

Abstract
Biosynthetic pathway evolution needs to consider the evolution of a group of genes that code for enzymes catalysing the multiple chemical reaction steps leading to the final end product. Tryptophan biosynthetic pathway has five chemical reaction steps that are highly conserved in diverse microbial genomes, though the genes of the pathway enzymes show considerable variations in arrangements, operon structure (gene fusion and splitting) and regulation. We use a combined bioinformatic and statistical analyses approach to address the question if the pathway genes from different microbial genomes, belonging to a wide range of groups, show similar evolutionary relationships within and between them. Our analyses involved detailed study of gene organization (fusion/splitting events), base composition, relative synonymous codon usage pattern of the genes, gene expressivity, amino acid usage, etc. to assess inter- and intra-genic variations, between and within the pathway genes, in diverse group of microorganisms. We describe these genetic and genomic variations in the tryptophan pathway genes in different microorganisms to show the similarities across organisms, and compare the same genes across different organisms to find the possible variability arising possibly due to horizontal gene transfers. Such studies form the basis for moving from single gene evolution to pathway evolutionary studies that are important steps towards understanding the systems biology of intracellular pathways.
Electronic supplementary material
The online version of this article (doi:10.1007/s11693-013-9127-1) contains supplementary material, which is available to authorized users.
Keywords: Pathway evolution, Tryptophan biosynthetic pathway, Relative synonymous codon usage (RSCU), Relative amino acid usage (RAAU), Codon adaptation index (CAI), Principal component analysis (PCA), Cluster analysis
Introduction
Sequencing large number of prokaryotic genomes from a variety of phyla and classes has offered new opportunities to study prokaryotic evolution. There are several methods to study molecular evolution of genomes through gene transfers. The commonly used one is through searching for sequence similarity using phylogenetic models (Felsenstein 2003), but a number of other approaches include identification of codon usage patterns, base composition (GC content) analysis, and nucleotide-pattern properties within genomes that differ from the genomic norm, as these are likely to represent acquired sequences (Moszer et al. 1999; Mrazek and Karlin 1999; Nakamura et al. 2004). In spite of many studies across genomes and for specific genes, a comprehensive understanding of how one can study such properties in groups of genes coding for the enzymes in the same metabolic pathway in different genomes, remain poorly understood.
Pathway evolution has been studied from the “origin of life” context, and two hypotheses have been put forward to explain the evolution of the sequence of steps in a pathway (Rison and Thornton 2002). In this study we attempt to study pathway evolution from a different perspective. Intracellular biosynthetic pathways consist of multiple reaction steps catalysed by enzymes, whose genes generally form a group (operon) in microorganisms. All genes of the enzymes involved in the pathway need to be concurrently functional for the end product to be available, so that the entire pathway can operate in the organisms for generations (Hao and Golding 2006). Thus this group of genes evolves under similar constraints determined by the functionality of the complete pathway, and hence they are expected to co-evolve. Hence, pathway evolution needs to consider the evolution of a group of genes, and this may entail all the genes in the same pathway to be under similar selection pressure. It is only recently that similar questions are being addressed as to how natural selection shapes the evolution of the group of genes for enzymes participating in specific biochemical pathways (Flowers et al. 2007; Invergo et al. 2013).
In this work, we have addressed if the systems-level constraints, operating at the whole pathway level (i.e., to produce the end product), influence the genetic and genomic features of the genes for the constituent enzymes in a correlated manner across different genomes. To achieve this we have studied the codon usage patterns, base composition and usage in third codon position, amino acid usage, and nucleotide-pattern properties of the constituent enzymes in a metabolic pathway. From the composition-based features of these genes, we examine the possibilities of gene transfer events within and between the genomes of different microorganism. It is known that organisms exhibit different preferred usage of the synonymous codons for the same amino acid, and the relative frequencies of different synonymous codons vary both within and between organisms (Grantham et al. 1980, 1981; Gouy and Gautier 1982; Lynn et al. 2002). Since codon usage varies from genome to genome, comparing the codon usage for all genes in a genome can help identify unusual nucleotide or codon usage in a gene and infer about lateral gene transfer (LGT) from another species (Kurland et al. 2003; Eisen 2000; Garcia-Vallve et al. 2003; Grocock and Sharp 2002; Gupta and Ghosh 2001; Kunin and Ouzounis 2003; Watt and Dean 2000). Other studies have indicated that diverse patterns of codon usage may arise from compositional constraints of the genomes as observed in the case of extremely GC- or AT-rich organisms (Karlin and Mrazek 1996; Ghosh et al. 2000).
We have considered the metabolic pathway for the biosynthesis of the amino acid Tryptophan as a case to study the above-mentioned features in a selected set of genomes spanning different phyla of bacteria and archaea. The choice of tryptophan biosynthetic pathway for our study is based on several issues. Tryptophan being the most energetically expensive amino acid to be synthesized in a cell (Bentley 1990; Akashi and Gojobori 2002), tryptophan biosynthesis is expected to be tightly controlled (Yanofsky et al. 1981, 2001). Though evolutionary studies on this pathway have been of interest for quite some time (Crawford 1975, 1989), the accumulation of genome sequences from a diverse group of organisms has enabled thorough and elaborate analysis of the evolution of the pathway in terms of the variation in gene arrangements, operon structure and regulation in a large number of microorganisms. A seminal review and a series of in-depth analysis of the trp operons in diverse groups of bacteria, archaea, and fungi genome sequences (Xie et al. 2001, 2002, 2003a, b, 2004) have demonstrated that the trp operon has been “organizationally reshuffled, invaded by insertion of apparently unrelated genes, disrupted by either partial or complete dispersal of genes to extra-operonic locations, or complicated by the seemingly unnecessary presence of additional operon-gene copies located outside of the operon”. These comprehensive studies analyzed the comparative organisation of the seven structural genes of the enzymes of the pathway, highlight the role of regulation, and elucidate some key organisms, which represent major evolutionary events in gene organization, and possible routes of lateral gene transfers. Needless to say, the tryptophan pathway gene clusters, though provide strong clues to vertical descent, has enough examples of mosaicism due to gene transfers by LGT or other events (Xie et al. 2003b, 2004; see Merino et al. 2008 for an excellent summary).
Genes in an operon typically have closely related functions, and consequently there is a strong selective pressure for the operon structure to be conserved between genomes (Lawrence and Roth 1996). In this study we have used a comparative genomic approach, complemented by statistical analysis, to study the evolution of the tryptophan biosynthetic pathway genes by analyzing their codon usage pattern along with the gene expressivity data in 15 microbes from different lineages (Table 1). The choice of organisms is based on several considerations—events of gene fusion/splitting in operon organization (Xie et al. 2003a, b), varying GC content of the genomes, variable number of genes in their trp operon, and organisms from taxonomically different lineages such as proteobacteria and archaea. Figure 1 gives the evolutionary tree (based on the 16 s rRNA genes for these organisms) showing their phylogenetic groupings. A comparison of Table 1 and Fig. 1 shows that organisms from same phylogenetic groups can have different base compositions and gene organisations.
Table 1.
Trp operon gene organization and whole genome GC content of 15 microbes from two lineages (12 Bacteria and 3 Archaea) and eight phyla. (*trpF in Mtu and Sco is a bifunctional (hisA-trpF) gene (Barona-Gómez and Hodgson 2003)
| Lineage | Organisms | Number of genes | % GC content |
|---|---|---|---|
| Bacteria | |||
| γ—proteobacteria | Escherichia coli (K-12 MG1655) (Eco) | 5 (trpE-GD-FC-B-A) | 50.8 |
| Salmonella typhi (CT18) (Sty) | 5 (trpE-GD-FC-B-A) | 52.2 | |
| Pseudomonas aeruginosa (PA01) (Pae) | 7 (trpE-G-D-F-C-B-A) | 66.6 | |
| Coxiella burnetti (RSA 493) (Cbu) | 5 (trpE-G-D-BF-A) | 42.6 | |
| α—proteobacteria | Brucella melitensis (16 M) (Bme) | 6 (trpEG-D-F-C-B-A) | 57.2 |
| Mesorhizobium loti (mlr2744) (Mlo) | 6 (trpEG-D-F-C-B-A) | 62.5 | |
| ε—proteobacteria | Helicobacter pylori (26695)(Hpy) | 6 (trpE-G-D-FC-B-A) | 38.9 |
| Thermotogae | Thermotoga maritima (Tma) | 6 (trpE-GD-F-C-B-A) | 46.2 |
| Actinobacteria | Mycobacterium tuberculosis (H37Rv) (Mtu)* | 5 (trpE-D-F-C-B-A) | 65.6 |
| Streptomyces coelicolor (Sco)* | 5 (trpE-G-D-F-C-B-A) | 72.2 | |
| Firmicutes | Bacillus subtilis (00750) (Bsu) | 7 (trpE-G-D-F-C-B-A) | 43.5 |
| Streptococcus pneumoniae (TIGR4) (Spn) | 7 (trpE-G-D-F-C-B-A) | 39.7 | |
| Archaea | |||
| Euryarchaeota | Archaeoglobus fulgidus (Afu) | 6 (trpE-G-C-F-B-A-D) | 48.6 |
| Methanobacterium thermoautotrophicum (Mth) | 7 (trpE-G-D-FC-B-A) | 49.5 | |
| Crenarchaeota | Sulfolobus solfataricus (Sso) | 7 (trpE-G-D-F-C-B-A) | 35.8 |
Fig. 1.

Phylogenetic tree of 16s rRNA of the organisms under study
The tryptophan branch of the aromatic amino acid group (Tyrosine, Phenylalanine and Tryptophan) proceeds from Chorismic acid, as shown in Fig. 2. It consists of a series of five chemical reactions (Crawford 1975) that involves five enzymes, encoded by 5–7 genes (trpEGDFCBA) in different microbes. The genes are usually arranged in a single cluster (trp operon), and codes for different chains for different enzymes as shown by the dashed lines. The first two genes trpE and trpG encode the two subunits of the enzyme Anthranilate Synthase (AS), which catalyses the chemical reaction step from the substrate Chorismate (1) to Anthranilate (2). Gene trpG is fused with trpD in some bacteria (such as, E. coli.), but except for two, are always split in Archaea Gene trpD codes for Anthranilate Phosphoglycerol Transferase (PRT), which catalyses the reaction step (2) to (3). Again, trpF and trpC, coding for N-(5′-Phospho-Ribosyl)-Anthranilate Isomerase (PRAI) and Indole-3-Glycerol Phosphate Synthase (IGPS), catalyze the reactions from steps (3) to (4) and steps (4) to (5) respectively, are found fused in some bacteria, but always are separate in Archaea. The genes trpB and trpA are never found fused in prokaryotes (Xie et al. 2003a, b), and both encode for the β and α subunits of the enzyme Tryptophan Synthase (TS), which catalyzes the reaction steps (5) to (6) finally making the end product amino acid l-Tryptophan.
Fig. 2.

Schematic diagram of substrates, genes, and gene-products (enzymes) catalyzing the reactions in Tryptophan biosynthetic pathway in Escherichia coli (Eco)
The chemical reactions of the pathway steps are highly conserved in a diverse group of organisms, but the genes corresponding to the five enzymes have considerable variation in their arrangements, operon structure and regulation in different bacteria, which continue to be an active area of study (Crawford 1975; Merino et al. 2008). The gene fusion/splitting events in the Trp operon organization are present in all lineages. Of the seven genes, the commonly observed fusions are G*D (e.g. Eco, Sty, and Tma), C*F (most of γ-proteobacteria, except Pae), even though D*C and E*G also exist. In the group of organisms chosen by us represents lineages that contain all the members of the trp gene “module”, but organized in different combinations. We have presented in this study a comprehensive analysis of the intra- and inter-genic variations in the base composition, amino acid usage, codon adaptation, gene expressivity, and multivariate analysis of their synonymous codon usage pattern, both within and between organisms of very different lineages. Given the fact that this “module” of genes underlies a specific function, and the organisms chosen lie at different branches of the microbial species tree, our study can shed light on the extent of gene-specific and species-specific variations in the same pathway genes, in the background of a significant vertical transmission of information though evolution. Examining the differences in these composition-based features may be indicative of differences in their evolutionary origin (vertical or lateral gene transfers) within the same module of genes underlying the same function.
Materials and methods
Source of sequence data
The nucleotide sequences for the Tryptophan biosynthetic pathway genes for 15 organisms (Table 1) from different groups of bacteria and archaea were retrieved from the KEGG pathway database Ortholog table (http://www.genome.jp/kegg). The nomenclature for the names of the genes is taken from KEGG (Kanehisa et al. 2007). The whole genome sequences, highly expressed genes and the 16S rRNA sequences were obtained from NCBI entrez genome project (http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi). The whole genome sequences were downloaded by selecting the corresponding organism’s REFSEQ displayed in cDNA/FASTA format. The highly expressed gene-set was made by taking only ribosomal genes separately for each organism from Tools/Protein table. (http://www.ncbi.nlm.nih.gov/genomes/lproks.cgi).
Phylogenetic analysis
The sequences for the 16 s rRNA tree and trpA were aligned using MUSCLE (Edgar 2004) and the phylogenetic tree was obtained using Phylip (Felsenstein 1989) using the kitsch program that implements the Fitch-Margoliash and Least Squares method. The trees were drawn using MEGA5 (Tamura et al. 2011).
Base composition analysis
The base composition of DNA refers to the content of the four nucleotides, Guanine (G), Cytosine (C), Adenine (A), and Thymine (T), in the sequence. Thus, GC content of a fragment of DNA refers to the content of the single nucleotides, G+C, and AT content refers to A+T content in that fragment. Organisms in Table 1 were chosen with a large range of variation of GC content—78–35 %. The frequency of the bases at the synonymous third position of the codon is indicated by A3 for Adenine, T3 for Thiamine and GC3 for Guanine+Cytosine. The average base composition of all the genes of enzymes involved in the tryptophan biosynthetic pathway is referred to as “pathway base content”. Mean whole-genome GC3 refers to the average base composition in the third position of the codons of all the genes in the genome.
Analysis of gene expressivities
Gene expressivities were measured using Codon Adaptation Index, CAI (Sharp and Li 1986; Carbone et al. 2003; Jansen et al. 2003).
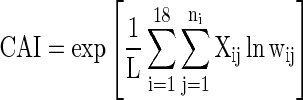 |
where, L is the sum total of the number of occurrences of all the codons for 18 amino acids present in the query gene and wij (adaptiveness) is the normalized RSCU value of the jth codon of ith amino acid of reference set of highly expressed genes.
This measure ranges between 0 and 1, where a value greater than 0.5 indicates a greater level of codon bias, and predicts a higher protein level. The CAI was calculated using CodonW.
Relative amino acid usage (RAAU)
RAAU represents the fractional content of a particular amino acid among all the 20 amino acid residues within a gene product, calculated using CodonW. The RAAU of all the pathway genes were compared individually with that of their whole genomes for each organism to find whether these pathway genes exhibit any difference in the pattern of amino acid usage. Amino acid usage are categorized as follows (Chanda et al. 2005): more than 10 %—Most Abundant; 6–10 %—Abundant; 4–6 %—Intermediate; 2–4 %—Rare; and less than 2 %—Very Rare. The RAAU values of trpG for Mtu was not plotted because the gene is listed as a putative gene in NCBI.
Relative synonymous codon usage (RSCU)
RSCU is defined as the ratio of the observed frequency of a codon to the expected frequency, if all the synonymous codons for those amino acids are used equally. It is used to study the overall codon usage variation among the genes. It is evaluated from the following mathematical expression (Sharp and Li 1987)
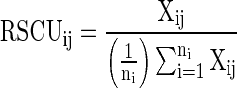 |
where, Xij is the number of occurrences of the jth codon for the ith amino acid, and ni is the number (from 1 to 6) of alternative codons for the ith amino acid. RSCU values were calculated for each gene in the pathway for the 15 organisms using CodonW (Peden 1999; http://codonw.sourceforge.net/) for 59 codons excluding 5 codons viz. AUG (methionine), UGG (tryptophan), and the three termination codons, as they give no bias.
To avoid biases due to gene length, amino acid usage, and codon degeneracy, codon usage data was normalized using the following formula (Suzuki et al. 2005).
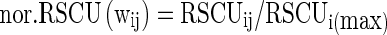 |
Correspondence analysis (COA)
COA is a method to factor categorical variables and display them in space so that association in two or more dimensions can be visualized. COA on RSCU (Benzecri 1992; Greenacre 1984; Das et al. 2005) of highly expressed genes (ribosomal genes) and trp genes were done to check the trend of codon usage preference between the two.
Multivariate analysis
Multivariate Analysis was done using Principal Component Analysis (PCA) and Cluster Analysis (CA) techniques.
Cluster Analysis is a method of classification of objects into groups, which reveal the relationships existing between the groups. The CA Plots are easier to interpret (Sharp et al. 1986). Cluster Analysis was done using MATLAB 2012b on the normalized RSCU for 59 codons of each gene. The two key steps considered within cluster analysis were the measurement of pair wise (Euclidean) distances between objects, and grouping the objects based upon the resultant linkage (unweighted pair group average) distances.
The purpose of Principal Component Analysis (PCA) is used to classify datasets. Principal Component Analysis was carried out on the normalized RSCU data of 59 codons, with 59 variables corresponding to the 59 degenerate codons (excluding the codons for Met, Trp and the stop codons), with 15 observations corresponding to the normalized codon usage values for the 15 organisms under study for each gene of the pathway. PCA was done using MATLAB 2012b. The genes C-F and G-D were manually concatenated and RSCU were calculated in cases where they were split in order to avoid the difference in number of genes in the pathway. In all the genes the first two PCs accounted for 50–60 % variance and the first three PCs accounted for 60–70 % variance in the data. Details of the analysis is given in Supplementary material S2.
Results
Analysis of base content at third position
Diverse patterns of codon usage may arise from compositional constraints of the genomes depending on if they are from GC- or AT-rich organisms (Karlin and Mrazek 1996). In amino acids with more than two fold codon degeneracy, it is the third position that decides the translational efficiency, and the total codon usage drive of an organism, as the third position bias positively correlates with gene expression (Carlini et al. 2001). The frequency of Adenine (A3), Thiamine (T3), Guanine+Cytosine (GC3) content for each pathway gene for all 15 organisms from different bacterial lineages are shown in Fig. 3. The organisms Afu, Sty, Bme, Mtu, Mlo, Pae and Sco, which have a GC content ranging from 49 to 72 % show comparatively higher GC3 bias (62–97 %) in the pathway genes. Almost equal usage (44–58 %) is seen in Bsu, Hpy, Tma, Mth and Eco, and Cbu, Spn and Sso exhibit higher AT3 bias.
Fig. 3.

Third position-specific bias in codons of trp genes in different organisms
Figure 4a shows a comparative study of each organism’s whole genome GC contents, and the mean GC and mean GC3 contents of the Trp pathway genes. Even though the average GC content of the pathway and whole genome is similar for all the organisms, significant variations exist in the GC3 preference of pathway genes among the organisms. The plot for mean whole genome GC3 and GC3 contents of the Trp pathway genes in different organisms are shown in Fig. 4b. It clearly shows that Sty, Afu, Spn, and Sco are GC3 skewed (>5 % difference), whereas Sso, Cbu and Mtu shows higher GC3 in the whole genome when compared to their mean pathway GC3 content. Thus, Figs. 3 and 4 indicate that, even though the average base composition of the Trp pathway genes are in accordance to their whole genome GC content, the GC3 usage varies among genes and show considerable skew in some organisms across phyla.
Fig. 4.

GC content analyses: a GC content of the whole genome, and mean GC and GC3 content of Trp pathway genes, b Mean GC3 content of whole genome and mean GC3 content of Trp pathway genes, in all organisms
Gene expressivity analysis
Every organism has a preferred set of codons, for each amino acid, that occur most frequently in genes that are translated at high abundance (e.g., the ribosomal proteins, or translation elongation factors) (Gouy and Gautier 1982). The patterns in the Codon Adaptation Index (CAI) are a useful measure to understand gene expressivity. In Table 2, we compare the CAI values of the Trp pathway genes to their ‘preferred’ codons—in this case, in the ribosomal proteins of each organism (Sharp and Li 1986).
Table 2.
CAI values of Trp pathway genes with respect to highly expressed genes in different organisms
| Org | Anthranilate synthase | Anthranilate phosphoribosyl transferase (TrpD) | Indole-3-glycerol phosphate synthase (TrpC) | Anthranilate isomerase (TrpF) | Tryptophan synthase | ||
|---|---|---|---|---|---|---|---|
| Comp I (TrpE) | Comp II (TrpG) | (TrpB) | (TrpA) | ||||
| Eco | 0.38 | 0.38 | 0.37 | 0.33 | 0.35 | 0.43 | 0.36 |
| Sty | 0.37 | 0.34 | 0.36 | 0.35 | 0.32 | 0.39 | 0.35 |
| Pae | 0.62 | 0.65 | 0.6 | 0.65 | 0.49 | 0.77 | 0.69 |
| Cbu | 0.8 | 0.72 | 0.78 | 0.79 | 0.75 | 0.79 | 0.76 |
| Bme | 0.58 | 0.64 | 0.62 | 0.57 | 0.47 | 0.67 | 0.63 |
| Mlo | 0.69 | 0.64 | 0.6 | 0.54 | 0.51 | 0.69 | 0.7 |
| Hpy | 0.69 | 0.74 | 0.71 | 0.65 | 0.71 | 0.7 | 0.7 |
| Tma | 0.76 | 0.72 | 0.72 | 0.75 | 0.73 | 0.72 | 0.75 |
| Mtu | 0.61 | – | 0.58 | 0.59 | 0.6 | 0.55 | 0.48 |
| Sco | 0.72 | 0.68 | 0.66 | 0.68 | 0.75 | 0.76 | 0.75 |
| Bsu | 0.38 | 0.38 | 0.37 | 0.4 | 0.41 | 0.41 | 0.37 |
| Spn | 0.43 | 0.48 | 0.5 | 0.38 | 0.41 | 0.53 | 0.49 |
| Afu | 0.76 | 0.67 | 0.72 | 0.72 | 0.72 | 0.74 | 0.71 |
| Mth | 0.69 | 0.7 | 0.67 | 0.66 | 0.61 | 0.71 | 0.65 |
| Sso | 0.68 | 0.66 | 0.67 | 0.64 | 0.67 | 0.68 | 0.7 |
Values in bold and regular indicate higher and, moderate expressivity, respectively. The italic values indicate the gene in the operon with comparatively low expression
It is clear from Table 2 that, with respect to the reference set of highly expressed ribosomal genes (HEGs), all the pathway genes show high codon adaptation (>0.5) in the three archaea (Afu, Mth and Sso) and seven bacterial species (Mlo, Mtu, Sco, Hpy, Cbu, and Tma). The three bacteria, Eco, Bsu, Sty and Spn, show moderate to low expressivity (0.3 < CAI < 0.5). All pathway genes in Sty have moderate but highly uniform CAI values.
In contrast, the two bacteria (Pae and Bme) show gene specific variability in CAI, most of the genes have higher expressivity except for trpF, which shows moderate expressivity.
The Correspondence Analysis (COA) plots (Supplementary material S1) are also in accordance with CAI analysis, where the Trp pathway genes in all archaea and few bacteria (Sco, Tma, Cbu) strongly cluster with the HEGs. These also show higher CAI (bold values in Table 2). The genes trpC, trpF and trpG are the three genes that remain outside the major cluster in several organisms, and these genes have lower CAI (shaded genes in Table 2). The COA plots of Eco, Bsu and Spn show that all the Trp pathway genes cluster separately from the HEGs. From these results, it can be inferred that the Trp pathway genes are generally highly expressed throughout all the lineages of bacteria and archaea, though few genes –trpC in Mlo, trpG in Hpy and trpF in Mlo and Pae and all the trp genes in Eco, Bsu and Spn show differential clustering.
Relative amino acid usage (RAAU) of Trp pathway genes
Biosynthetic cost of an amino acid is considered as the total sum of the energetic cost invested in the phosphate bonds in ATP and GTP molecules and the number of available hydrogen atoms carried in NADH, NADPH, and FADH2 molecules to metabolize an amino acid (Akashi and Gojobori 2002).Tryptophan, coded by a single codon (UGG), is a rarely used amino acid having the highest molecular weight and highest biosynthetic cost (Akashi and Gojobori 2002). Figure 5 compares the usage of Tryptophan in the whole genome (WG) and that in the Trp pathway genes, in all the organisms studied. Across all phyla, compared to its usage in WG, it is used very rarely (≪2 %) in the pathway genes, with the exception of trpF in Mtu (>2 %), and Sco and Bsu (both >1.5 %), and trpG in Cbu and Hpy.
Fig. 5.

Usage of Tryptophan by Trp pathway genes in different organisms. WG whole genome
Highly expressed proteins show increased abundance of energetically less costly amino acids, and avoid the usage of heavy amino acids (Seligmann 2003). A- or T-rich codons are known to encode for more costly amino acids (Akashi and Gojobori 2002). Hence amino acids encoded by AT-rich codons (Phe, Tyr, Met, Ile, Lys, Asn) and those encoded by GC-rich codons (Gly, Ala, Arg, Pro) do not show common trends related to gene expression. In Fig. 6, the percentage use of all the amino acids are shown by grouping them according to their biosynthetic cost for each gene of the pathway in all organisms. Around 55 % of total amino acid usages in the Trp pathway genes in all organisms are contributed by the low cost amino acids (Alanine, Glycine, Serine, Glutamine, Valine, Arginine, Leucine). Alanine is most abundantly used in the maximum number of pathway genes in most of the organisms (Eco, Sty, Pae, Bme, Mlo, Mtu, Sco), Valine is the most abundant in all pathway genes in Pae,Tma, Mtu, Sco, Spn, Bsu and Afu. Abundance of medium and higher cost amino acids (Lysine, Isoleucine, and Tyrosine) increases in the organisms with lower GC content (Cbu, Hpy, Bsu, Spn, Sso), because these amino acids are coded by AT rich codons. Usage of heavy and energetically most costly (>50 ATP) amino acids (Phenylalananine, Tyrosine, Tryptophan) by the trp genes was found to be very less (<2 %) in almost all organisms. Thus it is clear from Fig. 6 that the Trp pathway genes, in general, support the principle of cost minimization of amino acid usage in organisms where the genes are highly expressed, yet show extensive variability in the usage of amino acids indicating a lack of evolution through conserved vertical descent alone.
Fig. 6.

Relative amino acid usages by different organisms for Trp pathway genes. The energy cost in terms of ATP molecules required for the amino acids are shown in bracket as Black square High (>50)—Trp, Phe, Tyr, striped Medium (30–40)—His, Met, Ile, Lys; open square Low (12–30)—Asp, Asn, Glu, Gln, Thr, Pro, Val, Cys, Arg, Leu, gray square Very Low (11–7)—Ala, Gly, Ser
Variation of codon usage within Trp operon genes in different organisms
As mentioned earlier, every organism has a preferred set of codons, for each amino acid, that occur most frequently in genes that are translated at high abundance. We performed cluster analysis of the normalized RSCU for the Trp pathway genes, along with their whole genome, to reveal the similarities/dissimilarities in the codon usage pattern in different organisms. Figure 7a–c show the dendrograms of all pathway genes and its whole genome (WG), based on their RSCU values, revealing the inter-genic codon usage similarities/differences in three representative organisms. In 5 out of 9 organisms (Eco, Sty, Hpy, Mtu and Tma), having the fused-genes (trpGD or trpCF) in the pathway, trpA forms a distinct out-group, indicating that its codon usage is different from the other pathway genes and its whole genome (WG). This is shown in Fig. 7a. The organisms (Bsu, Pae, Spn, Mth, Sso) where all the pathway genes are in split condition i.e., trpE-G-D-F-C-B-A), the genes trpC, trpF, and trpG always form out-groups (Fig. 7b). For its codon usage preference, trpE clustered with its whole genome in 5 (Sty, Eco, Bme, Mtu,Bsu, and Mth) out of 15 organisms studied (Fig. 7c). This gene-specific analysis clearly shows that variability in codon usage pattern is observed in genes within the pathway in each organism, and there is no clear-cut relationship with respect to the classification of the microorganisms as is shown in their phylogenetic tree (Fig. 1).
Fig. 7.

Cluster Analysis on Normalized RSCU of Trp pathway genes and their WG in different organisms. See text for details
Variation of codon usage in different organisms for each Trp pathway
In order to find if particular genes in the Trp pathway have similar codon usage pattern in different organisms, the RSCU of 59 codons of each gene in 15 organisms were subjected to Principal Component Analysis (PCA). Figure 8 shows the principal component plot (PC1 vs PC2) of organisms for two representative Trp pathway genes—trpB and trpE. Organisms with similar loadings on PC1 and PC2 are encircled. The organisms Eco-Sty-Bme and Sco-Pae-Mlo-Mtu group together (Fig. 8b) for all pathway genes (trpA, trpB, trpCF, trpGD, and trpE). For most genes Tma-Afu-Mth also were found to be in close proximity, and Hpy-Bsu-Cbu always grouped together (Fig. 8a). The three unusual groups, different from the 16S rRNA tree (Fig. 1) (in Xie et al. 2003a, b; Woese 2000) are Tma-Afu-Mth, Mlo-Pae, and Hpy-Cbu-Bsu, where Tma from the bacterial group cluster with Afu, an archaea for most Trp pathway genes. It has been estimated that 24 % of the genes of the bacteria hyperthermophile Thermotoga maritima (Tma) are similar to archaeal genes and has been derived through horizontal gene transfer (Garcia-Vallve et al. 2000; Nelson et al. 1999), and our results on Trp pathway genes seem to provide evidence for gene transfer between Archaea and Bacteria. Similarly, Hpy, Bsu and Cbu are from two different bacterial groups but in terms of their codon usage, they separate out from their usual taxonomic grouping and show similarity with each other for all the Trp genes.
Fig. 8.

Principal Component plots of codon usage of organisms for different Trp pathway genes a trpB, b trpE. Open and filled circles represent Bacteria and Archaea respectively. Organisms with similar loadings are encircled
Sco, Pae and Mlo cluster together in the phylogentic trees of trpA, G and D and in the PCA of the same, but this clustering is not seen in the 16 s rRNA tree suggesting that the PCA based on the RSCU values do show clustering similar to that of gene trees. An example is shown in the Fig. 9a in which the gene tree of trpA and the PCA of the RSCU values (Fig. 9b) of the trpA show similar grouping for some organisms, e.g., Sco-Pae-Mlo. This group is also seen in other genes of the pathway in both phylogenetic tree and RSCU based groups (see Fig. 9). The archaea Sso is dissimilar in all features—phylogenetic and codon usages. Thus, the composition based features can also show differential grouping in organisms for the same pathway genes that indicate different evolutionary trends within pathway genes.
Fig. 9.

a trpA gene tree, b PCA plot of trpA
Discussion
Microevolution in prokaryotes is commonly regulated through the acquisition and loss of genetic material by several mechanisms, the most important being the horizontal/lateral gene transfer (H/L-GT). Even though they have been repeatedly linked with adaptation of lineages to new habitats, lifestyles, and pathogenicity, exceptions to the rules are also found in large abundance (Beiko et al. 2005; Dagan and Martin 2007; Ge et al. 2005). A bioinformatic study recently showed that a massive scale LGT may have transformed a strictly anaerobic, chemolithoautotrophic methanogen into the heterotrophic, oxygen-respiring, and bacteriorhodopsin-photosynthetic haloarchaeal common ancestor (Nelson-Sathi et al. 2012). Thus a clear-cut categorization between vertical and lateral transmission at the individual gene level is not always possible.
The relative impact of vertical transmission and lateral entry in microbial evolution is slowly giving way to the idea of extensive gene reorganization, even within groups of genes that belong to a set that perform a single function—say, production of an amino acid. Tryptophan, biochemically the most expensive amino acid to be synthesized in the cell, shows highly conserved reaction chemistry in all organisms. However, even though its biosynthetic pathway genes are frequently organized in whole-pathway operons, they have shown extensive fusion/splitting events for the same enzymes. Such an attribute is expected to facilitate multi-gene transfer in a single step. This has rendered the Trp pathway not only to be a good model system for studies in pathway evolution, it has also been used as a model system to assess the relative impact of the events of lateral gene transfer in an overall context of vertical genealogy (Xie et al. 2003a, b). The whole pathway trp operon analyses was also done using both phylogenetic and parametric approaches on 47 complete-genomes of Bacteria (Xie et al. 2004). The variety and frequency of rearrangements in the trp operons involving events of gene shuffling, fusion, operon splitting, total gene dispersal, and insertion of seemingly unrelated genes, raises questions about the forces of selection that enforce stability of the pathway operation. In this study, we have used a combination of comparative genomics and statistical analyses on a smaller, but carefully chosen, set of bacterial and archaeal genomes to identify signatures of diversification of genes within the Trp pathway. We selected organisms from prokaryote lineages that show trp gene organizations that have five pathway enzymes coded by 5–7 genes, which combine in different ways (through gene fusion and splitting) to facilitate Tryptophan biosynthesis. As has been shown in other studies (Moszer et al. 1999; Mrazek and Karlin 1999; Nakamura et al. 2004), to identify likely acquired sequences through lateral gene transfer, we have used four approaches—base composition analysis (AT, GC, and GC3 content), gene expressivity pattern, amino acid usage pattern, and codon usage patterns of the pathway genes that differ from the genomic norm of the organisms.
Our analysis shows that even though the Trp pathway genes follow the general trend of G+C content and is in equilibrium with the organism’s whole genome, there is differential GC3 bias observed independent of their taxonomic grouping—higher GC3 bias in Sco, Pae, Mlo and lesser GC3 bias in Sso, Cbu, Spn. The trp genes being part of organism’s regulatory and metabolic genes, are highly expressed in terms of their codon usage pattern in most organisms (Pae, Cbu, Bme, Mlo, Hpy, Tma, Mtu, Sco, Afu, Mth, Sso with CAI > 0.5). This reflects that they are highly adaptive to the codon usage pattern of the reference set of HEGs in their genomes. On the other hand, the codon usage pattern of the pathway genes in Eco, Bsu and Spn were found to be less adaptive (CAI < 0.5) and they exhibit moderate expression. The COA plots corroborate these results, where trp genes were found be clustering together with HEGs in all the organisms except Eco, Bsu and Spn. The principle of cost minimization in amino acid usage is supported, in general, by all pathway genes in all organisms, supporting the abundance of energetically less costly amino acids with proportional increase in the level of expression of the genes (Akashi and Gojobori 2002).
Cluster Analysis revealed that pathway genes, though work in tandem for functioning of the pathway, can evolve differentially. More variability is observed in genes, which are partners in gene fusion/splitting events (trpC, F, G, D). Our observations indicate that the events of gene fusion and gene splitting in the operon organization play a key role in promoting the variation in shaping the codon usage pattern in course of evolution of the pathway. The Principal Component Analysis of the codon usage patterns for all pathway genes consistently grouped Eco-Sty-Bme and Sco-Pae-Mlo-Mtu together. This is in accordance with the classification given by Xie et al. (2003a, b, 2004). But the two unusual groupings revealed from our multivariate analysis are—Tma-Mth-Afu and Bsu-Cbu-Hpy. These results clearly indicate that bacterial genomes are extremely dynamic and are continuously evolving to acquire novel ecological and pathogenic characters from distantly related species, thereby promoting diversification and speciation through lateral gene transfer (Ochman et al. 2000).
By taking microbial systems with different number of genes regulating the Trp biosynthetic pathway, we have attempted to show the variability in the pathway genes using a number of approaches. These are based on compositional features, such as base composition, usage of amino acids in the enzymes, expressivity, codon usage bias, etc. We have shown that the evolution of different genes in the same pathway in different organisms can exhibit organismal classifications that may not match with that of the 16S rRNA phylogenetic tree. Similar conclusions have been shown for large classes of organisms using phylogenetic methods (Jardine et al. 2002; Merrino et al. 2008; Nelson-Sathi et al. 2012). Our results corroborate the view that, even at the level of the genes regulating a pathway (operon module), a simple bifurcating tree of microbial phylogeny is an inadequate metaphor to represent the process of evolution (Doolittle 2004). With the bioinformatic and statistical analysis on codon usage data, we have quantified the genetic variation, which in turn can offer a different evolutionary relationship between organisms. Our overall analysis endorses a “synthesis” (Gogarten et al. 2002; Xie et al. 2003a, b; Kurland 2005) that acknowledges both the traditional vertical genealogy (tree-like behaviour) and web-like, reticulate behavior with lateral gene transfer for the evolution of Trp pathway genes. Studies that combine both phylogenetic and composition-based analyses form the basis for moving from single gene evolution to single pathway evolutionary studies, and finally to the evolution of complex network of intracellular biochemical pathways that regulate cellular functions at the systems level.
Electronic supplementary material
Acknowledgments
The authors thank Council for Scientific and Industrial Research, India for Senior Research Fellowship to Priya V K, and Department of Biotechnology, India for financial support to S. Sarkar. Somdatta Sinha thanks Department of Science and Technology India for the J. C. Bose Fellowship, and the Santa Fe Institute, USA for the Senior International Fellowship during early stages of the work.
References
- Akashi H, Gojobori T. Metabolic efficiency and amino acid composition in the proteomes of Escherichia coli and Bacillus subtilis. Proc Natl Acad Sci USA. 2002;99:3695–3700. doi: 10.1073/pnas.062526999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barona-Gómez F, Hodgson DA (2003) Occurrence of a putative ancient-like isomerase involved in histidine and tryptophan biosynthesis. EMBO Rep 4(3):296–300 [DOI] [PMC free article] [PubMed]
- Beiko RG, Harlow TJ, Ragan MA. Highways of gene sharing in prokaryotes. Proc Natl Acad Sci USA. 2005;102:14332–14337. doi: 10.1073/pnas.0504068102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley R. The shikimate pathway—a metabolic tree with many branches. Crit Rev Biochem Mol Biol. 1990;25:307–384. doi: 10.3109/10409239009090615. [DOI] [PubMed] [Google Scholar]
- Benzecri JP. The correspondence analysis handbook. Statistics: textbooks and monographs 125. New York: Marcel Dekker; 1992. [Google Scholar]
- Carbone A, Zinovyev A, Kepes F. Codon adaptation index as a measure of dominating codon bias. Bioinformatics. 2003;19:2005–2015. doi: 10.1093/bioinformatics/btg272. [DOI] [PubMed] [Google Scholar]
- Carlini DB, Chen Y, Stephan W. The relationship between third-codon position nucleotide content, codon bias, mRNA secondary structure and gene expression in the drosophilid alcohol dehydrogenase genes adh and adhr. Genetics. 2001;159:623–633. doi: 10.1093/genetics/159.2.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda I, Pan A, Dutta C. Proteome composition in Plasmodium falciparum: higher usage of GC-rich nonsynonymous codons in highly expressed genes. J Mol Evol. 2005;61:513–523. doi: 10.1007/s00239-005-0023-5. [DOI] [PubMed] [Google Scholar]
- Crawford IP. Gene rearrangements in the evolution of the tryptophan synthetic pathway. Bacteriol Rev. 1975;39:87–120. doi: 10.1128/br.39.2.87-120.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford IP. Evolution of a biosynthetic pathway: the tryptophan paradigm. Annu Rev Microbiol. 1989;43:567–600. doi: 10.1146/annurev.mi.43.100189.003031. [DOI] [PubMed] [Google Scholar]
- Dagan T, Martin W. Ancestral genome size specify the minimum rate of lateral gene transfer during prokaryote evolution. Proc Natl Acad Sci USA. 2007;104:870–875. doi: 10.1073/pnas.0606318104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Paul S, Chatterjee S, Dutta C. Codon and amino acid usage in two major human pathogens of genus Bartonella—optimization between replicational-transcriptional selection, translational control and cost minimization. DNA Res. 2005;12:91–102. doi: 10.1093/dnares/12.2.91. [DOI] [PubMed] [Google Scholar]
- Doolittle WF (2004) Microbial phylogeny and evolution: concepts and controversies. In: Sapp J (ed) Oxford Univ Press, New York, pp 119–133
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucl. Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen JA. Horizontal gene transfer among microbial genomes: new insights from complete genome analysis. Curr Opin Genet Dev. 2000;10:606–611. doi: 10.1016/S0959-437X(00)00143-X. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. PHYLIP–phylogeny inference package (version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- Felsenstein J. Inferring phylogenies. Massachusetts: Sinauer Associates Inc; 2003. [Google Scholar]
- Flowers JM, Sezgin E, Kumagai S, Duvernell DD, Matzkin LM, Schmidt PS, Eanes WF. Adaptive evolution of metabolic pathways in Drosophila. Mol Biol Evol. 2007;24(6):1347–1354. doi: 10.1093/molbev/msm057. [DOI] [PubMed] [Google Scholar]
- Garcia-Vallve S, Romeu A, Palau J. Horizontal gene transfer in bacterial and archaeal complete genomes. Genome Res. 2000;10:1719–1725. doi: 10.1101/gr.130000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Vallve S, Guzman E, Montero MA, Romeu A. HGT-DB: a database of putative horizontally transferred genes in prokaryotic complete genomes. Nucleic Acids Res. 2003;31:187–189. doi: 10.1093/nar/gkg004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge F, Wang LS, Kim J. The cobweb of life revealed by genome-scale estimates of horizontal gene transfer. PLoS Biol. 2005;3:e316. doi: 10.1371/journal.pbio.0030316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh TC, Gupta SK, Majumdar S. Studies on codon usage in Entamoeba histolytica. Int J Parasitol. 2000;30(6):715–722. doi: 10.1016/S0020-7519(00)00042-4. [DOI] [PubMed] [Google Scholar]
- Gogarten JP, Doolittle WF, Lawrence JG. Prokaryotic evolution in light of gene transfer. Mol.Biol.Evol. 2002;19:2226–2238. doi: 10.1093/oxfordjournals.molbev.a004046. [DOI] [PubMed] [Google Scholar]
- Gouy M, Gautier C (1982) Codon usage in bacteria: correlation with gene expressivity. Nucleic Acid Res 10(22):7055–7074 [DOI] [PMC free article] [PubMed]
- Grantham R, Gautier C, Gouy M, Mercier R, Pave A. Codon catalogue usage and genome hypothesis. Nucleic Acid Res. 1980;8:r49–r62. doi: 10.1093/nar/8.1.197-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham R, Gautier C, Gouy M, Jacobzone M, Mercier R. Codon catalog usage is a genome strategy modulated for gene expressivity. Nucleic Acid Res. 1981;9(1):r43–r74. doi: 10.1093/nar/9.1.213-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenacre MJ. Theory and applications of correspondence analysis. London: Academic; 1984. [Google Scholar]
- Grocock RJ, Sharp PM. Synonymous codon usage in Pseudomonas aeruginosa PA01. Gene. 2002;289:131–139. doi: 10.1016/S0378-1119(02)00503-6. [DOI] [PubMed] [Google Scholar]
- Gupta SK, Ghosh TC. Gene expressivity is the main factor in dictating the codon usage variation among the genes in Pseudomonas aeruginosa. Gene. 2001;273:63–70. doi: 10.1016/S0378-1119(01)00576-5. [DOI] [PubMed] [Google Scholar]
- Hao W, Golding GB. The fate of laterally transferred genes: life in the fast lane to adaptation or death. Genome Res. 2006;16:636–643. doi: 10.1101/gr.4746406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Invergo BM, Montanucci L, Laayouni H, Bertranpetit J (2013) A system-level, molecular evolutionary analysis of mammalian phototransduction. BMC Evol Biol 13:52 [DOI] [PMC free article] [PubMed]
- Jansen R, Bussemaker HJ, Gerstein M. Revisiting the codon adaptation index from a whole-genome perspective: analyzing the relationship between gene expression and codon occurrence in yeast using a variety of models. Nucleic Acids Res. 2003;31:2242–2251. doi: 10.1093/nar/gkg306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardine O, Gough J, Chothia C, Teichmann SA. Comparison of the small molecule metabolic enzymes of Escherichia coli and Saccharomyces cerevisiae. Genome Res. 2002;12(6):916–929. doi: 10.1101/gr.228002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, Yamanishi Y. KEGG for linking genomes to life and the environment Nucleic Acids Res. 2007;36:D480–D484. doi: 10.1093/nar/gkm882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin S, Mrazek J. What drives codon choices in human genes? J Mol Biol. 1996;262:459–472. doi: 10.1006/jmbi.1996.0528. [DOI] [PubMed] [Google Scholar]
- Kunin V, Ouzounis CA. The balance of driving forces during genome evolution in prokaryotes. Genome Res. 2003;13:1589–1594. doi: 10.1101/gr.1092603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurland CG. What tangled web: barriers to rampant horizontal gene transfer. BioEssays. 2005;27:741–747. doi: 10.1002/bies.20258. [DOI] [PubMed] [Google Scholar]
- Kurland CG, Canback B, Berg OG. Horizontal gene transfer: a critical view. Proc Natl Acad Sci USA. 2003;100:9658–9662. doi: 10.1073/pnas.1632870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence JG, Roth JR. Selfish operons: horizontal transfer may drive the evolution of gene clusters. Genetics. 1996;143:1843–1860. doi: 10.1093/genetics/143.4.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn DJ, Singer GAC, Hickey DA. Synonymous codon usage is subject to selection in thermophilic bacteria; Nucleic Acids Res. 2002;30:4272–4277. doi: 10.1093/nar/gkf546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino E, Jensen RA, Yanofsky C (2008) Evolution of bacterial trp operons and their regulation. Curr Opin Microbiol 11(2):78–86 [DOI] [PMC free article] [PubMed]
- Moszer I, Rocha EP, Danchin A. Codon usage and lateral gene transfer in Bacilus subtilis. Curr Opin Microbiol. 1999;2:524–528. doi: 10.1016/S1369-5274(99)00011-9. [DOI] [PubMed] [Google Scholar]
- Mrazek J, Karlin S. Detecting alien genes in bacterial genomes. Ann NY Acad Sci. 1999;870:314–329. doi: 10.1111/j.1749-6632.1999.tb08893.x. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Itoh T, Matsuda H, Gojobori T. Biased biological functions of horizontally transferred genes in prokaryotic genomes. Nat Genet. 2004;36:760–766. doi: 10.1038/ng1381. [DOI] [PubMed] [Google Scholar]
- Nelson KE, Clayton RA, Gill SR, Gwinn ML, Dodson RJ, Haft DH, Hickey EK, Peterson JD, Nelson WC, Ketchum KA, McDonald L, Utterback TR, Malek JA, Linher KD, Garrett MM, Stewart AM, Cotton MD, Pratt MS, Phillips CA, Richardson D, Heidelberg J, Sutton GG, Fleischmann RD, Eisen JA, White O, Salzberg SL, Smith HO, Venter JC, Fraser CM. Evidence for lateral gene transfer between Archaea and Bacteria from genome sequence of Thermotoga maritima. Nature. 1999;399:323–329. doi: 10.1038/20601. [DOI] [PubMed] [Google Scholar]
- Nelson-Sathi S, Dagan T, Landan G, Janssen A, Steel M, McInerney JO, Deppenmeier U, Martin WF. Acquisition of 1, 000 eubacterial genes physiologically transformed a methanogen at the origin of Haloarchaea. Proc Natl Acad Sci USA. 2012;109(50):20537–20542. doi: 10.1073/pnas.1209119109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405:299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- Peden JF (1999) CodonW, (http://sourceforge.net/projects/codonw/)
- Rison SCG, Thornton JM. Pathway evolution, structurally speaking. Curr Opin Struct Biol. 2002;12:374–382. doi: 10.1016/S0959-440X(02)00331-7. [DOI] [PubMed] [Google Scholar]
- Seligmann H. Cost minimization of amino acid usage. J Mol Evol. 2003;56:151–161. doi: 10.1007/s00239-002-2388-z. [DOI] [PubMed] [Google Scholar]
- Sharp PM, Li WH (1986) An evolutionary perspective on synonymous codon usage in unicellular organisms. J Mol Evol 24(1–2):28–38 [DOI] [PubMed]
- Sharp PM, Li WH. The codon adaptation index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987;15:1281–1295. doi: 10.1093/nar/15.3.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Tuohy TM, Mosurski KR (1986) Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res 14(13):5125–5143 [DOI] [PMC free article] [PubMed]
- Suzuki H, Saito R, Tomita M. A problem in multivariate analysis of codon usage data and a possible solution. FEBS Lett. 2005;579:6499–6504. doi: 10.1016/j.febslet.2005.10.032. [DOI] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol and Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt WB, Dean AM. Molecular functional studies of adaptive genetic variation in prokaryotes and eukaryotes. Annu Rev Genet. 2000;34:593–622. doi: 10.1146/annurev.genet.34.1.593. [DOI] [PubMed] [Google Scholar]
- Woese CR. Interpreting the universal phylogenetic tree. Proc Natl Acad Sci USA. 2000;97:8392–8396. doi: 10.1073/pnas.97.15.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Forst C, Bonner C, Jensen RA. Significance of two distinct types of tryptophan synthase beta chain in Bacteria, Archaea and higher plants. Genome Biol. 2001;3:0004.1–0004.13. doi: 10.1186/gb-2001-3-1-research0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Bonner CA, Jensen RA. Dynamic diversity of the tryptophan pathway in chlamydiae: reductive evolution and a novel operon for tryptophan recapture. Genome Biol. 2002;3(9):00511–005117. doi: 10.1186/gb-2002-3-9-research0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Keyhani NO, Bonner CA, Jensen RA. Ancient origin of the tryptophan operon and the dynamics of evolutionary change. Microbiol Mol Biol Rev. 2003;67:303–342. doi: 10.1128/MMBR.67.3.303-342.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Bonner CA, Brettin T, Gottardo R, Keyhani NO, Jensen RA. Lateral gene transfer and ancient paralogy of operons containing redundant copies of tryptophan-pathway genes in Xylella species and in heterocystous cyanobacteria. Genome Biol. 2003;4(R14):1–18. doi: 10.1186/gb-2003-4-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Bonner CA, Song J, Keyhani NO, Jensen RA. Inter-genomic displacement via lateral gene transfer of bacterial trp operons in an overall context of vertical genealogy. BMC Biol. 2004;2:15. doi: 10.1186/1741-7007-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanofsky C (2001) Advancing our knowledge in biochemistry, genetics, and microbiology through studies on tryptophan metabolism. Annu Rev Biochem 70:1–37 [DOI] [PubMed]
- Yanofsky C, Platt T, Crawford IP, Nichols BP, Christie GE, Horowitz H, Van Cleemput M, Wu AM. The complete nucleotide sequence of the tryptophan operon of Escherichia coli. Nucleic Acids Res. 1981;9(24):6647–6668. doi: 10.1093/nar/9.24.6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.