

Abstract
Lung cancer is primarily caused by exposure to tobacco smoke. Tobacco smoke contains numerous carcinogens, including Polycyclic Aromatic Hydrocarbons (PAH). The most common PAH studied is benzo[a]pyrene (B[a]P). B[a]P is metabolically activated through multiple routes, one of which is catalyzed by aldo-keto reductase (AKR) to B[a]P-7,8-dione (BPQ). BPQ undergoes a futile redox cycle in the presence of NADPH to generate reactive oxygen species (ROS). ROS, in turn, damages DNA. Studies with a yeast p53 mutagenesis system found that the generation of ROS by PAH o-quinones may contribute to lung carcinogenesis because of similarities between the patterns (types of mutations) and spectra (location of mutations) and those seen in lung cancer. The patterns were dominated by G to T transversions, and the spectra in the experimental system have mutations at lung cancer hotspots. To address repair mechanisms that are responsible for BPQ induced damage we observed the effect of mutating two DNA repair genes OGG1 and APE1 (APN1 in yeast) and tested them in a yeast reporter system for p53 mutagenesis. There was an increase in both the mutant frequency and the number of G:C/T:A transversions in p53 treated with BPQ in ogg1 yeast but not in apn1 yeast. Knocking out APN2 increased mutagenesis in the apn1 cells. In addition, we did not find a strand bias on p53 treated with BPQ in ogg1 yeast. These studies suggest that Ogg1 is involved in repairing the oxidative damage caused by BPQ, Apn1 and Apn2 have redundant functions and that the stand bias seen in lung cancer may not be due to impaired repair of oxidative lesions.
Keywords: PAH metabolism, Base excision repair, OGG1, APN1, APN2, Lung cancer
1. Introduction1
The major risk factor for lung cancer is exposure to tobacco smoke [1]. There are about 50 known carcinogens in tobacco smoke, but some of the most established are the polycyclic aromatic hydrocarbons (PAH). PAH are ubiquitous combustion products and are also found in charbroiled foods, coal smoke and car exhaust [2, 3]. The most common PAH found in tobacco smoke is benzo[a]pyrene (B[a]P). B[a]P does not react with DNA and must undergo metabolic activation to become mutagenic. There are three metabolic pathways that B[a]P can go through to become what are known as ultimate carcinogens, which are compounds that directly bind to and damage DNA. The first pathway involves the conversion of B[a]P to radical cations, which can form depurinating adducts by utilizing P450 peroxidases [4, 5]. The second and third pathways involve the formation of an intermediate product known as B[a]P-7,8-trans-dihydrodiol (Diol), through the combined action of cytochrome p4501A1 (CYP1A1) and epoxide hydrolases [6, 7]. B[a]P-7,8-trans-dihydrodiols can either be metabolized to to (±)anti-BPDE through the actions of CYP1A1 and CYP1B1 or metabolized by aldo-keto reductases (AKRs) to form another intermediate product, catechols [8]. (±)anti-BPDE is highly mutagenic and forms bulky adducts with DNA [9, 10]. Catechols can undergo two spontaneous oxidation reactions to form B[a]P-7,8-dione (BPQ) [8]. BPQ can form both stable and depurinating adducts [11, 12], however, based on measurements of oxidized macromolecules, the majority of the DNA damage that occurs from BPQ is through the production of reactive oxygen species (ROS), which is generated during a futile redox cycle in the presence of NADPH. Under redox cycling conditions, catechols spontaneously oxidize to o-quinones and are reduced back to catechols through enzymatic or non-enzymatic reduction in the presence of NADPH [8, 13–15].
Each pathway, diol epoxides, quinones and radical cations metabolizes PAH at comparable rates in cells [16]. The diol epoxide pathway is supported by studies showing that B[a]P-7,8-trans-dihydrodiol epoxide adducts are found in smokers lungs and the location of DNA adducts can be mapped to known hotspots on the tumor suppressor p53 [17]. Data showing that smoking causes oxidative stress, which can be measured by examining antioxidant levels [18] and elevated levels of the oxidative lesion 8-oxo-2′-deoxyguanosine (8-oxo-dGuo), support the PAH o-quinone pathway [19–24]. There have also been reports that products of radical cation damage and depurinating adducts are present in PAH treated mice and cells [5, 25]. Lesions caused by both anti-BPDE adducts and ROS cause G to T transversions on p53 [26], the major mutation found on p53 in lung cancer [27, 28], while the radical cation pathway is less mutagenic [29].
For mutations to lead to cancer, they must occur in key driver genes. The most commonly mutated gene in lung cancer is the tumor suppressor p53 [30]. p53 is a transcription factor responsible regulating cell cycle progression and apoptosis. Mutations in p53 result in unregulated cell cycle progression and may lead to carcinogenesis. Although p53 is mutated in many cancers, there are three features in lung cancer that result in a “signature” [27]. The first feature is that most of the mutations are G to T transversions. G to T transversions are rare in most other cancers. The second feature is that there is a strand bias seen on p53 in lung cancers. A strand bias occurs when there are more mutations on the coding strand compared to the transcribed strand. Specifically, there are more guanines that are mutated on the coding strand compared to the transcribed strand of p53 in lung cancers [31]. This is reflected in the observation that there are more G to T transversions than the reciprocal transversions C to A. The third feature of the p53 signature is that there are hotspot codons, in that about 23 codons account for about 50% of all mutations. The main hotspot codons include, but are not limited to, codon 157, 158, and 248 and the majority of these mutations are G to T transversions. However the hotspot codons on p53 are also mutated in other cancers so this attribute is not unique to lung cancers [30].
Reactive oxygen species may play a key role in the induction of lung cancer by generating 8-oxo-dGuo. 8-oxo-dGuo is usually repaired by the base excision repair (BER) pathway. Two DNA repair genes in the BER pathway that repair oxidative lesions are OGG1 and APE1 (APN1 in yeast). Ogg1 is a bi-functional glycosylase, in that it has both AP lyase and DNA glycosylase activity and one of its functions is to excise and remove 8-oxo-dGuo from DNA [32, 33]. It has been reported that there is a loss of heterozygosity of OGG1 in small cell lung cancers, and low levels of Ogg1 activity are associated with an increased risk of cancer [34, 35]. This suggests that not only is there an increase in ROS and oxidative damage during the induction of lung cancer, but also that reduced efficiency of oxidative damage repair is a contributing factor in carcinogenesis.
The other BER gene, APE1, is a type II apurinic/apyrimidinic (AP) endonuclease. Ape1 cleaves the 5′ end of an AP site after a damaged base is removed by a mono-functional glycosylase [36]. Ape1 works in conjunction with Ogg1, a bi-functional glycosylase, and other BER pathway enzymes to remove oxidative lesions such as 8-oxo-dGuo. Ape1, through its 3′ phosphodiesterase activity, removes the 3′ blocking end of the AP site, which is formed after Ogg1 removes the 8-oxo-dGuo formed during oxidative damage [37, 38]. Ape1 can enhance the glycosylase activity of Ogg1 suggesting the two genes work together to repair the oxidative damaged caused by ROS [39, 40].
In this study we addressed the role of the BER enzymes, Ogg1, Apn1 (yeast homologue of Ape1) and Apn2 in BPQ induced p53 mutagenesis. Using a yeast system that utilizes a red/white selection paradigm to test for mutations in p53 cDNA, we determined if knocking out OGG1, APN1 or APN1 and APN2 affected the mutant frequency, the mutation pattern and mutation spectrums of BPQ treated p53. To do this, we treated p53 cDNA with BPQ under redox cycling conditions and measured the mutant frequencies of p53 in wild type (yIG397), ogg1, apn1 and apn1apn2 yeast strains. We then isolated and sequenced the mutant p53 to determine the mutation patterns and spectrums of the mutations. The loss of Ogg1, but not Apn1, increased the mutant frequency and the incidence of G to T transversions in BPQ mutagenesis of p53. Although the loss of APN1 did not result in increase in mutant frequency, the loss of APN1 and APN2 increased mutant frequency of p53 approximately 3 fold compared to wild type. This suggests that Ogg1 plays a major role in BPQ induced DNA damage while Apn1 and Apn2 have a redundant role in oxidative damage caused by BPQ.
2. Materials and Methods
Caution: All PAHs are potentially hazardous and should be handled in accordance with NIH Guidelines for the Laboratory Use of Chemical Carcinogens.
2.1. Chemicals and Reagents
Adenine, L-leucine, L-tryptophan, cupric chloride and H2O2 were purchased from Sigma-Aldrich (St. Louis, MO). YEASTMAKER Yeast Transformation System Kit and all yeast culture media were obtained from CLONTECH (Palo Alto, CA). Yeast plasmid isolation kit was obtained from GE Healthcare Biosciences (Piscataway, NJ). NADPH tetrasodium salt was purchased from Calbiochem (San Diego, CA). B[a]P-7,8-dione was obtained from the National Cancer Institute, Chemical Carcinogen Standard Reference Repository (Midwest-Research Institute, Kansas City, Missouri). The purity of B[a]P-7,8-dione was assessed by LC/MS. All other reagents were the highest grade available.
2.2. Yeast Strains, Media and Plasmid
The ade reporter yeast strain (yIG397) and the pSS16 GAP repair vector was kindly provided by Dr. Richard Iggo (Swiss Institute for Experimental Cancer Research, 1066 Epalinges, Basel, Switzerland) [41]. To construct the mutant yeast strains, PCR was first performed on a pFA6a-kanMX6 vector, which contains the kanamycin gene, using modified open reading frame (ORF) deletion primers for OGG1 and APN1 published on the homepage of the Yeast Deletion Database website (http://www-sequence.stanford.edu/group/yeast_deletion_project/deletions3.html). The APN1 and APN2 double knock-out pcr constructs were generated using pFa6a-HIS3MX6 plasmid kindly provided by Erfei Bi (University of Pennsylvania). The expected 1 kb PCR product, containing homologous ends for each gene to be deleted, was purified by the PCR purification kit by QIAGEN (Valencia, CA). Approximately 1–5 μg of the purified PCR fragment was transformed into the yIG397 yeast strain using the lithium acetate procedure according to the Yeast Transformation System Kit by CLONTECH (Palo Alto, CA). After transformation the cells were plated on either YPD agar plates containing 200μg/mL G418 or –HIS plates. To confirm that the colonies that grew had indeed knocked out our gene of interest, genomic DNA was isolated from the selected yeast colonies using QIAGEN genomic DNA isolation kit and protocol (Valencia, CA). Next, PCR was performed on the genomic DNA isolated from our mutant and wild type yeast strains using conformation primers published on the Yeast Deletion Database website. The PCR revealed that the genes of interest, OGG1, APN1 and APN2, were indeed knocked out. To further confirm that we had knocked out each gene individually, we performed quantitative Real-time PCR using the Fast Taqman Assay obtained from Applied Biosystems (Carlsbad, CA) and determined the mRNA expression levels of OGG1 and APN1 in ogg1, apn1 and yIG397 (wild type) yeast strains. Our results confirmed that the OGG1 and APN1 message was completely knocked out in our ogg1 and apn1 yeast strains respectively (Fig.1B). The Fast Taqman Assay ids that were used are as follows: yeast OGG1 (Sc04151802_s1), yeast APN1 (Sc04141152_s1) and yeast tubulin1 (Sc04175846_s1).
Figure 1.
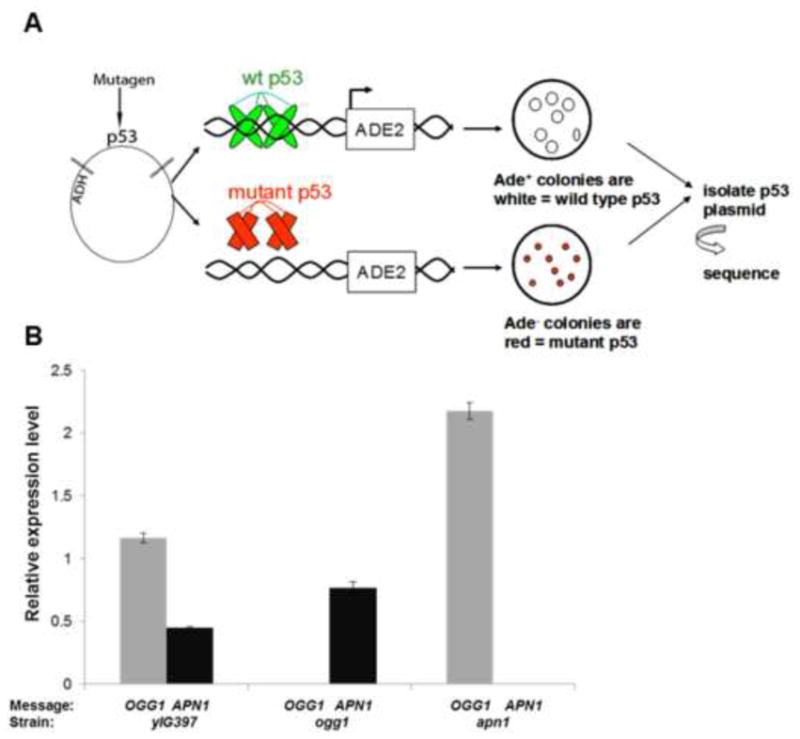
The yeast p53 assay and real-time PCR confirming the mutant yeast strains. A) Schematic diagram showing the assay used to determine p53 mutant frequency (based on red/white selection), mutation pattern and mutation spectrum in wild type (yIG397), ogg1 and apn1 yeast. B) Realtime PCR measuring expression levels of OGG1 and APN1 mRNA in yIG397, ogg1 and apn1 yeast strains.
2.3. p53 Mutagenesis Assay
Basic manipulation of yeast methods were carried out as described [42] and the p53 mutagenesis assay was carried out as described with minor modifications [13]. Approximately 4 μg of the plasmid DNA containing the p53 cDNA was treated with 100 μM of H2O2 and 100 μM CuCl2 in 100 mM of potassium phosphate buffer (pH 7.0) at a final volume of 50 μL for 2 hrs at 37°C. For BPQ treatment, approximately 4 μg of plasmid DNA was treated with .250 μM of BPQ with 100 μM of CuCl2 and 1mM of NADPH in 100 mM potassium phosphate buffer (pH. 7.0) at a final volume of 50 μL for 2 hrs at 37°C. NADPH and CuCl2 treatment alone, without BPQ, was used as a control to determine background mutant frequencies. After treatment the plasmid DNA was then precipitated by adding 1/10 the volume of 3M sodium acetate (pH. 5.5) and 2.5 volumes of 100% ethanol, frozen at −80°C for at least 2 hrs and spun in a centrifuge at 13000 rpm for 20 minutes at 4°C. The plasmid was then washed with 70% ethanol and re-centrifuged at 13000 rpm, air dried and suspended in 10 μL of sterile H2O. The plasmid DNA was then transformed into the yIG397, ogg1, apn1 or apn1apn2 yeast strains using the lithium acetate procedure according to the YEASTMAKER Yeast Transformation System Kit (CLONTECH). After transformation the yeast were plated on synthetic minimal medium minus leucine plus minimal adenine (5 μg/mL) and incubated at 30°C for 3–5 days. The yIG397 strain contains an ADE2 reporter linked to a p53 promoter stably integrated in its genome. Wild type p53 activates the ADE2 reporter and colonies appear white on low ADE plates while mutant p53 does not activate the ADE2 reporter and colonies appear red. The red colonies are more prominent after incubation of the plates at 4°C for 2–3 days. The mutant frequency is determined by number of red colonies divided by the total colonies on the plate.
2.4. Isolation and sequencing of p53 plasmids
The plasmid DNA was isolated using the Yeast Plasmid Isolation kit from GE Healthcare Biosciences (Piscataway, NJ). To amplify the plasmid for sequencing, we transformed the plasmid into Electromax DH10B electrocompetent cells (Life Technologies, San Diego, CA) using a Bio-Rad (Hercules, CA) Gene Pulser Electroporator according to the instruction manual. After transfections bacterial colonies were grown overnight in LB plus ampicillin (50 μG/mL) and plasmid DNA isolated using QIAGEN mini-prep kit. The DNA binding domain of p53 (codons 126 to 339) was sequenced on both strands with S6 (5′-dCTGGGACAGCCAAGTCTGT-3′) and R6 (5′-dCCTCATTCAGCTCTCGGAA-3′) primers. Sequencing was performed on an Applied Biosystems 373A automated sequencer in the DNA sequencing facility at the Perelman School of Medicine, University of Pennsylvania.
3. Results
3.1. Increase in the mutant frequency of p53 treated with B[a]P-7,8-dione (BPQ) and H2O2 in ogg1 and apn1apn2 yeast
To address the role of DNA repair genes in BPQ induced mutagenesis of p53, we first knocked out two major yeast repair genes that are responsible for repairing oxidative damage, OGG1 and APN1. To construct the knock-outs we used a modified PCR protocol with published open reading frame (ORF) deletion primers from the Yeast Deletion Database. We confirmed that our genes of interest were knocked out by PCR using published primers from the Yeast Deletion Database website (http://www.sequence.stanford.edu/group/yeast_deletion_project/deletions3.html). In addition to genomic PCR, we confirmed that the genes of interest were knocked-out by measuring the expression levels of OGG1 and APN1 mRNA by real-time PCR. Real-time PCR showed that OGG1 mRNA was not present in the ogg1 mutant strain and APN1 was not present in the apn1 mutant strain while both OGG1 and APN1 mRNA were present in the wild type (yIG397) yeast strain (Fig. 1B). Next, using the experimental paradigm illustrated in Figure 1, we tested the mutant frequency, which is measured by the number of red mutant p53 colonies versus the total colonies on a plate. There was a 2.5 fold increase in the percentage of red colonies in ogg1 yeast transformed with H2O2 treated p53 cDNA compared to the yIG397 strain (Fig. 2A), suggesting that OGG1 is required to repair oxidative damage caused by H2O2 in our system. In addition we found a statistically significant (p<.01) increase in mutant frequency in p53 treated with H2O2 in apn1apn2 yeast compared to wild type (Fig. 2B), but not in apn1 yeast. To determine if Ogg1, Apn1or Apn1 and Apn2 played a role in repairing quinone induced damage on p53, we treated the p53 plasmid cDNA with B[a]P-7,8-dione (BPQ) under redox cycling conditions (CuCl2+NADPH) and transformed the treated cDNA into our wild type, ogg1, apn1 or apn1apn2 yeast strains. We found that there was a 1.8 fold increase in the percentage of red colonies in ogg1 yeast strain compared to the wild type strain (Fig. 3A). We did not, however, find a significant change in the mutant frequency of p53 treated with BPQ in apn1 mutant yeast (Fig. 3B). We also conducted the same experiments with APN1 and APN2 double knock-out yeast strains and found that there was a three fold increase in the mutant frequency of p53 in apn1apn2 yeast compared to wild type (Fig. 3C). These data suggests that Ogg1 plays a direct role in repairing BPQ induced damage on p53 cDNA and that Apn1 alone may not play an essential role in repairing BPQ induced DNA damage. However, since we found an increase in mutant frequency of p53 in apn1apn2 yeast, these proteins may play a redundant role in repairing BPQ induced p53 DNA damage.
Figure 2.
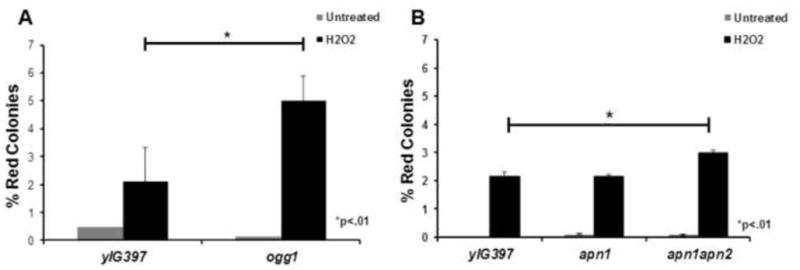
Increased mutant frequency of p53 treated with H2O2 in ogg1 and apn1apn2 yeast. A) Red/white yeast colony assay shows a 2.5 fold increase in the mutant frequency of p53 treated with H2O2 in ogg1 yeast compared to wild type control. B) Significant increase in the mutant frequency of p53 treated with H2O2 in apn1apn2 yeast compared to wild type yeast.
Figure 3.
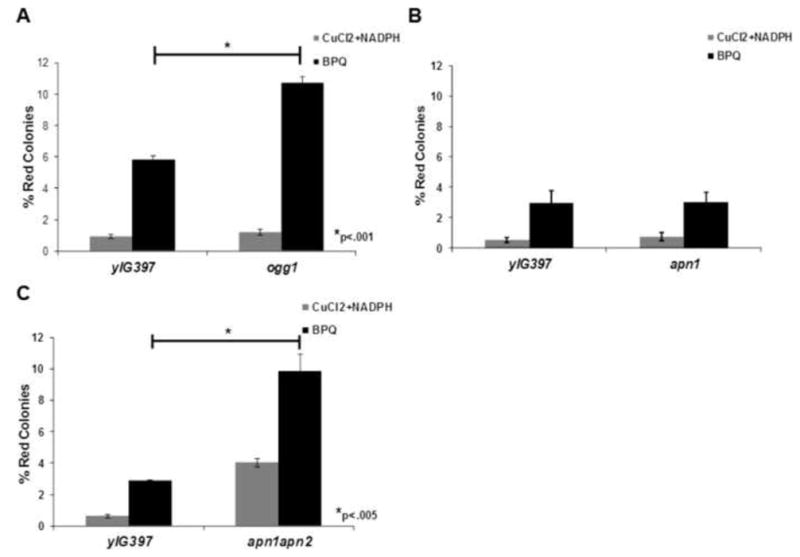
Increase in the mutant frequency of p53 treated with BPQ in ogg1 and apn1apn2 yeast. A) Graph showing a 1.8 fold increase in the mutant frequency of p53 treated with BPQ in ogg1 yeast. B) No difference in mutant frequency was observed in p53 treated with BPQ in apn1 yeast. C) Significant increase in mutant frequency was observed in p53 treated with BPQ in apn1apn2 yeast compared to wild type yeast.
3.2. Loss of Ogg1 increases the frequency of G to T transversions
After determining the mutant frequency of p53 cDNA in wild type, ogg1 and apn1 yeast strains, we isolated the mutant p53 plasmids from the red yeast colonies and sequenced the DNA. In the wild type yeast strain we found 34% of the mutations were G:C/T:A transversions, 40% were G:C/A:T transitions, 8% were G:C/C:G transversions and less than 5% for the other three possible changes (Fig 4A). Interestingly, in the ogg1 yeast strain we found that 62% were G:C/T:A transversions 30% were G:C/A:T transitions and less than 5% for the other four possible changes (Fig. 4B). There was an 1.8 fold increase of G:C/T:A transversions in the ogg1 yeast strain compared to the wild type strain. We did not, however, find a strand bias, more mutations on the coding strand than the transcribed strand, as reported previously in an ogg1 mouse model [43]. In apn1 yeast we found that 31% of the mutations were G:C/T:A transversions, 33% were G:C/A:T transitions, 15% were G:C/C:G transversions, 20% were A:T/G:C transitions and less than 5% for the other two possible changes (Fig. 4C). We did find a trend towards a strand bias in our apn1 yeast strain where 9 of the mutations were G to T transversions and 3 of the mutations were C to A transversions. This data was close to being statistically significant in that the two tailed p value was .08 in the Chi-square test (Data not shown). These data support a major role for Ogg1 in repairing BPQ induced DNA damage, but less so for Apn1.
Figure 4.
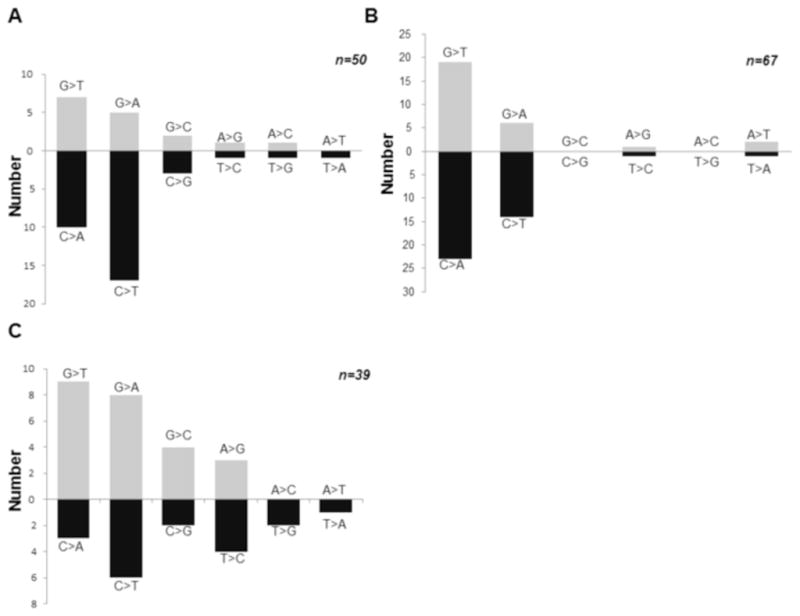
Mutation pattern of p53 treated with BPQ in wild type, oggl and apn1 yeast. A) Mutation pattern of p53 treated with BPQ in wild type yeast shows that 34% of the mutations were G:C/T:A transversions indicative of increased oxidative DNA damage. Data is plotted with complementary changes below which would reflect a similar change on the other DNA strand. B) Mutation pattern of p53 treated with BPQ in ogg1 yeast shows that 63% of the mutations were G:C/T:A transversions suggesting that Ogg1 is required to repair oxidative damaged caused by BPQ. C) Mutation pattern of p53 treated with BPQ in apn1 yeast shows a high percentage of G:C/T:A transversions as well as a trend toward a strand bias.
3.3. Mutation Spectrum of BPQ treated plasmid p53 cDNA in mutant yeast does not show preferential mutations at cancer hotspots
The mutation spectrum of BPQ treated p53 cDNA isolated from wild type yeast shows that the majority of the mutations did not occur at hotspot codons; however, they occurred near the hotspot codons (Fig. 5A). The spectrum from wild type yeast showed high number of hits at codons 283 and 276 which are near hotspot codons 282 and 273. Some hotspot codons were targeted for mutation such as codon 158 which had 2 hits. However, the overall spectrum was random and did not follow the p53 pattern seen in lung cancer. In the ogg1 strain the p53 spectrum also seemed to be random however one codon, codon 250, was targeted 11 times, however, it was not mutated when we repeated the experiment (Fig. 5B). The mutation spectrum from the apn1 yeast strain also showed no distinct pattern but as with the wild type and ogg1 mutant, many of the mutations occurred near codon hotspots such as codons 176, 250 and 280 (Fig. 6), which are near hotspot codons 175, 249 and 282. In addition there was no distinct pattern for amino acid changes in all three yeast mutants, except for a large number of proline to lysine changes seen on codon 250 in ogg1 yeast (Fig. S2). This amino acid change was mainly due to the C to T transition on nucleotide 749 in codon 250 consistently seen in ogg1 yeast.
Figure 5.
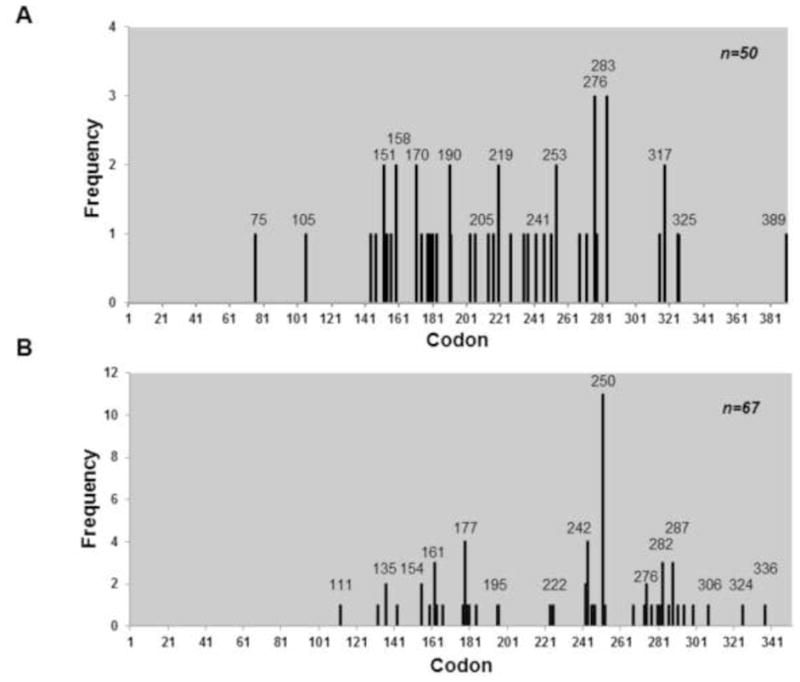
Mutation spectrum of p53 treated with BPQ in wild type and ogg1 yeast. A) Graph showing no distinct pattern in the mutation spectrum of p53 treated with BPQ in wild type yeast. B) Mutation spectrum of p53 treated with BPQ reveals no distinct signature in ogg1 yeast.
Figure 6.
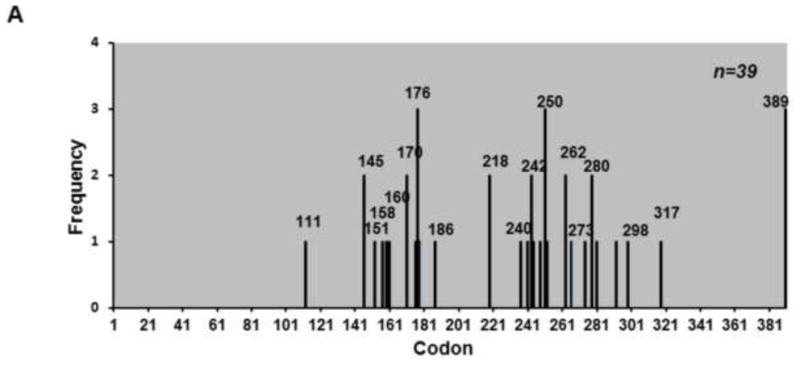
Mutation spectrum of p53 treated with BPQ in apn1 yeast. A) Mutation spectrum of p53 treated with BPQ in apn1 yeast is similar to mutation spectrum of p53 in wild type yeast.
4.0 Discussion
PAH o-quinones such as BPQ can damage DNA by forming either bulky adducts, apurinic sites or 8-oxo-dGuo. However, only 8-oxo-dGuo is likely to occur frequently enough to cause mutations. Bulky adducts occur infrequently, and while apurinic sites can be detected, they are not as frequent as 8-oxo-dGuo. Most lines of evidence suggest the primary route to DNA damage by BPQ is redox generated ROS, which then generates 8-oxo-dGuo. PAH o-quinone induced mutagenesis occurs only under redox cycling conditions, it is inhibited by antioxidants, shows a linear correlation with the production of 8-oxo-dGuo and 8-oxo-dGuo is formed at higher levels than other adducts, including apurinic sites [13–15]. This study provides additional support for the role of ROS in PAH o-quinone induced mutagenesis by finding that the loss of the repair enzyme Ogg1 increases both the frequency of mutagenesis and the frequency of G to T transversions.
Although oxidative damage through PAH o-quinones can cause a pattern of mutations similar to what is seen in smokers, the mutation spectra did not show a preference for known hotspots in p53. This is because the selection system used in this study identifies all nonfunctional mutations in p53. Hotspot enrichment requires additional selection pressure which is approximated in yeast by screening for dominant p53 mutations [14, 44–46].
We observed increases in the mutant frequency and G to T transversions in Ogg1 knockout cells suggesting that 8-oxo-dGuo is the primary DNA lesion caused by PAH o-quinones. As observed previously, we found that H2O2 induced mutagenesis was increased in the ogg1 strain [47, 48]. Ogg1 is a bi-functional DNA glycosylase that has both AP lyase and glycosylase activity, which remove 8-oxo-dGuo, the major lesion caused by ROS [32, 33, 49]. Through its DNA glycosylase activity Ogg1 first cleaves the oxidized base then cleaves the DNA strand 3′ to the abasic site by β-elimination [50]. Although Ogg1 can cleave the 3′ end, AP endonucleases are required to cleave the 5′ end of the damaged nucleotide to complete repair.
Oxidative damage, specifically 8-oxo-dGuo, is repaired primarily by the Base Excision Repair (BER) pathway [51, 52]. Ogg1 is specific for 8-oxo-dGuo, while Apn1 can repair oxidative lesions caused either by depurination as well as by assisting Ogg1 [53]. Apn1 is a type II apurinic/apyrimidinic (AP) endonuclease. It cleaves the 5′ end of an AP site created when a damaged base is removed by a monofunctional DNA glycosylase. However Apn1 also has 3′ phosphodiesterase and exonuclease activities and can remove and digest back the 3′ blocking end created when a bi-functional DNA glycosylase, such as Ogg1, removes a damaged base [38, 54]. This suggests that Apn1 may be required when removing 8-oxo-dGuo from DNA. However, we found that there was no change in the mutant frequency of p53 or the abundance of G:C to T:A transversions on p53 in apn1 yeast compared to wild type yeast. A likely explanation for this discrepancy is that other proteins that have endonuclease activity, such as Apn2, are redundant with Apn1 in 8-oxo-dGuo repair [55]. Apn2 is a strong candidate because it has both endonuclease activity and phosphodiesterase activity which can remove the 3′ block which may be left behind when Ogg1 cleaves a lesion [56]. In fact, our double knock out (apn1apn2) yeast showed a 3 fold increase in mutant frequency of p53 treated with BPQ compared to control, suggesting that Apn1 and Apn2 play a redundant role in repairing oxidative damage caused by BPQ.
There are several reports that suggest Apn1 has a role in oxidative repair, although the reports are somewhat contradictory. One study shows that APN1 knockout yeast are sensitive to H2O2 as well as other DNA damaging agents [57]. However, the spontaneous mutations show only a small increase in G to T transversions compared to A to C transversions [58]. Another study found that APN1 knock-out yeast are not sensitive to H2O2, but the double knockout of APN1 and APN2 were sensitive to H2O2 [56]. These data suggest only a minor role for Apn1 in repairing 8-oxo-dGuo. The minor role also extends to the human isoform as overexpression of Ape1, the human homologue of APN1, does not suppress repair of 8-oxo-dGuo in human cells, which result in G:C to T:A transversions. In the same cell model, overexpression of Ogg1 reduces transversions [59].
Transcription coupled repair (TCR) causes a strand bias. A strand bias is defined as more mutations on the coding strand compared to the transcribed strand of p53. In lung cancers there are about 10 times more G to T transversions on the coding strand than C to A transversions. The cause of the bias is believed to be the result of preferential repair of the transcribed strand. While the strand is being transcribed, the transcription is blocked by some adducts, and NER enzymes are recruited to the site, so the transcribed strand is more accurately repaired than the non-transcribed strand. Prior work has shown that BPQ induces 8-oxo-dG as the primary lesion with very few oxidized pyrimidines [14, 15]. Thus for G to T transversions, we can infer that the lesion is in the guanine and not the cysteine. Of note, we did not see a strand bias in either wild type cells or ogg1 cells. This may be due to several reasons. p53 may not be transcribed at robust enough levels for TCR. However, it is more likely that yeast RNA polymerase bypasses 8-oxo-dGuo lesions. Most studies find that TCR is observed primarily when the damage is caused by bulky adducts and apurinic sites, but rarely with oxidative damage. The former two lesions block transcription while oxidative damage either does not block RNA polymerase or weakly blocks it [60–64]. There is one report of a preference for mammalian Ogg1 to cleave oxidative damage on the transcribed strand, but the study did not perform mutagenesis, so it is not clear if the reported difference in cleavage would result in a strand bias [43]; in fact, a later study did not find a strand bias in mammalian cell mutagenesis experiments [65]. A study in E. coli also found evidence for TCR in oxidative repair, but this study was based on transcript analysis and not mutagenesis [66]. We found a trend for a strand bias in apn1 cells although it was not significant (Fig. 4C). Such would be expected if apurinic sites were generated frequently since they are strong blockers of transcription and replication [62, 63, 67].
In summary, we used a yeast reporter assay to study BPQ induced mutagenesis of p53 to address two repair genes. Our data shows that there is an increase in the mutant frequency of p53 treated with BPQ under redox conditions in ogg1 yeast and not in apn1 yeast. Although we did not observe a change in the mutant frequency of p53 in apn1 yeast, we did see that the mutant frequency of p53 treated with BPQ in apn1apn2 yeast increased by 3 fold, suggesting that these two proteins play a redundant role in repairing oxidative damage caused by BPQ. In addition we show a predominance of G:C/T:A transversions in p53 in three of our yeast strains and that number doubles in ogg1 yeast. Furthermore, we show that there is no strand bias in p53 treated with BPQ in ogg1 yeast. These data suggest that the major damage caused by BPQ is through the generation of ROS and Ogg1 is essential in repairing this damage while Apn1alone is not. In addition damage caused by ROS generated by BPQ is not repaired by transcription coupled repair in our system and that the strand bias seen in lung cancer on p53 may not be due to impaired repair of oxidative damage but rather due to repair of bulky adducts.
Supplementary Material
Highlights.
Repair of DNA damage caused by the PAH o-quinone B[a]P-7,8-dione (BPQ).
Increased mutant frequency of p53 treated with BPQ in yeast lacking OGG1.
Increase in G>T transversions on p53 treated with BPQ seen in OGG1 mutant yeast.
Yeast lacking APN1 show no increase in mutant frequency of p53 treated with BPQ
Increased mutant frequency of p53 treated with BPQ in yeast lacking APN1 and APN2
No strand bias on p53 treated with BPQ was observed in yeast lacking OGG1.
Acknowledgments
We wish to thank Elise Morocco and Josh Henkin for technical assistance.
Role of funding sources
National Institutes of Health grant R01 GM48241, R01 NIEHS 015662 and R25 ES016146 to J.F. and P30 ES013508. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIEHS, NIH.
Footnotes
Abbreviations: (±)-anti-BPDE, (±)anti-benzo[a]pyrene 7,8-diol 9,10-epoxide; B[a]P, benzo[a]pyrene; BER, base excision repair; BPQ, benzo[a]pyrene-7,8-dione; NER, nucleotide excision repair; PAH, polycyclic aromatic hydrocarbons; ROS, reactive oxygen species; TCR, transcription coupled repair.
Conflict of interest statement
The authors declare that there are no conflicts of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jemal A, Thun MJ, Ries LAG, Howe HL, Weir HK, Center MM, Ward E, Wu XC, Eheman C, Anderson R, Ajani UA, Kohler B, Edwards BK. Annual Report to the Nation on the Status of Cancer, 1975–2005, Featuring Trends in Lung Cancer, Tobacco Use, and Tobacco Control. J Nat Cancer Inst. 2008;100:1672–1694. doi: 10.1093/jnci/djn389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91:1194–1210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- 3.Loeb LA, Harris CC. Advances in Chemical Carcinogenesis: A Historical Review and Prospective. Cancer Res. 2008;68:6863–6872. doi: 10.1158/0008-5472.CAN-08-2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cavalieri EL, Rogan EG. Central role of radical cations in metabolic activation of polycyclic aromatic hydrocarbons. Xenobiotica. 1995;25:677–688. doi: 10.3109/00498259509061885. [DOI] [PubMed] [Google Scholar]
- 5.Chakravarti D, Pelling JC, Cavalieri EL, Rogan EG. Relating aromatic hydrocarbon-induced DNA adducts and c-H-ras mutations in mouse skin papillomas: the role of apurinic sites. Proc Natl Acad Sci USA. 1995:10422–10426. doi: 10.1073/pnas.92.22.10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gelboin HV. Benzo[a]pyrene metabolism, activation and carcinogenesis: Role and regulation of mixed function oxidases and related enzymes. Physiol Rev. 1980;60:1107–1166. doi: 10.1152/physrev.1980.60.4.1107. [DOI] [PubMed] [Google Scholar]
- 7.Conney AH. Induction of microsomal enzymes by foreign chemicals and carcinogenesis by polycyclic aromatic hydrocarbons. G.H.A. Clowes Memorial Lecture. Cancer Res. 1982;42:4875–4917. [PubMed] [Google Scholar]
- 8.Penning TM, Burczynski ME, Hung CF, McCoull KD, Palackal NT, Tsuruda LS. Dihydrodiol dehydrogenases and polycyclic aromatic hydrocarbon activation: Generation of reactive and redox active o-quinones. Chem Res Toxicol. 1999;12:1–18. doi: 10.1021/tx980143n. [DOI] [PubMed] [Google Scholar]
- 9.Jennette KW, Jeffery AM, Blobstein SH, Beland FA, Harvey RG, Weinstein IB. Nucleoside adducts from the in vitro reaction of benzo[a]pyrene-7,8-dihydrodiol-9,10-oxide or benzo[a]pyrene-4,5-oxide with nucleic acids. Biochemistry. 1977;16:932–938. doi: 10.1021/bi00624a019. [DOI] [PubMed] [Google Scholar]
- 10.Koreeda M, Moore PD, Wislocki PG, Levin W, Conney AH, Yagi H, Jerina DM. Binding of benzo[a]pyrene-7,8-diol-9,10-epoxides to DNA, RNA and protein of mouse skin occurs with high stereoselectivity. Science. 1978;199:778–781. doi: 10.1126/science.622566. [DOI] [PubMed] [Google Scholar]
- 11.McCoull KD, Rindgen D, Blair IA, Penning TM. Synthesis and characterization of polycyclic aromatic hydrocarbon o-quinone depurinating N7-guanine adducts. Chem Res Toxicol. 1999;12:237–246. doi: 10.1021/tx980182z. [DOI] [PubMed] [Google Scholar]
- 12.Shou M, Harvey RG, Penning TM. Reactivity of benzo[a]pyrene-7,8-dione with DNA. Evidence for the formation of deoxyguanosine adducts. Carcinogenesis. 1993;14:475–482. doi: 10.1093/carcin/14.3.475. [DOI] [PubMed] [Google Scholar]
- 13.Yu D, Berlin JA, Penning TM, Field J. Reactive oxygen species generated by PAH o-quinones cause change-in-function mutations in p53. Chem Res Toxicol. 2002;15:832–42. doi: 10.1021/tx010177m. [DOI] [PubMed] [Google Scholar]
- 14.Park JH, Gelhaus S, Vedantam S, Oliva AL, Batra A, Blair IA, Troxel AB, Field J, Penning TM. The pattern of p53 mutations caused by PAH o-quinones is driven by 8-oxo-dGuo formation while the spectrum of mutations is determined by biological selection for dominance. Chem Res Toxicol. 2008;21:1039–49. doi: 10.1021/tx700404a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park JH, Troxel AB, Harvey RG, Penning TM. Polycyclic Aromatic Hydrocarbon (PAH) o-Quinones Produced by the Aldo-Keto-Reductases (AKRs) Generate Abasic Sites, Oxidized Pyrimidines, and 8-Oxo-dGuo via Reactive Oxygen Species. Chem Res Toxicol. 2006;19:719–28. doi: 10.1021/tx0600245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu D, Harvey RG, Blair IA, Penning TM. Quantitation of Benzo[a]pyrene Metabolic Profiles in Human Bronchoalveolar (H358) Cells by Stable Isotope Dilution Liquid Chromatography–Atmospheric Pressure Chemical Ionization Mass Spectrometry. Chem Res Toxicol. 2011;24:1905–1914. doi: 10.1021/tx2002614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Denissenko MF, Pao A, Tang MS, Pfeifer GP. Preferential formation of Benzo[a]pyrene adducts at lung cancer mutation hotspots in P53. Science. 1996;274:430–432. doi: 10.1126/science.274.5286.430. [DOI] [PubMed] [Google Scholar]
- 18.Ames BN, Shigenaga MK, Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci USA. 1993;90:7915–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loft S, Vistisen K, Ewertz M, Tjonneland A, Overvad K, Poulsen HE. Oxidative DNA damage estimated by 8-hydroxydeoxyguanosine excretion in humans: influence of smoking, gender and body mass index. Carcinogenesis. 1992;13:2241–7. doi: 10.1093/carcin/13.12.2241. [DOI] [PubMed] [Google Scholar]
- 20.Prieme H, Loft S, Klarlund M, Gronbaek K, Tonnesen P, Poulsen HE. Effect of smoking cessation on oxidative DNA modification estimated by 8-oxo-7,8-dihydro-2′-deoxyguanosine excretion. Carcinogenesis. 1998;19:347–51. doi: 10.1093/carcin/19.2.347. [DOI] [PubMed] [Google Scholar]
- 21.Park JH, Mangal D, Tacka KA, Quinn AM, Harvey RG, Blair IA, Penning TM. Evidence for the aldo-keto reductase pathway of polycyclic aromatic trans-dihydrodiol activation in human lung A549 cells. Proc Natl Acad Sci USA. 2008;105:6846–51. doi: 10.1073/pnas.0802776105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Birtwistle J, Hayden RE, Khanim FL, Green RM, Pearce C, Davies NJ, Wake N, Schrewe H, Ride JP, Chipman JK, Bunce CM. The aldo-keto reductase AKR1C3 contributes to 7,12-dimethylbenz(a)anthracene-3,4-dihydrodiol mediated oxidative DNA damage in myeloid cells: Implications for leukemogenesis. Mutat Res/Fund Mol Mech Mutagen. 2009;662:67–74. doi: 10.1016/j.mrfmmm.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 23.Thorne D, Wilson J, Kumaravel TS, Massey ED, McEwan M. Measurement of oxidative DNA damage induced by mainstream cigarette smoke in cultured NCI-H292 human pulmonary carcinoma cells. Mutat Res/Gen Toxicol Environ Mutagen. 2009;673:3–8. doi: 10.1016/j.mrgentox.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 24.Abedin Z, Sen S, Field J. Aldo-Keto Reductases Protect Lung Adenocarcinoma Cells from the Acute Toxicity of B[a]P-7,8-trans-Dihydrodiol. Chem Res Toxicol. 2012;25:113–21. doi: 10.1021/tx200272v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang H, Gelhaus SL, Mangal D, Harvey RG, Blair IA, Penning TM. Metabolism of benzo[a]pyrene in human bronchoalveolar H358 cells using liquid chromatography-mass spectrometry. Chem Res Toxicol. 2007;20:1331–41. doi: 10.1021/tx700107z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen YM, Troxel AB, Vedantam S, Penning TM, Field J. Comparison of p53 Mutations Induced by PAH o-Quinones with Those Caused by anti-Benzo[a]pyrene Diol Epoxide in Vitro: Role of Reactive Oxygen and Biological Selection. Chem Res Toxicol. 2006;19:1441–1450. doi: 10.1021/tx0601206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 28.Hainaut P, Pfeifer GP. Patterns of p53 G-->T transversions in lung cancers reflect the primary mutagenic signature of DNA-damage by tobacco smoke. Carcinogenesis. 2001;22:367–74. doi: 10.1093/carcin/22.3.367. [DOI] [PubMed] [Google Scholar]
- 29.Sen S, Bhojnagarwala P, Francey L, Lu D, Penning TM, Field J. p53 Mutagenesis by Benzo[a]pyrene Derived Radical Cations. Chemical Research in Toxicology. 2012;25:2117–2126. doi: 10.1021/tx300201p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beroud C, Soussi T. p53 gene mutation: software and database. Nucleic Acids Res. 1998;26:200–204. doi: 10.1093/nar/26.1.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toyooka S, Tsuda T, Gazdar AF. The TP53 gene, tobacco exposure, and lung cancer. Hum Mutat. 2003;21:229–39. doi: 10.1002/humu.10177. [DOI] [PubMed] [Google Scholar]
- 32.Boiteux S, Radicella JP. The human OGG1 gene: structure, functions, and its implication in the process of carcinogenesis. Arch Biochem Biophys. 2000;377:1–8. doi: 10.1006/abbi.2000.1773. [DOI] [PubMed] [Google Scholar]
- 33.Shinmura K, Yokota J. The OGG1 gene encodes a repair enzyme for oxidatively damaged DNA and is involved in human carcinogenesis. Antioxid Redox Signal. 2001;3:597–609. doi: 10.1089/15230860152542952. [DOI] [PubMed] [Google Scholar]
- 34.Lu R, Nash HM, Verdine GL. A mammalian DNA repair enzyme that excises oxidatively damaged guanines maps to a locus frequently lost in lung cancer. Curr Biol. 1997;7:397–407. doi: 10.1016/s0960-9822(06)00187-4. [DOI] [PubMed] [Google Scholar]
- 35.Nishimura S. 8-hydroxyguanine: From its discovery in 1983 to the present status. Proceedings of the Japan Academy Series B-Physical and Biological Sciences. 2006;82:127–141. doi: 10.2183/pjab.82.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ho R, Rachek LI, Xu Y, Kelley MR, LeDoux SP, Wilson GL. Yeast apurinic/apyrimidinic endonuclease Apn1 protects mammalian neuronal cell line from oxidative stress. J Neurochem. 2007;102:13–24. doi: 10.1111/j.1471-4159.2007.04490.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Evans AR, Limp-Foster M, Kelley MR. Going APE over ref-1. Mutat Res. 2000;461:83–108. doi: 10.1016/s0921-8777(00)00046-x. [DOI] [PubMed] [Google Scholar]
- 38.Wilson DM, 3rd, Barsky D. The major human abasic endonuclease: formation, consequences and repair of abasic lesions in DNA. Mutat Res. 2001;485:283–307. doi: 10.1016/s0921-8777(01)00063-5. [DOI] [PubMed] [Google Scholar]
- 39.Hill JW, Hazra TK, Izumi T, Mitra S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 2001;29:430–8. doi: 10.1093/nar/29.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saitoh T, Shinmura K, Yamaguchi S, Tani M, Seki S, Murakami H, Nojima Y, Yokota J. Enhancement of OGG1 protein AP lyase activity by increase of APEX protein. Mutat Res. 2001;486:31–40. doi: 10.1016/s0921-8777(01)00078-7. [DOI] [PubMed] [Google Scholar]
- 41.Flaman JM, Frebourg T, Moreau V, Charbonnier F, Martin C, Chappuis P, Sappino AP, Limacher JM, Bron L, Benhattar J, Tada M, Van Meir EG, Estreicher A, Iggo RD. A simple p53 functional asay for screening cell lines, blood, and tumors. Proc Natl Acad Sci USA. 1995;92:3963–3967. doi: 10.1073/pnas.92.9.3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guthrie C, Fink GR. Methods in Enzymology. Academic Press; New York: 1991. Guide to yeast genetics and molecular biology. [PubMed] [Google Scholar]
- 43.Le Page F, Klungland A, Barnes DE, Sarasin A, Boiteux S. Transcription coupled repair of 8-oxoguanine in murine cells: the ogg1 protein is required for repair in nontranscribed sequences but not in transcribed sequences. Proc Natl Acad Sci USA. 2000;97:8397–8402. doi: 10.1073/pnas.140137297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brachmann RK, Vidal M, Boeke JD. Dominant-negative p53 mutations selected in yeast hit cancer hot spots. Proc Natl Acad Sci USA. 1996;93:4091–4095. doi: 10.1073/pnas.93.9.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rodin SN, Rodin AS. Human lung cancer and p53: the interplay between mutagenesis and selection. Proc Natl Acad Sci USA. 2000;97:12244–9. doi: 10.1073/pnas.180320897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoon JH, Lee CS, Pfeifer GP. Simulated sunlight and benzo[a]pyrene diol epoxide induced mutagenesis in the human p53 gene evaluated by the yeast functional assay: lack of correspondence to tumor mutation spectra. Carcinogenesis. 2003;24:113–9. doi: 10.1093/carcin/24.1.113. [DOI] [PubMed] [Google Scholar]
- 47.Thomas D, Scot AD, Barbey R, Padula M, Boiteux S. Inactivation of OGG1 increases the incidence of G. C-->T. A transversions in Saccharomyces cerevisiae: evidence for endogenous oxidative damage to DNA in eukaryotic cells. Mol Gen Genet. 1997;254:171–178. doi: 10.1007/s004380050405. [DOI] [PubMed] [Google Scholar]
- 48.Melo RG, Leitao AC, Padula M. Role of OGG1 and NTG2 in the repair of oxidative DNA damage and mutagenesis induced by hydrogen peroxide in Saccharomyces cerevisiae: relationships with transition metals iron and copper. Yeast. 2004;21:991–1003. doi: 10.1002/yea.1144. [DOI] [PubMed] [Google Scholar]
- 49.Karahalil B, Girard PM, Boiteux S, Dizdaroglu M. Substrate specificity of the Ogg1 protein of Saccharomyces cerevisiae: excision of guanine lesions produced in DNA by ionizing radiation- or hydrogen peroxide/metal ion-generated free radicals. Nucleic Acids Res. 1998;26:1228–33. doi: 10.1093/nar/26.5.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hu J, Imam SZ, Hashiguchi K, de Souza-Pinto NC, Bohr VA. Phosphorylation of human oxoguanine DNA glycosylase (alpha-OGG1) modulates its function. Nucleic Acids Res. 2005;33:3271–82. doi: 10.1093/nar/gki636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Demple B, Harrison L. Repair of oxidative damage to DNA: enzymology and biology. Annu Rev Biochem. 1994;63:915–48. doi: 10.1146/annurev.bi.63.070194.004411. [DOI] [PubMed] [Google Scholar]
- 52.Girard PM, Boiteux S. Repair of oxidized DNA bases in the yeast Saccharomyces cerevisiae. Biochimie. 1997;79:559–66. doi: 10.1016/s0300-9084(97)82004-4. [DOI] [PubMed] [Google Scholar]
- 53.Ishchenko AA, Yang X, Ramotar D, Saparbaev M. The 3′→5′ Exonuclease of Apn1 Provides an Alternative Pathway To Repair 7,8-Dihydro-8-Oxodeoxyguanosine in Saccharomyces cerevisiae. Mol Cell Biol. 2005;25:6380–6390. doi: 10.1128/MCB.25.15.6380-6390.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mol CD, Hosfield DJ, Tainer JA. Abasic site recognition by two apurinic/apyrimidinic endonuclease families in DNA base excision repair: the 3′ ends justify the means. Mutat Res/DNA Repair. 2000;460:211–229. doi: 10.1016/s0921-8777(00)00028-8. [DOI] [PubMed] [Google Scholar]
- 55.Boiteux S, Guillet M. Abasic sites in DNA: repair and biological consequences in Saccharomyces cerevisiae. DNA Repair. 2004;3:1–12. doi: 10.1016/j.dnarep.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 56.Swanson RL, Morey NJ, Doetsch PW, Jinks-Robertson S. Overlapping specificities of base excision repair, nucleotide excision repair, recombination, and translesion synthesis pathways for DNA base damage in Saccharomyces cerevisiae. Mol Cell Biol. 1999;19:2929–35. doi: 10.1128/mcb.19.4.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ramotar D, Popoff SC, Gralla EB, Demple B. Cellular role of yeast Apn1 apurinic endonuclease/3′-diesterase: repair of oxidative and alkylation DNA damage and control of spontaneous mutation. Mol Cell Biol. 1991;11:4537–4544. doi: 10.1128/mcb.11.9.4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kunz BA, Henson ES, Roche H, Ramotar D, Nunoshiba T, Demple B. Specificity of the mutator caused by deletion of the yeast structural gene (APN1) for the major apurinic endonuclease. Proc Natl Acad Sci USA. 1994;91:8165–9. doi: 10.1073/pnas.91.17.8165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yamane A, Shinmura K, Sunaga N, Saitoh T, Yamaguchi S, Shinmura Y, Yoshimura K, Murakami H, Nojima Y, Kohno T, Yokota J. Suppressive activities of OGG1 and MYH proteins against G:C to T:A mutations caused by 8-hydroxyguanine but not by benzo[a]pyrene diol epoxide in human cells in vivo. Carcinogenesis. 2003;24:1031–7. doi: 10.1093/carcin/bgg056. [DOI] [PubMed] [Google Scholar]
- 60.Larsen E, Kwon K, Coin F, Egly JM, Klungland A. Transcription activities at 8-oxoG lesions in DNA. DNA Repair. 2004;3:1457–1468. doi: 10.1016/j.dnarep.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 61.Tornaletti S, Maeda LS, Kolodner RD, Hanawalt PC. Effect of 8-oxoguanine on transcription elongation by T7 RNA polymerase and mammalian RNA polymerase II. DNA Repair (Amst) 2004;3:483–94. doi: 10.1016/j.dnarep.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 62.Tornaletti S, Maeda LS, Hanawalt PC. Transcription arrest at an abasic site in the transcribed strand of template DNA. Chem Res Toxicol. 2006;19:1215–20. doi: 10.1021/tx060103g. [DOI] [PubMed] [Google Scholar]
- 63.Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol. 2008;9:958–70. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 64.Pastoriza-Gallego M, Armier J, Sarasin A. Transcription through 8-oxoguanine in DNA repair-proficient and Csb /Ogg1 DNA repair-deficient mouse embryonic fibroblasts is dependent upon promoter strength and sequence context. Mutagenesis. 2007;22:343–351. doi: 10.1093/mutage/gem024. [DOI] [PubMed] [Google Scholar]
- 65.Thorslund T, Sunesen M, Bohr VA, Stevnsner T. Repair of 8-oxoG is slower in endogenous nuclear genes than in mitochondrial DNA and is without strand bias. DNA Repair. 2002;1:261–273. doi: 10.1016/s1568-7864(02)00003-4. [DOI] [PubMed] [Google Scholar]
- 66.Bregeon D, Doddridge ZA, You HJ, Weiss B, Doetsch PW. Transcriptional mutagenesis induced by uracil and 8-oxoguanine in Escherichia coli. Mol Cell. 2003;12:959–70. doi: 10.1016/s1097-2765(03)00360-5. [DOI] [PubMed] [Google Scholar]
- 67.Kim N, Jinks-Robertson S. Abasic sites in the transcribed strand of yeast DNA are removed by transcription-coupled nucleotide excision repair. Mol Cell Biol. 2010;30:3206–15. doi: 10.1128/MCB.00308-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.