

Significance
The crystallographic model of the Major Facilitator Superfamily (MFS) member, d-xylose permease XylE from Escherichia coli, a homologue of human d-glucose transporters, the GLUTs (SLC2), provides a structural framework for the identification and physical localization of crucial residues in transporters with medical relevance (i.e. the GLUTs). The mechanism and substrate specificity of human and prokaryotic sugar transporters are discussed by using homology modeling, molecular docking, and experimentation. Substrate-specificity determinants for XylE, GLUT1, and GLUT5 are proposed. Furthermore, concepts derived from other bacterial MFS transporters are examined for their relevance to the GLUTs by comparing conservation of critical residues. XylE mutants that mimic the characteristics of GLUT1 are tested, revealing that uniport and symport are mechanistically related.
Keywords: membrane proteins, sequence analysis, glucose transport, solute carrier transporter, ligand docking
Abstract
The Major Facilitator Superfamily (MFS) is a diverse group of secondary transporters with over 10,000 members, found in all kingdoms of life, including Homo sapiens. One objective of determining crystallographic models of the bacterial representatives is identification and physical localization of residues important for catalysis in transporters with medical relevance. The recently solved crystallographic models of the d-xylose permease XylE from Escherichia coli and GlcP from Staphylococcus epidermidus, homologs of the human d-glucose transporters, the GLUTs (SLC2), provide information about the structure of these transporters. The goal of this work is to examine general concepts derived from the bacterial XylE, GlcP, and other MFS transporters for their relevance to the GLUTs by comparing conservation of functionally critical residues. An energy landscape for symport and uniport is presented. Furthermore, the substrate selectivity of XylE is compared with GLUT1 and GLUT5, as well as a XylE mutant that transports d-glucose.
The exceptionally diverse Major Facilitator Superfamily (MFS), with over 10,000 sequenced members, includes 74 families with members from Archaea to Homo sapiens (1–3). One goal of determining crystallographic structures of bacterial representatives in this family is identification and physical localization of residues important for catalysis in transporters with medical relevance. The d-xylose permease XylE from Escherichia coli is recognized as a homolog of human d-glucose transporters, the GLUTs (SLC2) (4), although XylE catalyzes d-xylose/H+ symport, and uphill (i.e., active) transport is driven by the H+ electrochemical gradient (∆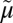 H+; interior negative or alkaline). Furthermore, d-glucose competitively inhibits d-xylose translocation (5). The recent high-resolution crystal structure of XylE with bound d-xylose or d-glucose (5) provides important detailed information about the organization of the bacterial transporter. Here we compare these transport proteins and describe functionally relevant similarities and differences.
H+; interior negative or alkaline). Furthermore, d-glucose competitively inhibits d-xylose translocation (5). The recent high-resolution crystal structure of XylE with bound d-xylose or d-glucose (5) provides important detailed information about the organization of the bacterial transporter. Here we compare these transport proteins and describe functionally relevant similarities and differences.
One or more of the 14 GLUT proteins are expressed in virtually every cell type in the human body (6), and all have well-established roles as transporters in various tissues and cell types (7). The GLUTs are grouped into three different classes based on sequence similarities (8), and XylE displays a bias to class III. Unlike XylE, most GLUTs are uniporters that catalyze equilibration of d-glucose across the cell membrane without a coupling ion (9). The sole known exception is GLUT13, the myo-inositol/H+ symporter (HMIT) (10). All GLUTs appear to transport hexoses or polyols, but the primary physiological substrate for many of the GLUTs remains uncertain (9). On the basis of mutational analyses, specific residues have been proposed to participate in substrate recognition by GLUT1 as well as other isoforms [e.g., Gln282, Tyr293 (11, 12)]. The QLS sequence motif in helix 7 is thought to be involved in d-glucose specificity based on its presence in GLUT1, -3, and -4, which transport d-glucose but not fructose, and its absence in GLUT2, which transports d-fructose as well as d-glucose (13). Other positions that have been implicated in the specificity of GLUT1–4 for d-glucose include the STSIF-motif in loop 7 (14), as well as Trp388, and Gln161 in helix 5 (15). However, a crystal structure with or without bound sugar has not been obtained yet for any GLUT.
XylE contains a remarkably conserved sugar-binding site. All residues in contact with d-glucose in the crystal structure are also suggested to be involved in d-glucose binding in GLUT1 (12). Based on the d-glucose–bound XylE structure, a more precise structural model of GLUT1 was generated that allows mapping of disease-related positions and localization of side-chains ligating d-glucose in GLUT1 (5). Although the side-chains postulated to be involved in binding in GLUT1 are conserved in XylE, d-glucose is not transported (16).
The paradigm of the MFS, lactose permease from E. coli (LacY), a galactoside/H+ symporter, is arguably the most intensively studied secondary transporter at present (17–19). Each of the 417 aminoacyl side-chains in LacY has been mutated (20). Remarkably, fewer than 10 residues are irreplaceable for active lactose transport: Glu126 (helix IV) and Arg144 (helix V), which are critical for substrate binding, as well as Trp151 (helix V), where an aromatic side-chain is essential; Glu269 (helix VIII), His322 (helix X), and Tyr236 (helix VII), which may be involved in coupling between protonation and sugar binding; and Arg302 (helix IX), and Glu325 (helix X), which are exclusively involved in H+ translocation (17, 19). It is striking that neutral replacement mutants for Glu325 are specifically defective in all steps involving net H+ translocation, but affinity is unaffected, and the mutants catalyze equilibrium exchange and counterflow as well or better than WT LacY (reviewed in ref. 21). Thus, Glu325 is clearly required for deprotonation of LacY. Although only a few residues are absolutely irreplaceable, Cys replacement of 82 additional residues has a significant effect on activity, inhibiting the steady-state level of accumulation by 50–80% (20, 22).
Converging lines of evidence demonstrate that LacY functions by an alternating access mechanism (18). The galactoside-binding site in LacY is located in a deep cavity in the approximate middle of the molecule, and LacY contains 12 mostly irregular transmembrane helices organized in two pseudosymmetrical six-helix bundles (23–26), a common structural feature in the MFS (5, 23, 27–35). Binding of galactosides to LacY induces a widespread conformational transition, increasing the open probability of a hydrophilic cleft on the periplasmic side of the molecule with closure of the cytoplasmic cavity in reciprocal fashion (18, 36). These coordinated conformational transitions are fundamental to secondary transport and represent the basis for the alternating access mechanism. Accordingly, the catalytic cycle of a transporter does not involve significant movement of sugar- and H+-binding sites relative to the membrane. Rather, the protein essentially moves around the substrate, reciprocally exposing the binding sites to either side of the membrane (i.e., alternating access in ref. 37). Recent X-ray structures of the bacterial l-fucose/H+ symporter FucP (29), d-xylose/H+ symporter XylE (5, 38), and d-glucose/H+ symporter GlcPSe (39) indicate that other MFS sugar transporters probably function in similar fashion.
Interspin distances obtained from double electron–electron resonance measurements suggest the presence of intermediates in the transport cycle of LacY, in addition to the inward- and outward-open conformations (40, 41). Further analysis of interspin distances combined with homology modeling (42) provide support for an occluded unprotonated, sugar-free apo intermediate, as well as a protonated, sugar-bound occluded intermediate. Moreover, de-convolution of the conformational distributions of the LacY molecule has allowed an assessment of relative energy levels of the respective conformations. A hypothetical energy landscape for the transport cycle was proposed in which the occluded intermediates accommodate a higher energy level relative to a moderately facile energy profile between the open conformers (42). Although all MFS symporters likely operate by an alternating access mechanism (43), almost incontrovertible evidence is available for LacY only (reviewed in refs. 18 and 36). Recently, crystallographic support for this hypothesis was obtained for LacY, showing the molecule in an outward-open conformation with an almost entirely occluded sugar derivative in the binding-pocket (44).
A principal difficulty in comparing MFS proteins is low sequence conservation. Despite a conserved fold and in some cases overlapping function, sequence identity ranges around 12–18% (22). Recently, we described an example of flexibility in design between LacY and the l-fucose permease (FucP), two MFS symporters (22). The order of the helix-triplets in FucP was permuted from their natural order relative to LacY to obtain better sequence conservation. The alignment was tested for conservation by comparing the 92 LacY mutants that impair function with 34 analogous functional mutations in FucP. In contrast to a conventional alignment, homology of the sugar- and H+-binding sites in the two proteins are observed. It was suggested that LacY and FucP (22), and many other MFS members (43), might have evolved from primordial noncovalently fused helix-triplets that formed functional transporters, and the functional segments assembled in a different consecutive order. Using this notion of evolution, we can begin to identify functionally related residues that do not correlate when primary sequences are compared, but do correlate in the tertiary structures of MFS transport proteins.
Results
Homology Modeling.
To draw functional conclusions with respect to the GLUTs, homology models of GLUT1, -5, and GlcPSe were generated based on the crystallographic coordinates of XylE (PDB ID codes 4GBY, 4GC0, and 4GBZ) (5). Because of relatively high sequence similarity and additional restraints resulting from the α-helical character of the proteins, the models cover all transmembrane segments in the GLUTs. Moreover, the positions and in certain instances the function of the side-chains that cause greater than 50% inhibition of d-glucose transport activity in GLUT1 upon replacement with Cys (12, 45–52) are included. The Z-DOPE scores of the top models are ≤−0.3, suggesting that ≥60% of the Cα atoms are within 3.5 Å of the correct position (Table S1) (53, 54).
Functional Correlation Between Residues of GLUT1, XylE, and LacY.
The X-ray structures of various MFS transporters reveal a common fold. Twelve transmembrane helices are organized into four symmetrically disposed triple-helix motifs that can be aligned individually and examined for conservation of functionally important residues. This combinatorial alignment of the triple-helices allows comparison of functionally homologous positions in different MFS transporters (22).
Similar to a previous study comparing LacY with FucP (22), and in more recent work comparing less closely related MFS proteins that transport a wide range of substrates (43), functionally related substrate-binding residues are not located in the same six-helix bundle as in LacY. For example, the aromatic constituents of the substrate binding site in LacY, Trp151 (helix triplet B), and Phe308 in FucP, (helix triplet C), correspond to Trp416 and Trp412 (helix triplet D) in XylE and GLUT1, respectively (Fig. 1 and Table 1). Polar sugar ligands are also involved in d-xylose binding in XylE, Glu168 and Gln175 (helix triplet B), and FucP, Gln159 and Asn162 (helix triplet B), and these residues superpose with Glu269 and Asn272 in LacY (helix triplet C). Glu269 in LacY corresponds to the conserved Gln161 in the GLUTs. The location of helix triplet C in XylE is inferred from the strong positional homology of Pro301 and Asp337 to the Pro31 and Asp68, respectively, in LacY helix triplet A. In GLUT1 these positions correspond to Thr295 and Glu329, respectively; both pairs of side-chains may have similar functional characteristics. By using these positions as markers, the consecutive order of the helix triplets in the GLUTs and in XylE is assigned as C-D-B-A relative to A-B-C-D in LacY. This order of the motifs results in inversion of helix triplets A and B in GLUT1 and XylE with respect to helix triplets C and D in LacY (Fig. 1C). Thus, the periplasmic loops of the first six-helix bundle (black flags) superpose on the cytoplasmic loops of the respective helix triplets. This superposition results in the apparent inversion of the C-terminal six-helix bundle of GLUT1 and XylE with respect to LacY. For most of the functionally significant residues from LacY, FucP, and XylE, corresponding residues in GLUT1 also show significantly decreased transport activities upon mutations (Fig. 1C, red boxes) (reviewed in ref. 12).
Fig. 1.

Alignment. (A) Helices represented by colored boxes are shown in consecutive order in the sequence and colored according to helix-triplets. Arrows of the same color indicate the positions of correlating residues. (B) Helix-triplets from XylE and LacY are aligned (helices 1–3, blue; helices 4–6, green; helices 7–9, orange; helices 10–12, yellow). The flags indicate the loops within triple-helix motifs. Helix-triplets from LacY and XylE are colored as in A. The alignments are oriented with the LacY cytoplasmic side at the top. The flags indicate loops within triple-helix motifs (white, cytoplasmic loop; gray, periplasmic loop). The numbers on the flags indicate the two helices that are connected by the respective loop. (C) Schematic superposition. Helix-triplets are colored as in A. Overlapping side-chain positions are shown in the same color for corresponding helices. Contacts between side-chains are indicated as broken lines. Red boxes indicate positions lowering the transport activity of GLUT1 Cys-mutants; gray boxes indicate positions not tested by Cys-scanning mutagenesis in GLUT1. Yellow background indicates positions implicated in a medical condition.
Table 1.
Functional side-chain correlations in GLUT1 concluded from positional homology of corresponding residues in XylE, GlcPSe, FucP, and LacY
| Function | GLUT1 | XylE | GlcPSe | FucP | LacY |
| Substrate ligand | F26 (Y32*) | F24 | N23 | N42 | H322 |
| Q161 | Q168 | Q137 | Q159 | E269 | |
| I168 | Q175 | I144 | N162 | N272 | |
| Q282 | Q288 | Q250 | — | F20 | |
| Q283 | Q299 | Q251 | — | — | |
| N288 | N294 | N256 | Y365 | — | |
| W388 (A396*) | W392 | W357 | Q274 | E126 | |
| N411 (H419*) | Q415 | N381 | R312 | R144 | |
| W412 | W416 | N381 | F308 | W151 | |
| H+ site | N29 (D79†) | D27 | D22 | D46 | E325 |
| Structural | G31 | T28 | N23 | P50 | P327 |
| G75/G76 | G67/C68 | G54/A55 | G73/– | A361/M362 | |
| G79/G134 | G71/G141 | G58/G110 | E135/Y74 | D237/K358 | |
| S80 | G72 | A59 | I79 | S366 | |
| G84/P401 | G76/G405 | G63/G370 | G402/G64 | G370/G159 | |
| G286/S414 | G292/A418 | G254/T384 | A359/C307 | G24/C154 | |
| S294 | P301 | S262 | P366 | P31 | |
| G384 (S392*) | G388 | G353 | G272 | G121 | |
| P385 | P389 | P354 | A276 | P123 | |
| Salt-bridged | D329/R400 | D337/R404 | D299/- | D406/R283 | D68/K131 |
| R126/–/N29 | R133/E206/D27 | R102/–/D22 | Y101/Y212/D45‡ | K319/D240/R302 |
An em dash (—) means homologous residue not found.
GLUT5.
GLUT13.
Polar interaction.
The H+ coupling site.
Glu325 in LacY, which is directly involved in H+ translocation (55), superposes with Asp27 in XylE, suggesting functional homology (Fig. 2A). Mutagenesis of Asp27 to either Ala or Asn results in complete loss of uphill d-xylose/H+ symport. However, binding of d-xylose to XylE mutant D27N (KD = 0.27 ± 0.02 mM) is similar to that of WT (KD = 0.35 ± 0.03 mM) (Fig. S1). As with neutral replacements for Glu325 in LacY, XylE mutant D27N or D27A retains full counterflow activity (i.e., reverse exchange). In contrast, neutral replacement of Glu206, which is tightly H-bonded to Asp27 and Arg133 in XylE, does not inhibit uphill d-xylose/H+ symport or counterflow (Fig. 2B). In most ∆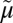 H+-independent GLUTs, a highly conserved Asn (Asn29 in GLUT1) or an Ala in GLUT6 and GLUT8 aligns with Asp27 of XylE or Glu325 in LacY. In GLUT13 (HMIT), which is ∆
H+-independent GLUTs, a highly conserved Asn (Asn29 in GLUT1) or an Ala in GLUT6 and GLUT8 aligns with Asp27 of XylE or Glu325 in LacY. In GLUT13 (HMIT), which is ∆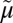 H+-dependent, this position corresponds to Asp79.
H+-dependent, this position corresponds to Asp79.
Fig. 2.

Transport activity of XylE mutants. (A) Superposition of helix-triplet D from LacY (beige) with helix-triplet A from XylE (blue) and GLUT1 (magenta). The position of Glu325, Asp27, and Asn29 is indicated with stick models colored according to the model. The helices are indicated with roman numbers, for LacY on a yellow circle and for XylE and GLUT1 on a magenta circle. (B) Levels of active transport (blue) and counterflow (green) of d-xylose by WT and mutants D27N, D27A, and E206Q. The transport activities of the mutants are normalized against the WT. “Control” refers to uptake by XylE-deficient E. coli transformed with empty vector in the active transport assay or liposomes without protein in the counterflow assay.
The sugar-binding site.
d-Glucose is not transported by XylE, but is a competitive inhibitor (5, 16). Moreover, the crystal structure of XylE with d-xylose or d-glucose demonstrates that all side-chains coordinating d-xylose also coordinate with d-glucose (see, for example, Fig. 4A). However, the C6-OH group of d-glucose is H-bonded to Gln175, the C1-OH group is H-bonded to the carbonyl oxygen of Gly388; these interactions are not observed with d-xylose (5).
Fig. 4.

Comparison of binding sites in MFS sugar transporters. (A) The X-ray structures of XylE (PDB ID code 4GBY, green, and 4GBZ, yellow), as well as the comparative models of (B) GLUT1 (orange) and (C) GLUT5 (blue), in the ligand-bound occluded conformation are shown. (A) Crystallographic coordinates of d-xylose (yellow) and d-glucose (green) are compared. Labels indicate polar contacts of the protein to the ligand. (B) The predicted binding pose for d-glucose in GLUT1 (orange) is compared with the binding conformation in XylE (same as in A; green). Labels are shown for GLUT1, bold labels indicate important differences. (C) The predicted poses for d-glucose in GLUT1 (same as in B; orange) and d-fructose in GLUT5 (blue) are compared. Labels are shown for GLUT5; bold labels indicate important differences to GLUT1. Broken lines indicate polar contacts to the ligand. See Fig. S2 for comparison of GlcPSe with XylE and Fig. S3 presenting the general orientation of the ligand-binding site.
Sequence comparison of XylE and GLUT1-4 reveals conservation of the sugar-binding site, with the exception of Gln175 in XylE, which corresponds to an invariant Ile in the GLUTs (Ile168 in GLUT1). Another position differing in XylE from the GLUTs is Leu297. In the GLUTs and in the bacterial d-glucose transporter GlcPSe (39), as well as in LacY, Leu297 aligns with a conserved Phe. Mutant Q175I or L297F in XylE exhibits a slight increase in d-glucose transport activity compared with WT (Fig. 3A). Although WT XylE shows little d-glucose transport activity, the double-mutant Q175I/L297F transports d-glucose to a significant level (Fig. 3A). Notably, although mutant Q175I transports d-glucose poorly, it retains relatively good d-xylose transport activity (∼75% of WT XylE) (Fig. 3B). However, the L297F mutation virtually abolishes the d-xylose transport activity, but the double mutant Q175I/L297F restores d-xylose transport activity. In brief, the XylE double mutant exhibits transport of both d-xylose and d-glucose, but transport of d-glucose is low relative to d-xylose transport (Fig. 3B).
Fig. 3.

Counterflow of d-xylose or d-glucose by XylE WT or mutants. (A) Counterflow of d-glucose with XylE WT and given mutants. (B) Counterflow of d-xylose with WT and given mutants. “Control” refers to liposomes without protein. (C) Crystallographic binding pose of d-glucose (blue) compared with predicted binding pose in the homology model of XylE mutant Q175I/L297F (violet). Green lines indicate potential H-bonding. Importantly, interactions with Gly388 in the mutant, fulfilling potential H-bonding criteria, are not found for the d-glucose docking poses to the theoretical model of the Q175I/L297F mutant.
Transport Specificity.
A remarkable feature of the human GLUTs is their differing substrate specificity (56). GLUTs -1 and -3 transport d-glucose and d-galactose, but not d-fructose, whereas GLUT4 transports d-glucose but not d-galactose. In addition to d-glucose and d-galactose, GLUT2 also transports d-fructose (57). Moreover, GLUT5 is specific for d-fructose (9). In addition, the common GLUT-inhibitor cytochalasin B does not ubiquitously inhibit all GLUTs but is specific, and GLUT2, -5, -7, or -9 are insensitive. To seek an explanation for this specificity, we screened computationally for the recurrence of substrate-binding patterns of substrates and inhibitors in transporters with distinctive characteristics.
d-Glucose is transported by most of the GLUTs. The residues implicated in substrate binding in well-characterized GLUT1 (12) overlap with residues found in the crystallographic structure of XylE with bound d-glucose (5), although two residues differ: the afore-mentioned Gln175, as well as Gln415, which corresponds to Asn411 in GLUT1. Docking of d-glucose to the homology model of GLUT1 (Fig. 4B) suggests that the shorter side-chain of Asn411 in GLUT1 results in closer distance of d-glucose to helix XI, and consequently the C1-OH of d-glucose is withdrawn from Gly384 (Fig. 4A) (Gly388 in XylE).
d-Fructose transport by GLUT5 and GLUT7 depends critically on the presence of Ile296 or Ile302. Mutation of either residue to the homologous Val in GLUT1 abrogates d-fructose transport in GLUT5 and GLUT7 without affecting d-glucose transport in GLUT7 (58). Docking d-fructose to the GLUT5 model suggests binding poses that involve contacts of residues in the binding site conserved exclusively in d-fructose transporters (Fig. 4C). For example, the conserved Asn411 in GLUT1 is homologous with His419 in GLUT5. Positions that provide only a hydrophobic interaction with the substrate in GLUT1, such as Phe26 or Phe397, are replaced with Thr32 and His387, respectively, which provide more specific interactions in GLUT5.
Inhibitors, such as cytochalasin B and STF-31 (a pyridyl anilino thiazole), are reported to exhibit specificity among sugar transporters. Cytochalasin B has no effect on GLUTs that transport d-fructose (GLUT2, -5, and -7 in ref. 7), whereas STF-31 is lethal selectively to cells lacking the von Hippel–Lindau tumor-suppressor gene by specifically targeting GLUT1 (59). Docking of STF-31 to GLUT1 produces related docking poses (Fig. 5) and suggests that the pyridyl-end of the molecule binds in the sugar-binding pocket and the phenyl-end is wedged in the extracellular vestibule of the central cavity. Interestingly, the sulfonyl group is coordinated by the Thr295, which is not conserved in GLUT2.
Fig. 5.

Docking of STF-31 to GLUT1 homology model. (A) Cut-away representation of binding position of the inhibitor in GLUT1. Two docking poses are shown in the cavity leading to the extracellular space (outside). (B) STF-31 interactions with specific side-chains. Residues making polar interactions with the ligand are illustrated as sticks; substrate binding residues are colored light blue, Thr30 and Thr295 are colored orange, Thr292 is colored gray; hydrogen atoms are colored in white, nitrogen atoms in blue, and oxygen atoms in red; H-bonds are represented by red lines. (C) Chemical structure of STF-31.
Discussion
Generally, clustering of sequences based on similarity reflects evolution and therefore provides a guide to structure, mechanism, and function. Sequence similarities of proteins with a common descent are expected to reflect homologous structures and functions. The broad sequence diversity of the MFS transporters presents a difficult problem for the identification and physical location of residues involved in substrate binding and conformational transitions. It has been suggested (60, 61) that MFS transporters may have arisen by intragenic multiplication of the triple-helix motif to two pseudosymmetrical six-helix bundles, the most common topological feature of the MFS transport proteins. We used those triple-helix motifs and functional information to align the four motifs in a combinatorial fashion (42, 43) (Fig. 1). A comparison of the MFS transport proteins discussed here exhibits functionally relevant similarities and conclusions are proposed for the related GLUTs.
Three key findings emerge from our study with regard to the H+-coupling site, the sugar-binding site, and mechanism.
The H+ Coupling Site.
Helix I in XylE (helix-triplet A) corresponds to helix X in LacY (helix-triplet D). These helices carry the carboxyl side-chain Asp27 in XylE and the well-characterized Glu325 in LacY, which overlap in space in the polypeptides aligned as described (Fig. 2C). Mutants with neutral replacements for Glu325 in LacY do not catalyze lactose/H+ symport, but binding, counterflow, and equilibrium exchange are totally unaffected (55). This and additional evidence (17, 62, 63) indicate that Glu325 is irreplaceable and is directly involved in H+ symport. Similar to the findings obtained with Glu325 mutants in LacY, replacement of Asp27 with Ala or Asn in XylE results in a similar phenotype (Fig. 2), and like findings have been reported for Asp46 in FucP (64) and Asp22 in GlcSe (39), supporting the conclusion that these residues are functionally homologous to Glu325 in LacY. Therefore, a similar kinetic mechanism is likely operative. The corresponding positions in the GLUTs, which catalyze facilitated diffusion, cannot be protonated and are mostly Asn. The exception to the rule is GLUT2, which has an Asp in the corresponding position but remains a uniporter. Recent experimental data support the proposal (39) that Asp27 is H-bonded to an adjacent Ser residue and therefore cannot be protonated.
Deprotonation of Glu325 in LacY depends on Arg302 (65). In the crystallographic structure of inward-facing LacY, Arg302 is in contact with the weakly salt-bridged residues Asp240 and Lys319 in helix VII and X, respectively (23, 25, 26). Although the precise function of Asp240 and Lys319 in LacY is unknown, it is clear that lactose/H+ symport depends on the presence of the interaction and its dissociation (66). In contrast to charged-paired Asp237/Lys358, Asp240 and Lys319 cannot be reversed. However, similar to Asp237/Lys358, single replacement of Asp240 or Lys319 with neutral side-chains leads to inactivation, whereas LacY mutants with double neutral replacement of both side-chains exhibits decreased but significant transport activity (67–69). Furthermore, cross-linking studies indicate that the impermanent interaction of Asp240 with Lys319 is required for activity because cross-linked D240C/K319C LacY is inactive (66).
Asp27, Arg133, and Glu206 in XylE superpose with Asp240, Arg302, and Lys319, repectively, in LacY as a result of inverting the polarity of the N-terminal six-helix bundle in XylE, and helices I, IV, and VI in XylE correspond to the helices VII, IX, and X in LacY (Fig. 1C). It is attractive to postulate that the interactions between these specific helices are functionally related. In this context, the three-residue motif observed in LacY, XylE, and FucP is absent from the GLUTs, which may be related to their function as uniporters rather than H+-coupled symporters.
The Sugar-Binding Site.
The crystallographic coordinates of XylE with bound substrate or inhibitor (5) are of exceptional value in that detailed structural information is provided regarding the organization of the sugar-binding site. An alignment of the triple-helix motifs in combinatorial fashion allows detection of functionally related residues in phylogenetically distant transporters that are correlated in the tertiary structures but uncorrelated in the primary structures.
Residues correlated with substrate binding in MFS symporters are located at the interface between the two six-helix domains in the approximate middle of the membrane (5, 23, 30, 32). A central residue in the sugar-binding sites of LacY, XylE, FucP, and most of the GLUTs is an aromatic residue located in a position that superposes upon Trp151 in LacY. GlcPSe seems to be an exception with Asn381 at the homologous position (Table 1). The unique sequence motif in the d-glucose binding-site is a Gln pair (e.g., Gln288-Gln289 in XylE), which coordinates the C2-OH and C3-OH of d-glucose (5).
In contrast to the structure with bound d-xylose, the substrate ligands observed in the d-glucose-bound XylE structure exhibit two contacts of d-glucose with the C6-OH group by an H-bond to Gln175 and with the C1-OH group to Gly388. Conventional sequence alignment of the GLUTs and the crystal structures of XylE reveal that two residues, Gln175 and Leu297, are involved in the substrate selectivity of XylE. These residues corresponding to Gln175 and Leu297 are Ile and Phe, respectively, in GLUTs1–4. Mutant Q175I/L297F in XylE exhibits significant d-glucose transport, although the activity is low relative to transport of d-xylose.
In the Q175I/L297F mutant, interaction of d-glucose with Gln175 is not present, and consequently the sugar is not restrained. The docking position most similar to the crystallographic model of d-glucose bound to the XylE mutant Q175I/L297F homology model (Fig. 3C) suggests that the interaction of the C1-OH with Gly388 is distorted enough to obviate an H-bond. Gly388 corresponds to Gly121 in LacY, where a Cys replacement alters the rate of active transport to <20% of the control (70). In GLUT1, the corresponding Gly384 is part of a conserved GlyProGlyPro cluster where Cys replacements lower transport activity to <25% (50). Clearly, restrictions to helix flexibility at this position cause slow transport. The position of d-glucose in the mutant XylE may allow enough helix flexibility to initiate detectible transport, but it is insufficient for maximal rates.
With the intention of carrying out a classification of unique substrate specificities among the GLUTs, we screened computationally for the recurrence of substrate-binding patterns of sugars and inhibitors with distinctive binding characteristics (7, 9). Docking poses of d-glucose in GLUT1 and d-fructose in GLUT5 homology models were compared (Fig. 4) to the location of d-xylose or d-glucose in the XylE X-ray structures (PDB ID codes 4GBY and 4GBZ). Most of the suggested substrate-binding residues in biochemically well-characterized GLUT1 (12) overlap with residues found in contact with d-glucose in the crystallographic structure of XylE (5). However, an important consequence arises from alternation of Gln415 in XylE to Asn411 in GLUT1. Docking of d-glucose to GLUT1 (Fig. 4B) suggests that the shorter side-chain of Asn411 in GLUT1 results in substrate positioning closer to helix XI, and consequently to the C1-OH of d-glucose, as opposed to Gly388 in XylE. Gln415 in XylE and Asn411 in GLUT1 correspond to the essential Arg144 (helix V) ligand in LacY. Even mutant R144K displays essentially no galactoside affinity and little or no transport activity (71).
Docking of d-fructose to the GLUT5 model reveals an important aspect of this site. At the position corresponding to Asn411 in GLUT1, His419 is found in contact with d-fructose. His419 and His387, which are also involved in d-fructose binding, are uniquely conserved in GLUTs -5 and in -7 (His425 and His393, respectively), which has also been shown to transport d-fructose (58). Another variation in the conserved residues in the MFS d-glucose transporters is Ala396 in GLUT5 or Ser402 in GLUT7, which replace the conserved Trp. Docking of d-fructose to GLUT5 suggests that a bulky residue in place of Ala396 would sterically block access to the 6-OH in d-fructose (Fig. 4C). Interestingly, the orientation of d-fructose in GLUT5 suggested by docking is remarkably consistent with the proposed orientation based on a structure-activity relationship study with conformationally locked d-fructose and l-sorbose derivatives in GLUT5 (72).
In a recent study, a pyridyl anilino thiazole (STF-31) was discovered to be selectively lethal to cells lacking the von Hippel–Lindau tumor-suppressor gene, which occurs in 80% of renal cell carcinomas, by specifically targeting d-glucose uptake through GLUT1 but not GLUT2 (59). In a docking screen to GLUT1, STF-31 exhibits related docking poses (Fig. 5) in a binding mode compatible with theoretical predictions (73). Accordingly, the induced transition fit for the single binding center in a transporter predicts that the substrate is not optimally liganded in the ground state as with enzymes. However, as opposed to enzymes, the unchanged substrate induces a major reorganization in the binding site, leading to formation of an intermediate with optimum fit (the occluded state). Increased substrate protein interactions provide a larger “intrinsic” binding energy to balance the energy barrier for the transporter to reconfigure its conformation (e.g., from inward- to outward-facing). In other words, the catalytic energy necessary for transport is recruited from the substrate–protein interaction, which reaches a maximum in the occluded state. Furthermore, Klingenberg (73) predicts that an inhibitor would provide additional contacts, thereby increasing binding energy and pushing the inhibitor–protein complex into an energy trap. Docking shows that the pyridyl-end of STF-31 interacts with substrate-binding residues and in addition, the benzamidyl moiety likely forms a π–π interaction with Tyr292, and the sulfonyl group forms two H-bonds with Thr30 and Thr295, which are not conserved in GLUT2, which is resistant to STF-31 (59).
Mechanism.
An overall mechanism for coupling in LacY is presented in Fig. 6A (17, 18, 74), and it has been suggested that a similar mechanism may be used by additional symporters in the MFS (43). The key features of the ordered mechanism are summarized as follows: (i) Lactose/H+ symport in the uphill or downhill energetic modes is precisely the same reaction. The difference is in the rate-limiting step. For downhill symport, deprotonation is rate-limiting; for uphill transport, deprotonation is no longer limiting, and either dissociation of sugar or a conformational change that leads to deprotonation becomes limiting. (ii) Sugar-binding and dissociation, not ∆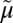 H+, are the driving force for alternating access (∆
H+, are the driving force for alternating access (∆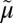 H+ has no effect on equilibrium exchange or counterflow). (iii) LacY must be protonated to bind sugar (the pKa for sugar binding is ∼10.5). (iv) Galactoside binds by an induced-fit mechanism, which powers transition to an occluded state. (v) Sugar dissociates first. (vi) Upon sugar dissociation, there is a conformational change that causes Arg302 (helix IX) to approximate Glu325, leading to deprotonation of LacY.
H+ has no effect on equilibrium exchange or counterflow). (iii) LacY must be protonated to bind sugar (the pKa for sugar binding is ∼10.5). (iv) Galactoside binds by an induced-fit mechanism, which powers transition to an occluded state. (v) Sugar dissociates first. (vi) Upon sugar dissociation, there is a conformational change that causes Arg302 (helix IX) to approximate Glu325, leading to deprotonation of LacY.
Fig. 6.

Transport cycle of MFS symporter. (A) Overview of the postulated steps in the transport model. Inward-facing (green) and outward-facing (blue) conformations are separated by the apo-intermediate conformations (orange) or by the occluded-intermediate conformations (gray). Steps are numbered consecutively: 1: Opening of the H+ site; 2: Deprotonation to inside and reorientation to the apo-intermediate with a central cavity closed to either side of the membrane; 3: Opening of the outward-facing cavity and reprotonation from the outside; 4: Formation of outward-open, substrate-free conformation; 5 and 6: Substrate binding and induced fit to the occluded conformation; 7: Opening of the inward-facing cavity and release of the sugar; 8: Formation of the protonated, substrate-free conformation. (B) Hypothetical energy profile for the transport cycle. Conformational shown in A are translated into relative energy states (indicated by the icons of conformations defined in A with the cytoplasmic side of the symporter facing up). The schemes are cycles read by following the arrowheads. The red part of the cycle represents the transitions between steps 1 and 4 of the empty pathway. The blue line corresponds to steps 5–8 for net sugar transporting steps (and for the exchange reaction). The free energy of the putative rate-limiting step in absence of ∆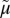 H+ (opening of the periplasmic cavity) is indicated by the vertical red arrow (ΔGn*). The hypothetical effect of an imposed ∆
H+ (opening of the periplasmic cavity) is indicated by the vertical red arrow (ΔGn*). The hypothetical effect of an imposed ∆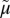 H+ is shown as a broken black vertical arrow, and the broken red lines show the resulting energy profile. (C) Postulated steps in the transport cycle of a uniporter. Steps 1–4 are similar to steps 1–4 in A but without release of a H+. Steps 5–8 are the same as in A. (D) Hypothetical energy profile for the transport cycle of a uniporter. The colors of the lines correspond to parts of the transport reaction equivalent to A. The energy trap for binding an inhibitor is indicated by the broken blue line.
H+ is shown as a broken black vertical arrow, and the broken red lines show the resulting energy profile. (C) Postulated steps in the transport cycle of a uniporter. Steps 1–4 are similar to steps 1–4 in A but without release of a H+. Steps 5–8 are the same as in A. (D) Hypothetical energy profile for the transport cycle of a uniporter. The colors of the lines correspond to parts of the transport reaction equivalent to A. The energy trap for binding an inhibitor is indicated by the broken blue line.
Based on interspin-distance measurements from double electron-electron resonance and site-directed alkylation, combined with homology modeling, the existence of two structurally distinct intermediates was suggested in which the central cavity is occluded: a sugar-free apo-intermediate and a protonated, sugar-bound occluded-intermediate (40–42). Moreover, analysis of the conformational distributions of LacY in the presence and absence of substrate sheds light on the relative energy levels of the respective conformations. A hypothetical energy landscape for the transport cycle is suggested in which the occluded intermediates accommodate a higher energy level relative to a comparatively facile energy profile between the open conformers (Fig. 6B) (42).
As stated above, the pKa for galactoside binding is ∼10.5. Therefore, it is evident that LacY is protonated at physiologic pH before substrate binding, and recent experiments supporting this conclusion more directly have been presented (75). Critically, neutral replacements for Glu325 yield mutants that are totally unable to carry out any reaction involving net H+ transport but catalyze equilibrium exchange and counterflow as well as or better than the WT (55). As indicated in Fig. 6A, a good explanation for the behavior of Glu325 neutral replacement mutants that provides convincing evidence for the ordered mechanism shown is that the mutants can oscillate between inward- and outward-open states in the substrate-bound protonated form (steps 5–8), but cannot deprotonate, and therefore cannot form the apo-intermediate and complete the cycle. In other words, the order of release in the WT must be dissociation of the galactoside first, followed by deprotonation. Because XylE, FucP, and GlcPSe, have carboxyl groups corresponding to Glu325 in LacY that display a similar phenotype when neutralized by mutagenesis, it is apparent that these symporters and perhaps many other MFS symporters may operate by a similar ordered kinetic mechanism.
d-Glucose transport by most human GLUTs occurs by facilitated diffusion and does not rely on H+ symport. However, the individual steps in the kinetic cycle are similar to those in a symport mechanism (Fig. 6C), and the empty uniporter undergoes similar conformational transitions as symporters; however, it is on a more facile energy landscape (Fig. 6D).
In summary, it is concluded that MFS transporters, including the GLUT uniporters, operate in a fashion analogous to enzymes with the exception that the intermediate is a conformer(s) of the protein rather than a transition-state intermediate of the substrate (76). By this means, an intermediate occluded conformation is induced by sugar binding, and binding leads to lowering of the activation energy barriers for the transition between inward- and outward-facing conformers.
Materials and Methods
XylE Purification.
The cDNA of full-length XylE from E. coli strain O157:H7 was subcloned into pET15b (Novagen). For overexpression of XylE proteins, E. coli BL21 (DE3) cells were grown at 37 °C in flasks each containing 1 L of LB medium and induced by 0.2 mM isopropyl−β-d-thiogalactoside (IPTG) when the cell density reached an OD600 of 1.5. After growth for 4 h at 37 °C, cells were harvested, homogenized in 25 mM Tris⋅HCl (pH 8.0)/150 mM NaCl, and disrupted by two passes through a French pressure cell at 10,000–15,000 psi. Cell debris was removed by low-speed centrifugation for 10 min. The supernatant was collected and ultracentrifuged at 150,000 × g for 1 h. The membrane fraction was harvested and incubated for 1 h with 1.5% (wt/vol) dodecyl-β-d-maltopyranoside (DDM; Anatrace) at 4 °C. After another ultracentrifugation step at 150,000 × g for 30 min, the supernatant was collected and loaded onto Ni(II)-nitrilotriacetate affinity resin (Ni-NTA; Qiagen) and rinsed with 25 mM Tris⋅HCl (pH 8.0)/150 mM NaCl/20 mM imidazole/0.02% DDM. The protein was eluted from the affinity resin with 25 mM Tris⋅HCl (pH 8.0)/150 mM NaCl/250 mM imidazole/0.02% DDM. After removal of the hexa-His tag, the protein was concentrated to about 10 mg/mL before further purification by gel filtration (Superdex-200; GE Healthcare) in 0.02% DDM/25 mM Tris-HCl (pH 8.0)/150 mM NaCl. The peak fractions were collected and flash-frozen in liquid nitrogen.
All XylE mutants were generated with a standard PCR-based protocol and were subcloned, overexpressed and purified in the same manner as the WT protein.
Preparation of Liposomes and Proteoliposomes.
Liposomes were prepared using a standard protocol as described (77). Proteoliposomes for d-[3H]xylose or d-[3H]glucose (American Radiolabeled Chemicals) counterflow assays were prepared in 50 mM potassium phosphate (KPi; pH 6.5)/2 mM MgSO4 (KPM)/20 mg/mL pre-extruded phospholipids (E. coli polar lipids; Avanti)/1% n-octyl-β-d-glucoside (β-OG; Anatrace)/20 mM d-xylose or d-glucose/10 μg per milligram lipid of WT or mutant XylE protein. β-OG was removed by incubation overnight with 400 mg/mL Bio-Beads SM2 (Bio-Rad). After the removal of β-OG, the proteoliposomes were frozen and thawed for five cycles. After extrusion through a 400-nm membrane filter, the proteoliposomes were harvested by ultracentrifugation at 100,000 × g for 1 h and washed twice with ice-cold KPM (pH 6.5) to remove extra sugar. The proteoliposomes were resuspended in ice-cold KPM (pH 6.5) to a final concentration of 100 mg/mL immediately before the counterflow assay.
Counterflow Assay.
All counterflow assays were performed at 25 °C. For each assay, 2 μL of concentrated proteoliposomes preloaded with a given substrate at 20 mM were diluted into 100 μL of KPM at indicated pH containing 1 μCi of d-[3H]d-xylose (specific radioactivity 12 Ci mmol−1) d-[3H]glucose (specific radioactivity 20 Ci/mmol). Uptake of radiolabeled substrates was terminated at 30 s (unless otherwise indicated) by rapid filtration through 0.22 μm filters (Millipore). The filters were washed rapidly with 2 mL of ice-cold reaction buffer without sugar and assayed for radioactivity by liquid scintillation spectrometry.
Active Transport.
The xylE-deficient E. coli strain Keio Collection JW3991 used in this assay was purchased from the National BioResource Project (NIG). WT and XylE mutants containing given missense point mutations were prepared as previously described (5).
Transport in intact cells was assayed by the following protocol: XylE-deficient E. coli cells transformed with plasmids were grown in LB medium at 37 °C and induced with 50 μM IPTG for 30 min, when the cell density reached an OD600 of ∼1.5. Cells were then harvested by centrifugation. After washing twice with 150 mM KCl/5 mM MES (pH 6.5) (MK buffer), the cells were resuspended in the same buffer to an OD600 of 2.0. Before reaction, cells were energized by addition of glycerol to a final concentration of 20 mM. To compare the transport activity of the XylE mutants, d-[3H]xylose was applied at 0.14 μM, and each reaction was terminated at 30 s. The samples were then rapidly filtered through 0.45-μm cellulose acetate filter (Sartorius) and washed immediately with 2 mL of ice-cold MK buffer, dried, and assayed for radioactivity by liquid scintillation spectrometry. Control experiments were performed with cells transformed with an empty pQLINK vector. All experiments were repeated at least three times. Error bars represent SD. All of the reactions were performed at 25 °C. All of the XylE mutants were expressed and quantified by following the same protocol as described for WT XylE. Homogeneity of purified XylE mutants was examined by size-exclusion chromatography, which showed profiles similar to that of WT protein.
Isothermal Titration Calorimetry.
Binding affinity between ligands and XylE mutants was measured with an ITC200 microcalorimeter (MicroCal). Purified proteins at 0.1 mM in 0.015% DDM/25 mM MES (pH 6.5)/150 mM NaCl were used for isothermal titration calorimetry titration. The protein was titrated with 10 mM of a given sugar dissolved in identical solution. Data were collected at at 22 °C and fitted using Origin 7.0 (MicroCal) software.
Structure and Sequence Alignments.
The helix triplets were generated from the crystallographic coordinates of LacY (A, Thr7-Asn102; B, Leu104-Phe187; C, Lys220-Ser309; D, Ala311-Leu400) (25) according to Radestock and Forrest (78). XylE helix triplets were generated from the crystallographic coordinates choosing three consecutive helices and omitting IC1, IC2, and IC3 with neighboring residues (A, Tyr5-Ile112; B, Tyr125-Pro221; C, Gly276-Thr365; D, Gly369-Glu465) (5), the helix triplets in GLUTs were inferred from the homology to XylE, and the superimposition of the helix triplets was carried out as previously described (22). In brief, computer programs COOT v0.7 (79) and UCSF-Chimera (80) were used for the structure-guided sequence alignment. No positional restraints were applied to functionally significant residues in the sequence or structure alignments. The superposition was visually inspected for the conservation of LacY functional markers (Dataset S1). As functional markers, 22 polar residues in LacY were used where mutations to Cys cause greater than 50% or 75% inhibition of the transport rate with Cys-less LacY (22, 81).
Homology Model Construction of the GLUTs.
Homology modeling was performed using the default-modeling schedule of MODELER (version 9v12) with “Thorough Variable Target Function Schedule” and “Slow MD Annealing” (82). The X-ray structure of the d-xylose/H+ symporter, (PDB ID code 4GBZ or 4GBY) (5) were used as a template. The initial sequence alignment was generated by aligning sequences of GLUTs 1–14 on XylE, GlcPSe (39), and then improved manually by removing gaps within the helices. For each template structure and alignment, 100 models were generated. Using Z-DOPE, a normalized atomic distance-dependent statistical potential based on known protein structures (83), the quality of the initial models was assessed. The final models were inspected visually and unmodeled loop regions were removed.
Ligand Docking.
The flexible-ligand sampling algorithm in AutoDock Vina was used to predict binding poses (84). The input files were generated using AutoDockTools (ADT v1.5.7rc1) (85). Partial charges from the united-atom AMBER force field were used for all receptor atoms (86). Internally calculated atomic affinity grids of the protein are used for the substrate molecule to perform a random walk in the space around the search box. At each step in the annealing, a random displacement is applied to each of the degrees-of-freedom to the center of gravity of the substrate. The displacement results in a new energy, which is evaluated using the grid interpolation procedure against the energy of the preceding step (84). A maximum energy range of 6 kcal/mol was set where binding modes with scores out of this range were discarded. The best-scoring conformation of each docked molecule was then inspected visually to prioritize binding modes similar to conformations of the substrate or inhibitor observed in the crystallographic structures (PDB ID code 4GBZ or 4GBY) (5, 54, 87).
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants DK51131, DK069463, and GM073210; National Science Foundation Grant MCB-1129551 (to H.R.K.); Ministry of Science and Technology Grant 2011CB910501 and National Natural Science Foundation of China Projects 31125009 and 91017011 (to N.Y.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1400336111/-/DCSupplemental.
References
- 1.Saier MH., Jr Families of transmembrane sugar transport proteins. Mol Microbiol. 2000;35(4):699–710. doi: 10.1046/j.1365-2958.2000.01759.x. [DOI] [PubMed] [Google Scholar]
- 2.Saier MH, Jr, et al. The major facilitator superfamily. J Mol Microbiol Biotechnol. 1999;1(2):257–279. [PubMed] [Google Scholar]
- 3.Reddy VS, Shlykov MA, Castillo R, Sun EI, Saier MH., Jr The major facilitator superfamily (MFS) revisited. FEBS J. 2012;279(11):2022–2035. doi: 10.1111/j.1742-4658.2012.08588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maiden MC, Davis EO, Baldwin SA, Moore DC, Henderson PJ. Mammalian and bacterial sugar transport proteins are homologous. Nature. 1987;325(6105):641–643. doi: 10.1038/325641a0. [DOI] [PubMed] [Google Scholar]
- 5.Sun L, et al. Crystal structure of a bacterial homologue of glucose transporters GLUT1-4. Nature. 2012;490(7420):361–366. doi: 10.1038/nature11524. [DOI] [PubMed] [Google Scholar]
- 6.Thorens B, Mueckler M. Glucose transporters in the 21st century. Am J Physiol Endocrinol Metab. 2010;298(2):E141–E145. doi: 10.1152/ajpendo.00712.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Augustin R. The protein family of glucose transport facilitators: It’s not only about glucose after all. IUBMB Life. 2010;62(5):315–333. doi: 10.1002/iub.315. [DOI] [PubMed] [Google Scholar]
- 8.Joost HG, et al. Nomenclature of the GLUT/SLC2A family of sugar/polyol transport facilitators. Am J Physiol Endocrinol Metab. 2002;282(4):E974–E976. doi: 10.1152/ajpendo.00407.2001. [DOI] [PubMed] [Google Scholar]
- 9.Mueckler M, Thorens B. The SLC2 (GLUT) family of membrane transporters. Mol Aspects Med. 2013;34(2–3):121–138. doi: 10.1016/j.mam.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Uldry M, et al. Identification of a mammalian H(+)-myo-inositol symporter expressed predominantly in the brain. EMBO J. 2001;20(16):4467–4477. doi: 10.1093/emboj/20.16.4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hruz PW, Mueckler MM. Structural analysis of the GLUT1 facilitative glucose transporter (review) Mol Membr Biol. 2001;18(3):183–193. doi: 10.1080/09687680110072140. [DOI] [PubMed] [Google Scholar]
- 12.Mueckler M, Makepeace C. Model of the exofacial substrate-binding site and helical folding of the human Glut1 glucose transporter based on scanning mutagenesis. Biochemistry. 2009;48(25):5934–5942. doi: 10.1021/bi900521n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seatter MJ, De la Rue SA, Porter LM, Gould GW. QLS motif in transmembrane helix VII of the glucose transporter family interacts with the C-1 position of D-glucose and is involved in substrate selection at the exofacial binding site. Biochemistry. 1998;37(5):1322–1326. doi: 10.1021/bi972322u. [DOI] [PubMed] [Google Scholar]
- 14.Doege H, et al. Serine-294 and threonine-295 in the exofacial loop domain between helices 7 and 8 of glucose transporters (GLUT) are involved in the conformational alterations during the transport process. Biochem J. 1998;329(Pt 2):289–293. doi: 10.1042/bj3290289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mueckler M, Weng W, Kruse M. Glutamine 161 of Glut1 glucose transporter is critical for transport activity and exofacial ligand binding. J Biol Chem. 1994;269(32):20533–20538. [PubMed] [Google Scholar]
- 16.Henderson PJ, Maiden MC. Homologous sugar transport proteins in Escherichia coli and their relatives in both prokaryotes and eukaryotes. Philos Trans R Soc Lond B Biol Sci. 1990;326(1236):391–410. doi: 10.1098/rstb.1990.0020. [DOI] [PubMed] [Google Scholar]
- 17.Guan L, Kaback HR. Lessons from lactose permease. Annu Rev Biophys Biomol Struct. 2006;35:67–91. doi: 10.1146/annurev.biophys.35.040405.102005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smirnova I, Kasho V, Kaback HR. Lactose permease and the alternating access mechanism. Biochemistry. 2011;50(45):9684–9693. doi: 10.1021/bi2014294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Madej MG, Kaback HR (2014) The life and times of Lac permease: Crystals ain’t enough. Membrane transporter function: To structure and beyond. Springer Series in Biophysics: Trasporters, eds Ziegler C, Krämer R, in press.
- 20.Frillingos S, Sahin-Tóth M, Wu J, Kaback HR. Cys-scanning mutagenesis: A novel approach to structure function relationships in polytopic membrane proteins. FASEB J. 1998;12(13):1281–1299. doi: 10.1096/fasebj.12.13.1281. [DOI] [PubMed] [Google Scholar]
- 21.Kaback HR. Permease on parade: Application of site-directed mutagenesis to ion-gradient driven active transport. Bioessays. 1987;7(6):261–265. doi: 10.1002/bies.950070608. [DOI] [PubMed] [Google Scholar]
- 22.Madej MG, Dang S, Yan N, Kaback HR. Evolutionary mix-and-match with MFS transporters. Proc Natl Acad Sci USA. 2013;110(15):5870–5874. doi: 10.1073/pnas.1303538110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abramson J, et al. Structure and mechanism of the lactose permease of Escherichia coli. Science. 2003;301(5633):610–615. doi: 10.1126/science.1088196. [DOI] [PubMed] [Google Scholar]
- 24.Chaptal V, et al. Crystal structure of lactose permease in complex with an affinity inactivator yields unique insight into sugar recognition. Proc Natl Acad Sci USA. 2011;108(23):9361–9366. doi: 10.1073/pnas.1105687108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guan L, Mirza O, Verner G, Iwata S, Kaback HR. Structural determination of wild-type lactose permease. Proc Natl Acad Sci USA. 2007;104(39):15294–15298. doi: 10.1073/pnas.0707688104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mirza O, Guan L, Verner G, Iwata S, Kaback HR. Structural evidence for induced fit and a mechanism for sugar/H+ symport in LacY. EMBO J. 2006;25(6):1177–1183. doi: 10.1038/sj.emboj.7601028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang Y, Lemieux MJ, Song J, Auer M, Wang DN. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science. 2003;301(5633):616–620. doi: 10.1126/science.1087619. [DOI] [PubMed] [Google Scholar]
- 28.Yin Y, He X, Szewczyk P, Nguyen T, Chang G. Structure of the multidrug transporter EmrD from Escherichia coli. Science. 2006;312(5774):741–744. doi: 10.1126/science.1125629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dang S, et al. Structure of a fucose transporter in an outward-open conformation. Nature. 2010;467(7316):734–738. doi: 10.1038/nature09406. [DOI] [PubMed] [Google Scholar]
- 30.Doki S, et al. Structural basis for dynamic mechanism of proton-coupled symport by the peptide transporter POT. Proc Natl Acad Sci USA. 2013;110(28):11343–11348. doi: 10.1073/pnas.1301079110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Newstead S, et al. Crystal structure of a prokaryotic homologue of the mammalian oligopeptide-proton symporters, PepT1 and PepT2. EMBO J. 2011;30(2):417–426. doi: 10.1038/emboj.2010.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pedersen BP, et al. Crystal structure of a eukaryotic phosphate transporter. Nature. 2013;496(7446):533–536. doi: 10.1038/nature12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Solcan N, et al. Alternating access mechanism in the POT family of oligopeptide transporters. EMBO J. 2012;31(16):3411–3421. doi: 10.1038/emboj.2012.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan H, et al. Structure and mechanism of a nitrate transporter. Cell Rep. 2013;3(3):716–723. doi: 10.1016/j.celrep.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 35.Zheng H, Wisedchaisri G, Gonen T. Crystal structure of a nitrate/nitrite exchanger. Nature. 2013;497(7451):647–651. doi: 10.1038/nature12139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaback HR, Smirnova I, Kasho V, Nie Y, Zhou Y. The alternating access transport mechanism in LacY. J Membr Biol. 2011;239(1–2):85–93. doi: 10.1007/s00232-010-9327-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mitchell P. A general theory of membrane transport from studies of bacteria. Nature. 1957;180(4577):134–136. doi: 10.1038/180134a0. [DOI] [PubMed] [Google Scholar]
- 38.Quistgaard EM, Löw C, Moberg P, Trésaugues L, Nordlund P. Structural basis for substrate transport in the GLUT-homology family of monosaccharide transporters. Nat Struct Mol Biol. 2013;20(6):766–768. doi: 10.1038/nsmb.2569. [DOI] [PubMed] [Google Scholar]
- 39.Iancu CV, Zamoon J, Woo SB, Aleshin A, Choe JY. Crystal structure of a glucose/H+ symporter and its mechanism of action. Proc Natl Acad Sci USA. 2013;110(44):17862–17867. doi: 10.1073/pnas.1311485110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang X, et al. Evidence for an intermediate conformational state of LacY. Proc Natl Acad Sci USA. 2012;109(12):E698–E704. doi: 10.1073/pnas.1201107109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smirnova I, et al. Sugar binding induces an outward facing conformation of LacY. Proc Natl Acad Sci USA. 2007;104(42):16504–16509. doi: 10.1073/pnas.0708258104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Madej MG, Soro SN, Kaback HR. Apo-intermediate in the transport cycle of lactose permease (LacY) Proc Natl Acad Sci USA. 2012;109(44):E2970–E2978. doi: 10.1073/pnas.1211183109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Madej MG, Kaback HR. Evolutionary mix-and-match with MFS transporters II. Proc Natl Acad Sci USA. 2013;110(50):E4831–E4838. doi: 10.1073/pnas.1319754110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kumar H, et al. 2014. Structure of sugar-bound LacY. Proc Natl Acad Sci USA 111(5):1784–1788.
- 45.Mueckler M, Makepeace C. Transmembrane segment 12 of the Glut1 glucose transporter is an outer helix and is not directly involved in the transport mechanism. J Biol Chem. 2006;281(48):36993–36998. doi: 10.1074/jbc.M608158200. [DOI] [PubMed] [Google Scholar]
- 46.Mueckler M, Makepeace C. Cysteine-scanning mutagenesis and substituted cysteine accessibility analysis of transmembrane segment 4 of the Glut1 glucose transporter. J Biol Chem. 2005;280(47):39562–39568. doi: 10.1074/jbc.M509050200. [DOI] [PubMed] [Google Scholar]
- 47.Mueckler M, Roach W, Makepeace C. Transmembrane segment 3 of the Glut1 glucose transporter is an outer helix. J Biol Chem. 2004;279(45):46876–46881. doi: 10.1074/jbc.M408632200. [DOI] [PubMed] [Google Scholar]
- 48.Mueckler M, Makepeace C. Analysis of transmembrane segment 8 of the GLUT1 glucose transporter by cysteine-scanning mutagenesis and substituted cysteine accessibility. J Biol Chem. 2004;279(11):10494–10499. doi: 10.1074/jbc.M310786200. [DOI] [PubMed] [Google Scholar]
- 49.Alisio A, Mueckler M. Relative proximity and orientation of helices 4 and 8 of the GLUT1 glucose transporter. J Biol Chem. 2004;279(25):26540–26545. doi: 10.1074/jbc.M402303200. [DOI] [PubMed] [Google Scholar]
- 50.Mueckler M, Makepeace C. Analysis of transmembrane segment 10 of the Glut1 glucose transporter by cysteine-scanning mutagenesis and substituted cysteine accessibility. J Biol Chem. 2002;277(5):3498–3503. doi: 10.1074/jbc.M109157200. [DOI] [PubMed] [Google Scholar]
- 51.Hruz PW, Mueckler MM. Cysteine-scanning mutagenesis of transmembrane segment 11 of the GLUT1 facilitative glucose transporter. Biochemistry. 2000;39(31):9367–9372. doi: 10.1021/bi000821g. [DOI] [PubMed] [Google Scholar]
- 52.Hruz PW, Mueckler MM. Cysteine-scanning mutagenesis of transmembrane segment 7 of the GLUT1 glucose transporter. J Biol Chem. 1999;274(51):36176–36180. doi: 10.1074/jbc.274.51.36176. [DOI] [PubMed] [Google Scholar]
- 53.Eramian D, Eswar N, Shen MY, Sali A. How well can the accuracy of comparative protein structure models be predicted? Protein Sci. 2008;17(11):1881–1893. doi: 10.1110/ps.036061.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schlessinger A, et al. Structure-based discovery of prescription drugs that interact with the norepinephrine transporter, NET. Proc Natl Acad Sci USA. 2011;108(38):15810–15815. doi: 10.1073/pnas.1106030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carrasco N, et al. Characterization of site-directed mutants in the lac permease of Escherichia coli. 2. Glutamate-325 replacements. Biochemistry. 1989;28(6):2533–2539. doi: 10.1021/bi00432a028. [DOI] [PubMed] [Google Scholar]
- 56.Uldry M, Thorens B. The SLC2 family of facilitated hexose and polyol transporters. Pflugers Arch. 2004;447(5):480–489. doi: 10.1007/s00424-003-1085-0. [DOI] [PubMed] [Google Scholar]
- 57.Uldry M, Ibberson M, Hosokawa M, Thorens B. GLUT2 is a high affinity glucosamine transporter. FEBS Lett. 2002;524(1-3):199–203. doi: 10.1016/s0014-5793(02)03058-2. [DOI] [PubMed] [Google Scholar]
- 58.Manolescu A, Salas-Burgos AM, Fischbarg J, Cheeseman CI. Identification of a hydrophobic residue as a key determinant of fructose transport by the facilitative hexose transporter SLC2A7 (GLUT7) J Biol Chem. 2005;280(52):42978–42983. doi: 10.1074/jbc.M508678200. [DOI] [PubMed] [Google Scholar]
- 59.Chan DA, et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci Transl Med. 2011;3(94):94ra70. doi: 10.1126/scitranslmed.3002394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hvorup RN, Saier MH., Jr Sequence similarity between the channel-forming domains of voltage-gated ion channel proteins and the C-terminal domains of secondary carriers of the major facilitator superfamily. Microbiology. 2002;148(Pt 12):3760–3762. doi: 10.1099/00221287-148-12-3760. [DOI] [PubMed] [Google Scholar]
- 61.Pao SS, Paulsen IT, Saier MH., Jr Major facilitator superfamily. Microbiol Mol Biol Rev. 1998;62(1):1–34. doi: 10.1128/mmbr.62.1.1-34.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Garcia-Celma JJ, Ploch J, Smirnova I, Kaback HR, Fendler K. Delineating electrogenic reactions during lactose/H+ symport. Biochemistry. 2010;49(29):6115–6121. doi: 10.1021/bi100492p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garcia-Celma JJ, Smirnova IN, Kaback HR, Fendler K. Electrophysiological characterization of LacY. Proc Natl Acad Sci USA. 2009;106(18):7373–7378. doi: 10.1073/pnas.0902471106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sugihara J, Sun L, Yan N, Kaback HR. Dynamics of the L-fucose/H+ symporter revealed by fluorescence spectroscopy. Proc Natl Acad Sci USA. 2012;109(37):14847–14851. doi: 10.1073/pnas.1213445109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sahin-Toth M, Kaback HR. Arg-302 facilitates deprotonation of Glu-325 in the transport mechanism of the lactose permease from Escherichia coli. Proc Natl Acad Sci USA. 2001;98(11):6068–6073. doi: 10.1073/pnas.111139698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang W, Guan L, Kaback HR. Helices VII and X in the lactose permease of Escherichia coli: Proximity and ligand-induced distance changes. J Mol Biol. 2002;315(1):53–62. doi: 10.1006/jmbi.2001.5206. [DOI] [PubMed] [Google Scholar]
- 67.Dunten RL, Sahin-Tóth M, Kaback HR. Role of the charge pair aspartic acid-237-lysine-358 in the lactose permease of Escherichia coli. Biochemistry. 1993;32(12):3139–3145. doi: 10.1021/bi00063a028. [DOI] [PubMed] [Google Scholar]
- 68.Sahin-Tóth M, Dunten RL, Gonzalez A, Kaback HR. Functional interactions between putative intramembrane charged residues in the lactose permease of Escherichia coli. Proc Natl Acad Sci USA. 1992;89(21):10547–10551. doi: 10.1073/pnas.89.21.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sahin-Tóth M, Kaback HR. Properties of interacting aspartic acid and lysine residues in the lactose permease of Escherichia coli. Biochemistry. 1993;32(38):10027–10035. doi: 10.1021/bi00089a019. [DOI] [PubMed] [Google Scholar]
- 70.Jung K, Jung H, Colacurcio P, Kaback HR. Role of glycine residues in the structure and function of lactose permease, an Escherichia coli membrane transport protein. Biochemistry. 1995;34(3):1030–1039. doi: 10.1021/bi00003a038. [DOI] [PubMed] [Google Scholar]
- 71.Frillingos S, Gonzalez A, Kaback HR. Cysteine-scanning mutagenesis of helix IV and the adjoining loops in the lactose permease of Escherichia coli: Glu126 and Arg144 are essential. off. Biochemistry. 1997;36(47):14284–14290. doi: 10.1021/bi972314d. [DOI] [PubMed] [Google Scholar]
- 72.Girniene J, et al. Inhibition of the D-fructose transporter protein GLUT5 by fused-ring glyco-1,3-oxazolidin-2-thiones and -oxazolidin-2-ones. Carbohydr Res. 2003;338(8):711–719. doi: 10.1016/s0008-6215(03)00007-7. [DOI] [PubMed] [Google Scholar]
- 73.Klingenberg M. Transport catalysis. Biochim Biophys Acta. 2006;1757(9–10):1229–1236. doi: 10.1016/j.bbabio.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 74.Kaback HR, Sahin-Tóth M, Weinglass AB. The kamikaze approach to membrane transport. Nat Rev Mol Cell Biol. 2001;2(8):610–620. doi: 10.1038/35085077. [DOI] [PubMed] [Google Scholar]
- 75.Smirnova I, Kasho V, Sugihara J, Vázquez-Ibar JL, Kaback HR. Role of protons in sugar binding to LacY. Proc Natl Acad Sci USA. 2012;109(42):16835–16840. doi: 10.1073/pnas.1214890109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mitchell P. Translocations through natural membranes. Adv Enzymol Relat Areas Mol Biol. 1967;29:33–87. doi: 10.1002/9780470122747.ch2. [DOI] [PubMed] [Google Scholar]
- 77.Veenhoff LM, Poolman B. Substrate recognition at the cytoplasmic and extracellular binding site of the lactose transport protein of Streptococcus thermophilus. J Biol Chem. 1999;274(47):33244–33250. doi: 10.1074/jbc.274.47.33244. [DOI] [PubMed] [Google Scholar]
- 78.Radestock S, Forrest LR. The alternating-access mechanism of MFS transporters arises from inverted-topology repeats. J Mol Biol. 2011;407(5):698–715. doi: 10.1016/j.jmb.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 79.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 80.Pettersen EF, et al. UCSF Chimera—A visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 81.Frillingos S, Sahin-Tóth M, Persson B, Kaback HR. Cysteine-scanning mutagenesis of putative helix VII in the lactose permease of Escherichia coli. Biochemistry. 1994;33(26):8074–8081. doi: 10.1021/bi00192a012. [DOI] [PubMed] [Google Scholar]
- 82.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 83.Shen MY, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15(11):2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Trott O, Olson AJ. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Morris GM, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Duan Y, et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24(16):1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 87.Schlessinger A, Khuri N, Giacomini KM, Sali A. Molecular modeling and ligand docking for solute carrier (SLC) transporters. Curr Top Med Chem. 2013;13(7):843–856. doi: 10.2174/1568026611313070007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.