

Abstract
Motivation: Inferring lengths of inherited microsatellite alleles with single base pair resolution from short sequence reads is challenging due to several sources of noise caused by the repetitive nature of microsatellites and the technologies used to generate raw sequence data.
Results: We have developed a program, GenoTan, using a discretized Gaussian mixture model combined with a rules-based approach to identify inherited variation of microsatellite loci from short sequence reads without paired-end information. It effectively distinguishes length variants from noise including insertion/deletion errors in homopolymer runs by addressing the bidirectional aspect of insertion and deletion errors in sequence reads. Here we first introduce a homopolymer decomposition method which estimates error bias toward insertion or deletion in homopolymer sequence runs. Combining these approaches, GenoTan was able to genotype 94.9% of microsatellite loci accurately from simulated data with 40x sequence coverage quickly while the other programs showed <90% correct calls for the same data and required 5∼30× more computational time than GenoTan. It also showed the highest true-positive rate for real data using mixed sequence data of two Drosophila inbred lines, which was a novel validation approach for genotyping.
Availability: GenoTan is open-source software available at http://genotan.sourceforge.net.
Contact: garner@vbi.vt.edu
Supplementary Information: Supplementary data are available at Bioinformatics online
1 INTRODUCTION
Since many inherited variants of microsatellite loci are known to be associated with several human diseases such as colorectal cancers and various neurological disorders (Sutherland and Richards, 1995), the variants can act as important biomarkers to identify risk of disease development. The advent of next generation sequencing (NGS) technologies allows for a robust comparison of large numbers of microsatellite loci from several individuals in a quick and cost-effective way (McIver et al., 2011) which then allows the discovery and use of microsatellites as biomarkers or causative agents of disease. Unfortunately, inferring precise lengths of inherited microsatellite alleles with single base-pair resolution from short sequence reads remains a challenge due to the repetitive nature of microsatellites and the technologies used to generate raw sequence data. Homopolymer sequences, which are the most dominant class in the microsatellite family, are especially difficult to analyze. In this article, we discovered that >60% of microsatellite loci showing variants in a human genome contain homopolymer sequences (>6 bases), which underscores the importance of properly calling the genotype for this class of polymorphism. Repetitive sequences often result in insertion/deletion (INDEL) sequencing errors at homopolymers and incorrect mapping of sequence reads (Minoche et al., 2011). Compounding the issue, biological or technological mutants at microsatellite loci due to individual cell mutation or PCR amplification artifacts are also frequently observed. Substitution errors (sequencing or biologically derived) in the repeat sequences themselves also greatly affect the quality of alignment. A few methods such as SAMtools (Li et al., 2009), GATK (McKenna et al., 2010) and Dindel (Albers et al., 2011) can be used to genotype microsatellite loci with single base-pair resolution without paired-end information, but they have been principally designed to identify short INDELs in non-repeat sequences. Although lobSTR (Gymrek et al., 2012) has been developed to profile short tandem repeat loci, it is limited to only analyzing 2∼6-mer motif repeat loci. Recently, RepeatSeq (Highnam et al., 2012) has been developed to genotype microsatellite loci, but it does not provide a training module to adjust parameters to different sequence data conditions. The genotyping programs, including Dindel, employ local alignment-based methods and assume mainly sequencing errors cause the INDEL errors if the mapping quality is high. However distant variants at microsatellite loci often fool mapping programs into assigning high quality scores to incorrectly mapped reads when the sequence reads from the repeat loci are very different from the reference sequence, which results in incorrect genotyping. Especially, accuracies of genotyping programs using a Bayesian approach are significantly affected by a few incorrectly mapped reads (Supplementary Material). Individual cell mutations and polymerase chain reaction (PCR) amplification errors also frequently alter lengths of reported microsatellites resulting in falsely calling genotypes different from the inherited alleles. In addition, as homopolymer sequences induce not only INDEL errors, but also substitution errors during sequencing (Supplementary Fig. S1) and INDEL errors in reads containing homopolymers are frequently recurrent in other reads mapped to the same locus (Supplementary Fig. S2), local alignment-based approaches often fail to correctly identify inherited alleles. Further, the programs weight the final call towards the reference sequence, which may result in an artificial selection against novel (non-reference) allele calls since mapping or alignment algorithms are mostly biased to the reference sequence.
Long homopolymers frequently induce false-positive allele calls and it is challenging to distinguish them from true variants. The reasons for homopolymer error occurrence are different for each sequencing technology. The pyrosequencing technology utilized by 454 Life Sciences generates high-signal intensity for a repeating sequence of a single-base type then base-calling software estimates the number of bases in the sequence proportional to that intensity (Supplementary Fig. S3A). As a result, this technology shows a higher rate of INDEL sequencing errors than other sequencing technologies. Sanger sequencing and Illumina sequencing technologies employ a dye-terminator-sequencing method which shows a significantly lower INDEL error rate. However, this sequencing approach also shows increased INDEL error rate as homopolymer length in sequence reads increases. In Sanger sequencing, intensities from repetitive bases often merge to make a continuously high-signal peak or affect intensities of neighborhood peaks, thus the boundaries or heights of peaks for individual bases that compose the repeat become ambiguous (Supplementary Fig. S3B). Base-calling programs (Ewing et al., 1998) for this method use spacing information estimated from unambiguous peaks to identify the number of repetitive bases from the merged intensities and can predict more or fewer bases than the actual number of bases. Ilumina sequencing also has been reported to generate homopolymer-related errors in the study of this article (Supplementary Fig. S4) and other studies (Albers et al., 2011), and it may be caused by several noise factors (Erlich et al., 2008) such as lagging or leading of nucleotide extension.
In this article, we utilize a Gaussian distribution to compensate for a common characteristic of several different sequencing technologies, in which INDEL events at a single nucleotide repeat are caused by incorrect signal intensities and the chances of insertion errors and deletion errors in a repeat are proportional (when the rate of insertion errors increases, deletion errors also increases) (Supplementary Fig. S5). Bidirectional INDELs have also been observed from mutations at microsatellite loci caused by polymerase slippage during DNA replication (Xu et al., 2000). To fully address this issue, we have combined the discretized Gaussian mixture model with a rules-based approach to genotype length variation of microsatellites loci from short-sequence reads. The genotyping method is composed of two regression steps (Fig. 1A and C) and a homopolymer decomposition step (Fig. 1B), which effectively filters out noise reads resulting in low false-positive and false-negative rates.
Fig. 1.

Two regression steps to identify inherited genotypes of microsatellite loci. (A) The first regression step using the NLS (non-linear least square) fitting method. GenoTan tests all available genotype candidates at each locus using the regression method with the observed read counts and chooses one producing the least square error. If the chosen genotype candidate is heterozygous, it is tested using a rule-based approach with the estimated parameters; θ, σ1 and σ2 to determine whether one of two peaks is due to noise. (B) From the called genotypes of all loci, αa, αb, βa and βb, which are used to calculate ω and υ, are estimated using the homopolymer decomposition method. At each locus, each allele candidate has a new length adjusted by ω, and the length is used as a mean of the second regression step; υ is used to estimate pseudo counts to be added to the training vector with the observed read counts. (C) The second regression step. All genotype candidates at each locus are tested again with the adjusted means and pseudo counts; the rule-based method is used to choose a genotype for the locus.
2 METHODS
2.1 Discretized Gaussian mixture distribution
When three or more allele candidates are detected from sequence reads mapped to a microsatellite locus in a reference sequence of a diploid genome, the simplest approach to decide the genotype of the locus is to choose two alleles with the highest read frequencies. The frequencies of reads containing the two different alleles are then compared to test whether the genotype of the locus is homozygous or heterozygous. If the ratio of the read count of the lower read frequency allele to the read count of the higher read frequency allele is larger than a given cutoff ratio [generally 0.25 (= 0.2/0.8)], the locus is determined to be heterozygous. Otherwise, the locus is determined as homozygous and the reads containing the lower read frequency allele are regarded as noise from sequencing or PCR amplification errors, individual cell mutation, misalignment or mis-mapping. The most frequently observed noise source is homopolymer errors. Homopolymer errors are observed not only at single long-homopolymers sequences, but also at repeats of motifs containing short homopolymers. Since the homopolymer error in microsatellite sequences alters their lengths in the sequence reads, the observed ratio of read counts supporting two different alleles at a single locus containing a long homopolymer may be different from the ratio of genomic DNA fragments derived from the alleles. This inconsistency can be addressed by using a discretized Gaussian distribution if we assume that the probability of invalid signal intensities generated by sequencing machines for target nucleotides follows a continuous distribution and we only observe values discretized by base-calling programs. Since probabilities of insertion and deletion errors in a sequence are proportional, they can be calculated from the cumulative distribution function of the Gaussian distribution. Let lL be the length of a candidate allele L at a target locus and let x be the observed length of the microsatellite sequence with INDEL errors in a read mapped to the locus with an assumption in which the length x is derived from the original length lL. Let FL(t) and fL(t) denote the distribution and the density functions of a Gaussian random variable with mean lL and variance σL2, respectively. Then the probability mass function pL(x) of x is
| (1) |
where x = 0, 1, 2, … , and 1/(1 – FL(0.5)) is a scale factor (Supplementary Material, Rescaling the Gaussian cumulative distribution function section).
For the heterozygous loci with allele lengths, lL1 and lL2, we can use the mixture distribution of the Equation (1) as follows
| (2) |
Where θ is the unknown mixture proportion parameter for reads derived from one of the two alleles, regardl555ess of the repeat sequence length x. It is also assumed that the associated parameters σL12 and σL22 are both unknown. These parameters can be estimated by an NLS regression function (Supplementary Material, Pseudo code applying the Nonlinear Least-Squares regression section).
If the sequence reads mapped to a same microsatellite locus contain INDEL errors, the number of observed lengths of the microsatellite at the locus would be ≥2. Because the inherited alleles are unknown, all observed lengths are allele candidates. We then apply the g(x) function for each combination of two allele candidates (two same candidates for homozygous genotype), calculate the squared error of each combination, and select the allele pair, L1* and L2*, that generates the minimum squared error as follows
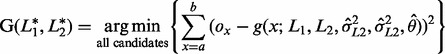 |
(3) |
where ox is an observed proportion of reads containing a length x microsatellite sequence, a is the minimum observed length minus a fixed amount k, and b is the maximum observed length plus k, where k is set to be five as default value. This is necessary because the g(x) function generates output values for all possible sequence lengths, the comparison between observed proportions and expected proportions need to be extended beyond the minimum and maximum observed lengths. Therefore, the boundaries of the calculation are extended by an additional value k.
As an example, suppose that we have 2, 8 and 4 mapped reads containing microsatellite sequences with lengths 14, 15 and 16 bases, respectively, at a locus. The list of possible genotype candidates G(lL1, lL2) for the locus are G(14, 14), G(14, 15), G(14, 16), G(15, 15), G(15, 16) and G(16, 16). In the example, the observed minimum and maximum lengths are 14 and 16, respectively, and the observed and expected values from the Equation (3) are compared for x ranging from 9 to 21. While the observed ratio of read counts between the highest read frequency allele (lL1 = 15) and the second highest read frequency allele (lL2 = 16) is 0.5 (= 4/8), the read ratio of those two alleles estimated by the NLS function was 0.163 (= (1 – θ)/θ = 0.14/0.86) (Fig. 1A). The difference between the two estimated ratios may result in a different decision for the genotype calls, depending on the cutoff ratio to determine if the second highest read frequency allele candidate is noise.
2.2 Preprocessing
We provide two additional PERL scripts along with GenoTan for the preprocessing of data. The first script searches for microsatellite loci and the second script realigns sequence reads mapped to the microsatellite loci. GenoTan takes a reference file, a list of microsatellite loci and BAM/SAM format files containing mapping results as input. If users do not submit a microsatellite list, GenoTan searches pure microsatellite loci from the reference. For users who want to use TRF (Benson 1999), an additional PERL script to convert the TRF results to the microsatellite list is available in our software package. For each locus, it then chooses allele candidates which satisfy three conditions: (i) at least two reads supporting the same allele candidate overlap at least three bases for both flanking sequences and they are not technical duplications (same mapping position and same sequence); (ii) microsatellite sequences of at least two reads supporting the same allele candidate have fewer than 10% mismatches in their length; and (iii) a consensus sequence of the reads span at least five bases at both flanking sequences. GenoTan then compares the genotype candidates to find the most likely genotype.
2.3 Two-step estimation
The whole process of genotyping consists of a two-step estimation (Fig. 1). In the first step, rough estimates find the candidate genotypes of microsatellite loci using the regression model described by the previous section and the method at ‘decision process to finalize genotyping call’ decides genotypes. In the second step, the regression method requires two additional parameters which are estimated from the results of the first regression step. The first parameter, ωL, represents error bias toward deletion or insertion depending on the homopolymer length in an allele candidate L. Since the Gaussian distribution has a symmetric form, the Equation (1) generates symmetric probabilities for deletion and insertion errors for any allele, which does not fit real data. It can be adjusted by adding additional parameters ωL1 and ωL2 to μ1 and μ2, respectively, as follows (Supplementary Fig. S6)
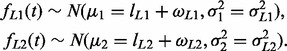 |
(4) |
Then, Equations (1) and (2) can generate different probabilities for deletion and insertion errors depending on the homopolymer length in L1 or L2. To estimate ωL for each allele candidate L, we use a homopolymer decomposition method which decomposes a given microsatellite sequence into a set of homopolymers and then estimates parameters from the set (Supplementary Material, Homopolymer decomposition section for detail).
The second parameter, υL, represents a variance of the prior probability distribution of read proportions for x derived from an allele candidate L. To apply the homopolymer effect to the NLS regression, we use different pseudo counts for different repeats based on the parameter. A data vector for the NLS regression is initialized to 0 and pseudo counts (positive fractions) estimated from the g(x; lL1, lL2, υL1, υL2, 0.5) function in which the parameters are {σ12 = υL1, σ22 = υL2, θ = 0.5} are added to the vector (Supplementary Material, Pseudo code applying the Nonlinear Least-Squares regression section). The parameter υL for each allele candidate L is also estimated by the homopolymer decomposition method. (The NLS regression function to estimate σL1, σL2 and θ requires as input a data vector containing the observed read proportions for length x microsatellite sequences. These estimated parameters are then used to calculate the probability of each x to be observed in a read at a locus. Recall that, the probability varies depending on the length of the homopolymer in the microsatellite sequence. Since the first regression step uses only the read proportions to estimate σL1, σL2 and θ, the estimated values of the parameters are always the same regardless of the lengths of homopolymers in alleles, if two or more different loci have different repeat sequences but contain the same proportions of reads. However, we have observed that the probability of the INDEL error increases with long homopolymer repeats, so we use pseudo counts for different repeats.)
Instead of the numbers of reads, sums of mapping probabilities of reads containing length x microsatellite sequences are added to the vector. If mapping probabilities of reads are high, their sum is near the number of the reads. Then, the values in the vector are converted to the proportions. If υL1 and υL2 are large and the number of total reads is small, the values in the vector get dispersed and the NLS function estimates large σL1 and σL2. But when the number of total reads is big, the effect of υL1 and υL2 becomes small.
2.4 Decision process to finalize genotyping call
The most probable genotype for a given set of sequence reads mapped to a locus is decided by the Equation (3). But the equation shows a tendency to call heterozygous genotypes, because the Gaussian mixture model is a better fit to the training data when more distributions are mixed. However, since reads supporting one or both predicted alleles may be from noise including individual cell mutation, PCR amplification error, sequencing error and mis-mapping, an evaluation method is necessary.
We use a rule-based approach to choose alleles and to decide the homozygosity of each locus because the frequencies of INDEL error reads derived from mis-mapping, PCR amplification error and individual cell mutation are more difficult to measure than that from the sequencing error. The rule-based approach also offers users an opportunity to adjust the boundary parameters to filter ambiguous loci, while most statistical approaches, including Bayesian approaches, adhere to strict decisions with limited user control (Supplementary Material, Limitation of the Bayesian approach considering only sequencing errors for INDEL genotyping section). For this approach, we assign a confidence score to each allele instead of calculating the probability of a genotype (a two allele set) for a locus. The probability of each allele can be generated by the Equation (1) as pL1(lL1) or pL2(lL2) if we assume the read frequencies from two different alleles at the heterozygotic locus are not correlated. However DNA fragments from two paired chromosomes have the same probability of being sequenced and the read frequencies of two alleles would tend to be similar. If the proportion of reads for an allele candidate Llow with lower read frequency is too small compared to that for another allele candidate Lhigh with higher read frequency (e.g. 0.1 versus 0.9), we may conclude that the reads for the allele candidate Llow are from noise and the locus is homozygous. Considering this condition, we multiply the ratio of θlow to θhigh and the output of pLlow (lLlow), where θlow is the output of MIN{θ, 1 – θ} and θhigh is the output of MAX{θ, 1 – θ}. The confidence scores of two allele candidate are then defined by
| (5) |
In the final tabulation, an allele candidate from the predicted genotype is removed when its confidence score is lower than a given cutoff value (0.35 for Lhigh and 0.25 for Llow) (Supplementary Fig. S7). When only confidence score of Llow is lower than the cutoff value, GenoTan generates a partial genotype call for the locus in which only one allele is called while the other allele is reported as unknown. GenoTan only reports the genotype of the locus as homozygous when the number of reads supporting the selected allele is >4 and its confidence score is ≥0.9. The confidence score of the second allele, Lhigh2, at a homozygous locus is calculated by
| (6) |
where [0.5n] represents the probability of the other unobserved allele exists when n reads support the selected allele.
3 RESULTS
Our method (GenoTan) was compared to GATK (v1.6.9 with ‘-T UnifiedGenotyper -glm INDEL’), Dindel (v1.01 with options in Supplementary Materials), SAMtools (v0.1.18 with the ‘varFilter’ post-filter) and RepeaSeq (v0.7.2) in genotyping performance using four different test sets including simulated data, mixed sequence data of two Drosophila inbred lines, sequence data of a single Drosophila inbred line and sequence data of a human sample. For the performance test, we compared INDEL lengths in microsatellite loci identified by the programs ([allele length] = [reference length] – [INDEL length]). In simulated datasets, true positive alleles do not have INDELs at multiple positions in their sequences. Partial genotypes called by GenoTan were counted as the homozygous calls to follow the traditional classification of the other programs. lobSTR could not be included in most tests since its target loci were limited (only analyzing 2∼6-mer motif repeat loci).
3.1 Performance test with simulated data
The first dataset was simulated data created as sequence reads of a single individual diploid genome. The reference sequence for this data was created from the human chromosome 1 (build 37) after removing all repeat sequences identified by RepeatMasker (http://repeatmasker.org), and microsatellites of 1∼8-mer motifs with 8∼48 bases in length (4∼25 repeats of a motif in a sequence) were inserted at 20 700 loci, one for every 140 bases of the reference sequence. The loci included 300 replicates of each microsatellite in which each 100 replicates were used for non-reference alleles of homozygous loci, reference/non-reference alleles of heterozygous loci and two different non-reference alleles of heterozygous loci, respectively. For each locus, three different read sets in 10, 20 and 40x sequence coverage were generated, and all reads overlapped at least 7 bases with both flanking sequences of the microsatellite locus. Then, INDEL/substitution errors depending on the homopolymer contexts of alleles were inserted at random positions of read sequences (Supplementary Table S1).
GenoTan (with ‘-L off’ which turns off the normalization of read counts using the allele lengths) achieved the highest overall performances in all coverages (Fig. 2A and Supplementary Table S2), most notably for the non-reference homozygous loci (Fig. 2B). In this test, GATK and SAMtools did not predict NR/NR (non-reference/non-reference) heterozygous genotypes and RepeatSeq did not predict NR homozygous genotypes correctly, while GenoTan identified homozygous, heterozygous R/NR (reference/non-reference) and heterozygous NR/NR genotypes with very high correct rate (97.4%, 93.5% and 93.7% with 40x coverage, respectively) (Fig. 2B, C and D). Dindel showed slightly higher correct rate (94.6 % versus 93.5%) than GenoTan in calling R/NR heterozygous loci with 40x coverage presumably due to Dindel’s algorithm favoring reference allele calls. For allele level comparison, GenoTan had very low rates of missed and wrong allele calls for both homozygous (Fig. 2E) and heterozygous loci (Fig. 2F) at all coverage depths. At error abundant loci, GenoTan showed low rates of missed and wrong allele calls compared to the other programs (Supplementary Figures S8, S9 and S10).
Fig. 2.

Performance test with simulated data. (A) The proportions of no-calls, incorrect calls and correct calls for all simulated datasets at different sequence coverage. (B) The proportions of calls for non-reference homozygous loci. (C) The proportions of calls for reference/non-reference (R/NR) heterozygous loci. (D) The proportions of calls for non-reference/non-reference (NR/NR) heterozygous loci. (E) The numbers of correct, missed and wrong allele calls at homozygous loci. The numbers of missed and wrong allele calls are shown in negative numbers. (F) The numbers of correct, missed and wrong allele calls at heterozygous loci
3.2 Performance test with mixed sequence data of two Drosophila inbred lines
To evaluate the performance of GenoTan for diploid genome datasets, the second dataset was created by merging sequence reads of two different Drosophila inbred lines RAL-365 and RAL-375 [SRA: SRX000537, SRA:SRX000538] (Illumina 36 cycle single-end sequencing) downloaded from the SRA website (http://www.ncbi.nlm.nih.gov/sra). From the datasets, the sequence coverage averaged 23x, which was sufficient to make reliable allele calls for many loci. The reference genome sequence was NCBI release_5_30 of Drosophila melanogaster. A list of microsatellite loci was generated from the reference by searching microsatellites of 1∼8-mer motifs with a minimum three in repeat number and a minimum 10 bases (10 bases for 1- and 2-mer, and 12 bases the other motifs) in length. When a distance of two loci was <10 bases, they were merged into a single locus. The original Drosophila inbred line study (Mackay et al., 2012) also analyzed microsatellite genotypes, but we did not use the data since many allele calls from the study for our target loci were inconsistent with our read alignments (the data is not shown).
The number of sequence reads in SRX000538 was reduced so that an equal number of reads was used for both samples (the end of SRR001962.fastq was clipped). To create the input for GenoTan, BWA (Li and Durbin, 2009) and GATK were used to map the sequence reads to the reference and to realign the reads, respectively. And microsatellite loci satisfying the following three conditions were chosen for the performance comparison. First, at each locus for both samples, the minimum number of reads supporting an allele and overlapping at least 3 bases to both flanking sequences of a microsatellite locus was 2. Second, at each locus for both samples, the number of reads supporting the second highest read frequency allele candidate was no more than half of the read supporting the major allele candidate at a locus. The second condition reduced the number of loci from possible replicated sequences such as transposon elements, which might result in incorrect validation. Third, alleles from one or both samples at the same locus were non-reference alleles. A total of 3300 loci (770 homozygous and 2530 heterozygous loci) were selected and the average number of reads completely covering repeat sequences per locus was 10.9.
Since we filtered out the microsatellite loci in which the numbers of reads supporting major allele candidates of each inbred were ambiguous (see Methods section for the details), we assumed that all alleles at the test microsatellite loci were clearly identified. The data does not include reference/reference homozygous loci because we selected loci containing at least one non-reference allele. GenoTan identified 90.2% of microsatellite loci correctly while RepeatSeq, GATK and Dindel called 61.7%, 45.9% and 77.9% of loci correctly (Fig. 3A). SAMtools and GATK had the highest no-call rates (59.9% and 45.4%) presumably because they required a high number of sequence reads which were consistent with their consensus to call an allele. GenoTan showed the highest proportion of correct genotype calls for both homozygous and heterozygous loci in all sequence coverage (Fig. 3B), but called a slightly higher proportion (12.3%) of incorrect genotypes for low sequence coverage (5∼10x) heterozygous loci than Dindel (10.6%). Most false-positive alleles called by the genotyping programs, except GenoTan and RepeatSeq, were reference allele calls (no-INDEL call) in the incorrect heterozygous genotype calls for homozygous loci. To prevent other programs calling genotypes with sequence reads not completely covering microsatellites, we filtered out the reads and tested RepeatSeq, GATK and Dindel again (Supplementary Fig. S11), but the programs did not show significant improvement.
Fig. 3.

The performance comparison for the mixed sequence data of two different Drosophila inbred lines. All microsatellite loci contain at least one non-reference allele. (A) The proportions of no-calls, incorrect calls and correct calls for the mixed sequence data. (B) The detailed proportions of calls at different sequence coverage for homozygous and heterozygous loci
3.3 Performance test with sequence data of a single Drosophila inbred line
The third dataset was sequence reads of a single Drosophila inbred line RAL-301 [SRA: SRX000530] (Illumina 36-cycle single-end sequencing). This dataset was used to compare the incorrect genotype calls of the genotyping programs for the homozygous loci. The reference genome sequence was the same as that use in the second dataset and the list of microsatellite loci included additional loci for 8 and 9 base homopolymers. After BWA mapping and GATK realignment, microsatellite loci satisfying the following three conditions were chosen for the comparison. First, the minimum number of reads supporting a same allele length and overlapping at least three bases to both flanking sequences of a microsatellite locus was 4. Second, at least one read completely covering the locus contained INDEL errors or misalignments in the microsatellite. Third, the number of reads supporting the second highest read frequency allele candidate was not more than half of the reads supporting the major allele candidate at a locus. As a result, a total of 1304 loci were selected. The average sequence coverage of the whole genome was 21x and the average number of reads completely covering a microsatellite (three base overlapping with both flanking sequences of the microsatellite) per locus was 14.9.
GenoTan showed superior performance at the loci covered by reads supporting multiple allele candidates (Fig. 4A and Supplementary Fig. S13). Notably, when the number of allele candidates was ≥3 at a locus, the proportion of incorrect genotypes called by the other programs increased while that by GenoTan decreased. This indicated that the discretized Gaussian mixture model improved performance at noise-abundant loci. Figure 4B shows proportions of incorrect calls at microsatellite loci with different motif lengths. We visually inspected the incorrectly called loci of 2,3-mer motifs and observed the presence of a few noise reads at each locus, which might be derived from mis-mapping, PCR amplification error or individual cell mutation. It was also possible that the reads supporting the major allele candidates of the microsatellite loci were mis-mapped, but we could not distinguish between these possible scenarios. The length of a microsatellite allele is also an important factor that affects the accuracy of genotyping (Fig. 4C), as the length of the allele gets longer, there are fewer reads completely spanning the repeat sequence. Especially, because sequencing machines frequently fail to generate sequence reads including long homopolymers, the rate of reads covering the homopolymer sequence decreases significantly (Supplementary Fig. S13). When the length of an allele was ≥14 bases in the test data, 2-mer motif microsatellites became dominant because most homopolymer microsatellites were not covered at a sufficient read depth to call genotypes. Since polymer motif microsatellites produced much fewer sequencing errors and higher sequence coverage than homopolymers, the rate of ‘no-call’ loci dramatically decreased, but most programs became more sensitive to noise reads (due to PCR amplification error or individual cell mutation) at polymer motif microsatellite loci.
Fig. 4.

Analysis of the incorrect genotype calls for non-reference homozygous loci in a single Drosophila inbred line. An allele supported by the highest number of reads at a locus was assumed as the inherited allele. ‘No-calls’ of SAMtools (thin gray) were analyzed together. (A) Proportions of incorrect calls at the loci with several different number of allele candidates. (B) Proportions of incorrect calls at the loci with different lengths of motifs. (C) Proportions of incorrect calls at the loci with different lengths of the highest read frequency allele (assumed as inherited alleles)
3.4 Comparison with two sequencing datasets from a single human individual
As a final test, we used two sequencing datasets from blood and saliva samples from a single human individual [SRA: SRX097307, SRA: SRX097312] (Illumina 101-cycle paired-end sequencing), which contains 1 499 021 500 and 3 040 306 840 reads, respectively. The reads were aligned to the human genome reference NCBI build 37 by BWA and realigned by GATK. To create a list of microsatellite loci, TRF (Benson, 1999) was used to search repeat sequences including incomplete repeat sets. Loci containing at least 10 bases for 1-mer, 12 bases for 2-mer and 15 bases for 3-, 4-, 5-, 6-mer of pure repeat sequences were selected for evaluation. When two microsatellite loci were within 40 bases, they were merged into one locus. To check for uniqueness of microsatellite flanking sequences, a 30-base sequence which included a 25-base flanking sequence and a 5-base microsatellite sequence was extracted from each side of the microsatellite and mapped by BWA to a reference sequence. When BWA did not assign the maximum mapping scores (Q37, Phred quality score) to both flanking sequences of the microsatellite, the microsatellite was removed from the target microsatellite list. As results, we obtained 58 245 microsatellite loci in chromosome 1. Among them, 57 603 and 57 693 loci were completely covered by 1 810 477 reads of the blood sample and 3 373 598 reads of the saliva samples, respectively. Most of the reads (1 396 921 reads of blood and 2 610 205 of saliva) supported reference alleles. Since it was difficult to distinguish true positive calls from false-positive calls in genotyping results for the microsatellites especially for loci containing homopolymers from a human genome, we simply measured the number of loci consistent and inconsistent within genotyping results for the two different samples using RepeatSeq, GATK, Dindel and GenoTan (Fig. 5). To analyze the difference, microsatellite loci containing at least one non-reference allele identified by each program were compared. The numbers of loci called for only one sample were not directly compared because we could not know whether the genotypes for the same loci in the other sample were called as homozygous reference allele loci or were not called. RepeatSeq, GATK, Dindel and GenoTan identified heterozygous alleles from 11 596, 13 640, 16 074 and 14 277 loci in both samples, respectively. Though Dindel genotyped the highest number of loci (12 368 loci) consistently, it also had high number of loci (3706 loci) inconsistently called. The consistency rate of the genotyping results was measured by [(number of consistent calls)/(number of consistent calls + number of inconsistent calls)] and GenoTan showed the highest consistency rate (0.85) while RepeatSeq, GATK and Dindel showed 0.39, 0.82 and 0.77, respectively.
Fig. 5.

Comparison of microsatellite loci containing non-reference alleles identified in two sequence datasets from a single human individual. Two sequencing datasets from blood and saliva samples from a single human individual were used to measure the consistency of genotyping results generated by RepeatSeq, GATK, Dindel and GenoTan. The numbers in the diagrams are the numbers of microsatellite loci containing at least one non-reference allele identified by each program for two different samples. The consistency of the genotyping results generated by each program was measured by (number of consistent calls)/(number of consistent calls + nummber of inconsistent calls)
3.5 Other comparisons
GenoTan is computationally fast which provides a significant competitive performance advantage. While GenoTan could genotype 20 700 loci in the 40x sequence coverage simulated data in 9 m 20 s, GATK and Dindel genotyped them in 57 m 40 s and 4 h 53 m 4 s, respectively. RepeatSeq archived the fastest speed (15 s), but it showed high false-negative rates in all tests. The additional comparisons of computational speeds are available in Supplementary Table S3.
The performance of genotyping programs were compared for different mapping results generated by two different mapping programs, BWA and Novoalign (http://novocraft.com). Simulated sequence reads for the Drosophila reference sequence were generated by pIRS (Hu et al., 2012) (Supplementary Material, Performance test with two different mapping program for simulated data generated by pIRS from the Drosophila reference section for detail). The genotyping results from lobSTR for target microsatellite loci were also compared and evaluated. GATK, DIndel, GenoTan and RepeatSeq had correct percentages of 79.8%, 92.4%, 91.8% and 53.7% with BWA mapping, respectively, and 84.3%, 95.6%, 95.4% and 55.0% with Novoalign mapping. Not relying on these mapping programs, lobSTR had 2.8% of correct calls. It should be noted that for RepeatSeq and lobSTR, many genotypes were not wrong, but were uncalled. High false-negative rate of lobSTR is consistent with results in the RepeatSeq study (Highnam et al., 2012). All loci in our test set contained at least one non-reference allele but lobSTR called only reference alleles for many loci.
4 DISCUSSION
Here we presented GenoTan, a method using a discretized Gaussian mixture model combined with a rules-based approach to identify inherited alleles of microsatellite loci from NGS data, which often contain noise reads caused by substitution/INDEL sequencing error, PCR amplification error or individual cell mutation, without paired-end information. It also employs an additional novel approach, homopolymer decomposition, to estimate error bias toward deletion or insertion in homopolymer runs. Combining these approaches, we were able to successfully genotype microsatellite loci from both simulated data and real data quickly without biased calls taken to be references alleles, while other approaches required 5∼30× more computational time than GenoTan and favored calling reference alleles. In our experiments, the rule-based approach had better accuracy in distinguishing noise from inherited alleles at the microsatellite loci than the Bayesian approach used by GATK and Dindel. This is because noise reads at the microsatellite loci were derived not only from sequencing errors, but also from mis-mapping, PCR amplification error and individual cell mutation, of which error frequencies are very difficult to measure using statistical methods. The discretized Gaussian mixture also showed enhanced performance in reducing noise at noise-abundant loci covered by sequence reads containing long homopolymers which often induce substitution and INDEL sequencing errors. Two programs, lobSTR and RepeatSeq, have been developed to genotype specifically microsatellites, but since they did not call genotypes for many loci in our test sets, of which all loci contained at least one non-reference allele and true alleles were clearly identified, their performance is still unknown. In addition, RepeatSeq frequently calls more than two alleles for a locus and may perform better with sequencing data of tumor samples to search for multiple alleles.
Even though GenoTan has improved performance in controlling homopolymer errors, it, like other methods, has limitations. GenoTan has been designed to detected microsatellite variants shorter than read lengths, but long microsatellite sequences are frequently observed and are not addressed by this method. And it requires high sequence coverage to reliably estimate genotypes. These two limitations are common to many genotyping programs and may be reduced by advanced sequencing technologies producing longer reads and higher coverage than the current technologies, even if the homopolymer error problems found in various technologies continue.
Mismapping is also still problematic for genotyping. We visually inspected the loci incorrectly called by genotyping programs from Drosophila inbred line sequencing data and observed several loci covered by reads which appeared to be mismapped, in which the true alleles could not be identified. Most genotyping programs including GanoTan incorporate the mapping quality scores generated by mapping programs into their methods to control for incorrectly mapped reads. However relying on the mapping quality scores could result false positive prediction in INDEL genotypes, since mapping programs often fail to map reads containing microsatellite variants and generate incorrect quality scores. Reducing the effect of incorrectly mapped reads remains a challenge for genotyping programs.
Since genotyping more complex genomes, such as human, is significantly more difficult due to the abundance of transposable elements that interfere with mapping and are commonly associated with microsatellite loci, we indirectly compared the performance of genotyping programs for two matched sequencing datasets (blood and saliva) from a single human individual. With the human data, GenoTan showed the highest concordance for these two genotyping tests.
Lastly, GenoTan was designed to call only two alleles as paternal and maternal alleles, while many alleles could be acquired after birth especially in tumor cells. The current version treats the acquired alleles as noise (individual cell mutation), but next version should be available to call more than two alleles to address this issue.
Supplementary Material
ACKNOWLEDGEMENTS
DAC (Data Analysis Core) at the Virginia Bioinformatics Institute helped sequence data analysis.
Funding: The Medical Informatics and Systems Division director’s funds; the National Human Genome Research Institute (of the National Institute of Health) [The 1000 Genomes Project Dataset Analysis Grant; grant numbers U01 HG005719-01, U01 HG005719-02].
Conflict of Interest: none declared.
REFERENCES
- Albers CA, et al. Dindel: accurate indel calls from short-read data. Genome Res. 2011;21:961–973. doi: 10.1101/gr.112326.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlich Y, et al. Alta-Cyclic: a self-optimizing base caller for next-generation sequencing. Nat. Methods. 2008;5:679–682. doi: 10.1038/nmeth.1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing B, et al. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Gymrek M, et al. lobSTR: a short tandem repeat profiler for personal genomes. Genome Res. 2012;22:1154–1162. doi: 10.1101/gr.135780.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Highnam G, et al. Accurate human microsatellite genotypes from high-throughput resequencing data using informed error profiles. Nucleic Acids Res. 2012;41:e32. doi: 10.1093/nar/gks981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, et al. pIRS: Profile-based Illumina pair-end reads simulator. Bioinformatics. 2012;28:1533–1535. doi: 10.1093/bioinformatics/bts187. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay TF, et al. The Drosophila melanogaster Genetic Reference Panel. Nature. 2012;482:173–178. doi: 10.1038/nature10811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIver LJ, et al. Evaluation of microsatellite variation in the 1000 Genomes Project pilot studies is indicative of the quality and utility of the raw data and alignments. Genomics. 2011;97:193–199. doi: 10.1016/j.ygeno.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minoche AE, et al. Evaluation of genomic high-throughput sequencing data generated on Illumina HiSeq and Genome Analyzer systems. Genome Biol. 2011;12:R112. doi: 10.1186/gb-2011-12-11-r112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland GR, Richards RI. Simple tandem DNA repeats and human genetic disease. Proc. Natl Acad. Sci. 1995;92:3636–3641. doi: 10.1073/pnas.92.9.3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, et al. The direction of microsatellite mutations is dependent upon allele length. Nat. Genet. 2000;24:396–399. doi: 10.1038/74238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.