

Abstract
Exosomes (EXOs) are secreted, nano-sized membrane vesicles that contain potent immunostimulatory materials. We have recently demonstrated that insulinoma-released EXOs can stimulate the autoimmune responses in nonobese diabetic (NOD) mice, a spontaneous disease model for type 1 diabetes. To investigate whether primary islet cells can produce EXOs, we isolated cells from the islet of Langerhans of NOD mice and cultured them in vitro. Interestingly, cultured islets release fibroblast-like, fast-replicating cells that express mesenchymal stem cell (MSC) markers, including CD105 and stem-cell antigen-1. These islet MSC–like cells release highly immunostimulatory EXOs that could activate autoreactive B and T cells endogenously primed in NOD mice. Serum EXO levels and EXO-induced interferon-γ production were positively correlated with disease progression at the early prediabetic stage. Consistent with these observations, immunohistological analysis of pancreata showed that CD105+ cells are restricted to the peri-islet area in normal islets but penetrate into the β-cell area as lymphocyte infiltration occurs. Immunization with EXOs promoted expansion of transferred diabetogenic T cells and accelerated the effector T cell–mediated destruction of islets. Thus, EXOs could be the autoantigen carrier with potent adjuvant activities and may function as the autoimmune trigger in NOD mice.
Introduction
Type 1 diabetes (T1D) is caused by the infiltration of islet antigen–specific autoreactive T cells into the pancreatic islets and autoimmune-mediated destruction of insulin-producing β-cells. In nonobese diabetic (NOD) mice, a loss of tolerance to islet self-antigens occurs spontaneously early in life, and the early peri-insulitis and later intraislet insulitis caused by lymphocyte infiltration are well-known characteristics that represent human T1D. However, the reason for the loss of tolerance to islet antigens and the activation of autoreactive T cells is still unknown. In the absence of lymphocyte infiltration, islet physiological abnormalities including vascular pathology (1) and increased β-cell endoplasmic reticulum stress (2) are detectible in the NOD strain. Also, inflammatory cytokines are upregulated first in the islets before they are detected systemically (3). These suggest that the early inflammatory triggers are present in the pancreas. Consequently, these cytokines and other cytolytic components may lead to β-cell death and the release of the islet antigens required for priming the autoreactive T cells (4). Therefore, understanding the cellular composition of islets and their functional relationships with insulin production and inflammation are of the utmost importance in order to identify the initial triggers for the lymphocyte activation and infiltration in islets. Peri-islet Schwann cells have been suggested as the early autoimmune targets associated with the initial peri-insulitis (5), and the presence of autoreactive T cells specific for Schwann cell antigens have been reported (6). Islet endothelial cells are essential for revascularization of islet transplants and are also believed to contribute to the early phase of T1D, possibly via facilitating the entry of lymphocytes into the islets (7). In addition, lymphatic vessel endothelial cells are required for islet inflammation (8). Interestingly, some islet-derived fibroblast-like cells can expand in culture, and these cells do not originate from β-cells and have characteristics of mesenchymal stem cells (MSCs) (9,10), which have potent immune regulatory functions. Thus, instead of endocrine cells, islet precursor and/or stromal cells might be the key elements triggering the local inflammatory responses in the islets and thus β-cell–specific autoimmunity.
Exosomes (EXOs) are small-sized (30–100 nm), biologically active entities that are secreted as microvesicles by many different types of cells (11). EXOs can be found in body fluids, including blood, saliva, breast milk, urine, and bronchoalveolar lavage fluid, under physiological or pathological conditions (12,13). They are stable structures, due to enriched lipid raft, cholesterol, and sphingomyelin (14,15), and can be isolated from body fluids frequently by ultracentrifugation or density gradient centrifugation. Exosomal proteomics has been a subject of interest in recent research (16). Presumably, novel disease biomarkers unique to EXOs and/or their cellular origins might be identified in biological fluids. The molecular pathway of EXO biogenesis is unclear, but it is believed to share a common pathway involving the formation of multivesicular bodies (17). Multivesicular bodies can fuse with plasma membrane, releasing EXOs into the extracellular space, or can fuse with lysosomes for degradation (11).
EXOs may display immunostimulatory or immunoregulatory functions (11,12,18). Vaccination with tumor antigen-loaded EXOs resulted in tumor rejection in an antigen-specific manner (19). Intriguingly, tumor-derived EXOs also activate regulatory T cells (20,21). We have studied immune responses in an autoimmune-prone condition in NOD mice, in which effector rather than regulatory T cells are preferentially generated. This approach may lead to further understanding why EXOs function in both immunostimulation and immunoregulation. We have demonstrated that insulinoma-released EXOs contain candidate diabetes-causing autoantigens that may stimulate autoreactive T cells in NOD mice (22). We also observed that these EXOs could stimulate autoreactive marginal zone-like B cells accumulated in prediabetic NOD mice (23). In this study, we demonstrate that cultured islet MSC–like cells (iMSC) can produce immunostimulatory EXOs that can activate autoreactive T cells and B cells in NOD mice. We propose that abnormal or excess EXOs released by these MSC-like precursor cells in islets may trigger tissue-specific autoimmunity in the NOD mouse strain.
Research Design and Methods
Mice
NOD/ShiLtJ (NOD), NOD.mip-green fluorescent protein (GFP) (stock #008173), and C57BL/6J (B6) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). NOD.Thy1.1+.BDC2.5 (BDC2.5) transgenic mice were provided by Linda Sherman of the Scripps Research Institute (TSRI; La Jolla, CA). All different mouse strains were maintained as inbred strains at the animal facility of the Torrey Pines Institute for Molecular Studies. Splenocytes from the NOD.MyD88−/− mouse strain were provided by Li Wen of Yale University (New Haven, CT). Splenocytes from B6.lpr−/−.Unc93b1 3d mutant and B6.lpr−/− mouse strains were provided by Dwight Kono of TSRI. Experimental protocols were conducted with approvals from the Ethical Review Committee of Torrey Pines Institute for Molecular Studies.
Isolation and Expansion of iMSC
Individual islets of NOD or B6 mice (8–10 weeks old) were handpicked after internal digestion of pancreata with collagenase (0.46 mg/mL) (24). A minimum of four mice per group was used for the islet isolation, and a total of 300–500 islets was collected. Islets were seeded on cell culture flask with complete high-glucose Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% FCS, 100 units/mL penicillin, 100 µg/mL streptomycin, 6 mmol/L l-glutamine. During the first week of culture, cells from the islets were released and adhered on the plastic. To remove serum EXOs, FCS was diluted in DMEM (1:1 volume) and precentrifuged at 100,000g for 90 min. For flow cytometry analysis, freshly isolated islets were digested by 0.25% trypsin-EDTA for 2 min at 37°C followed by pipetting to produce single cells for antibody staining. Cultured iMSC were detached by 0.25% trypsin-EDTA (3–5 min) and analyzed by flow cytometry for surface molecules.
EXO Preparation
A homogenous (≥90%) iMSC population was achieved after two passages, and culture supernatant was collected in the following passages until the 10th passage. Culture supernatants were harvested every 3 days while the iMSC were growing in the 150-cm2 flasks. After reaching confluence (80%), trypsin was applied and one-third of cells were inoculated back to allow growth for next passage. The supernatants were centrifuged at 300g for 20 min, followed by 10,000g for 20 min at 4°C. EXOs were collected by spinning the final supernatant in an ultracentrifuge (Sorvall Discovery 90SE; Hitachi) at 100,000g for 60 min. After washing once with an equal volume of PBS, the EXO pellet was resuspended in PBS (1:200 concentrated from the original volume of culture supernatant). Protein concentration was determined by Bradford assay (Bio-Rad, Hercules, CA). One liter of culture supernatant from an iMSC cell line yields 0.5–1.0 mg of EXOs.
Flow Cytometry and Intracellular Interferon-γ Staining
Flow cytometry analysis was performed on a FACScalibur flow cytometer (BD Biosciences). Fluorescent antibodies against CD105-Biotin or CD105-Alexa Fluor 488 (clone MJ7/18), stem-cell antigen-1 (Sca-1)-Biotin (D7), and CD31-Biotin (MEC13.3), CD31-fluorescein isothiocyanate (390), and CD81-phycoerythrin (PE) (Eat-2) were purchased from BioLegend (San Diego, CA). CD44-fluorescein isothiocyanate (FITC) (1M7), CD45-PE (30-F11), and CD90.2-PE (53–2.1) were purchased from BD Biosciences (San Jose, CA). Common antibodies for IgM, B220/CD45R, and CD4 were from BD Biosciences or eBioscience (San Diego, CA). For staining intracellular interferon-γ (IFN-γ), a single-cell suspension of mouse splenocytes were stimulated with anti-CD3/28–coated beads (Invitrogen, San Diego, CA) (cell:bead = 1:1) for 3 h in the presence of Brefeldin A (10 µg/mL) followed by surface staining with anti-CD4. After fixation and permeabilization with Cytofix/Cytoperm solution (BD Biosciences), cells were stained with anti–IFN-γ-PE (BD Biosciences) and analyzed by FACScalibur flow cytometer.
Cytometric Bead Array Cytokine Assay
A cytometric bead array (CBA)–based flow cytometry method (BD Biosciences) was used to analyze the following six different cytokines or chemokines: interleukin (IL)-6, IL-10, MCP-1, IFN-γ, tumor necrosis factor-α, and IL-12p70, according to the manufacturer’s protocol. The concentration was extrapolated from the standard curves by testing with the respective recombinant proteins of each cytokine or chemokine.
Carboxyfluorescein Succinimidyl Ester Cell Proliferation Assay
Total splenocytes (107 cells/mL PBS) were labeled with 5 μmol/L carboxyfluorescein succinimidyl ester (CFSE) (Sigma-Aldrich, St. Louis, MO) at 37°C for 10 min, and then cultured in a 96-well flat bottom plate at 8 × 105 cells/well in 200 µL RPMI complete medium with or without EXOs. After 72 h, the cells were harvested and stained with fluorescence-labeled B220 (or CD19) and CD4 antibodies to identify CFSE-lo, proliferating B220+ (or CD19+) B cells and CD4+ T cells by flow cytometry.
Immunohistochemistry and Electron Microscopy
Whole pancreas tissue was embedded in Cryo-OCT-compound (Sakura Finetek, Torrance, CA) and kept frozen at −80°C. After cryo-sectioning (8 µm in thickness), tissue slides were fixed in 4% formaldehyde. To stain the slides, tissue sections were first blocked with avidin followed by incubation with biotin to reduce background caused by endogenous biotin, biotin receptors, or avidin-binding sites in the tissues (Vector Laboratories, Burlingame, CA); and then the sections were incubated with biotin-conjugated primary antibodies overnight. After washing, tissue sections were incubated with streptavidin-horseradish peroxidase. A color reaction was developed using the standard ABC kit (Vector Laboratories), according to the instructions provided by the manufacturer. Tissue slides were counterstained with hematoxylin, which stains cell nuclei blue. Positively stained cells appeared red against a blue background. For electron microscopy, EXOs were fixed in 2% glutaraldehyde, washed and contrasted in 2% uranyl acetate, and embedded in a mixture of uranyl acetate (0.8%) and methyl cellulose (0.13%); and then were examined by Malcom Wood at the Microscopy Core Facility of the TSRI.
Flow Cytometry of EXOs
Isolated EXOs were analyzed by flow cytometry according to the protocol published by Qazi et al. (25) with small modifications. Briefly, EXOs in PBS (total 50 µg) were allowed to bind to 10 µL of 4 µm aldehyde latex beads (Invitrogen), precoated with anti-CD63. The coating was performed by incubating 10-µL beads with 5–10-µg CD63 antibody (BioLegend) overnight at 4°C followed by blocking with 100 mmol/L glycine for 30 min at room temperature. After washing, the coated beads were resuspended in 100-µL PBS. Unbound EXOs were removed by two washes with PBS via centrifugation. EXOs bound on the beads were detected with anti-mouse CD81-PE (BioLegend).
Measuring Serum EXOs by ELISA
Serum EXO concentration was measured using an ExoQuick kit (System Biosciences, Mountain View, CA) with some modifications. Briefly, 250-µL serum was incubated with 63-µL ExoQuick polymer solution for 30 min to precipitate EXOs. EXO pellets were lysed in 200-µL EXO-binding buffer, followed by centrifugation at 1,500g for 5 min. Then, 50-µL lysed supernatant was coated on Microtiter plates overnight at 37°C. After three washes, plates were incubated with anti-CD81 for 1 h, and then with a horseradish peroxidase–conjugated secondary antibody. Substrate 3,3′,5,5′-tetramethylbenzidine was used for color reaction. The optical density (OD) was read at 450 nm. The levels of CD81 protein in serum EXOs are shown as OD values.
In Vivo EXO Treatment and Adoptive BDC2.5 Cell Transfer
One-week-old NOD mice were treated with EXOs (2.5–5-µg in 50-µL PBS/mouse/injection) intraperitoneally three times 1 week apart. At the age of 4 weeks, the treated mice or untreated controls were killed, and total spleen cells were labeled with CFSE and stimulated in vitro with EXOs for 72 h to assess B-cell proliferation. Some mice (both treated and untreated) received CFSE-labeled Thy1.1+ BDC2.5 effector cells (2 × 106/mouse) at the age of 4 weeks via intravenous injection. BDC2.5 effector cells were produced after 3-day culture of BDC2.5 transgenic splenocytes with 10 µg/mL mimotope, which has homology to a peptide of mouse glutamic acid decarboxylase (GAD) 65 kDa p526–541, but contains a single altered residue K534W (LSKVAPVIWARMMEYG) that can activate BDC2.5 splenocytes to proliferate at a range of 1–10 µg/mL (Y.D.D., H. Sheng, I. Marrero, Y. Li, S. Hassanali, D.B. Wilson, and E.E. Sercarz, unpublished data). The recipient mice were killed at 7 days post-transfer to evaluate the proliferation of the donor cells.
Pancreatic Islet Histology and Scoring
Paraffin-embedded mouse pancreata were sectioned (5 µm in thickness) and stained in the facility of Pacific Pathology (San Diego, CA). Tissue sections were stained with hematoxylin-eosin to examine lymphocyte infiltration. A total of 50–100 individual islets were examined by microscopy to calculate the different levels of islet infiltration. Scoring criteria for individual islets were 0, no infiltration; 1, <20%; 2, 20–50%; and 3, >50% of the islet area with lymphocyte infiltrated, and performed in a blinded manner.
Results
Detection of CD105+ Stromal or Stem Cells in the Islets of Langerhans
To examine the islet stem cells by flow cytometry, single–islet cell preparation was performed. As shown in Fig. 1A, islet CD105+ cell frequency within CD45− nonhematopoietic cells was 16% in 8- to 10-week-old NOD mice; almost all islet CD45−CD105+ cells coexpressed a second MSC marker, Sca-1, confirming a stem cell phenotype of these CD105+ cells. By immunohistochemistry, we found that CD105+ cells locate at the surrounding periphery of normal islets but penetrate into the β-cell area as lymphocyte infiltration occurs (Fig. 1B). Interestingly, they appear to lead the lymphocyte infiltration and are present in the area without lymphocytes. In comparison, CD31+ endothelial cells do not have a peri-islet distribution in normal islets; instead of leading to lymphocyte infiltration, they were apparently recruited/expanded in the area infiltrated by lymphocytes (Fig. 1B). Thus, different types and/or differentiation stages of stem/stromal cells may be associated with lymphocyte entry, migration, or expansion.
Figure 1.

CD105+ cells in NOD islets. A: Single-cell suspension of handpicked islets was prepared from four to six NOD mice (8 weeks old) and was analyzed by flow cytometry. CD45− stromal cells are shown at the right. B: Immunohistochemical staining for CD105 (top) and CD31 (bottom) in mouse pancreata isolated from 8-week-old NOD mice. Islets were grouped based on their levels of lymphocyte infiltration: normal, peri-insulitis, and insulitis. Data represent three to four independent experiments. SSC-H, side scatter height.
In Vitro Cultured Islet Stem Cells Express MSC Markers
To study the relationship of islet stem/stromal cells with lymphocytes, we successfully expanded islet fibroblast–like cells in vitro following a previously described method (9) by culturing isolated pancreatic islets in regular high-glucose complete DMEM. We observed that cells were released from the islets during the first week in culture and appeared in a small blast, which slowly formed large aggregates as the cells multiplied (Fig. 2A). To confirm that cells were not derived or contaminated with insulin-producing β-cells, we used NOD.mip-GFP mice for islet isolation, in which a fluorescent GFP protein is introduced into β-cells under the control of mouse insulin I promoter (26). GFP+ β-cells could not proliferate in the culture condition and disappeared after two to three passages (Supplementary Fig. 1). We next examined the cell surface molecules expressed on the cultured islet cells. After the second passage, cells were >90% homogeneous, and expressed MSC markers CD105, Sca-1, CD90, and CD44, but were negative for endothelial cell markers CD31 and CD202b (data not shown) (Fig. 2B). Though freshly isolated islet cells from adult NOD mice contain CD45+ cells, which are likely islet-infiltrating lymphocytes, only CD105+ stromal cells can grow, and there were no CD45+ cells present in the culture after two passages. Cells collected from P3-P10 passages, remained negative for CD45 and CD31, but positive for those MSC markers. We thus name these cultured cell populations iMSCs.
Figure 2.

In vitro culture and immunophenotyping of islet MSC-like cells. A: Islet stromal cells isolated from NOD mice (pooled from four to five mice) and cultured in vitro for different periods. B: After two to three passages, cells were trypsinized and analyzed by flow cytometry using CD105, Sca-1, CD31, CD44, CD90, CD45, and CD81 antibodies. Broken line is for isotype control, and shaded area represents positive marker expression. Data represent three (A) or five to six (B) independent experiments. Max, maximum.
iMSCs Produce EXOs That Are Immunostimulatory
EXOs were collected from the culture supernatants of the iMSCs via ultracentrifugation. Electron microscopic observation showed that EXO particles were round, vesicle shaped with a size <100 nm (Supplementary Fig. 2). The expression of tetraspanins such as CD81 and other exosomal signature proteins such as heat shock proteins was detected by mass spectrometry, confirming that EXOs are the major microparticles released by the iMSC.
To test whether the EXOs are immunostimulatory, we incubated NOD spleen cells (8 weeks old) with EXOs in vitro and evaluated innate cytokine production as well as T-cell and B-cell activation. As shown in Fig. 3A, EXOs stimulated spleen cells to release large amounts of IL-6, IFN-γ, tumor necrosis factor-α, and MCP-1. The percentage of major histocompatibility complex class II+CD86+ cells was increased twofold (data not shown), suggesting antigen-presenting cell (APC) activation. Similar to MIN6 insulinoma-released EXOs (22), iMSC-derived EXOs also required a myeloid differentiation factor 88–mediated innate pathway to induce cytokine production and APC activation (data not shown). Also, the innate response to these iMSC-derived EXOs did not require endosomal Toll-like receptors (TLRs) for cell activation since a 3d mutation of Unc93b1 adaptor (27) did not affect the EXO-induced cytokine production. Thus, iMSC-EXOs also function via stimulating surface TLRs or other innate receptors. To examine the B-cell response, we performed a CFSE assay using splenocytes from 6- to 8-week-old NOD mice, as shown in Fig. 3B, after EXO stimulation. CFSE-lo-proliferating B cells were increased by fivefold to sixfold compared with nonstimulated controls; whereas, B cells from young NOD mice (4–5 weeks old) or B6 mice (8–10 weeks old) failed to proliferate in response to the EXOs (data not shown). These results suggest that iMSC-derived EXOs are highly immunostimulatory, likely via binding TLRs, and may contain unique antigens stimulating autoreactive B cells.
Figure 3.

iMSC-derived EXOs are immunostimulatory. EXOs obtained from iMSCs were used to stimulate splenocytes (8 × 105/200 µL/well) from 6- to 8-week-old NOD mice at 5 µg/mL concentration for 72 h. A: CBA assay was performed to determine cytokine and chemokine levels in the culture supernatants. B: B-cell proliferation was measured by CFSE dilution assay after EXO stimulation. The decreasing CFSE signal was recorded as dividing B cells after EXO stimulation. Averages of dividing B cells are shown on the right bar diagram. Data represent 10 mice per group in three experiments. The P value was calculated using the Student t test. TNF-α, tumor necrosis factor-α.
EXO-Specific IFN-γ Responses Correlated With Increased Serum EXO Levels in Prediabetic NOD Mice
We have previously shown that prediabetic NOD mice accumulate EXO-reactive IFN-γ–secreting T cells (22). Thus, measuring the IFN-γ response to EXOs may help to identify autoreactive T-helper type 1 (Th1) cells developed in these mice. To this end, we examined spleen cells from NOD mice at different ages. EXO-specific IFN-γ responses increased >10-fold between 4 and 8 weeks of age (Fig. 4A). Interestingly, this increase in EXO-reactive Th1 cells was accompanied by increased serum EXO levels; as shown in Fig. 4B, serum EXO levels were low in all 4-week-old young mice, but increased over twofold in older prediabetic NOD mice (8 week old) and were maintained at a high level in 12-week-old mice (Fig. 4B). In contrast, serum EXO levels in B6 mice at all ages were consistently lower than that in 4-week-old NOD mice (data not shown). These results indicate that serum EXO levels correlate with the induction of autoreactive Th1 cells.
Figure 4.

Correlation of EXO-induced IFN-γ secretion with serum EXO levels in prediabetic NOD mice. A: Splenocytes were isolated from NOD mice (three to four mice/group) at 4 and 8 weeks of age and stimulated for 72 h in vitro with iMSC-EXOs (5 µg/mL). Culture supernatants were tested for released IFN-γ using CBA assay. B: Serum EXOs were isolated by the ExoQuick kit. EXO concentration was determined by measuring CD81 protein by ELISA. Data show OD values for individual mice at different ages, with the horizontal line indicating mean values. Experiments were repeated four times (A) or three times (B) with similar results.
EXOs Released by NOD iMSC Are More Effective in Inducing IFN-γ Production Than EXOs From B6-Derived iMSCs
To further dissect whether the EXO-specific IFN-γ responses in NOD mice are due to unique antigens expressed by the NOD-derived EXOs, we compared EXOs released by NOD- or B6-derived iMSCs. Both NOD and B6 iMSCs grow similarly in culture and express the same MSC markers. B6-derived EXOs expressed the exosomal signature protein CD81 to a level similar to that of NOD-derived EXOs (Fig. 5A). Surprisingly, NOD EXOs induced a stronger IFN-γ response than B6 EXOs when cultured with splenocytes from prediabetic NOD mice (Fig. 5B). The variation of IFN-γ response among individual NOD mice ranged from undetectable (<10 pg/mL) to 1,500 pg/mL. We noticed that different EXO preparations from both NOD and B6 iMSCs varied in their ability to induce IFN-γ production from NOD spleen cells, which may explain the insignificant difference in Fig. 5B. However, in all three independent experiments, the averages of IFN-γ induced by NOD EXOs were consistently higher than those induced by B6 EXOs. Thus, NOD EXOs may contain some unique and/or more Th1-stimulating antigens than those expressed by B6 EXOs.
Figure 5.
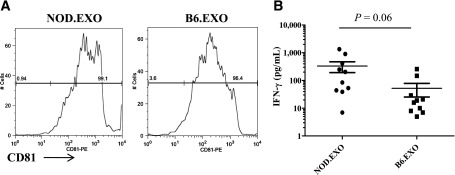
NOD-derived but not B6-derived EXOs are more effective in inducing IFN-γ production. A: EXOs isolated from cultured NOD and B6 iMSCs contain comparable levels of the CD81 molecule as detected by flow cytometry analysis of EXO-coated beads. B: Spleen cells from 6- to 8-week-old NOD mice were stimulated for 72 h with the NOD- or B6-derived EXOs, and IFN-γ levels were measured by CBA assay. Data show the concentration for individual mice, with horizontal line indicating mean values. The P value was calculated based on data from three independent experiments using the Student t test, where P < 0.05 was considered significant.
Immunization With EXOs Induced EXO-Specific Memory T and B Cells
Because in vitro stimulation with iMSC EXOs can activate spontaneously developed autoreactive T and B cells in NOD mice, we next investigated whether EXO injections in young NOD mice (<3 weeks old), before the onset of insulitis, could have any effects on the development of autoreactive T and B cells. Two weeks after the last EXO injection (at day 17 after birth), pancreatic lymph nodes (PanLNs) and inguinal lymph nodes (iLNs) were taken from EXO-treated or age-matched control mice (4–5 weeks old) and examined ex vivo for their cytokine secretion. We found that cells, including splenocytes from mice treated with either MIN6 or NOD EXOs, released a large number of cytokines and chemokines in culture even in the absence of any additional antigens, whereas cells from noninjected control mice were inactive (Fig. 6A), suggesting an expansion of autoreactive memory cells in the treated mice at the age of 4–5 weeks. We next assessed the autoreactive memory T-cell and B-cell responses in the EXO-treated mice. By short-term (3 h) anti-CD3/28 stimulation, we detected a robust increase of IFN-γ –secreting memory Th1 cells in the EXO-injected mice (9% vs. 2%) compared with the control mice (Fig. 6B). Without stimulation, but with culture for 48 h, splenocytes from EXO-treated mice released a 10- to 20-fold greater IFN-γ than cells from control mice (Fig. 6C). Similar to this increased memory T-cell response, memory B cells were also induced in the EXO-injected mice as detected by increased B-cell proliferation in vitro after restimulation with EXOs (Fig. 6D). This is consistent with our recent observation that splenic B cells from normal young NOD mice (4 weeks old) exhibited a marginal increase in proliferation after EXO stimulation compared with unstimulated splenocytes, but increased in number after early EXO immunization (23). Collectively, these data reveal a pathway to expand and/or modulate the autoreactive T and B cells in young NOD mice by injecting EXOs.
Figure 6.

EXO injections induce systemic inflammatory and/or autoimmune responses in young NOD mice. A: NOD mice (1 week old) were injected intraperitoneally three times (waiting for 7 days between injections) with either MIN6-EXOs or NOD-EXOs at 5 µg per animal per injection (2.5 µg for the first injection) or left untreated (Control). Cells were collected at the age of 4 weeks from PanLNs or iLNs and were cultured ex vivo for 72 h without EXO stimulation. Release of cytokines and chemokines was measured by CBA assay. B: Spleen cells from the control or treated mice were stimulated with anti-CD3/28 beads for 3 h in the presence of Brefeldin A (10 µg/mL), followed by CD4 and intracellular IFN-γ staining. C: Splenocytes from the 4-week-old treated or control mice (three mice per group) were cultured for 72 h without adding EXOs. IFN-γ concentration in the culture supernatants was determined by CBA assay. The experiment was repeated once with similar observations. D: CFSE-labeled splenocytes from the treated or control mice were stimulated with iMSC EXOs for 72 h. Proliferating B cells were identified as B220+/CFSE-lo. EXOinj, EXO-treated; TNF-α, tumor necrosis factor-α.
EXO-Injected Young NOD Mice Are Highly Susceptible to BDC2.5 Effector Cell–Induced Early Insulitis
To test whether EXO-treated young NOD mice (4 weeks old) were susceptible to the early development of insulitis, we adoptively transferred effector cells from Thy1.1+ BDC2.5 transgenic mice into 4-week-old NOD mice that were previously injected with EXOs three times within the first 3 weeks of their life. After 1 week of the transfer, we compared the division of donor BDC2.5 cells in PanLNs and the levels of insulitis in the recipient mice. As shown in Fig. 7A, EXO-treated NOD mice had a twofold higher rate of dividing BDC2.5 cells (59% of donor cells) in the PanLNs compared with those in nontreated control recipients (32%). The division of BDC2.5 cells was fourfold to fivefold less in iLNs for both treated and control mice, suggesting that the EXO treatment promoted islet-specific expansion of the effector T cells in the PanLNs. Similar results were obtained when we cotransferred EXOs and BDC2.5 T cells into nontreated 4-week-old NOD female mice (data not shown). Thus, EXOs are sufficient to stimulate BDC2.5, likely via activating unique APCs in vivo. This observation is further supported by the enhanced insulitis in the EXO-treated mice compared with the nontreated control NOD mice after transferring BDC2.5 effector cells (Fig. 7B); in fact, transferring 2 × 106 BDC2.5 effector cells to 4-week-old untreated NOD mice did not cause insulitis in two of three recipients. Diabetes development in these EXO-treated mice with or without transferring BDC2.5 cells has not been evaluated.
Figure 7.
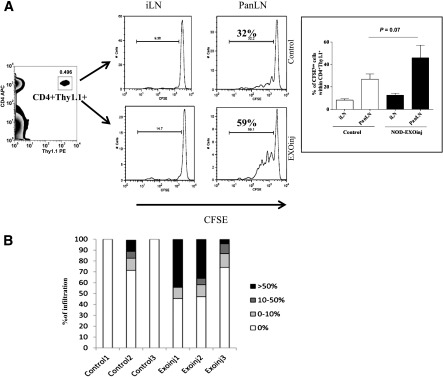
EXO-treated NOD mice are highly susceptible to early insulitis upon BDC2.5 cell transfer. A: EXO-treated 4-week-old NOD mice (similar to Fig. 6) received BDC2.5 transgenic effector T cells (2 × 106 cells/mouse) via intravenous route. BDC2.5 cells were prelabeled with CFSE. Cell division in both PanLN and iLN was measured 7 days after transfer by flow cytometry to gate on CD4+/Thy1.1+ donor cells for CFSE dilution. Experiments were repeated at least three times, and averages are shown on the right bar diagram. B: Insulitis levels after receiving BDC2.5 cells were scored in a blinded manner, and the data showed the percentages of islets within each level of lymphocyte infiltration for individual mice in control and EXO-treated NOD mice. EXOinj, EXO-treated.
Discussion
MSCs are self-renewing, multipotent progenitors that can be isolated from various tissues. Bone marrow MSCs are the most extensively used in clinical trials because of their high plasticity. They are able to differentiate into bone, cartilage, fat, tendon, muscle, adipocytes, chondrocytes, or osteocytes (28). In addition to these tissue regeneration or repair functions, bone marrow MSCs are potent immunoregulatory cells that can delay the onset of T1D in mice (29). Islets of Langerhans contain various progenitor cells, including precursor cells for insulin-producing β-cells, which can restore normoglycemia in mice treated with streptozotocin (30), and endothelial progenitor cells, which are responsible for forming blood or lymphatic vessels under inflammatory situations. These types of stem or precursor cells can be found both inside and outside the islets (31). In this study, we found that cultured mouse islets release iMSCs that can grow for at least 20 passages and consistently express MSC surface markers. Immunohistochemical data from pancreata revealed that CD105+ cells have a particular peri-islet location in normal islets, but penetrate into the center of islets when lymphocyte infiltration starts at the peri-islet area, consistent with a possible role of these stem-like cells in triggering and spreading islet pathology. More importantly, these iMSCs released EXOs into culture supernatant. Consistent with our previous findings when studying immune responses to insulinoma-derived EXOs (22), these iMSC-derived EXOs were also highly immunostimulatory and could activate the autoreactive T and B cells spontaneously developed in prediabetic NOD mice. Collectively, these data suggest a possible causative role of the islet stem cells and their EXOs in triggering the islet-specific autoimmunity in the NOD mouse strain.
Under physiological stress situations such as islet development and β-cell damage, various intercellular mediators including proinflammatory factors can be released from islet stromal or β-cells, which can recruit phagocytes to clear the damaged β-cells and increase lymphocyte trafficking or infiltration in the islets. This initial inflammatory response may recruit and stimulate islet precursor cells or stem cells to release excess and/or abnormal EXOs to cause autoimmunity in susceptible individuals. Future studies are needed to clarify how iMSCs and their EXOs interact with different APC populations and activate pathogenic effector T cells in vivo. The conditions causing EXO release, the cell types and their hierarchy of being recruited to the islet, and the cellular sources of islet EXOs are key questions that remain to be addressed. We speculate that during β-cell apoptosis in the islet, which might be a spontaneous process (32) in the early developmental stage of life or triggered by islet inflammation (33), islet stem cells might be activated or recruited to repair the damage. Whether the stem cells can release any candidate autoantigens such as GAD65, proinsulin, or tyrosine phosphatase IA-2 via EXO secretion is unknown, although we have confirmed that GAD65 is expressed in MIN6-derived EXOs (22). It is possible that the expression of these autoantigens might be restricted to a certain differentiating stage of the stem cells and require an inflammatory environment.
Though naïve T cells can migrate to the islet, they cannot stay unless encountering APCs that are displaying specific antigens. More likely, priming of naïve T cells preferentially occurs in the pancreas-draining lymph nodes (34) after migration of activated APCs from tissue to draining lymph nodes. Lennon et al. (35) showed that GAD antigen expression in the islets is a prerequisite for the GAD-specific T cells to infiltrate the islets. Also, the inhibition of major histocompatibility complex class I transport in the β-cells impaired CD8+ T-cell infiltration to the islet (36). Thus, local expression of autoantigens is a necessity to initiate and/or sustain the insulitis. Conversely, other studies have demonstrated that immune cell migration to the islets could be a bystander process since transfer of BDC2.5 cells initiated recruitment of a diverse population of T cells that were not necessarily involved in islet β-cell destruction or diabetes development (35). This recruitment is not antigen-specific, and an inflammatory environment could be involved in such cell migration. Nevertheless, a hierarchy of individual diabetogenic clones clearly exists (37), which might be determined by the accessibility of their specific antigens and peptides to the T cells in the thymus and periphery or in the islet (35). Given the suggested possibility that only islet antigen–specific T cells are truly responsible for the destruction of insulin-producing β-cells (38,39), the initial triggering events that cause the recruitment and activation of the specific autoreactive T cells remain unclear. Our findings indicate a potential triggering mechanism, via the iMSC-produced EXOs, which carry innate stimuli and may harbor unique antigens, and are sufficient to induce early inflammation and activate autoreactive T and B cells in the islets. Because major histocompatibility complex class II alleles have been demonstrated as the most important locus associated with the susceptibility to T1D (40), and DQ8 and I-Ag7 have significant similarities in the binding to T-cell peptides of insulin and GAD65 (41), the likely mechanism of EXO-induced autoimmunity may involve the presentation of unique exosomal antigens to activate β-cell–reactive T cells, although it cannot be excluded that islet-specific EXOs may express β-cell antigens under stressed or inflammatory situations.
IFN-γ–secreting effector T cells reactive to target autoantigens appear in young NOD mice (3–4 weeks old), and increase in older prediabetic mice (42). A sequential activation of autoreactive T cells against candidate peptides of GAD, insulin, and heat shock protein (43,44) has been suggested as a consequence of epitope spreading (45). Consistent with this increased frequency of Th1 cells in prediabetic NOD mice, we also observed increased immune responses to EXOs in these mice, as measured by the higher levels of IFN-γ secretion and B-cell expansion compared with younger or resistant mice. Earlier studies from our group found increased numbers of EXO-reactive T cells (22) and marginal zone–like B cells (23) in prediabetic NOD mice, but the EXOs were derived from the insulinoma cell line MIN6, which does contain candidate β-cell antigens, including GAD65 and insulin. In this current study, we found that the EXOs released by the cultured iMSC can also induce comparable levels of IFN-γ and B-cell expansion, but did not express these two autoantigens. The role of candidate autoantigens in the EXO-induced T-cell and B-cell responses remains to be determined. We also observed that MIN6 EXOs could reactivate memory autoreactive T and B cells collected from mice that were primed by the iMSC EXOs in vivo. This suggests that autoreactive T and B cells might recognize some common or cross-reactive exosomal antigens. Additional studies are required to understand whether and how these EXO-specific autoreactive T and B cells contribute to the disease.
Interestingly, EXOs isolated from diabetes-resistant B6 mice were not as potent as NOD EXOs in their ability to activate the autoreactive cells, particularly the IFN-γ–producing T cells. This means that though EXOs can be released from the cultured iMSCs of both NOD and B6 origins, NOD-derived cells intrinsically bear unique antigenic stimuli that are not expressed or are weakly expressed on B6 cells. It is important to mention herein that EXOs from NOD or B6 cells were able to induce comparable levels of innate immune responses (data not shown), which suggests that the strain difference might mainly affect the antigens that specifically stimulate adaptive immune responses. Expression of candidate autoantigens by EXOs could be one possible explanation, but we failed to detect insulin and GAD65 in both NOD and B6 iMSCs. It should be noted that the iMSCs underwent several passages in vitro, and some antigens may be lost and others upregulated during cell culture. Further, gene expression and proteomics analysis of noncultured islet stem/stromal cells are required to identify possible unique autoantigens that may be differentially expressed between NOD and B6 cells by islet MSCs and their EXOs.
EXOs could be useful disease biomarkers in biological fluids because protein and RNA contents may be unique to one cell type. Tumor-derived EXOs could be used as cancer diagnostic markers because they contain tumor-specific antigens and microRNA, and could be easily detected in blood plasma and urine (46). We found increased serum levels of EXOs in prediabetic NOD mice (8–10 weeks old), which dropped slightly in mice close to the age of diabetes onset (12–14 weeks old). The source of serum EXOs in NOD mice is unknown, and their contents might vary depending on the stage of disease progression. In addition to MSCs, dendritic cells (11), endothelial cells (47), and T and B cells can also produce EXOs (48,49), but likely with different biological functions. We found that serum EXOs could stimulate NOD splenocytes to release innate inflammatory cytokines, although B-cell proliferation was insignificant in the CFSE-labeling assay (data not shown). Whether serum EXOs also contain autoantigens similar to MIN6- or iMSC-released EXOs remains to be studied. Nevertheless, serum EXO levels clearly reflect the chronological inflammatory response in the islets during the prediabetic stage (6–12 weeks old) in NOD mice. Although it is thought that islet inflammation may be initiated locally via abnormal EXO release, the ultimate effect of this autoimmune response is not restricted to the islets; the effect can be detected systemically in the prediabetic stage either due to a spread from the initial local inflammation or to some systemic genetic defects in the NOD strain. Possibly, this increase of serum EXO levels may further compromise the immune regulatory system and accelerate the local islet inflammation.
In summary, EXO-specific T and B cells in NOD mice are autoreactive immune cells that contribute to islet immunopathology. Islet-derived MSCs are a possible source of EXO production to initiate local autoimmune response. Thus, the results from this study may provide insights into the triggering events that could occur after abnormal stem cell activity and the release of highly immunostimulatory EXOs in this autoimmune-prone mouse strain.
Supplementary Material
Article Information
Acknowledgments. The authors thank Drs. Linda Wicker (University of Cambridge, Cambridge, U.K.) and Linda Sherman (The Scripps Research Institute) for critical discussion and editing of the manuscript. The authors thank Linda Sherman for providing the Thy1.1+ BDC2.5 transgenic mouse strain. The authors thank Dr. Qazi Khaleda (Karolinska Institute, Stockholm, Sweden) for sharing protocols for the flow cytometry–based EXO detection.
Funding. This work was supported by a grant from the National Institutes of Health (R01-DK-091663).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. M.J.R. designed the experiments, performed the experiments and the analysis, interpreted the data, and wrote the manuscript. D.R. and R.B. performed the experiments and the analysis. Y.D.D. conceived the project, designed the experiments, performed the experiments and the analysis, interpreted the data, and wrote the manuscript. Y.D.D. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db13-0859/-/DC1.
See accompanying article, p. 835.
References
- 1.Chaparro RJ, Konigshofer Y, Beilhack GF, Shizuru JA, McDevitt HO, Chien YH. Nonobese diabetic mice express aspects of both type 1 and type 2 diabetes. Proc Natl Acad Sci USA 2006;103:12475–12480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tersey SA, Nishiki Y, Templin AT, et al. Islet β-cell endoplasmic reticulum stress precedes the onset of type 1 diabetes in the nonobese diabetic mouse model. Diabetes 2012;61:818–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Toyoda H, Formby B, Magalong D, et al. In situ islet cytokine gene expression during development of type I diabetes in the non-obese diabetic mouse. Immunol Lett 1994;39:283–288 [DOI] [PubMed] [Google Scholar]
- 4.Turley S, Poirot L, Hattori M, Benoist C, Mathis D. Physiological beta cell death triggers priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J Exp Med 2003;198:1527–1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Winer S, Tsui H, Lau A, et al. Autoimmune islet destruction in spontaneous type 1 diabetes is not beta-cell exclusive. Nat Med 2003;9:198–205 [DOI] [PubMed] [Google Scholar]
- 6.Tsui H, Chan Y, Tang L, et al. Targeting of pancreatic glia in type 1 diabetes. Diabetes 2008;57:918–928 [DOI] [PubMed] [Google Scholar]
- 7.Nyqvist D, Speier S, Rodriguez-Diaz R, et al. Donor islet endothelial cells in pancreatic islet revascularization. Diabetes 2011;60:2571–2577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yin N, Zhang N, Lal G, et al. Lymphangiogenesis is required for pancreatic islet inflammation and diabetes. PLoS One 2011;6:e28023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carlotti F, Zaldumbide A, Loomans CJ, et al. Isolated human islets contain a distinct population of mesenchymal stem cells. Islets 2010;2:164–173 [DOI] [PubMed] [Google Scholar]
- 10.Chase LG, Ulloa-Montoya F, Kidder BL, Verfaillie CM. Islet-derived fibroblast-like cells are not derived via epithelial-mesenchymal transition from Pdx-1 or insulin-positive cells. Diabetes 2007;56:3–7 [DOI] [PubMed] [Google Scholar]
- 11.Théry C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol 2009;9:581–593 [DOI] [PubMed] [Google Scholar]
- 12.Admyre C, Johansson SM, Qazi KR, et al. Exosomes with immune modulatory features are present in human breast milk. J Immunol 2007;179:1969–1978 [DOI] [PubMed] [Google Scholar]
- 13.Qazi KR, Torregrosa Paredes P, Dahlberg B, Grunewald J, Eklund A, Gabrielsson S. Proinflammatory exosomes in bronchoalveolar lavage fluid of patients with sarcoidosis. Thorax 2010;65:1016–1024 [DOI] [PubMed] [Google Scholar]
- 14.Simons M, Raposo G. Exosomes—vesicular carriers for intercellular communication. Curr Opin Cell Biol 2009;21:575–581 [DOI] [PubMed] [Google Scholar]
- 15.Trajkovic K, Hsu C, Chiantia S, et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008;319:1244–1247 [DOI] [PubMed] [Google Scholar]
- 16.Choi DS, Kim DK, Kim YK, Gho YS. Proteomics, transcriptomics and lipidomics of exosomes and ectosomes. Proteomics 2013;13:1554–1571 [DOI] [PubMed] [Google Scholar]
- 17.Denzer K, Kleijmeer MJ, Heijnen HF, Stoorvogel W, Geuze HJ. Exosome: from internal vesicle of the multivesicular body to intercellular signaling device. J Cell Sci 2000;113:3365–3374 [DOI] [PubMed] [Google Scholar]
- 18.Wang GJ, Liu Y, Qin A, et al. Thymus exosomes-like particles induce regulatory T cells. J Immunol 2008;181:5242–5248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zitvogel L, Regnault A, Lozier A, et al. Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat Med 1998;4:594–600 [DOI] [PubMed] [Google Scholar]
- 20.Wieckowski EU, Visus C, Szajnik M, Szczepanski MJ, Storkus WJ, Whiteside TL. Tumor-derived microvesicles promote regulatory T cell expansion and induce apoptosis in tumor-reactive activated CD8+ T lymphocytes. J Immunol 2009;183:3720–3730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y, Xiang X, Zhuang X, et al. Contribution of MyD88 to the tumor exosome-mediated induction of myeloid derived suppressor cells. Am J Pathol 2010;176:2490–2499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sheng H, Hassanali S, Nugent C, et al. Insulinoma-released exosomes or microparticles are immunostimulatory and can activate autoreactive T cells spontaneously developed in nonobese diabetic mice. J Immunol 2011;187:1591–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bashratyan R, Sheng H, Regn D, Rahman MJ, Dai YD. Insulinoma-released exosomes activate autoreactive marginal zone-like B cells that expand endogenously in prediabetic NOD mice. Eur J Immunol 2013;43:2588–2597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leiter EH. The NOD mouse: a model for insulin-dependent diabetes mellitus. Curr Protoc Immunol 2001;15:Unit 15.9 [DOI] [PubMed] [Google Scholar]
- 25.Qazi KR, Gehrmann U, Domange Jordö E, Karlsson MC, Gabrielsson S. Antigen-loaded exosomes alone induce Th1-type memory through a B-cell-dependent mechanism. Blood 2009;113:2673–2683 [DOI] [PubMed] [Google Scholar]
- 26.Melli K, Friedman RS, Martin AE, et al. Amplification of autoimmune response through induction of dendritic cell maturation in inflamed tissues. J Immunol 2009;182:2590–2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tabeta K, Hoebe K, Janssen EM, et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol 2006;7:156–164 [DOI] [PubMed] [Google Scholar]
- 28.Le Blanc K, Mougiakakos D. Multipotent mesenchymal stromal cells and the innate immune system. Nat Rev Immunol 2012;12:383–396 [DOI] [PubMed] [Google Scholar]
- 29.Fiorina P, Jurewicz M, Augello A, et al. Immunomodulatory function of bone marrow-derived mesenchymal stem cells in experimental autoimmune type 1 diabetes. J Immunol 2009;183:993–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guz Y, Nasir I, Teitelman G. Regeneration of pancreatic beta cells from intra-islet precursor cells in an experimental model of diabetes. Endocrinology 2001;142:4956–4968 [DOI] [PubMed] [Google Scholar]
- 31.Chen LB, Jiang XB, Yang L. Differentiation of rat marrow mesenchymal stem cells into pancreatic islet beta-cells. World J Gastroenterol 2004;10:3016–3020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trudeau JD, Dutz JP, Arany E, Hill DJ, Fieldus WE, Finegood DT. Neonatal beta-cell apoptosis: a trigger for autoimmune diabetes? Diabetes 2000;49:1–7 [DOI] [PubMed] [Google Scholar]
- 33.Mathis D, Vence L, Benoist C. beta-Cell death during progression to diabetes. Nature 2001;414:792–798 [DOI] [PubMed] [Google Scholar]
- 34.Höglund P, Mintern J, Waltzinger C, Heath W, Benoist C, Mathis D. Initiation of autoimmune diabetes by developmentally regulated presentation of islet cell antigens in the pancreatic lymph nodes. J Exp Med 1999;189:331–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lennon GP, Bettini M, Burton AR, et al. T cell islet accumulation in type 1 diabetes is a tightly regulated, cell-autonomous event. Immunity 2009;31:643–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yamanouchi J, Verdaguer J, Han B, Amrani A, Serra P, Santamaria P. Cross-priming of diabetogenic T cells dissociated from CTL-induced shedding of beta cell autoantigens. J Immunol 2003;171:6900–6909 [DOI] [PubMed] [Google Scholar]
- 37.Burton AR, Vincent E, Arnold PY, et al. On the pathogenicity of autoantigen-specific T-cell receptors. Diabetes 2008;57:1321–1330 [DOI] [PubMed] [Google Scholar]
- 38.Bergman B, Haskins K. Islet-specific T-cell clones from the NOD mouse respond to beta-granule antigen. Diabetes 1994;43:197–203 [DOI] [PubMed] [Google Scholar]
- 39.Haskins K. Pathogenic T-cell clones in autoimmune diabetes: more lessons from the NOD mouse. Adv Immunol 2005;87:123–162 [DOI] [PubMed] [Google Scholar]
- 40.Todd JA, Wicker LS. Genetic protection from the inflammatory disease type 1 diabetes in humans and animal models. Immunity 2001;15:387–395 [DOI] [PubMed] [Google Scholar]
- 41.Suri A, Walters JJ, Gross ML, Unanue ER. Natural peptides selected by diabetogenic DQ8 and murine I-A(g7) molecules show common sequence specificity. J Clin Invest 2005;115:2268–2276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tisch R, McDevitt H. Insulin-dependent diabetes mellitus. Cell 1996;85:291–297 [DOI] [PubMed] [Google Scholar]
- 43.Tian J, Dang H, von Boehmer H, Jaeckel E, Kaufman DL. Transgenically induced GAD tolerance curtails the development of early β-cell autoreactivities but causes the subsequent development of supernormal autoreactivities to other β-cell antigens. Diabetes 2009;58:2843–2850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tian J, Gregori S, Adorini L, Kaufman DL. The frequency of high avidity T cells determines the hierarchy of determinant spreading. J Immunol 2001;166:7144–7150 [DOI] [PubMed] [Google Scholar]
- 45.Lehmann PV, Forsthuber T, Miller A, Sercarz EE. Spreading of T-cell autoimmunity to cryptic determinants of an autoantigen. Nature 1992;358:155–157 [DOI] [PubMed] [Google Scholar]
- 46.Yang C, Robbins PD. The roles of tumor-derived exosomes in cancer pathogenesis. Clin Dev Immunol 2011;2011:842849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Balkom BW, de Jong OG, Smits M, et al. Endothelial cells require miR-214 to secrete exosomes that suppress senescence and induce angiogenesis in human and mouse endothelial cells. Blood 2013;121:3997–4006, S1–S15 [DOI] [PubMed] [Google Scholar]
- 48.Bryniarski K, Ptak W, Jayakumar A, et al. Antigen-specific, antibody-coated, exosome-like nanovesicles deliver suppressor T-cell microRNA-150 to effector T cells to inhibit contact sensitivity. J Allergy Clin Immunol 2013;132:170–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vallhov H, Gutzeit C, Johansson SM, et al. Exosomes containing glycoprotein 350 released by EBV-transformed B cells selectively target B cells through CD21 and block EBV infection in vitro. J Immunol 2011;186:73–82 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.