elsdenii. J. Biol. Chem. 244, 794–802 [PubMed] [Google Scholar]
- 19. Engel P. C., Massey V. (1971) The purification and properties of butyryl-coenzyme A dehydrogenase from Peptostreptococcus elsdenii. Biochem. J. 125, 879–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nakos G., Mortenson L. (1971) Purification and properties of hydrogenase, an iron sulfur protein, from Clostridium pasteurianum W5. Biochim. Biophys. Acta 227, 576–583 [DOI] [PubMed] [Google Scholar]
- 21. Lehman T. C., Hale D. E., Bhala A., Thorpe C. (1990) An acyl-coenzyme A dehydrogenase assay utilizing the ferricenium ion. Anal. Biochem. 186, 280–284 [DOI] [PubMed] [Google Scholar]
- 22. Ziegenhorn J., Senn M., Bücher T. (1976) Molar absorptivities of β-NADH and β-NADPH. Clin. Chem. 22, 151–160 [PubMed] [Google Scholar]
- 23. Kabsch W. (2010) Xds. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Djordjevic S., Pace C. P., Stankovich M. T., Kim J. J. (1995) Three-dimensional structure of butyryl-CoA dehydrogenase from Megasphaera elsdenii. Biochemistry 34, 2163–2171 [DOI] [PubMed] [Google Scholar]
- 25. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 26. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 27. Chang Y. J., Pukall R., Saunders E., Lapidus A., Copeland A., Nolan M., Glavina Del Rio T., Lucas S., Chen F., Tice H., Cheng J. F., Han C., Detter J. C., Bruce D., Goodwin L., Pitluck S., Mikhailova N., Liolios K., Pati A., Ivanova N., Mavromatis K., Chen A., Palaniappan K., Land M., Hauser L., Jeffries C. D., Brettin T., Rohde M., Göker M., Bristow J., Eisen J. A., Markowitz V., Hugenholtz P., Kyrpides N. C., Klenk H. P. (2010) Complete genome sequence of Acidaminococcus fermentans type strain (VR4). Stand. Genomic. Sci. 3, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Whitfield C. D., Mayhew S. G. (1974) Purification and properties of electron-transferring flavoprotein from Peptostreptococcus elsdenii. J. Biol. Chem. 249, 2801–2810 [PubMed] [Google Scholar]
- 29. Dawson R. M. C., Elliott D. C., Elliott H. C., Jones K. M. (1986) Data for Biochemical Research, 3 Ed., Clarendon Press, Oxford [Google Scholar]
- 30. Aboulnaga el-H., Pinkenburg O., Schiffels J., El-Refai A., Buckel W., Selmer T. (2013) Butyrate production in Escherichia coli. Exploitation of an oxygen tolerant bifurcating butyryl-CoA dehydrogenase/electron transferring flavoprotein complex from Clostridium difficile. J. Bacteriol. 195, 3704–3713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Herrmann T. G. E. (2008) Enzymes of two clostridial amino-acid fermentation pathways. Ph.D. thesis, Philipps-Universität, Marburg, Germany [Google Scholar]
- 32. Kölzer S. (2008) Aufreiningung und Charakterisierung des butyryl-CoA dehydrogenase/ETF komplexes aus Clostridium tetanomorphum. Master thesis, Philipps-Universität, Marburg, Germany [Google Scholar]
- 33. Hans M., Bill E., Cirpus I., Pierik A. J., Hetzel M., Alber D., Buckel W. (2002) Adenosine triphosphate-induced electron transfer in 2-hydroxyglutaryl-CoA dehydratase from. Acidaminococcus fermentans. Biochemistry 41, 5873–5882 [DOI] [PubMed] [Google Scholar]
- 34. Roberts D. L., Frerman F. E., Kim J. J. (1996) Three-dimensional structure of human electron transfer flavoprotein to 2.1-Å resolution. Proc. Natl. Acad. Sci. U.S.A. 93, 14355–14360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roberts D. L., Salazar D., Fulmer J. P., Frerman F. E., Kim J. J. (1999) Crystal structure of Paracoccus denitrificans electron transfer flavoprotein. Structural and electrostatic analysis of a conserved flavin binding domain. Biochemistry 38, 1977–1989 [DOI] [PubMed] [Google Scholar]
- 36. Leys D., Basran J., Talfournier F., Sutcliffe M. J., Scrutton N. S. (2003) Extensive conformational sampling in a ternary electron transfer complex. Nat. Struct. Biol. 10, 219–225 [DOI] [PubMed] [Google Scholar]
- 37. Sato K., Nishina Y., Shiga K. (2003) Purification of electron-transferring flavoprotein from Megasphaera elsdenii and binding of additional FAD with an unusual absorption spectrum. J. Biochem. 134, 719–729 [DOI] [PubMed] [Google Scholar]
- 38. Sato K., Nishina Y., Shiga K. (2013) Interaction between NADH and electron-transferring flavoprotein from Megasphaera elsdenii. J. Biochem. 153, 565–572 [DOI] [PubMed] [Google Scholar]
- 39. Williamson G., Engel P. C., Mizzer J. P., Thorpe C., Massey V. (1982) Evidence that the greening ligand in native butyryl-CoA dehydrogenase is a CoA persulfide. J. Biol. Chem. 257, 4314–4320 [PubMed] [Google Scholar]
- 40. Murthy H. M., Hendrickson W. A., Orme-Johnson W. H., Merritt E. A., Phizackerley R. P. (1988) Crystal structure of Clostridium acidi-urici ferredoxin at 5-Å resolution based on measurements of anomalous X-ray scattering at multiple wavelengths. J. Biol. Chem. 263, 18430–18436 [PubMed] [Google Scholar]
- 41. Toogood H. S., van Thiel A., Basran J., Sutcliffe M. J., Scrutton N. S., Leys D. (2004) Extensive domain motion and electron transfer in the human electron transferring flavoprotein/medium chain acyl-CoA dehydrogenase complex. J. Biol. Chem. 279, 32904–32912 [DOI] [PubMed] [Google Scholar]
- 42. Moser C. C., Keske J. M., Warncke K., Farid R. S., Dutton P. L. (1992) Nature of biological electron transfer. Nature 355, 796–802 [DOI] [PubMed] [Google Scholar]
- 43. Toogood H. S., van Thiel A., Scrutton N. S., Leys D. (2005) Stabilization of non-productive conformations underpins rapid electron transfer to electron-transferring flavoprotein. J. Biol. Chem. 280, 30361–30366 [DOI] [PubMed] [Google Scholar]
- 44. Massey V., Palmer G. (1966) On the existence of spectrally distinct classes of flavoprotein semiquinones. A new method for the quantitative production of flavoprotein semiquinones. Biochemistry 5, 3181–3189 [DOI] [PubMed] [Google Scholar]
- 45. Müh U., Cinkaya I., Albracht S. P., Buckel W. (1996) 4-Hydroxybutyryl-CoA dehydratase from Clostridium aminobutyricum. Characterization of FAD and iron-sulfur clusters involved in an overall non-redox reaction. Biochemistry 35, 11710–11718 [DOI] [PubMed] [Google Scholar]
- 46. Cinkaya I., Buckel W., Medina M., Gomez-Moreno C., Cammack R. (1997) Electron-nuclear double resonance spectroscopy investigation of 4-hydroxybutyryl-CoA dehydratase from Clostridium aminobutyricum. Comparison with other flavin radical enzymes. Biol. Chem. 378, 843–849 [DOI] [PubMed] [Google Scholar]
- 47. Näser U., Pierik A. J., Scott R., Cinkaya I., Buckel W., Golding B. T. (2005) Synthesis of 13C-labeled γ-hydroxybutyrates for EPR studies with 4-hydroxybutyryl-CoA dehydratase. Bioorg. Chem. 33, 53–66 [DOI] [PubMed] [Google Scholar]
- 48. Toogood H. S., Leys D., Scrutton N. S. (2007) Dynamics driving function. New insights from electron transferring flavoproteins and partner complexes. FEBS J. 274, 5481–5504 [DOI] [PubMed] [Google Scholar]
- 49. Zhu J., Egawa T., Yeh S. R., Yu L., Yu C. A. (2007) Simultaneous reduction of iron-sulfur protein and cytochrome bL during ubiquinol oxidation in cytochrome bc1 complex. Proc. Natl. Acad. Sci. U.S.A. 104, 4864–4869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sheldrick G. M. (2008) A short history of SHELX. Acta Crystallogr. A 64, 112–122 [DOI] [PubMed] [Google Scholar]
- 51. De la Fortelle E., Bricogne G. (1997) Maximum-likelihood heavy-atom parameter refinement for multiple isomorphus replacement and multiwavelength anomalous diffraction method. In Methods of Enzymology (Carter C. W., ed.) Vol. 276, pp. 472–494, Academic Press, San Diego: [DOI] [PubMed] [Google Scholar]
- 52. Abrahams J. P., Leslie A. G. (1996) Methods used in the structure determination of bovine mitochondrial F1 ATPase. Acta Crystallogr. D Biol. Crystallogr. 52, 30–42 [DOI] [PubMed] [Google Scholar]
- 53. Afonine P. V., Grosse-Kunstleve R. W., Chen V. B., Headd J. J., Moriarty N. W., Richardson J. S., Richardson D. C., Urzhumtsev A., Zwart P. H., Adams P. D. (2010) phenix.model_vs_data. A high-level tool for the calculation of crystallographic model and data statistics. J. Appl. Crystallogr. 43, 669–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Langer G., Cohen S. X., Lamzin V. S., Perrakis A. (2008) Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]