

Abstract

Proton uptake accompanies the reduction of all known substrates by nitrogenase. As a consequence, a higher pH should limit the availability of protons as a substrate essential for turnover, thereby increasing the proportion of more highly reduced forms of the enzyme for further study. The utility of the high-pH approach would appear to be problematic in view of the observation reported by Pham and Burgess [(1993) Biochemistry 32, 13725–13731] that the MoFe-protein undergoes irreversible protein denaturation above pH 8.65. In contrast, we found by both enzyme activity and crystallographic analyses that the MoFe-protein is stable when incubated at pH 9.5. We did observe, however, that at higher pHs and under turnover conditions, the MoFe-protein is slowly inactivated. While a normal, albeit low, level of substrate reduction occurs under these conditions, the MoFe-protein undergoes a complex transformation; initially, the enzyme is reversibly inhibited for substrate reduction at pH 9.5, yet in a second, slower process, the MoFe-protein becomes irreversibly inactivated as measured by substrate reduction activity at the optimal pH of 7.8. The final inactivated MoFe-protein has an increased hydrodynamic radius compared to that of the native MoFe-protein, yet it has a full complement of iron and molybdenum. Significantly, the modified MoFe-protein retains the ability to specifically interact with its nitrogenase partner, the Fe-protein, as judged by the support of ATP hydrolysis and by formation of a tight complex with the Fe-protein in the presence of ATP and aluminum fluoride. The turnover-dependent inactivation coupled to conformational change suggests a mechanism-based transformation that may provide a new probe of nitrogenase catalysis.
Substrate reduction by the molybdenum-dependent nitrogenase involves two protein components, the molybdenum iron protein (MoFe-protein), containing the active site for substrate reduction, and the iron protein (Fe-protein), serving as the unique ATP-dependent reductant for the MoFe-protein.1−3 A striking feature of the nitrogenase-catalyzed reaction is that the electron flux through the system is independent of the substrate that is being reduced;1 under a given set of conditions, the number of electrons transferred to substrate per active site per unit time is the same for the reduction of dinitrogen to ammonia (the physiological reaction), the reduction of acetylene to ethylene (commonly used to assay nitrogenase activity), or the reduction of protons to dihydrogen, which occurs in the absence (or sufficiently low concentrations) of other reducible substrates. An important characteristic of the nitrogenase reaction is that substrates can bind only to forms of the MoFe-protein reduced by two or three electrons relative to the “as-isolated” form, which can be efficiently generated only in the presence of the reduced Fe-protein and ATP.4,5 Efforts to generate significant populations of more highly reduced forms of the MoFe-protein for biophysical or structural characterizations are confounded by the ubiquitous presence of protons that are reduced to dihydrogen with the concomitant return to the initial stages of the catalytic cycle.
While protons cannot be eliminated from the aqueous solution environment, their concentration can be reduced by working at higher pHs. The pH dependence of nitrogenase activity may be described as a bell-shaped curve,6−10 with optimal activity occurring around pH 7–8 and decreasing at both lower and higher pHs (half-maximal pHs of ∼6.5 and 8.5, respectively). While it is not surprising that nitrogenase activity decreases at high pH, Pham and Burgess8 reported that the MoFe-protein is “irreversibly damaged by preincubation above pH 8.65” in a 3 min incubation, through a process suggested to arise from “a critical group on the MoFe protein with a pK in that range whose deprotonation leads either to cluster destruction or to an irreversible change in the structure of some critical part of the protein.” Indeed, the idea that the MoFe-protein denatures at pHs outside the optimal range has been reflected in the subsequent design of pH-dependent experiments of nitrogenase activities (see ref (11)). However, while attempting to trap substrates and inhibitors of nitrogenase for crystallization studies, we observed a distinct type of high-pH inactivation of the MoFe-protein that was not simple denaturation. As we report here, inactivation of the MoFe-protein at higher pHs is not due to protein instability but is rather the consequence of a complex, mechanism-based reaction with the potential to provide insight into the mechanism(s) of nitrogenase.
Materials and Methods
Preparation of Nitrogenase Proteins
The MoFe-protein and Fe-protein from Azotobacter vinelandii (designated Av1 and Av2, respectively) were isolated under anaerobic conditions as previously described.12 The specific activities for acetylene reduction were ∼2200 nmol of ethylene min–1 mg–1 for Av1 and ∼1800 nmol of ethylene min–1 mg–1 for Av2. Unless otherwise noted, all operations were conducted anaerobically by appropriate manipulations using a Schlenk line connected to oxygen-scrubbed argon or in an anaerobic chamber.
The component ratio (CR) of Fe-protein to MoFe-protein is defined as the moles of Av2 per mole of Av1 active site (with two active sites per Av1 tetramer). The CR may be calculated from the concentrations of Av1 and Av2 through the relationship CR = 1.82(cAv2/cAv1), where cAv1 and cAv2 are the protein concentrations in the final reaction mixture, given in milligrams per milliliter, based on molecular masses of 233 and 64 kDa for Av1 and Av2, respectively. The protein concentration was determined by absorbance at 410 nm using extinction coefficients of 76 and 9.4 mM–1 cm–1 for Av1 and Av2, respectively.
Tribuffer Systems
Because the kinetics of reduction of the substrate by nitrogenase are sensitive to both pH6−10 and ionic strength,13,14 it is critical to use solution conditions under which the two parameters may be varied independently. For the pH range of 7.8–10 utilized in this study, a tribuffer system developed by Ellis and Morrison15 was employed, composed of three components: 100 mM N-(2-acetamido)-2-aminoethanesulfonic acid (ACES), 50 mM tris(hydroxymethyl)aminomethane (Tris), and 50 mM ethanolamine, with pKa values of 6.67, 8.0, and 9.5 at 30 °C, respectively. Over the pH range from 8 to 10, the ionic strength of this buffer ranges from 0.096 to 0.100 M. We note that this is a buffer system different from that employed in the study by Pham and Burgess {75 mM [bis(2-hydroxyethyl)amino] tris(hydroxymethyl)methane (Bis-Tris), 38 mM N-(2-hydroxyethy1)piperazine-N′-propanesulfonic acid (HEPPS), and 38 mM 2-(cyclohexylamino)ethanesulfonic acid (CHES)8} that exhibits a calculated ionic strength variation over the same pH range from 0.023 to 0.070 M.
Nitrogenase Activity Assays
The activity of the nitrogenase proteins was determined by the reduction of acetylene to ethylene as expressed in the head space gas equilibrated with the reaction solution. The assay contained 20 mM sodium dithionite (the source of reducing equivalents and also to maintain anaerobic conditions) and an ATP regeneration system (5 mM MgCl2, 5 mM ATP, 20 mM creatine phosphate, and 23 units/mL creatine phosphokinase).12 For the standard assay, the reaction mixture was buffered with 50 mM Tris-HCl (pH 7.8), while for reactions above pH 8, the tribuffer system described above was utilized. The pH of the tribuffer with ATP and creatine phosphate was adjusted by adding 6 M NaOH to a value higher than the target pH, so that the final pH was 9.5 (or other target pH) after processing, in order, addition of the kinase and MgCl2, multiple cycles of degassing, addition of dithionite, and finally introduction of the component proteins. Because these common buffers are strongly dependent on temperature, the final pH was verified by measurement at 30 °C for a test vial with all components. No inhibitory effects on acetylene reduction by nitrogenase were observed for the tribuffer when compared to the standard assay. For the kinetic studies, the reduction of acetylene was initiated by the addition to the reaction assay of premixed Av1 and Av2 to achieve the desired concentrations and component ratio. The volume of the added Av1/Av2 mixture was kept constant by addition of 50 mM Tris-HCl (pH 7.75), 200 mM NaCl, and 5 mM sodium dithionite, so that the final ionic strength was independent of the CR. The anaerobic assay reactions were conducted at 30 °C and for 10 min in 8.9 mL serum vials with a 1.0 mL assay solution and an argon atmosphere containing 1.0 mL of acetylene gas. The reaction was terminated by the addition of 0.25 mL of 1 M citric acid, and ethylene in the gas phase was quantified by gas chromatography.
In addition to the standard assay described above, two types of activity measurements were used: the “head space” and “specific activity” assays. In the head space assay, the activity was monitored by the rate of appearance of ethylene in the gas phase of the reaction vial while allowing the reaction to continue. Ethylene and acetylene were quantified by gas chromatography of gas aliquots (50 μL) removed from the head space at the designed time points. The specific activity of Av1 in the reaction vial was monitored by removing liquid samples from the assay mixture at specified time points, for immediate transfer into a pH 7.8 standard assay solution containing excess additional Av2 (CR of ∼60). The transfer time was <10 s, and the transfer initiated the standard 10 min, pH 7.8 assay. SigmaPlot version 11.0 (Systate Software, Inc.) was used for curve fitting and display of the experimental data.
Analytical Column Chromatography
The proteins in a reaction mixture were analyzed by size exclusion chromatography on a 1 cm × 30 cm column of Superdex S-200, run under anaerobic conditions. The elution profile was monitored by the absorbance at either 390 or 410 nm, as indicated.
ATP Hydrolysis As Measured by Creatine
Because 1 mol of creatine is produced by the creatine kinase-catalyzed phosphorylation of 1 mol of ADP by creatine phosphate, the amount of ATP hydrolyzed was established by measuring the change in creatine concentration in the reaction mixture.16,17 These reactions were identical to the acetylene reduction assay, except the reaction was quenched by 0.25 mL of 0.5 M Na2EDTA (pH 8.0).
Formation of the Av1–Av2–ADP-AlF4– Complex
The ADP-AlF4–-stabilized Av1–Av2 complex was prepared following the protocols of Renner and Howard.17 Inactive Av1 at a final concentration of 0.066 mg/mL and Av2 at a CR of ∼4 were incubated in 100 mM MOPS, 50 mM Tris, and 100 mM NaCl (pH 7.3) with 10 mM sodium dithionite, 4 mM NaF, and 0.2 mM AlCl3. For the reaction in the presence of ATP, 1 mM ATP, 8 mM MgCl2, and the creatine kinase-based ATP regenerating system were added. For reactions with ADP, 1 mM ADP and 8 mM MgCl2 were added, without an ATP regenerating system. The reactions were allowed to proceed for 30 min at 30 °C and the products analyzed by analytical column chromatography.
Metal Analysis
The molybdenum and iron contents of Av1 were determined by inductively coupled plasma mass spectrometry (ICP-MS) using a Hewlett-Packard 4500 ICP-MS system. Iron and molybdenum standards (Ultra Scientific Analytical solutions for ICP-MS) over concentration ranges comparable to and overlapping with those of the protein were analyzed in tandem with the protein samples. Proteins and standards were prepared in ultrapure H2O containing 1% nitric acid (ICP-MS grade, Fluka) with appropriate reagent and buffer blanks. The protein concentration was determined by absorbance at 410 nm prior to 1:400 dilution in nitric acid to give a concentration of 35–50 μg/mL for aspiration by the ICP-MS instrument. The Fe and Mo concentrations were determined using 56Fe and 96Mo isotopes and normalized for the Av1 concentration. The values were verified using the other natural abundance isotopes for both elements.
Determination of the Crystal Structure for Native Av1 at pH 9.5
Av1 was crystallized at 22 °C in an anaerobic chamber using the sitting drop method with mixed drops containing 2 μL of a 42 mg/mL Av1 solution and 4 μL of the precipitant. The precipitant contained 150 mM ACES, 75 mM Tris, 75 mM ethanolamine (pH 9.79), 17–18% PEG 3350, 0.8 M NaCl, and 1 mM sodium dithionite. Upon addition of the protein, the final pH of the mixed drop was 9.5. For cryoprotection, crystals of native Av1 grown at pH 9.5 were successively transferred into the reservoir solution with increasing concentrations of 2-methyl-2,4-pentanediol (MPD) up to 20%. Diffraction data were collected on beamline 12-2 at the Stanford Synchrotron Radiation Lightsource (SSRL), processed with XDS,18 and scaled with SCALA.19 The structure was determined by molecular replacement with MOLREP,20 using as a search model the Av1 structure determined at 1.16 Å resolution [Protein Data Bank (PDB) entry 1M1N]. The refinement was performed with REFMAC21 and PHENIX,22 while graphics program COOT23 was used for displaying maps and rebuilding the model. Data and refinement statistics are summarized in Table 1.
Table 1. Data and Refinement Statistics for the Determination of the Crystal Structure of Native Av1 at pH 9.5.
| Crystal Parameters | |
| space group | P21 |
| a (Å) | 76.53 |
| b (Å) | 127.92 |
| c (Å) | 107.08 |
| β (deg) | 108.9 |
| Data Processing | |
| resolution range (Å) | 39.3–2.0 (2.11–2.00) |
| total no. of observations | 356532 (52041) |
| no. of unique observations | 124962 (18522) |
| completeness (%) | 95.3 (96.8) |
| multiplicity | 2.9 (2.8) |
| Rmerge (%)a | 10.7 (0.733) |
| mean I/σ(I) | 8.3 (1.7) |
| Refinement | |
| resolution range (Å) | 36.7–2.0 (2.023–2.00) |
| Rwork | 0.156 (0.255) |
| Rfree | 0.204 (0.300) |
| root-mean-square deviation for bond lengths (Å) | 0.012 |
| root-mean-square deviation for bond angles (deg) | 1.69 |
| Ramachandran plot [favored, allowed, outliers (%)] | 96.5, 3.3, 0.2 |
Rmerge(I) = ∑hkl[(∑i|Ihkl,i – ⟨Ihkl⟩|)/∑iIhkl,i], where Ihkl is the intensity of an individual measurement of the reflection with indices hkl and ⟨Ihkl⟩ is the mean intensity of that reflection.
Residue 440 of the α-chain was corrected to Gln rather than the Glu reported in all previous structures to reflect the correct A. vinelandii nifD gene sequence;24 this discrepancy was highlighted by a recent analysis of 95 MoFe-protein sequences demonstrating that all but two had Gln and Asn at this position, with no examples of Glu.25
Results
Time Course of Acetylene Reduction at High pH
The initial test of nitrogenase activity well above its pH optimum was conducted at pH 9.5 using the head space measure of ethylene formation with time as shown in Figure 1A. The rate curve indicated a progressive loss of product formation with complete cessation well before depletion of any component of the assay. Supplementing the reaction mixture at longer times with fresh aliquots of dithionite, Av2, or components of the ATP regenerating system did not result in further substrate reduction, which confirmed that the loss of acetylene reduction activity was not due to depletion of any of these components. Together, these studies implicated progressive inhibition of the Av1 that can be described by a first-order process characteristic of slow inhibitors26 such as the aluminum fluoride inhibition of nitrogenase activity:17
| 1 |
where v0, ki, and P(t) are the initial rate of product formation (the enzyme catalytic activity expressed in nanomoles per minute), the observed first-order inactivation rate constant (inverse minutes) of Av1, and the time-dependent amount of ethylene in the head space (nanomoles), respectively. v0/ki is the total amount of product produced at the limit t = ∞. The solid lines in Figure 1 represent the nonlinear curve fitting of the data points based on eq 1. For this type of inhibition, the cessation of product formation at long time points (the plateau in Figure 1) implies a quasi-irreversible state of inhibition of the enzyme, at least under the pH 9.5 conditions of the assay.17,26 For a slowly reversible reaction, a flat line plateau would not be observed and the kinetic expression would have additional elements to include the slower back reaction.26
Figure 1.

Acetylene reduction activity as measured in the head space assay. Assays contained 0.25 mg of Av1 with a CR of 2.5 at 30 °C. (A) Time course of ethylene production in the head space generated at pH 9.5. The solid line was generated by curve fitting time points using eq 1. (B) Time course of ethylene production at various pHs over the range of 8.80–9.68. (C) Initial rate (v0) and inhibition rate constant (ki) calculated according to eq 1 for the assays that generated the data depicted in panel B. Error bars show standard errors estimated from the curve fitting.
The generality of the progressive inhibition as a consequence of pH was demonstrated by a series of experiments using the head space assay in the upper range of pH 8.8–9.8 that has been previously reported for various nitrogenase studies.6−10 The data were fit using eq 1 shown as the solid lines in Figure 1B, with the calculated ki and v0 values given in Figure 1C. Consistent with the accepted nitrogenase pH activity dependence, the initial velocity, v0, decreases with an increased pH. More significantly, the observed inactivation rate, ki, increases with an increased pH, suggesting that a deprotonated state of the enzyme is a precursor for the inhibition. This clearly separates the two phenomena: the enzymatic activity follows a pH-dependent decline, while the inhibition process increases with a higher pH.
Turnover Conditions Are Required for High-pH Inactivation of Av1
Inhibition of the enzymatic activity as a consequence of Av1 inactivation was established by the specific activity assay that determines the amount of active Av1 at a saturating Av2 concentration in the pH 7.8 assay. Following initiation of the reaction at pH 9.5, aliquots of the reaction mixture were sampled at designated time points and the Av1 specific activity was determined. As shown in Figure 2, Av1 from the pH 9.5 incubation was progressively inactivated as indicated by the loss of specific activity measured at pH 7.8. The time-dependent loss of Av1 specific activity at pH 9.5 required turnover of the complete system that was established by a series of changes in the pH 9.5 reaction conditions. If Av2 or the ATP regenerating system was omitted or if AMP-PNP, a nonhydrolyzable ATP analogue, was used, no acetylene reduction was detected at pH 9.5 (as expected), and through the first 2 h of incubation at pH 9.5, there was an only ∼10% decrease in Av1 specific activity as determined at pH 7.8, a time in which there was ∼90% inactivation under the full turnover conditions. Most importantly, incubation of Av1 or Av2 alone at pH 9.5 showed only minimal activity loss even after 4 h. Although acetylene reduction was the most convenient way to monitor enzyme activity, the inactivation proceeded equally well when dinitrogen replaced the acetylene or when there was no added substrate beyond the protons in the solution (see Figure S1 of the Supporting Information). Furthermore, the inhibitor of all substrates except protons, carbon monoxide, did not block the inactivation of Av1.
Figure 2.

Specific activity of Av1 measured by the standard assay at pH 7.8 following incubation at pH 9.5 and 30 °C for the indicated times. The reactions at pH 9.5 were variations of the full turnover conditions with 0.25 mg of Av1: (●) Av1 and Av2 at a CR of 2.5 with the ATP regenerating system, (◆) Av1 and Av2 at a CR of 2.5 with the nonhydrolyzable ATP analogue AMP-PNP, (▲) Av1 with the ATP regenerating system (without Av2), and (■) Av1 and Av2 at a CR of 2.5 without nucleotide. Error bars indicate the standard deviation for duplicate experiments.
Component Ratio and Protein Concentration Effects on the pH 9.5 Reaction
Central to the understanding of the complex kinetic properties of the two-protein component system of nitrogenase is that the activity is dependent on the total protein concentration and component ratio as important factors controlling the electron flux during turnover.27,28 As one example, for a fixed concentration of the MoFe-protein, the initial velocity for acetylene reduction increases with an increasing CR until a plateau level of activity is obtained, indicative of saturation kinetics; the specific activity of the MoFe-protein is determined from the plateau level of activity upon titration with the Fe-protein at pH 7.8. The same general behavior was also observed upon titration of a fixed amount of Av1 with different CRs of Av2 at pH 9.5, although the amounts of product formed per time are much smaller than at pH 7.8 (Figure 3). By fitting the head space data (Figure 3A) to eq 1, we can determinie initial velocity v0 as a function of CR, and v0 is presented in the form of an Av1 titration curve. As shown in Figure 3B, the initial velocity exhibits a strong CR dependence similar in form to that observed at pH 7.8, suggesting the initial rate correctly reflects the nitrogenase reaction and its pH dependence. At the saturation of Av1 by Av2, the specific activity of Av1 at pH 9.5 was estimated to be 100 nmol min–1 mg–1, or ∼5% of the value at pH 7.8. As also observed at the optimal pH,29−31 the Av1 titration curve at pH 9.5 is not fully hyperbolic and has a “lag” at low concentrations of Av2 (CR < ∼1). This is consistent with a similar process of interactions between the MoFe-protein and Fe-protein that is observed at the optimal pH and is maintained at the higher pH. One measure of the association of Av2 and Av1 is the CR at half-saturation that at pH 9.5 is ∼4 and similar to that at pH 7.8 (an absolute comparison is obviated by the necessary protein concentration differences with the approximately 2 order of magnitude difference in v0). An additional test that shows that the enzymatic substrate reduction at pH 9.5 reflects a turnover mechanism similar to that at pH 7.8 is the protein concentration dependence of the reaction rate. As shown in Figure 3C, over an 8-fold protein concentration range and at a constant CR, v0 increased ∼6-fold, a relative increase comparable to that observed at pH 7.8.32−34
Figure 3.

Component ratio and protein concentration dependence of Av1 inactivation monitored by the head space assay at pH 9.5 and 30 °C. (A) Time-dependent ethylene formation at various CRs with the Av1 concentration fixed at 0.25 mg per reaction. Solid lines are nonlinear curve fits to the data points by eq 1. (B) Initial rate (v0) and inhibition rate (ki) calculated according to eq 1. Error bars indicate standard errors from the curve fitting. (C) Initial rate (v0) and inhibition rate (ki) determined from progress curves at various protein concentrations and at a constant component ratio of 2.5. The data are plotted in terms of the Av1 concentration using an average of two determinations for each data point.
In contrast to the clear similarity of the patterns in turnover parameters at pH 9.5 and at the optimal pH, the inactivation rate constant, ki, showed a fully different pattern with these variables (Figure 3B,C). Namely, ki is substantially independent of CR and protein concentration and was calculated to be ∼0.07 min–1 (corresponding to a half-life of ∼10 min). The small variance of ∼20% for ki is in contrast to ≥1 order of magnitude changes in v0 values for the same CR. The implication is that whatever mechanism of inhibition is occurring, it is a first-order process that is independent of the overall nitrogenase turnover rate as measured by product formed.
Effects of the Component Ratio on Specific Activity
In contrast to the apparent first-order inhibition kinetics observed for the head space assay at pH 9.5, the inactivation kinetics monitored by the specific activity measured at pH 7.8 cannot be modeled by a single kinetic phase. As shown in Figure 4, the kinetics of inactivation measured by specific activity are quite sensitive to the CR used in the pH 9.5 reaction; e.g., for a CR of 0.15, incubation for ∼3 h is required to eliminate 50% of the specific activity, while for CRs of >1, the corresponding time is ∼30 min. Most importantly, the loss of specific activity is significantly slower than the inhibition observed at pH 9.5 based on the direct measure of product in the head space. As seen via comparison of Figures 3 and 4, for all CRs at 60 min, acetylene reduction at pH 9.5 has stopped while the specific activity ranges from 25 to 75%. This strongly implies that the inhibited state at pH 9.5 is partially reversible at pH 7.8. The time of incubation at pH 7.8 before initiating the specific activity assay showed no effect on the measured value, which suggests the reversible step must be fast compared to the minimal time for sampling of the pH 9.5 reaction into the pH 7.8 assay.
Figure 4.

Time course of the specific activity measured at pH 7.8 of Av1 from the inactivation reactions at pH 9.5 with various CRs. At the indicated times, aliquots were removed from the inactivation reaction mixture at pH 9.5 and the specific activity of the Av1 was measured at pH 7.8 with saturating concentrations of Av2. The pH 9.5 inactivation reaction mixtures contained 0.25 mg of Av1. Error bars indicate the standard deviation from triplicate reactions.
ATP Hydrolysis during Turnover
The kinetic properties of the reaction at pH 9.5 versus the loss of specific activity measured at pH 7.8 suggested that some level of interactions between Av1 and Av2 proceeds after cessation of the observed product formation in the head space gas. ATP hydrolysis occurs only in the complex between Av1 and Av2, although ATP hydrolysis is independent of the oxidation state of the two proteins.7,35,36 Indeed, while Av2 can bind MgATP, a conformational change stabilized by complex formation with Av1 is required for hydrolysis.17,37 The number of ATP equivalents turned over by the enzyme can be conveniently measured by following the formation of creatine in the ATP regenerating system.16,17 The results for the correlation of ATP equivalents used and product formation at pH 9.5 are plotted in Figure 5A for CRs of 0.3 and 1.0. The clear result is that ATP hydrolysis initially tracks the substrate reduction yet continues long after product formation has reached completion. Even for those low component ratios (e.g., CR = 0.3) where the level of ethylene production is below the threshold of detection at pH 9.5, ATP hydrolysis readily occurs long into the time course for inactivation and provides evidence that the Av2 is still active. Indeed, the ability of the various forms of Av1 generated during turnover at pH 9.5 to support ATP hydrolysis is unmistakably shown in Figure 5B for the changes in specific activity over the time course of inactivation. For a CR of 1.0 where at 240 min the Av1 is nearly fully inactivated, ATP hydrolysis is still supported at ∼40% of the rate of native Av1 at saturation with Av2. The initial time points show an ATP:ethylene ratio of ∼5, in keeping with the accepted value of 4–5 with dithionite as the terminal reducing agent, but the ratio rapidly increases to >50 for the later time points as the Av1 is inactivated. These high ratios represent the small fraction of active enzyme plus the increasing amount of inactive enzyme that is still capable of supporting substantial ATP hydrolysis.
Figure 5.

Time course of ethylene production and ATP hydrolysis during inactivation at pH 9.5. (A) Formation of ethylene and ATP hydrolysis (measured as the production of creatine from the creatine kinase-based ATP regenerating system) in the inactivation reaction at pH 9.5 for CRs of 0.3 and 1.0. Assays contained 0.5 mg of Av1. (B) Ethylene production and ATP hydrolysis in specific activity assays at pH 7.8 for samples from the time course of the pH 9.5 inactivation. Values are normalized to the values at time zero. Error bars indicate the standard deviation from duplicate reactions.
Characterization of Native Av1 Incubated at pH 9.5
The activity studies clearly indicated that Av1 was not denatured with loss of activity at pH 9.5. To directly assess the structural consequences of incubation of Av1 at pH 9.5 (in the absence of turnover) and to provide a control structure for future work at higher pHs, the crystal structure at pH 9.5 was determined. Single crystals were obtained and reached their maximal size in 3 days. X-ray diffraction data were collected to 2.0 Å resolution, and the resulting structure of native Av1 at pH 9.5 was found to adopt the same overall conformation as that crystallized at pH 8.038,39 (Figure 6). The root-mean-square deviation (rmsd) in positions of main chain atoms between the pH 9.5 structure and that refined at 1 Å resolution (PDB entry 3U7Q(39)) was 0.3 Å, with little differences observed in the metalloclusters (Figure 6). Some repositioning of side chains was indicated that may reflect deprotonation at high pH, including the C1 carboxyl group of the FeMo-cofactor homocitrate. This carboxyl group interacts with Glnα191, and intriguingly, diastereomers of fluorohomocitrate substituted at the methylene group of this arm of homocitrate support significantly different nitrogen reduction activities;40 a previous analysis of the diastereomer results in terms of the Av1 structure suggested a possible role for the C1 arm of homocitrate in proton transfer reactions.41
Figure 6.
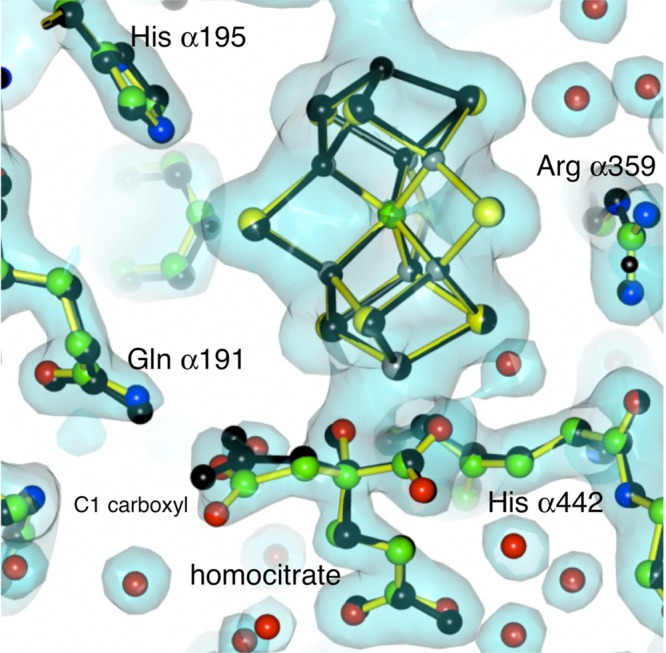
Electron density map around the FeMo-cofactor and homocitrate of the A. vinelandii MoFe-protein crystallized at pH 9.5. The map is calculated at 2.0 Å resolution and contoured at 1.5 times the standard deviation. The yellow bonds and colored atoms represent the Av1 structure at pH 9.5, while the black bonds and atoms indicate the structure of Av1 at pH 8.0 as determined by Spatzal et al. at 1.0 Å resolution (PDB entry 3U7Q(39)). Overall, the two structures are quite similar (rmsd of 0.3 Å), although a displacement of the C1 carboxyl arm of homocitrate is indicated in the pH 9.5 structure compared to the pH 8.0 structure.
Characterization of Modified (inactive) Av1
To characterize the inactive protein, we isolated Av1 from the pH 9.5 reaction mixture after incubation for 4 h (specific activity of <10%) using the size exclusion chromatography column at pH 7.3. Intriguingly, as shown in Figure 7A, the peak position of inactive Av1 shifted to an earlier elution time, relative to the position observed for native Av1 or for Av1 incubated at pH 9.5 in the absence of turnover components. The shift in elution position indicates that the hydrodynamic radius of inactive Av1 has increased relative to that of native Av1. The observed shift in the hydrodynamic radius was a consequence of the pH 9.5 turnover as shown in Figure 7A; incubation of Av1 alone or in combination with Av2 without ATP did not cause a shift in the elution position. Indeed, the complete recovery of both Av1 and Av2 (as measured by the absorbance at 410 nm) from a 4 h incubation at pH 9.5, either alone or under turnover conditions, further substantiates the stability of the proteins at pHs as high as 9.5. This form of Av1, hereafter termed Av1-mod, is stable to multiple cycles of rechromatography at pH 7.3–8.0 in 100–200 mM NaCl.
Figure 7.

Anaerobic size exclusion chromatography of Av1 on a 1 cm × 30 cm column of Superdex S-200. The flow rate was 0.5 mL/min at 22 °C. (A) Reaction mixtures (1.0 mL) containing 1.0 mg of Av1, with or without Av2 at a CR of 3.4 and with or without the ATP regenerating system, were incubated at pH 9.5 in tribuffer and 50 mM NaCl at 30 °C for 3.5 h; 0.8 mL of the reaction mixture was injected from the reaction mixture onto the column that was equilibrated with 50 mM Tris buffer (pH 7.5) containing 200 mM NaCl and 5 mM sodium dithionite: elution line 1 (red), Av1 and Av2 incubated without the ATP regenerating system; elution line 2 (blue), Av1 alone; elution line 3 (black), complete turnover conditions, Av1 and Av2 with the ATP regenerating system. Elution was monitored at 410 nm. (B) Same column as in panel A equilibrated with 50 mM MOPS buffer (pH 7.3) containing 100 mM NaCl, 1 mM MgCl2, 5 mM NaF, 0.25 mM AlCl3, and 5 mM sodium dithionite. Av1-mod was prepared as described for line 3 of panel A and re-isolated in MOPS buffer (pH 7.3) without AlCl3 or NaF. Av1-mod (∼0.2 mg) was incubated with Av2 (CR = 4.6) in 2.4 mL of MOPS buffer containing 5 mM NaF, 0.25 mM AlCl3, and either no nucleotide [elution line 1 (black)], 1 mM ATP regenerating system [elution line 2 (purple)], or 1 mM ADP [elution line 3 (green)]. After incubation at 25 °C for 30 min to form the complex, 0.9 mL of each reaction mixture was applied to the column. The absorption of the eluate was monitored at 390 nm.
The absorption at 410 nm arises from the metalloclusters, and the integrated peak areas for Av1 were similar for all samples, irrespective of pH and precise conditions of the turnover (the same total amounts of Av1 were initially used in the compared chromatographs). This observation implies that the metalloclusters in Av1-mod are not significantly changed through either loss of iron, rearrangement of clusters, or other changes in the metal environment. This latter conclusion was substantiated by the results of Fe and Mo analysis of native and Av1-mod as given in Table 2. Within the experimental uncertainty of the analysis, there are two important findings: incubation of native Av1 at pH 9.5 does not lead to metal loss as might be expected for a denatured protein, and likewise, inactivation of Av1 is not due to general denaturation with the loss of either iron or molybdenum.
Table 2. Fe and Mo Contents of Native and Modified Av1a.
| sample | Fe | Mo | Fe:Mo ratio |
|---|---|---|---|
| native Av1, 1 | 29.7 ± 0.5 | 1.99 ± 0.03 | 14.9 ± 0.5 |
| native Av1, 2 | 29.7 ± 0.5 | 1.98 ± 0.03 | 15.0 ± 0.5 |
| Av1-mod, 1 | 29.1 ± 0.5 | 1.99 ± 0.03 | 14.6 ± 0.5 |
| Av1-mod, 2 | 29.0 ± 0.5 | 1.99 ± 0.04 | 14.6 ± 0.5 |
Av1-mod was generated at pH 9.5 by incubation of Av1 and Av2 (CR = 4.0) under turnover conditions at 30 °C for 4 h. The protein was isolated by size exclusion chromatography at pH 9.5 using the same tribuffer that was used for the inactivation. The control sample of Av1 alone was isolated by chromatography at pH 7.5. Full duplicate reactions and isolations were prepared for both proteins. The metal content was determined by ICP-MS performed in triplicate for each sample. The standard deviation for 56Fe was ±0.56% and for 96Mo was ±0.22%, while for the protein, the concentration varied from ±1.5 to ±1.8%. The results are normalized to the protein concentration for each individual sample and given with the standard deviation based upon multiple determinations of an individual sample.
The results of monitoring ATP hydrolysis during the two assay procedures (see the preceding section) clearly demonstrated that the inactive Av1 retained the ability to interact with Av2 in a way that induces the Av2 conformation necessary for nucleotide hydrolysis. To further evaluate the integrity of Av1-mod with respect to binding of ATP-bound Av2, the established ability of Av1 and Av2 to form a complex stabilized by ADP-AlF4– was investigated.17,42 For native Av1 and Av2, the complex can be formed by reaction of either MgADP or MgATP as the nucleotide component with AlF4–.17 The reaction with MgATP is faster than with MgADP but leads to the same final complex.17,43,44 As shown in Figure 7B, Av1-mod forms a faster-migrating species when it is incubated with Av2, ATP, and AlF4–. On the basis of this earlier elution time compared to that of Av1-mod, a stable complex is inferred. This peak contained Av2 (by sodium dodecyl sulfate–polyacrylamide gel electrophoresis) and is estimated to be a 2:1 complex (assuming the tetramer of Av1-mod). Under similar conditions, MgADP also induced a stable complex that was intermediate in elution time between Av1-mod and the putative 2:1 Av1-mod–2Av2[ADP-AlF4–]4 complex. However, in contrast to the similar study with the native Av1, formation of the complex with MgADP appears to give only a 1:1 Av2–Av1 complex (based upon elution position). The assumption for the native proteins is that AlF4– stabilizes a hypothetical transition state in electron transfer between Av2 and Av1 and that ATP hydrolysis leads to this state; by microscopic reversibility, ADP reaches the same state. For Av1-mod, the path by microscopic reversibility for ADP appears to be much slower, and only partial conversion to the complex is observed even after reaction for 30 min.
Discussion
Protons have multiple roles in the catalytic mechanism of enzymes as reflected in the variation of activity with pH. Among these roles for nitrogenase are the proper protonation state of catalytic residues, the ionic state of residues in the nucleotide binding site, ionic states of residues involved in binding the Fe-protein to the MoFe-protein, and, most importantly, protons as a cosubstrate for all known nitrogenase reactions, including the direct reduction to dihydrogen. The latter reaction poses a particular challenge for mechanistic studies of nitrogenase because the ubiquitous presence of protons in aqueous solutions necessarily precludes the preparation of more reduced states of the MoFe-protein. Although protons cannot be completely removed from nitrogenase-containing solutions, the use of higher-pH conditions can significantly reduce their concentration. However, the conclusion of Pham and Burgess that Av1 is irreversibly inactivated by protein denaturation at higher pHs8 would preclude this potentially promising approach.
We have reinvestigated the effect of increased pH on Av1 with significantly different results and conclusions. First among our results is the finding that Av1 is neither denatured nor inactivated when it is incubated alone at pHs as high as 9.5. Av1 exhibited only minimal change in specific activity after incubation for several hours at pH 9.5 and 30 °C and could be crystallized at pH 9.5 with no important changes in the structure even after the several days needed to form the crystals.
As Pham and Burgess8 found, we did observe a pH-dependent inactivation of Av1, but in contrast to their conclusions, we found that the inactivation is a consequence of catalytic turnover resulting in a modified but structurally intact protein; inactivation of Av1 requires both Av2 and ATP hydrolysis. At pH 9.5, the model condition studied here, the enzyme activity, while turning over, decreased with time with a pattern similar to that of “slow inhibitors” such that both the initial rate, v0, and the inhibition rate constant, ki, could be determined by following the product in the gas phase of the reaction. Evaluation of the changes in v0 with protein concentration and component ratio indicated this rate was measuring the true enzyme reaction and was consistent with a decrease in catalytic activity expected over a measured pH range. Hence, we find that some forms of activity can be reliably studied at higher pHs when they are limited to appropriately determined initial rates, a condition generally applicable to all steady state enzyme kinetics.
While the inhibition at pH 9.5 requires enzyme turnover and, hence, is related to turnover, the inactivation was distinctly different from the enzyme activity. The inhibition rate increased as the rate of enzyme turnover decreased; that is, the inhibition rate increases with an increase in pH as activity decreases. The following characteristics summarize the observations with some of the implications.
(i) The inactivation reaction, being turnover-dependent, suggests a process involving states of the MoFe-protein that are distinct from the as-isolated form, mostly likely a different redox state. Because the inactivation is independent of which added substrate is present, one of the first enzymatically reduced states of the MoFe-protein may be sufficient to lead to inactivation.
(ii) The inhibition at pH 9.5 appears to be first-order when it is monitored by the decrease in the rate of ethylene formation with time at pH 9.5 in the head space assay.
(iii) The rate of inhibition, ki (eq 1), appears to be independent of the component ratio, protein concentration, and the amount of product produced (at least for CRs above ∼1). This indicates that the probability of inhibition is distinct from the probability of product formation.
(iv) Although there are insufficient data to assign reliable apparent pKa values for the groups associated with the v0 and the ki, the results in Figure 1C clearly show them to be different and, hence, derived from different functional groups.
(v) The decrease in specific activity upon turnover at pH 9.5 as measured at pH 7.8 follows a more complex kinetic pattern involving multiple phases and requiring significantly longer times (hours) for completion versus the time needed for the pH 9.5 inhibition.
(vi) The observed difference in the two inactivation/inhibition rates suggests that the inhibited state of Av1 at pH 9.5 is different from, although likely on the path to, the ultimately inactive Av1-mod. The first state must be somewhat reversible for activity to be seen at pH 7.8. For any CR used in Figure 3 at pH 9.5 and at 60 min, product formation has ceased, while at the same time point and pH 7.8, the specific activity retains 25–75% of the initial value, depending on the CR at pH 9.5. The final state, however, is irreversibly inactive in substrate reduction.
(vii) Turnover at pH 9.5 continues past the cessation of product formation as determined by continued ATP hydrolysis. For the fully inactive Av1-mod, the rate of associated ATP hydrolysis is ∼40% of that of the native protein with a saturating Av2 concentration.
Av1-mod has an expanded conformation (increased hydrodynamic radius) that maintains structural integrity. The new conformation is stable over time, retains metal content within the limits set by the analytical methods, and has the ability to associate with Av2. The latter is demonstrated by supporting ATP hydrolysis and the formation of a tight complex mediated by nucleotide-AlF4–. Together, these observations suggest a mechanism-based reaction with multiple forms of Av1 that can interconvert during the turnover at pH 9.5, some of which lead to inactivation. Although a comprehensive model that describes the kinetic behavior of this system under all these conditions would be desirable, we have demurred in such a construction because there are an enormous number of potential pH-dependent ionic states relating the multiple components. To assign pKas, activity levels, inhibition rates, and association constants for the two components and for the nucleotides would be outside the number of parameters that we have collected. Indeed, although the pH 9.5 inhibition rate is first-order and follows “slow inhibitor” rate law, we have not found a simple, rational kinetic measure of the specific activity decrease that would follow from the pH 9.5 inhibition rate.
Most importantly, our work identifies some of the conditions promoting a mechanism-based inactivation of the MoFe-protein. This should provide both a new direction for probing the catalytic mechanism(s) and the conditions to evaluate studies at pH >8.5, e.g., the reported changes in electron allocation with pH or the trapping of intermediates.8,10,45 While the focus of this paper has been on the inactivation reaction at pH 9.5, the process carries to lower pHs; it is only that the inhibition at the higher pH is sufficiently fast to facilitate kinetic characterization. One may anticipate that the phenomenon of inactivation would also take place at more physiologically relevant pHs, albeit with a slower rate. For example, the inactivation process may be particularly sensitive to amino acid residues in the cofactor environment such that differences in apparent substrate reduction rates for mutant nitrogenase with amino acid substitutions may be a consequence of changes in inactivation rates even when they are studied at the native protein pH optimum. Just as the generation of highly oxidizing species in photosystem II during water oxidation leads to protein inactivation with subsequent adaptations for protein turnover,46 the generation of highly reducing species in nitrogenase during substrate reduction is also associated with protein inactivation, which may require protein turnover adaptations in vivo.
Acknowledgments
The assistance of Dr. Jens Kaiser and Dr. Nathan Dalleska is appreciated. We acknowledge the Gordon and Betty Moore Foundation, the Beckman Institute, and the Sanofi–Aventis Bioengineering Research Program at Caltech for their generous support of the Molecular Observatory at Caltech. This project benefited from the use of instrumentation made available by the Caltech Environmental Analysis Center. Portions of this research were conducted at the Stanford Synchrotron Radiation Lightsource (SSRL), a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the Department of Energy Office of Biological and Environmental Research and by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program (P41RR001209), and the National Institute of General Medical Sciences.
Glossary
Abbreviations
- Av1 and Av2
MoFe-protein and Fe-protein from A. vinelandii, respectively
- Av1-mod
stable form of the inactive MoFe-protein that can be isolated after incubation under turnover conditions at pH 9.5
- ACES
N-(2-acetamido)-2-aminoethanesulfonic acid
- Bis-Tris
[bis(2-hydroxyethyl)amino]tris(hydroxymethyl)methane
- CHES
2-(cyclohexylamino)ethanesulfonic acid
- CR
component ratio or molar ratio of active sites in Fe-protein and MoFe-protein
- HEPPS
N-(2-hydroxyethyl)piperazine-N′-propanesulfonic acid
- ICP-MS
inductively coupled plasma mass spectrometry
- MPD
2-methyl-2,4-pentanediol
- Tris
tris(hydroxymethyl)aminomethane.
Supporting Information Available
Inactivation rates for Av1 with different substrates and inhibitors at pH 9.5 (Figure S1). This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
Coordinates and structure factors have been deposited in the Protein Data Bank as entry 4ND8.
Author Present Address
∥ K.-Y.Y.: 1676 S. Blaney Ave., San Jose, CA 95129.
Author Present Address
⊥ C.A.H.: Booz Allen Hamilton, Washington, DC 20024.
This work was supported by National Institutes of Health Grant GM045162.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Burgess B. K.; Lowe D. J. (1996) Mechanism of molybdenum nitrogenase. Chem. Rev. 96, 2983–3011. [DOI] [PubMed] [Google Scholar]
- Howard J. B.; Rees D. C. (2006) How many metals does it take to fix N2? A mechanistic overview of biological nitrogen fixation. Proc. Natl. Acad. Sci. U.S.A. 103, 17088–17093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seefeldt L. C.; Hoffman B. M.; Dean D. R. (2009) Mechanism of Mo-nitrogenase. Annu. Rev. Biochem. 78, 701–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorneley R. N. F., and Lowe D. J. (1985) Kinetics and mechanism of the nitrogenase enzyme system. In Molybdenum Enzymes (Spiro T. G., Ed.) pp 221–284, John Wiley & Sons, Inc., New York. [Google Scholar]
- Thorneley R. N. F.; Lowe D. J. (1996) Nitrogenase: Substrate binding and activation. JBIC, J. Biol. Inorg. Chem. 1, 576–580. [Google Scholar]
- Hadfield K. L.; Bulen W. A. (1969) Adenosine triphosphate requirement of nitrogenase from Azotobacter vinelandii. Biochemistry 8, 5103–5108. [DOI] [PubMed] [Google Scholar]
- Imam S.; Eady R. R. (1980) Nitrogenase of Klebsiella pneumoniae: Reductant-independent ATP hydrolysis and the effect of pH on the efficiency of coupling of ATP hydrolysis to substrate reduction. FEBS Lett. 110, 35–38. [DOI] [PubMed] [Google Scholar]
- Pham D. N.; Burgess B. K. (1993) Nitrogenase reactivity: Effects of pH on substrate reduction and CO inhibition. Biochemistry 32, 13725–13731. [DOI] [PubMed] [Google Scholar]
- Schneider K.; Müller A.; Krahn E.; Hagen W. R.; Wassink H.; Knüttel K.-H. (1995) The molybdenum nitrogenase from wild-type Xanthobacter autotrophicus exhibits properties reminiscent of alternative nitrogenases. Eur. J. Biochem. 230, 666–675. [DOI] [PubMed] [Google Scholar]
- Igarashi R. Y.; Laryukhin M.; Dos Santos P. C.; Lee H. I.; Dean D. R.; Seefeldt L. C.; Hoffman B. M. (2005) Trapping H– bound to the nitrogenase FeMo-cofactor active site during H2 evolution: Characterization by ENDOR spectroscopy. J. Am. Chem. Soc. 127, 6231–6241. [DOI] [PubMed] [Google Scholar]
- Han J. H.; Newton W. E. (2004) Differentiation of acetylene-reduction sites by stereoselective proton addition during Azotobacter vinelandii nitrogenase-catalyzed C2D2 reduction. Biochemistry 43, 2947–2956. [DOI] [PubMed] [Google Scholar]
- Wolle D.; Kim C.; Dean D.; Howard J. B. (1992) Ionic interactions in the nitrogenase complex. Properties of Fe-protein containing substitutions for Arg-100. J. Biol. Chem. 267, 3667–3673. [PubMed] [Google Scholar]
- Burns A.; Watt G. D.; Wang Z. C. (1985) Salt inhibition of nitrogenase catalysis and salt effects on the separate protein components. Biochemistry 24, 3932–3936. [Google Scholar]
- Deits T. L.; Howard J. B. (1990) Effect of salts on Azotobacter vinelandii nitrogenase activities. J. Biol. Chem. 265, 3859–3867. [PubMed] [Google Scholar]
- Ellis K. J.; Morrison J. F. (1982) Buffers of constant ionic strength for studying pH-dependent processes. Methods Enzymol. 87, 405–426. [DOI] [PubMed] [Google Scholar]
- Ennor A. H. (1957) Determination and preparation of N-phosphates of biological origin. Methods Enzymol. 3, 850–861. [Google Scholar]
- Renner K. A.; Howard J. B. (1996) Aluminum fluoride inhibition of nitrogenase: Stabilization of a nucleotide·Fe-protein·MoFe-protein complex. Biochemistry 35, 5353–5358. [DOI] [PubMed] [Google Scholar]
- Kabsch W. (2010) XDS. Acta Crystallogr. D66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D62, 72–82. [DOI] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. (1997) MOLREP: An automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025. [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. (2010) PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigle K. E.; Newton W. E.; Dean D. R. (1985) Complete nucleotide sequence of the Azotobacter vinelandii nitrogenase structural gene cluster. Gene 37, 37–44. [DOI] [PubMed] [Google Scholar]
- Howard J. B.; Kechris K. J.; Rees D. C.; Glazer A. N. (2013) Multiple amino acid sequence alignment nitrogenase component 1: Insights into phylogenetics and structure-function relationships. PLoS One 8, e72751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison J. F.; Walsh C. T. (1988) The behavior and significance of slow-binding enzyme inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol. 61, 201–301. [DOI] [PubMed] [Google Scholar]
- Hageman R. V.; Burris R. H. (1978) Kinetic studies on electron transfer and interaction between nitrogenase components from Azotobacter vinelandii. Biochemistry 17, 4117–4124. [DOI] [PubMed] [Google Scholar]
- Wherland S.; Burgess B. K.; Stiefel E. I.; Newton W. E. (1981) Nitrogenase reactivity: Effects of component ratio on electron flow and distribution during nitrogen fixation. Biochemistry 20, 5132–5140. [DOI] [PubMed] [Google Scholar]
- Bergersen F. J.; Turner G. L. (1973) Kinetic studies of nitrogenase from soya-bean root-nodule bacteroids. Biochem. J. 131, 61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orme-Johnson W. H., Davis L. C., Henzl M. T., Averill B. A., Orme-Johnson N. R., Munck E., and Zimmerman R. (1977) Components and pathways in biological nitrogen fixation. In Recent Developments in Nitrogen Fixation (Newton W. E., Postgate J. R., and Rodriguez-Barrueco C., Eds.) pp 131–178, Academic Press, London. [Google Scholar]
- Johnson J. L.; Nyborg A. C.; Wilson P. E.; Tolley A. M.; Nordmeyer F. R.; Watt G. D. (2000) Analysis of steady state Fe and MoFe protein interactions during nitrogenase catalysis. Biochim. Biophys. Acta 1543, 24–35. [DOI] [PubMed] [Google Scholar]
- Silverstein R.; Bulen W. A. (1970) Kinetic studies of the nitrogenase-catalyzed hydrogen evolution and nitrogen reduction reactions. Biochemistry 9, 3809–3815. [DOI] [PubMed] [Google Scholar]
- Thorneley R.; Lowe D. J. (1983) Nitrogenase of Klebsiella pneumonia: Kinetics of the dissociation of oxidized iron protein from molybdenum-iron protein: Identification of the rate-limiting step for substrate reduction. Biochem. J. 215, 393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J. L.; Nyborg A. C.; Wilson P. E.; Tolley A. M.; Nordmeyer F. R.; Watt G. D. (2000) Mechanistic interpretation of the dilution effect for Azotobacter vinelandii and Clostridium pasteurianum nitrogenase catalysis. Biochim. Biophys. Acta 1543, 36–46. [DOI] [PubMed] [Google Scholar]
- Bui P. T.; Mortenson L. E. (1969) The hydrolysis of adenosine triphosphate by purified components of nitrogenase. Biochemistry 8, 2462–2465. [DOI] [PubMed] [Google Scholar]
- Ljones T.; Burris R. H. (1972) ATP hydrolysis and electron transfer in the nitrogenase reaction with different combinations of the iron protein and the molybdenum-iron protein. Biochim. Biophys. Acta 275, 93–101. [DOI] [PubMed] [Google Scholar]
- Wolle D.; Dean D. R.; Howard J. B. (1992) Nucleotide-iron-sulfur cluster signal transduction in the nitrogenase iron-protein: The role of Asp125. Science 258, 992–995. [DOI] [PubMed] [Google Scholar]
- Einsle O.; Tezcan F. A.; Andrade S. L. A.; Schmid B.; Yoshida M.; Howard J. B.; Rees D. C. (2002) Nitrogenase MoFe-protein at 1.16 Å resolution: A central ligand in the FeMo-cofactor. Science 297, 1696–1700. [DOI] [PubMed] [Google Scholar]
- Spatzal T.; Aksoyoglu M.; Zhang L.; Andrade S. L. A.; Schleicher E.; Weber S.; Rees D. C.; Einsle O. (2011) Evidence for interstitial carbon in nitrogenase FeMo cofactor. Science 334, 940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden M. S.; Kindon N. D.; Ludden P. W.; Shah V. K. (1990) Diastereomer-dependent substrate reduction properties of a dinitrogenase containing 1-fluorohomocitrate in the iron-molybdenum cofactor. Proc. Natl. Acad. Sci. U.S.A. 87, 6517–6521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard J. B.; Rees D. C. (1994) Nitrogenase: A nucleotide-dependent molecular switch. Annu. Rev. Biochem. 63, 235–264. [DOI] [PubMed] [Google Scholar]
- Duyvis M. G.; Wassink H.; Haaker H. (1996) Formation and characterization of a transition state complex of Azotobacter vinelandii nitrogenase. FEBS Lett. 380, 233–236. [DOI] [PubMed] [Google Scholar]
- Schindelin H.; Kisker C.; Schlessman J. L.; Howard J. B.; Rees D. C. (1997) Structure of ADP-AlF4– stabilized nitrogenase complex and its implications for signal transduction. Nature 387, 370–376. [DOI] [PubMed] [Google Scholar]
- Schmid B.; Einsle O.; Chiu H.-J.; Willing A.; Yoshida M.; Rees D. C.; Howard J. B. (2002) Biochemical and structural characterization of the crosslinked complex of nitrogenase: Comparison to the ADP-AlF4– stabilized structure. Biochemistry 41, 15557–15565. [DOI] [PubMed] [Google Scholar]
- Hageman R. V.; Burris R. H. (1980) Electron allocation to alternative substrates of Azotobacter nitrogenase is controlled by the electron flux through dinitrogenase. Biochim. Biophys. Acta 591, 63–75. [DOI] [PubMed] [Google Scholar]
- Adir N.; Zr H.; Shochat S.; Ohad I. (2003) Photoinhibition: A historical perspective. Photosynth. Res. 76, 343–370. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.