

Abstract
This article describes 3,3-difluoroxindole (HOFox) – mediated glycosylation. The uniqueness of this approach is that both the in-situ synthesis of 3,3-difluoro-3Hindol-2-yl (OFox) glycosyl donors and activation thereof can be conducted in a regenerative fashion as is a typical reaction performed under nucleophilic catalysis. Only a catalytic amount of the OFox imidate donor and a Lewis acid activator are present in the reaction medium. The OFox imidate donor is constantly regenerated upon its consumption until all glycosyl acceptor has reacted.
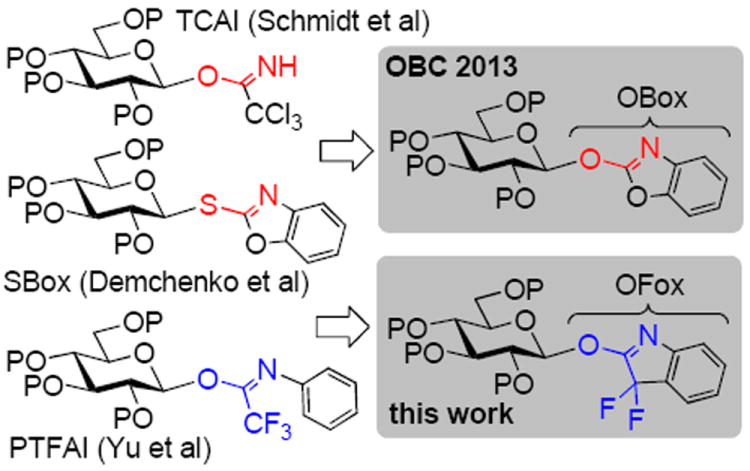
Practically all complex carbohydrates have an oligomeric sequence wherein monosaccharide residues are linked via glycosidic linkages.1 Both simple methods and sophisticated strategies for glycoside synthesis and oligosaccharide assembly exist.2-3 However, the complexity of glycosylation reactions4-7 is responsible for many drawbacks that all current methods experience. In spite of significant progress, chemical glycosylation remains challenging due to the requirement to achieve complete stereocontrol and to suppress side reactions.8 Amongst a plethora of leaving groups developed, a vast majority of glycosylations make use of thioglycosides9-12 and O-trichloroacetimidates (TCAI).13-15 Our laboratory has also been developing new leaving groups for chemical glycosylation. 16-17 For instance, we developed S-benzoxazolyl (SBox) donors18 and more recently introduced O-benzoxazolyl (OBox) imidates,19 which represent a hybrid structure between SBox and TCAI (Scheme 1), but it is more reactive than either.
Scheme 1.
O-Imidates, established and new.
N-Phenyl trifluoroacetimidates (PTFAI) introduced by Yu20-21 have recently gained a considerable niche amongst methods used for chemical glycosylation. Excellent results have been achieved with these donors, and we decided to investigate whether the 3,3-difluoro-3H-indol-2-yl (OFox) leaving group (Scheme 1) that represents a potentially cheaper, cyclic analog of PTFAI would provide an efficient alternative. While developing the new class of O-imidates we noticed a feature of the OFox leaving group that differentiates it from all others developed for and used in chemical glycosylation. The aglycone structure needed to introduce OFox and that of the departed leaving group are essentially the same, cyclic amide 3,3-difluoroxindole (HOFox).22-23 The significance of this observation is that, in principle, one should be able to conduct both the introduction and activation of this leaving group in the catalytic “donor-regenerative” fashion, which is routinely done in enzymatic glycosylations,24-25 but represents a new direction in the field of chemical glycosylation. This new method falls into the general classification of nucleophilic (covalent) catalysis that is broadly used in synthetic26 and enzymatic transformations.27 Examples wherein nucleophilic catalysis is used to obtain glycomimetics have been reported.28-29 The effect of stoichiometric nucleophilic additives on the outcome of O-glycosylation has been investigated. 30-34
Prior to establishment of the regenerative approach, we first validated that OFox imidate 3 performs similarly to that of common O-imidate donors 1 and 2.20,35 As depicted in Table 1, coupling of donors 1-3 with acceptor 4 produced disaccharide 536 in 5 min either at -78 °C or at ambient temperature in comparable yields. We further conducted an exploratory study of OFox imidates and investigated a number of factors that affect stereoselectivity and reactivity of these glycosyl donors. This study is presented as a part of the SI and details the effect of promoter, solvent, protecting groups, leaving group orientation, primary and secondary acceptors, glycosyl donors of other sugar series, etc. Excellent yields have been achieved in a majority of cases, and some reactions provided high stereoselectivity (see the SI)
Table 1.
Comparative investigation of O-imidates 1-3.
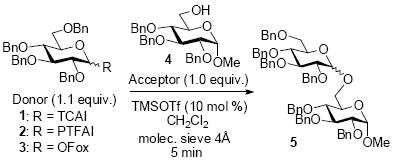
| ||||
|---|---|---|---|---|
| Entry | Donor | Temperature | Yield of 5 | Ratio α/β |
| 1 | 1 | -78 °C | 92% | 1/15 |
| 2 | 1 | rt | 94% | 4.0/1 |
| 3 | 2 | -78 °C | 70% | 1/4.4 |
| 4 | 2 | rt | 91% | 1/2.2 |
| 5 | 3 | -78 °C | 94% | 1/24 |
| 6 | 3 | rt | 93% | 1/1.2 |
Having established basic principles of the synthesis and activation of OFox imidates, we turned our attention to the investigation of regenerative glycosylation, a concept that sets use of OFox donors apart from other known methods for chemical glycosylation. Both TCAI and PTFAI are obtained from hemiacetal precursors, using either inexpensive trichloroacetonitrile or rather costly 2,2,2-trifluoro-N-phenylacetimidoyl chloride, respectively, but their syntheses often originate from thioglycosides (Scheme 2). The use of thioglycosides as general building blocks is very broad. Thioglycosides are also excellent glycosyl donors, but their relatively low reactivity profile and the requirement for stoichiometric promoters sometimes hinders their application in direct glycosidations. Conversely, O-imidates are very reactive and require only a catalytic amount of the activator. Many O-imidates can be purified, but none can be stored and hence have to be used in glycosidations right away. Leaving groups in the previously studied imidates TCAI or PTFAI depart as unreactive amides, trichloroacetamide or 2,2,2-trifluoro-N-phenylacetamide, respectively, and cannot be reused directly (Scheme 2).
Scheme 2.
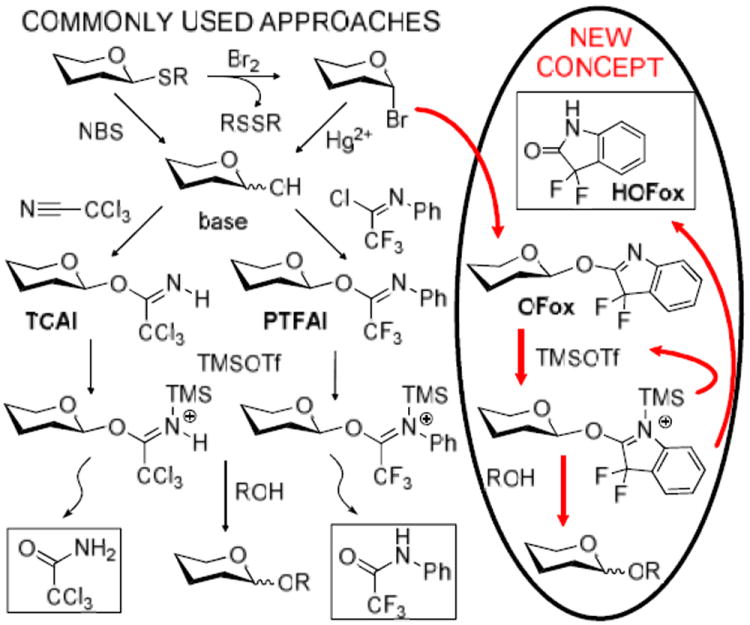
Common approaches and the new concept
Uniquely, since both the reagent for the introduction of the OFox leaving group and the departed leaving group are essentially the same (HOFox), both the synthesis and glycosidation of OFox imidates can be conducted using catalytic amounts of the HOFox aglycone. HOFox aglycone will first react with a stable precursor to form highly reactive OFox imidate donor. The latter will then react with the acceptor while regenerating HOFox aglycone, which will be available for the next catalytic cycle to regenerate the OFox donor. Resultantly, only a relatively small amount of the reactive donor is present in the reaction mixture, but it can vary based on the amount of added HOFox. It should be emphasized that the OFox donor can be regenerated only upon consumption of the first batch (Scheme 2).
In our preliminary study of the regenerative concept disclosed herein, we first reacted S-ethyl glycoside 637 with stoichiometric bromine to form glycosyl bromide 7. The latter is stable under the reaction condition, but gets readily converted into OFox imidate 3 in the presence of HOFox. The amount of the reactive glycosyl donor present in the reaction medium can be controlled by the amount of HOFox added (range studied 0.1-1.0 equiv.). OFox imidates have reasonable shelf-life but are readily activated with various Lewis acids (5-10 mol %, see the SI). Thus, an attempt to activate bromide 7 in the presence of BF3-Et2O showed very sluggish conversion into disaccharide 5 (9% in 5 h, Entry 1, Table 2). Since this reaction was performed in the presence of Ag2O with the primary purpose of scavenging HBr released in this reaction, bromide 7 would eventually react with the acceptor 4 and is likely to require more than 24-48 h to react completely. We managed to achieve reasonable glycosylation rate (3 h) with as little as 10 mol % of HOFox aglycone. As a result disaccharide 5 was isolated in a commendable yield of 84% (Entry 2, Table 2). With the increase of the amount of HOFox, the reaction rate steadily increased (entries 3-6). This culminated in a swift 10 min reaction in the presence of 1.0 equiv. of HOFox that produced disaccharide 5 in 90% (entry 6).
Table 2.
Regenerative glycosylation
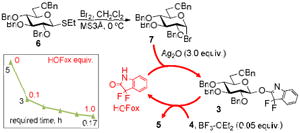
| ||||
|---|---|---|---|---|
| Entry | HOFox (equiv.) | Reaction time | Yield of 5 | Ratio α/β |
| 1 | 0 | 5 h | 9% | 1/1.1 |
| 2 | 0.1 | 3 h | 84% | 1/1.9 |
| 3 | 0.25 | 2 h | 79% | 1/1.9 |
| 4 | 0.5 | 40 min | 86% | 1/1.2 |
| 5 | 0.75 | 30 min | 88% | 1/1.2 |
| 6 | 1.0 | 10 min | 90% | 1/1.2 |
In the further expansion of the regenerative glycosylation we investigated secondary glycosyl acceptors and OFox imidates of other common sugar series (see the SI). A model NMR study of the reaction conducted in the absence of glycosyl acceptor showed the presence of OFox glycosides in ratios comparable to the amount of HOFox additive in respect to the starting bromide (see the SI).
In conclusion, we discovered a new regenerative concept for chemical glycosylation that allows for generating reactive OFox glycosyl donors and activation thereof in the catalytic fashion in situ. The concept has some similarities with the two-step activation by Nicolaou38-40 and Danishefsky41-44 and preactivation strategy by Huang-Ye,45-48 but differs in the catalytic conversion and continuous regeneration of the glycosyl donor. In the previous methods, the entire precursor (thioglycoside or glycal) is first converted into reactive intermediates (Br, F, OTf/NTf2 or epoxide) and then acceptor is added. This regenerative approach is expected to help reduce side reactions commonly found in conventional glycosylations wherein excess of the highly reactive or stoichiometrically preactivated glycosyl donor is present from the beginning.
Supplementary Material
Acknowledgments
This work was supported by awards from the NIGMS (GM090254 and GM077170). We thank Dr. Rensheng Luo (UM – St. Louis) for aquiring spectral data using 600 MHz NMR spectrometer that was purchased thanks to the NSF (award CHE-0959360). Dr. Winter and Mr. Kramer (UM – St. Louis) are thanked for HRMS determinations.
Footnotes
Additional experimental details and characterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Varki A, Cummings RD, Esko JD, Freeze HH, Bertozzi CR, Stanley P, Hart GW, Etzler ME. Essentials of Glycobiology. Second. CSH Laboratory Press; New York: 2009. [PubMed] [Google Scholar]
- 2.Zhu X, Schmidt RR. Angew Chem Int Ed. 2009;48:1900–1934. doi: 10.1002/anie.200802036. [DOI] [PubMed] [Google Scholar]
- 3.Smoot JT, Demchenko AV. Adv Carbohydr Chem Biochem. 2009;62:161–250. doi: 10.1016/S0065-2318(09)00005-5. [DOI] [PubMed] [Google Scholar]
- 4.Crich D. Acc Chem Res. 2010;43:1144–1153. doi: 10.1021/ar100035r. [DOI] [PubMed] [Google Scholar]
- 5.Crich D. J Org Chem. 2011;76:9193–9209. doi: 10.1021/jo2017026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ranade SC, Demchenko AV. J Carbohydr Chem. 2013;32:1–43. [Google Scholar]
- 7.Mydock LK, Demchenko AV. Org Biomol Chem. 2010;8:497–510. doi: 10.1039/b916088d. [DOI] [PubMed] [Google Scholar]
- 8.Demchenko AV, editor. Handbook of Chemical Glycosylation: Advances in Stereoselectivity and Therapeutic Relevance. Wiley-VCH; Weinheim, Germany: 2008. [Google Scholar]
- 9.Zhong W, Boons G-J. In: Handbook of Chemical Glycosylation. Demchenko AV, editor. Wiley-VCH; Weinheim, Germany: 2008. pp. 261–303. [Google Scholar]
- 10.Codee JDC, Litjens REJN, van den Bos LJ, Overkleeft HS, van der Marel GA. Chem Soc Rev. 2005;34:769–782. doi: 10.1039/b417138c. [DOI] [PubMed] [Google Scholar]
- 11.Oscarson S. In: Carbohydrates in Chemistry and Biology. Ernst B, Hart GW, Sinay P, editors. Vol. 1. Wiley-VCH; Wein-heim, New York: 2000. pp. 93–116. [Google Scholar]
- 12.Garegg PJ. Adv Carbohydr Chem Biochem. 1997;52:179–205. doi: 10.1016/s0065-2318(08)60091-8. [DOI] [PubMed] [Google Scholar]
- 13.Zhu X, Schmidt RR. In: Handbook of Chemical Glycosylation. Demchenko AV, editor. Wiley-VCH; Weinheim, Germany: 2008. pp. 143–185. [Google Scholar]
- 14.Schmidt RR, Jung KH. In: Carbohydrates in Chemistry and Biology. Ernst B, Hart GW, Sinay P, editors. Vol. 1. Wiley-VCH; Weinheim, New York: 2000. pp. 5–59. [Google Scholar]
- 15.Schmidt RR, Jung KH. In: Preparative Carbohydrate Chemistry. Hanessian S, editor. Marcel Dekker, Inc.; New York: 1997. pp. 283–312. [Google Scholar]
- 16.Hasty SJ, Demchenko AV. Chem Heterocycl Compd. 2012;48:220–240. doi: 10.1007/s10593-012-0984-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yasomanee JP, Demchenko AV. Trends Glycosci Glycotechnol. 2013;25:13–42. [Google Scholar]
- 18.Demchenko AV, Malysheva NN, De Meo C. Org Lett. 2003;5:455–458. doi: 10.1021/ol0273452. [DOI] [PubMed] [Google Scholar]
- 19.Nigudkar SS, Parameswar AR, Pornsuriyasak P, Stine KJ, Demchenko AV. Org Biomol Chem. 2013;11:4068–4076. doi: 10.1039/c3ob40667a. [DOI] [PubMed] [Google Scholar]
- 20.Yu B, Tao H. Tetrahedron Lett. 2001;42:2405–2407. [Google Scholar]
- 21.Yu B, Sun J. Chem Commun. 2010;46:4668–4678. doi: 10.1039/c0cc00563k. [DOI] [PubMed] [Google Scholar]
- 22.Zhu J, Zhang W, Hu J. J Org Chem. 2010;75:5505–5512. doi: 10.1021/jo1005262. [DOI] [PubMed] [Google Scholar]
- 23.Torres JC, Garden SJ, Pinto AC. Tetrahedron. 1999;55:1881–1892. [Google Scholar]
- 24.Ichikawa Y, Liu JLC, Shen GJ, Wong CH. J Am Chem Soc. 1991;113:6300–6302. [Google Scholar]
- 25.Chen X, Fang J, Zhang J, Liu Z, Shao J, Kowal P, Andreana P, Wang PG. J Am Chem Soc. 2001;123:2081–2082. doi: 10.1021/ja005738v. and references therein. [DOI] [PubMed] [Google Scholar]
- 26.Fu GC. Acc Chem Res. 2000;33:412–420. doi: 10.1021/ar990077w. [DOI] [PubMed] [Google Scholar]
- 27.Vocadlo DJ, Davies GJ, Laine R, Withers SG. Nature. 2001;412:835–838. doi: 10.1038/35090602. [DOI] [PubMed] [Google Scholar]
- 28.Loskot SA, Zhang J, Langenhan JM. J Org Chem. 2013;78:12189–12193. doi: 10.1021/jo401688p. [DOI] [PubMed] [Google Scholar]
- 29.Thygesen MB, Munch H, Sauer J, Clo E, Jorgensen MR, Hindsgaul O, Jensen KJ. J Org Chem. 2010;75:1752–1755. doi: 10.1021/jo902425v. [DOI] [PubMed] [Google Scholar]
- 30.Park J, Kawatkar S, Kim JH, Boons GJ. Org Lett. 2007;9:1959–1962. doi: 10.1021/ol070513b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chu A-HA, Nguyen SH, Sisel JA, Minciunescu A, Bennett CS. Org Lett. 2013;15:2566–2569. doi: 10.1021/ol401095k. [DOI] [PubMed] [Google Scholar]
- 32.Liu C-YI, Mulani S, Mong K-KT. Adv Synth Catal. 2012;354:3299–3310. [Google Scholar]
- 33.Lu SR, Lai YH, Chen JH, Liu CY, Mong KK. Angew Chem Int Ed. 2011;50:7315–7320. doi: 10.1002/anie.201100076. [DOI] [PubMed] [Google Scholar]
- 34.Nogueira JM, Issa JP, Chu A-HA, Sisel JA, Schum RS, Bennett CS. Eur J Org Chem. 2012;2012:4927–4930. [Google Scholar]
- 35.Schmidt RR, Stumpp M. Liebigs Ann Chem. 1983:1249–1256. [Google Scholar]
- 36.Garcia BA, Gin DY. J Am Chem Soc. 2000;122:4269–4279. [Google Scholar]
- 37.Andersson F, Fugedi P, Garegg PJ, Nashed M. Tetrahedron Lett. 1986;27:3919–3922. [Google Scholar]
- 38.Randall JL, Nicolaou KC. ACS Symp Ser. 1988;374:13–28. [Google Scholar]
- 39.Nicolaou KC, Dolle RE, Papahatjis DP, Randall JL. J Am Chem Soc. 1984;106:4189–4192. [Google Scholar]
- 40.Nicolaou KC, Mitchell HJ. Angew Chem Int Ed. 2001;40:1576–1624. [PubMed] [Google Scholar]
- 41.Halcomb RL, Danishefsky SJ. J Am Chem Soc. 1989;111:6661–6666. [Google Scholar]
- 42.Williams LJ, Garbaccio RM, Danishefsky SJ. In: Carbohydrates in Chemistry and Biology. Ernst B, Hart GW, Sinay P, editors. Vol. 1. Wiley-VCH; Weinheim, New York: 2000. pp. 61–92. [Google Scholar]
- 43.Danishefsky SJ, Bilodeau MT. Angew Chem Int Ed Engl. 1996;35:1380–1419. [Google Scholar]
- 44.Friesen RW, Danishefsky SJ. J Am Chem Soc. 1989;111:6656–6660. [Google Scholar]
- 45.Zeng Y, Wang Z, Whitfield D, Huang X. J Org Chem. 2008;73:7952–7962. doi: 10.1021/jo801462r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang L, Huang X. Chem Eur J. 2007;13:529–540. doi: 10.1002/chem.200601090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang X, Huang L, Wang H, Ye XS. Angew Chem Int Ed. 2004;43:5221–5224. doi: 10.1002/anie.200460176. [DOI] [PubMed] [Google Scholar]
- 48.Huang L, Wang Z, Huang X. Chem Commun. 2004:1960–1961. doi: 10.1039/b405886k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.