

Abstract
Protein glycosylation is a ubiquitous post-translational modification found in all domains of life. Despite their significant complexity in animal systems, glycan structures have crucial biological and physiological roles, from contributions in protein folding and quality control to involvement in a large number of biological recognition events. As a result, they impart an additional level of ‘information content’ to underlying polypeptide structures. Improvements in analytical methodologies for dissecting glycan structural diversity, along with recent developments in biochemical and genetic approaches for studying glycan biosynthesis and catabolism, have provided a greater understanding of the biological contributions of these complex structures in vertebrates.
Glycans associated with cell surface and intracellular proteins and lipids contribute to numerous biological functions in animal systems1. Oligosaccharide structures at the cell surface influence interactions with the extracellular environment by providing ligands for cell adhesion, macromolecule interactions and pathogen invasion1,2. The glycans associated with cell surface receptors and proteins also directly modulate protein function and signalling, as well as altering the dynamics of glycoprotein endocytosis and cell surface half-life through binding to multivalent lectins3. Glycan structures on newly synthesized glycoproteins are crucial for protein secretion, as they influence protein folding, provide ligands for lectin chaperones, contribute to quality control surveillance in the endoplasmic reticulum (ER) and mediate transit and selective protein targeting throughout the secretory pathway4,5. More general roles of glycan structures include contributions to the regulation of cytosolic and nuclear functions, immune surveillance, inflammatory reactions, auto-immunity, hormone action and tumour metastasis1,6. It is clear that these post-translational modifications confer an additional information content at the molecular and cellular level that extends beyond the simple information flow from genome-derived transcripts to encoded protein functions7.
Protein glycosylation is broadly used by cell biologists to monitor protein transit through the secretory pathway (for example, endoglycosidase H-resistant glycan structures are used to monitor protein translocation in the Golgi8). Moreover, glycans detected by specific antibodies9 and ectopically expressed fluorescently tagged glycosylation enzymes10 are used as markers for intracellular compartments. Glycan structures also distinguish cell types in developing and mature animal tissues2. Despite this limited use of mammalian glycan structures or glycosylation machinery as tools for cell biology studies, few cell biologists have truly embraced the diversity and complexity of glycan modifications for their contributions and roles in biological systems. This results from an intimidating collection of challenges for studying the functions of glycan structures at the molecular, cellular and organismal level.
It has been estimated that approximately 700 proteins are required to generate the full diversity of mammalian glycans (estimated to be ≥7,000 structures), which are assembled from only ten monosaccharides: fucose (Fuc), galactose (Gal), glucose (Glc), N-acetylgalactosamine (GalNAc), N-acetylglucosamine (GlcNAc), glucuronic acid (GlcA), iduronic acid (IdoA), mannose (Man), sialic acid (SA) and xylose (Xyl)7,11,12. Among these proteins, ~200 are glycosyltransferases, which are enzymes that extend acceptor glycan structures using nucleotide or lipid-linked sugars as activated donor substrates. Competition between glycosyltransferases that possess overlapping glycan acceptor preferences but different donor specificities can strongly influence the relative abundance of glycan structural features in the total glycome of a cell or tissue. Thus, the glycan biosynthetic potential of a given cell is determined by multiple factors, including relative enzyme abundance and localization, abundance and trafficking of glycoprotein substrates, and the availability of activated sugar donors in appropriate secretory compartments.
Glycans can be attached to polypeptide structures through amide linkages to Asn side chains (N-glycosylation), through glycosidic linkages (O-glycosylation) to side chains of Ser/Thr, hydroxylysine (collagen) or Tyr (glycogenin), or through C-C linkages to the C2 position of Trp (C-mannosylation). Alternatively, they be can attached as a linker structure bridging glycosylphosphatidylinositol anchors to protein backbones13 (TABLE 1). Even among the O-linked structures to Ser/Thr, at least six distinct glycan types are known in animal systems (BOX 1; TABLE 1), with most found on extracellular, cell surface or intracellular membrane compartments. An O-linked monosaccharide addition (GlcNAc-β-Ser/Thr) is also found on a large number of cytosolic and nuclear proteins, where it contributes to the regulation of numerous cellular functions and can compete with Ser/Thr phosphorylation6. Monosaccharides and complex extended glycan structures are also found attached to many other macromolecule classes in animal systems, from glycolipids to secondary metabolites, but these latter structures are beyond the scope of this Review.
Table 1.
Consensus motifs and enzymes responsible for various glycosylation reactions*
| Type | Linkage | Enzyme | Consensus sequence | Domain | Examples |
|---|---|---|---|---|---|
| N-glycosylation | GlcNAc-β-Asn | OST | N-X-(S/T)‡ (standard sequons) | Nascent polypeptides | |
| N-X-C‡,§, N-G§, N-X-V‡,§ (non-standard sequons) | |||||
| O-glycosylation | GalNAc-α-Ser/Thr | ppGALNTs | Isoform specific|| | Mucins | |
| GlcNAc-β-Ser/Thr | OGT | No set consensus¶ | Cytosolic, nuclear proteins | ||
| GlcNAc-β-Ser/Thr | EOGT | Unknown | EGF | Notch, Dumpy | |
| Xyl-β-Ser | XYLT1, XYLT2 | a-a-a-a-G-S-G-a-(a/G)-a (‘a’ represents Asp or Glu)# | Heparin, proteoglycan core proteins | ||
| Fuc-α-Ser/Thr | POFUT2 | C-X-X-(S/T)-C-X-X-G | TSR | Thrombospondin, ADAMTS family | |
| POFUT1 | C2-X-X-X-X-(S/T)-C3 | EGF | Notch, clotting factors | ||
| Glc-β-Ser | POGLUT1 | C1-X-S-X-(A/P)-C2 | EGF | Notch, clotting factors | |
| Man-α-Ser/Thr | POMT1, POMT2 | I-X-P-T-(P/X)-T-X-P-X-X-X-X-P-T-X-(T/X)-X-X** | α-dystroglycan | ||
| Gal-β-Hyl | GLT25D1, GLT25D2 | X-Hyl-Gly (collagen repeats)‡‡ | Collagen, adiponectin | ||
| Glc-α-Tyr | GYG | Tyr194 of GYG§§ | GYG (autoglycosylation during glycogen formation) | ||
| C-mannosylation | Man-α-Trp | Unknown | W-X-X-W|||| | TSR | Thrombospondin, ADAMTS family |
| Glypiation | Pr-C(O)EthN-6-P-Man | Transamidase | No set consensus¶¶ | THY1, NCAM1 |
References provided in the text except where noted.
X cannot be Pro.
Determined by high-throughput proteomic analysis of lectin-enriched glycopeptides21. Other non-standard sequons have been observed for recombinant immunoglobulin G2 antibodies expressed in Chinese hamster ovary cells23 or mouse laminin22, but these structures have not been confirmed more widely in animal systems.
Variation in ppGALNT specificities was determined empirically154.
Prediction software available from the YinOYang WWW server155.
The specificity for the initiation of proteoglycan core synthesis is defined empirically156.
Collagen domains are modified at Lys residues to form hydroxylysine (Hyl) and galactosylated, then they are extended with an α1,2-Glc residue prior to triple helix formation144.
Autoglucosylation and extension of the initial glycogen polymer on the glycogenin backbone occurs on the Tyr194 hydroxyl group143.
Mannose liked to the C2 of the indole ring of the tryptophan residue based on the corresponding consensus sequence111.
Prediction of consensus sequence has been defined based on hidden Markov model and prediction software158. ADAMTS, a disintegrin and metalloproteinase with thrombospondin motifs; EOGT, EGF domain-specific O-linked GlcNAc transferase; GLT25D1/2, glycosyltransferase 25 family member 1/2; GYG, glycogenin; NCAM1, neural cell adhesion molecule 1; OGT, O-linked GlcNAc transferase; OST, oligosaccharyltransferase; POFUT, protein O-fucosyltransferase; POGLUT, protein O-glucosyltransferase; POMT, protein O mannosyltransferase; ppGALNTs, polypeptide GalNAc transferases; TSR, thrombospondin type 1 repeat; XYLT, xylosyltransferase.
Box 1. Major classes of vertebrate glycan structures.
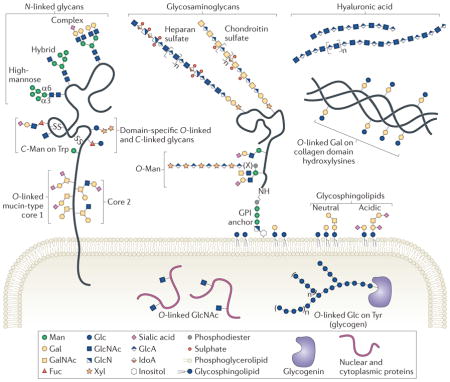
Most glycans on membrane and secreted proteins are found in N-linkage to Asn or in O-linkage to Ser/Thr. N-linked glycans that have undergone minimal mannosidase processing are called ‘high-mannose’ glycans. Addition of N-acetylglucosamine (GlcNAc) to the α3 arm of an acceptor containing five Man residues produces a hybrid glycan14,18. Hybrid glycans can be extended on the α3 arm with galactose (Gal), N-acetylgalactosamine (GalNAc), fucose (Fuc) and sialic acid (SA). Further processing of hybrid glycans produces complex glycans, which can also be decorated with Gal, GalNAc, Fuc and SA.
Glycosaminoglycans (GAGs) are O-linked glycans initiated by a conserved tetrasaccharide (GlcA-β1,3-Gal-β1,3-Gal-β1,4 Xyl-β) and classified by the composition of their disaccharide repeat. GAG chains can be post-synthetically modified by sulphation and epimerization (GlcA conversion to IdoA), producing substantial heterogeneity140. Glycoproteins carrying one or more GAG chains are called proteoglycans, and can be secreted or transmembrane or glycosylphosphatidiylinositol (GPI)-anchored. Hyaluronic acid, a GAG-like polymer of the extracellular matrix, is the only glycan that is not linked to a protein or lipid141.
Other O-linked glycans are classified by their initiating monosaccharide. Addition of GalNAc to Ser/Thr initiates mucin-type O-linked glycans. Extension with Gal, GlcNAc or GalNAc produces eight different core structures142. For example, addition of a branching GlcNAc to a core 1 disaccharide (Gal β1,3-GalNAc) produces core 2. Man initiates another class of O-linked glycan (O-Man glycans). A subset of O-Man glycans are extended with a repeating disaccharide — (-3-Xyl-α1,3-GlcA-β-)n — in phosphodiester linkage to an incompletely defined bridging moiety (X)121,127. Two types of O-linked Fuc glycan and one type of O-linked Glc glycan can be added to specific Cys-rich domains, as well as C-Man residues on Trp side chains94,118. O-linked GlcNAc is found on the extracellular domains of some proteins and on numerous cytosolic and nuclear proteins, but different enzymes mediate extracellular and nucleocytoplasmic O-GlcNAcylation6. O-linked glycan structures attached to other amino acids include Gal on hydroxylysine of collagen domains that is extended by the addition of an α1,2-Glc residue and the addition of a Glc on Tyr of glycogenin that is extended and branched with additional Glc residues to form glycogen143,144. In addition to proteins, sphingolipids can be modified by glycosylation. Eukaryotic cell surfaces are enriched in glycosphingolipids, which are ceramide-linked glycans that can be capped and branched with Fuc and SA145.
Glycan structures attached to proteins can be highly complex, with numerous possibilities for branching and anomeric linkage, and accordingly they have much greater structural diversity than linear nucleic acid or polypeptide structures7. Additional complexity and diversity arises from the fact that mammalian glycans are synthesized in intricate biosynthetic pathways, and the resulting products depend on potential competition among multiple enzymes for the same glycan substrates12,14. This competition, when combined with the relative inefficiency of the enzymatic reactions, often results in a diverse ensemble of branched glycan structures (termed ‘microheterogeneity’), even at the same glycosylation site of otherwise identical proteins15. The diversity of glycan modification can also produce complex pleiotropy, where unique modifications on one glycosylation site may alter function or recognition within a specific cellular context, but may cause other effects or be functionally silent in other contexts. Finally, the diversity of glycan structures found in a typical cell or tissue sample presents considerable challenges for structural elucidation, even when using the most contemporary analytical approaches16. Thus, the goal of determining the roles and contributions of individual oligosaccharides in various biological contexts is equivalent to hunting for a handful of discrete glycan functions within a forest of cellular carbohydrate structures. This complexity commonly dissuades non-glycobiologists from exploring the functional information content embedded within these glycan modifications.
To facilitate broader exploration by non-glycobiologists of the functional information content embedded within glycan structures, this Review article focuses on four key areas of contemporary glycobiology research that have a great impact within the cell biology community: the roles of glycans in quality control; the mechanisms of glycoprotein transport and modification through the Golgi complex; protein domain-specific glycosylation; and high-throughput analytical approaches for glycan structural profiling. The overall goals are to highlight the latest developments in the glycobiology field while emphasizing the unique information content and functions that glycans have in animal systems.
N-glycan synthesis and quality control
Transfer of glycan precursor to nascent polypeptides
N-glycosylation is the most highly studied form of protein glycosylation in eukaryotic organisms. It has been estimated that approximately half of all human proteins are glycoproteins, and most of those contain N-glycan structures17. N-glycans are initially synthesized as a lipid-linked oligosaccharide (LLO) precursor, and the glycans are transferred from LLO to a nascent polypeptide chain during translation. Although the general details of co-translational glycan transfer have been known for decades18, the mechanistic details of oligosaccharide transfer have only recently been revealed19. Eukaryotic organisms generally use a multi-subunit oligosaccharyltransferase (OST) on the lumenal face of the ER membrane to catalyse glycan transfer to acceptor peptide sequons20, which are comprised of an Asn-X-(Ser/Thr) tripeptide (and less frequently of Asn-X-Cys21 and other non-standard sequons22,23), where X can be any amino acid except for Pro (FIG. 1a; TABLE 1). One OST subunit, STT3, contains the catalytic site of the enzyme24 and the mechanism of its interaction with the tripeptide sequon has long been hypothesized on the basis of theoretical and biochemical studies25.
Figure 1. Protein N-glycosylation and quality control of protein folding.

a | During glycoprotein biosynthesis, the translation of nascent polypeptides is followed by their translocation through the SEC61 pore and the simultaneous transfer of a glycan from a lipid-linked intermediate to peptide acceptor sequons by the oligosaccharyltransferase (OST). One cleft in the STT3 subunit of OST scans for Asn-X-Ser/Thr acceptor sequons, while an adjacent cleft binds the glycan donor. b | Glycan trimming through Glc removal occurs immediately after transfer by α-glucosidase I (GIsI) and the α-glucosidase II α–β heterodimer (GIsIIα/β). Folding intermediates containing Glc1Man9GlcNAc2 structures are ligands for the lectins calnexin or calreticulin, which function in complex with ERp57. Dissociation from the lectins is followed by further Glc cleavage. Additional chaperone assistance is provided by the ATP-driven chaperone BiP (also known as GRP78). Correctly folded glycoproteins are packaged for transport to the Golgi. c | Incompletely folded glycoproteins are recognized by the folding sensor UDP-Glc:glycoprotein glucosyltransferase (UGGT1). They are then re-glucosylated through the addition of a Glc residue back to the glycan structure and are reintegrated into the calnexin cycle. d | Terminally misfolded glycoproteins are subjected to endoplasmic reticulum (ER) disposal by Man trimming (through the activity of ER α-mannosidase I (ERManI) or GolgiManIA, GolgiManIB and GolgiManIC (not shown), followed by the activity of ER degradation-enhancing α-mannosidase-like 1 (EDEM1), EDEM2 and EDEM3 (which are homologues of Htm1 (also known as Mnl1) in yeast)). The trimmed glycans bind the ER lectins OS9 or XTP3B (not shown) and are translocated into the cytosol via a complex of derlin 1 (DER1), DER2 and DER3 (the SEL1L complex; Hrd3 in yeast) using the driving force of the cytosolic ubiquitin binding protein and ATPase functions of valocin-containing protein (VCP; also known as TER ATPase; known as Cdc48, Ufd1 or Npl4 in yeast). The peptide is deglycosylated by a cytosolic PNGase and degraded by the proteasome149.
Recent work has identified a homologous form of N-glycosylation on similar acceptor sequons of periplasmic and secreted glycoproteins in a restricted set of Gram-negative bacteria26–28. Bacterial glycosylation also relies on the transfer of glycans from an LLO precursor to nascent polypeptide chains, but the enzyme responsible is a single polypeptide, PglB, with high sequence similarity to eukaryotic STT3 (REFS 27–29). The structure of Campylobacter lari PglB with a bound peptide acceptor was recently solved19, and the co-complex provides interesting insights into the mechanistic details of sugar transfer for both bacteria and eukaryotes. PglB is anchored in the bacterial membrane via 13 transmembrane helical segments while a large globular domain faces into the periplasm. Two clefts are found at the junction between the transmembrane and globular domains, and the acceptor peptide was found to be positioned across one of the clefts with its Asn side chain extending through a hole into the adjacent cleft, which is presumed to bind the LLO donor. The nature of the interactions between the peptide acceptor and the active site cleft, the indirect catalytic role of a protein-bound divalent cation, and the conformational change in a loop region required for product release all provide novel insights into the mechanism of glycan transfer25. The additional subunits required for eukaryotic OST activity presumably act to facilitate coupling to the Sec61 protein translocation pore and to select full-length Glc3Man9GlcNAc2 LLO glycan substrates, as truncated glycans are transferred with greatly reduced efficiency in animal systems20.
N-glycosylation in quality control of protein folding
Following transfer of Glc3Man9GlcNAc2 glycans to nascent polypeptide chains on the lumenal face of the ER in eukaryotes, trimming of two Glc residues results in the formation of a GlcMan9GlcNAc2 structure that acts as a ligand for the membrane-bound or soluble lectin chaperones, calnexin and calreticulin30 (FIG. 1b). These lectins are tethered to ERp57 (also known as protein disulphide-isomerase A3 (PDIA3)), and they protect nascent polypeptides from hydrophobic aggregation and non-productive disulphide bonding during their folding itinerary31. Iterative cycles of Glc removal by glucosidase II and Glc re-addition by UDP-Glc:glycoprotein glucosyltransferase (UGGT1), followed by re-binding to the lectin chaperones, help to facilitate efficient folding of newly synthesized glycoproteins in the ER lumen30,31 (FIG. 1c). Glucosidase II is comprised of an α (catalytic) and a β (lectin) subunit that facilitate substrate interactions, but neither glucosidase subunits nor the ER lectins calnexin and calreticulin appear to recognize the polypeptide backbone of the folding intermediate. However, UGGT1 clearly recognizes glycan structures in proximity to incompletely folded ‘molten globule’ peptide structures, and it acts as a folding sensor that drives re-entry into the ‘calnexin cycle’30,32.
Control of misfolded glycoprotein disposal from the ER
In addition to recruiting chaperones during protein folding, glycan structures define the ER residence time for the downstream quality control of newly synthesized glycoproteins. Glycoproteins with slow folding kinetics owing to mutation or incomplete oligomeric assembly of subunits are targeted for degradation by the activity of Man trimming enzymes, which is the basis for the ‘mannose timer’ model of ER quality control30. Glycan trimming is followed by recognition of the trimmed structures by components of a multiprotein complex at the ER membrane that facilitates ‘retro-translocation’ and subsequent proteasomal degradation in a process termed ER-associated degradation (ERAD)30,31. Recent studies on ERAD in Saccharomyces cerevisiae demonstrated that two related ER mannosidases (Mns1 and Htm1 (also known as Mnl1)) are required for sequential trimming of terminal Man residues33, with Htm1 working in a complex with Pdi1 (REF. 34). The resulting trimmed glycan is recognized by the ERAD lectin Yos9 (OS9 in humans along with its homologue XTP3B (also known as ERLEC1)), which mediates transfer to the retrotranslocon complex (FIG. 1d).
In mammals, the components involved in glycan trimming and recognition are more complex. Seven enzyme isoforms related to the yeast trimming man-nosidases are found in mammalian cells, where they have roles in both ERAD and glycan maturation to complex type structures in the secretory pathway35. Mannosidase inhibitor and overexpression studies indicate that Man trimming is the proximal and rate-limiting step in ERAD and is tightly coupled to components of the calnexin cycle to assure effective quality control surveillance and clearance of misfolded proteins from the lumen of the ER36. However, the subcellular location of the ERAD compartment, the substrate specificities and the localizations of the trimming enzymes, as well as their relative contributions in ERAD versus glycan maturation, remain controversial36,37.
Neither the trimming mannosidases nor the Yos9 homologues are believed to recognize the folding state of the underlying polypeptide, and only some glycans on ERAD substrates are required for effective disposal38,39. An intriguing model has recently been developed suggesting that some protein glycosylation sites mark secondary structure elements that are sensitive to the overall folding state of the protein38. A bipartite signal that combines trimmed glycans and underlying protein conformation would allow the selection of ERAD substrates while sparing fully folded intermediates and ER-resident proteins from disposal. The identity of the ERAD components that couple the glycan and peptide recognition components still remains unclear38. Thus, protein glycosylation has key roles in assisting in protein folding as well as in defining the timing for protein disposal as a part of the quality control function in the early secretory pathway.
Golgi organization and glycomic diversity
Glycan processing in the post-ER secretory pathway
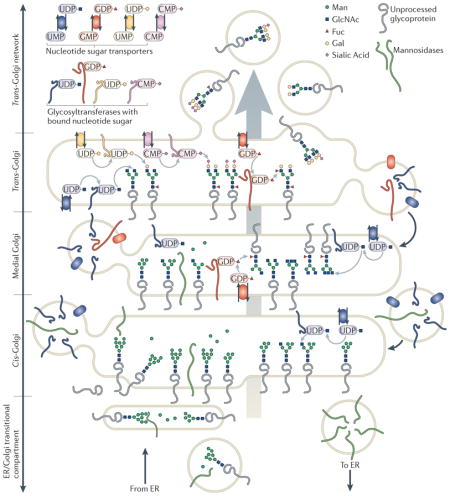
After leaving the ER, glycoproteins traverse the Golgi apparatus, where their glycans are susceptible to further processing. Generally, few ‘high-mannose’ glycans reach the cell surface of most differentiated vertebrate cell types40. Almost all glycoprotein glycans are subject to trimming and extension as they transit the Golgi. Localization studies have demonstrated that glycan processing enzymes are distributed across the Golgi in a cis-to-trans topography that correlates successive glycan maturation steps with vectorial traffic through the organelle41,42 (BOX 2). However, this generic Golgi architecture, where glycan processing enzymes serve as defining markers for specific Golgi subcompartments, is an oversimplification. Overlapping or altered enzyme distributions are common, and different cell types or organisms frequently present unique Golgi organizations43–47. Thus, Golgi heterogeneity, coupled with an incomplete understanding of the mechanisms that regulate membrane trafficking and sub-Golgi enzyme targeting, complicate the standard definitions of Golgi compartmentalization.
Box 2. Distribution of glycan processing enzymes across a generic Golgi apparatus.
Transitional endoplasmic reticulum (ER) and cis-Golgi compartments contain enzymes that initiate O-linked glycosylation and mannosidases for trimming high-mannose N-linked glycans. Medial compartments contain enzymes that branch O-linked glycans and that initiate complex N-linked glycan formation, branching and core fucosylation9,74. The N-linked glycans on glycoproteins that achieve this level of processing become resistant to removal by endoglycosidase H, and studies of trafficking frequently rely on this characteristic to report the transit of a protein through the Golgi. Trans compartments elaborate additional branching and capping reactions (galactosylation, sialylation, sulphation and external fucosylation) on complex N-linked and O-linked antennae42. Beyond the trans-Golgi, capping reactions are continued in the trans-Golgi network (TGN), a tubulovesicular compartment that stages glycoprotein cargoes for secretion or sorting to specific cell surface or subcellular membrane domains. Bulk flow of Golgi contents from cis-to-trans is counterbalanced by retrograde transport that returns resident Golgi proteins to appropriate cisterna61. Glycoprotein substrates carrying appropriate glycan acceptors, glycosyltransferases possessing appropriate enzymatic activities, and nucleotide sugar transporters of the appropriate specificities must all be brought together within the same functional unit of the Golgi apparatus in order to achieve glycan maturation.
It is easy to envision that alterations in the glycan assembly line could affect glycan expression. However, it is not always possible to predict the precise effects of altered Golgi morphology on glycan production. Pharmacologic interventions that disperse the Golgi stack produce glycosylation changes that range from relatively minor glycan modifications (after low doses of nocodozole) to extreme under-processing of N-linked glycans (after brefeldin A administration)48–53. More subtle changes to the Golgi architecture — such as changes that cells might routinely use to modulate their glycan expression — have not been investigated in any significant detail in terms of their impact on the cellular glycome.
Current models of Golgi trafficking offer many intriguing candidates for proteins that may influence cellular glycan expression. Many excellent reviews have presented the evolution of evidence supporting and contradicting the three major models for Golgi trafficking — cisternal maturation, vesicular transport and rapid partitioning — and have also discussed several alternative models54–61. Each of these models makes specific predictions regarding the mechanisms by which glycan biosynthetic enzymes are brought into contact with their substrates (BOX 3). From a glycomic viewpoint, the most important consideration is whether these models can account for the diversity of cellular glycans and whether they accommodate the known biochemical and localization characteristics of the glycan processing machinery.
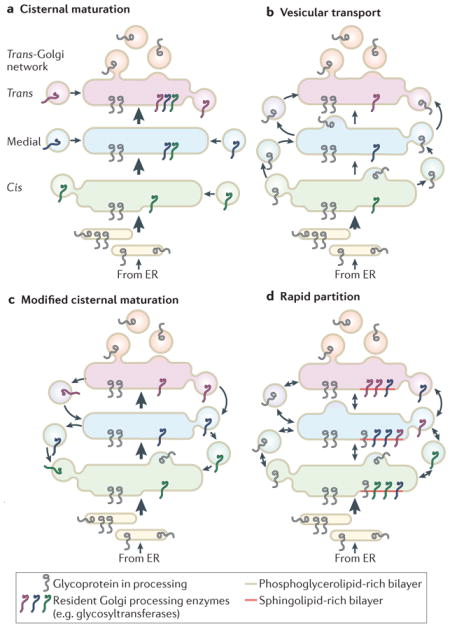
Box 3. Models of Golgi trafficking in animal cells.
The original cisternal maturation model proposed that glycoproteins exit the endoplasmic reticulum (ER) in vesicles or vesiculotubular structures that coalesce into cis-cisternae, which subsequently receive enzymes that function in the early stages of glycan processing. Cisternal maturation would then be achieved by the sequential delivery of enzymes responsible for successively terminal glycan modifications. Ongoing delivery of new ER cargo vesicles would drive forward the translocation of maturing cisternae, which would eventually bud transport vesicles at the trans-Golgi146 (part a of the figure).
The vesicular transport model supposes the existence of stable Golgi cisternae that possess characteristic sets of glycan processing enzymes58. Glycoprotein cargos are moved by targeted anterograde vesicular transport from early to late cisternae (part b of the figure). However, the vesicular transport model is inconsistent with the enrichment of glycosyltransferases in transport vesicles and with the trafficking of macromolecular complexes that are too large to fit within vesicles54,60. Subsequent reassessments of the cisternal maturation model introduced retrograde transport of glycan processing enzymes as a mechanism to retrieve processing capacity into appropriate cisternae Both the cisternal maturation and vesicular transport models share the assumption that transported proteins progress through the Golgi as a wavefront. However, photobleaching measurements of glycoprotein progression through the Golgi in live cells have recently challenged this conveyer belt assumption by demonstrating that product disappears uniformly from all cisternae and that new protein arriving at the Golgi quickly accesses all cisternae63 (part c of the figure). These results have led to the formulation of the rapid partitioning model, which proposes a major role for Golgi lipid composition in establishing and regulating the polarity of the organelle and the distribution of its resident proteins147,148. The rapid partitioning model proposes the coexistence of two distinct lipid domains in each stack of the Golgi, one enriched in sphingolipid and the other enriched in phosphoglycerolipid. The phosphoglycerolipid domains carry out transport functions. The sphingolipid domains are home to Golgi resident proteins, such as glycan processing enzymes that may partition into these domains based on their sphingolipid composition62,64. Glycoprotein processing and transport occur as substrate proteins partition between these two domains. Although the central tenants of the major models are mutually exclusive, many of the proposed mechanisms are not, and the most useful conceptualization of Golgi trafficking allows for the simultaneous existence of aspects of each model (part d of the figure).
Compartmentalization of Golgi glycosylation enzymes
Several mechanisms have been proposed for the targeting and capturing of specific glycosylation enzymes within Golgi sub-compartments that ensures orderly glycan processing. The biophysical characteristics of the transmembrane domains of some processing enzymes may stabilize their distribution within membrane domains that possess specific ratios of sphingolipids, glycosphingolipids and phosphoglycerolipids62–64. It is not currently clear whether sufficient resolution exists in the matching of transmembrane domains with lipid micro-environments to account for specific cisternal sorting or whether this matching serves primarily as a means of excluding enzymes from transport to the cell surface. Essentially all Golgi glycosyltransferases are type 2 transmembrane proteins with very small NH2-terminal cytoplasmic domains. In yeast, a consensus sequence in the NH2-terminal cytoplasmic tail that confers binding of vacuolar protein sorting 74 (Vps74) has been identified in a subset of glycosyltransferases; in turn, this sequence interacts with coatomer protein complex I (COPI), suggesting a mechanism for segregating transferases into transport vesicles65,66. Golgi phosphoprotein 3 (GOLPH3) has been identified as the animal homologue of Vps74, but the yeast consensus sequence does not appear in the cytoplasmic tails of any human glycosyltransferase66, and little evidence supports a dominant role for the cytoplasmic tail in localization of animal glycosyltransferases. However, biochemical, mutagenesis and domain-swapping studies have demonstrated that amino acid sequences within the cytoplasmic, transmembrane and stalk domains of individual transferases, as well as disulphide bond formation between the lumenal domains of glycosyltransferase monomers, can contribute to targeting67–72.
In the absence of a single clear retention or sorting mechanism for glycosyltransferases, it is reasonable to assume that these enzymes probably use a combination of multiple, weak sorting signals for localization73,74. The kin recognition and oligomerization model, which was originally based on co-immunoprecipitation and mis-localization studies, proposed that glycosyltransferases form homo-oligomers with each other and hetero-oligomers with functionally related enzymes75. As larger aggregates are formed, they reach a size at which they are excluded from transport vesicles, thus stabilizing their localization within a specific cisterna. In the context of the vesicular transport model, the aggregation hypothesis was useful. However, in the cisternal maturation and rapid diffusion models, localization by aggregation is problematic, as it does not allow for dynamic changes in cisternal enzyme content. Nonetheless, elegant recent studies using bi-molecular fluorescence complementation have demonstrated that specific transferases form homodimers and heterodimers in living cells76. The organization of transferases into biosynthetic complexes has also been described for N-linked glycan precursor biosynthesis, yeast mannan extension and glycosaminoglycan and glycosphingolipid processing, but not for mucin-type O-linked glycosylation74,77–79. Oligomeric organization is thought to confer increased efficiency, as the product of one enzyme is immediately accessible as substrate for the next. Further work will be required to determine the extent to which glycosyltransferase localization and oligomerization are useful mechanisms for regulating cellular glycomic composition.
Golgi trafficking sculpts glycan profiles
Extensive testing of the three major models of Golgi organizational dynamics has driven substantial progress towards identifying molecular components of the Golgi trafficking machinery. Key proteins involved in intra-Golgi trafficking include vesicle COPI, tethering factors of the Golgi matrix (golgins and multiprotein complexes), small GTPases (the RAB family, ADP-ribosylation factor (ARF) and ARF-like GTPases, and associated GTPase activating proteins (GAPs) and guanine exchange factors (GEFs)) and the membrane fusion machinery (NSF (N-ethylmaleimide-sensitive factor), SNAP (soluble NSF attachment protein) and vesicle- and target-SNAREs (SNAP receptors)) (FIG. 2a–c). Among these components, the conserved oligomeric Golgi complex (COG complex) has been directly implicated in the regulation of cellular glycan profiles (FIG. 2d). The COG complex is composed of eight highly conserved protein subunits. To date, mutations in three human COG subunits (COG1, COG7 and COG8) have been identified, and each causes altered protein glycosylation and severe multi-systemic developmental defects, forming a subgroup of a growing class of inherited diseases known as congenital disorders of glycosylation (CDGs)80. Fibroblasts from these patients with CDG have proven to be useful experimental tools, shedding considerable light on the organization and function of the COG complex61,81–84. Knockdown of COG components in wild-type cells identified a set of proteins comprised of Golgi glycosylation enzymes and other related proteins (Golgi mannosidase II, four glycosyltransferases, the Golgi SNARE protein syntaxin 5 and several golgins). These proteins are collectively termed GEARs, and their stability and localization requires an intact COG complex10,61,85. Experimentally validated interactions between these GEARs and COG subunits support the hypothesis that Golgi vesicle tethering may occur in two stages: initial capture by golgins followed by COG-mediated stabilization that enhances vesicle association with the fusion machinery61. Thus, the COG complex may drive retrograde transport steps to completion and may also provide an opportunity for a final quality check before the commitment to fusion. Reduced function of the COG complex would be predicted to decrease retrograde transport, leading to mislocalization and degradation of the GEARs that mediate glycan processing.
Figure 2. Control points for Golgi trafficking and cellular glycomic diversity.

Intra-Golgi transport mechanisms provide regulatory points for glycome modulation. Two idealized Golgi compartments are depicted. Guanine nucleotide exchange cycles acting on small GTPases regulate trafficking steps important for glycosylation. a | ADP ribosylation factor 1 (ARF1) activation recruits coatomer protein complex I (COPI) coats to nascent transport vesicles. b,c | Golgin interactions with cisternal and vesicular GTPases influence vesicle capture150,151. At least 15 small GTPases of the RAB family and three of the ARF and ARF-like (ARL) family are associated with specific cisternae, vesicles and tethering factors56,152. Golgin tethering factors exist as transmembrane or peripheral membrane proteins that are retained at Golgi cisternae through binding interactions with myristoylated Golgi reassembly-stacking protein of 55 kDa (GRASP55; also known as GRS2) or GRASP65 (also known as GRS1) or small GTPases (prenylated RABs, acylated ARF1 or ARL1). d | The conserved oligomeric Golgi (COG) complex, a multiprotein-tethering complex, interacts with a subset of proteins called GEARs (including golgins), facilitating the fusion of appropriately tethered transport vesicles and modulating the stability of processing enzymes. e,f,g | Sphingolipid and cholesterol content, as well as glycosphingolipid complexity (that is, glycan size and charge) increase from early to late Golgi148,153. Glycosyltransferases catalysing sequential reaction sets may congregate into selective Golgi processing domains on the basis of preferences for specific lipid content imposed by the biophysical characteristics of their transmembrane domains. Therefore, altered lipid biosynthesis may affect glycosylation by contracting or expanding facilitative lipid domains within Golgi cisternae. t SNARE, target soluble N-ethylmaleimide-sensitive factor attachment protein receptor; v-SNARE, vesicle SNARE.
The major models for Golgi trafficking universally require a concerted process for retention and partitioning of glycosylation enzymes across the Golgi stacks in the face of considerable anterograde flow. The glycan structures delivered to the cell surface should reflect the length of time necessary for maturation within a given cisterna, which, in turn, should be coupled to antero-grade and retrograde transport efficiency. However, there is currently no reason to suspect that these transport events occur at constant rates across the organelle. Differential control of intra-organellar transport may provide cells with regulatory points for expanding, contracting or otherwise shifting their glycome. These regulatory points may include COPI coat dynamics, COG complex availability, RAB protein cycling, golgin expression and lipid biogenesis (FIG. 2a–g). Furthermore, a handful of paradigms have been described in which cellular responses to external stimuli affect Golgi form and function, presumably by altering trafficking. For example, administration of epithelial growth factor (EGF) and platelet-derived growth factor (PDGF) to HeLa cells activates a Golgi-localized SRC kinase that induces a redistribution of the enzymes responsible for initiating O-linked glycosylation from the cis-Golgi to the ER, where they drive increased mucin glycosylation86. Other Tyr and Ser/Thr kinases have recently been implicated as regulators of Golgi morphology and, in some cases, protein glycosylation87–90. These emerging paradigms, in which growth factors, cytokines, chemokines and other signalling molecules stimulate changes in Golgi dynamics, promise to reveal stronger mechanistic links between cell signalling and glycomic diversity.
Protein domain-specific glycosylation
Ubiquitous forms of N- and mucin-type peptide-O-GalNAc modifications have been extensively studied for decades. In addition, the selective modification of N-glycans on restricted families of proteins has been recognized for many years (for example, selective polysialylation of N-glycans on neural cell adhesion molecule 1 (NCAM1)91 and a handful of other proteins, or GlcNAc-1-PO4 addition to terminal Man residues on lysosomal enzymes5). Unconventional glycan additions have also been described for a small number of proteins21–23. An emerging field of glycobiology has recently focused on the selective glycosylation of distinct protein domains with specific classes of glycan structures (TABLE 1; BOX 1). These novel forms of glycosylation have been shown to have crucial roles in modulating biological recognition events in development and physiology.
The first of these domain-specific modifications was identified on the Cys-rich EGF domains of the cell surface transmembrane signalling proteins of the Notch family, where the glycans were found to modulate Notch–ligand interactions in developmental cell fate decisions92. Subsequent studies identified other protein domain-specific modifications, including modifications of thrombospondin type 1 repeats (TSRs) and other selective protein modifications such as those found on the mucin-type domains of α-dystroglycan93–97 (TABLE 1; BOX 1). In each case, initial peptide modification occurs in the ER, but further extension of the glycan structure continues in the Golgi complex.
EGF domain-specific glycosylation
EGF repeats are Cys-rich domains of ~30–40 amino acids that are found in many cell surface and secreted proteins and are frequently involved in protein–protein interactions98. The Notch family of single-pass, transmembrane receptors has served as a paradigm for studying glycosylation of EGF repeats, but modification of other EGF domain-containing proteins has not been well studied. In contrast with classical O-GalNAc glycosylation, Ser and Thr residues between the second and third Cys residues within EGF domains are modified by O-Fuc addition by the enzyme protein O-fucosyltransferase 1 (POFUT1; also known as OFUT in Drosophila melanogaster)99,100 (TABLE 1). The Fuc residue can be extended by additional glycosyltransferases to form the tetrasaccharide SA-α2,6-Gal-β1,4-GlcNAc-β1,3-Fuc-α1-O-Ser. Pofut1 is essential in mice101, as is Ofut1 in D. melanogaster102, and conditional knockouts of Pofut1 in adult mice revealed roles of O-Fuc modification in haematopoietic homeostasis103. The Fringe family of β3-GlcNAc transferases initiate elongation of glycans on O-Fuc residues and are modulators of Notch activity92.
EGF repeats of Notch have also been found to contain modifications with O-Glc residues added to Ser side chains before the second Cys residue of the EGF repeat by the action of protein O-glucosyltransferase 1 (POGLUT1; also known as O-glucosyltransferase Rumi in D. melanogaster104) (TABLE 1). This glycan can also be extended by the addition of two Xyl residues105,106. Most of the O-Glc consensus sites in mouse NOTCH1 appear to be occupied, but variability exists among cell types and depending on motif locations within the protein107. Poglut1-knockout mice do not survive past day 9.5, whereas heterozygous Poglut1+/− mice develop normally, suggesting an essential role for O-glucosylation in embryonic development108.
A third type of novel glycosylation of EGF repeats was recently described in D. melanogaster, where a single O-GlcNAc residue was found to be added to a Ser/Thr residue following the fifth Cys residue in an EGF domain of the extracellular matrix protein Dumpy109 and some EGF domains of NOTCH1 (REF. 110) (TABLE 1). Extension of the O-GlcNAc residue has not been detected. Previous studies had identified O-GlcNAc linkages exclusively on a large collection of cytosolic and nuclear proteins6, but the detection of EGF domain-specific O-GlcNAc modification was the first example of such a linkage on an extra-cellular protein109. O-GlcNAc addition to extracellular proteins is catalysed through the action of a recently identified protein called EGF domain-specific O-linked GlcNAc transferase (EOGT)109. D. melanogaster Eogt mutants exhibit aberrant cell–matrix interactions and are embryonic lethal in a Notch-independent manner109. Mammalian EOGT orthologues have also been identified. In summary, EGF domains can act as acceptors of at least three unique types of glycosylation that all have crucial roles in metazoan development.
TSR domain-specific glycosylation
Similar to EGF repeats, TSR domains are found in numerous cell surface and secreted proteins as tandem repeats of 50–60 amino acids with disulphide bonds formed by six conserved Cys residues. Two forms of domain-specific glycosylation are found in TSRs that are distinct from those found in EGF domains (TABLE 1). Similar to EGF domains, O-linked Fuc can be added to Ser/Thr residues of TSRs by the action of POFUT2 (REFS 111,112), which is a distantly related homologue of the EGF repeat-modifying POFUT1 protein113, and the O-Fuc is extended by the addition of a Glc residue through the enzymatic action of β-3-glycosyltransferase-like (B3GALTL)114,115. RNA interference knockdown of Pofut2 transcripts or mutagenesis of the acceptor sequence in a model TSR-containing protein resulted in reduced secretion, suggesting a functional role for this modification116. Recently, mouse knockouts in the Pofut2 locus revealed several developmental defects in epithelial to mesenchymal transition (EMT), as well as defects in cell localization and patterning117.
The second type of unique TSR glycosylation is the addition of Man to a Trp residue through a C-C bond (C-mannosylation)111. The C-Man residue is added to the TSR co-translationally, using dolichol-phosphate-Man as the sugar donor, before protein folding118,119. Mutagenesis to replace Trp residues within a model TSR-containing protein suggests a role for C-mannosylation in the control of protein secretion120, but the mannosyltransferase that creates this structure has yet to be identified.
Protein-selective glycosylation of dystroglycan
Finally, a distinct class of protein domain-specific glycosylation that contributes to cell–matrix interactions has been found predominantly on the cell surface protein α-dystroglycan (αDG), although other substrates may exist. Dystroglycan consists of two subunits, α and β, which are part of the membrane-spanning dystrophin glycoprotein complex that links the extracellular matrix with the cellular cytoskeleton. The α subunit is comprised of globular N- and C-terminal domains containing N-glycosylation sites and an extended central domain rich in classical O-GalNAc mucin-like structures, as well as glycans initiated with an O-Man linkage121 that are essential for binding of dystroglycan with its ligands (laminin, agrin, perlecan and neurexin) or to viruses121. Defects in O-Man addition and extension on αDG lead to several forms of congenital muscular dystrophy collectively known as dystroglycanopathies95.
The initial synthesis of the O-Man linkage is catalysed by protein O-mannosyltransferase 1 (POMT1) and POMT2, and the structure is extended by protein O-linked-Man β-1,2-N-acetylglucosaminyltransferase 1 (POMGNT1), which adds a β1,2-GlcNAc onto the O-Man core (TABLE 1). The most predominant O-Man structures of rabbit skeletal muscle and mouse brain αDG are linear tetrasaccharides containing terminal Gal and SA residues122,123, and defects in Pomt1, Pomt2 or Pomgnt1 cause loss of laminin binding and subsequent dystrogly-canopathies. However, the linear tetrasaccharide structure is not necessary for αDG ligand binding124.
The products of several additional genes, including LARGE1, LARGE2 (also known as GYLTL1B), fukutin (FKTN) and fukutin-related protein (FKRP), contribute to O-Man glycan modifications on αDG. Defects in these genes result in underglycosylation of αDG, disrupted ligand interactions, reduced stability of muscle fibres, disrupted basement membrane, altered neuronal migration and characteristic dystroglycanopathies121,125. However, the exact functions of these gene products remain unclear. Several lines of evidence indicate that functional O-Man structures are comprised of large, branched glycans that have resisted complete structural characterization (BOX 1). Mass spectrometry and NMR have detected a phosphodiester linkage to an unknown substituent at the 6-hydroxyl position of the core O-Man residue on αDG126, but the identity of the responsible phosphotransferase or kinase is currently unknown. In addition, LARGE1 has recently been shown to contain two glycosyltransferase domains that catalyse the extension of a novel (-3-Xyl-α1,3-GlcA-β1-) homopolymer in vitro127. Although the nature of the bridging moiety that might link the proteoglycan-like homopolymer and the O-Man core is still unclear, the size of this product of the glycosyltransferase activity of LARGE1 is consistent with the high molecular weight of the laminin binding forms of αDG isolated from natural sources. Recent work with αDG-null cells and tissues also revealed that there is an approximately equal abundance of O-Man oligosaccharides in wild-type and αDG-null animals122, and overexpression of LARGE1 in these cells was shown to restore laminin binding128. Therefore, other proteins besides αDG also carry O-Man structures, providing additional targets for understanding the role of LARGE1 in laminin binding.
Protein domain-specific glycosylation is an emerging field of study with great potential for elucidating important biological recognition functions. As many glycosyltransferase-related coding regions in animal systems remain uncharacterized, we anticipate that additional classes of protein glycosylation will be characterized in the future that have key regulatory roles in biological systems.
Glycan profiling and global regulation
The diversity of glycan structures in animal systems presents a huge analytical challenge for determining detailed glycan structural profiles in complex biological systems16. As glycan structures are commonly altered in response to biological signalling pathways, there is a major effort to develop broad analytical strategies that identify glycan signatures for cellular transitions129 or biomarkers for disease states130,131. In addition, detailed snapshots of glycan structural diversity can provide insights into potential biological recognition events and regulatory mechanisms for glycan biosynthesis12. Limitations in structural analysis depend on the glycan class, the complexities of the sample workup and the analytical tools that are available for structural inquiry132.
Numerous approaches are now available for highly sensitive, broad N-glycan analysis16. These approaches include mass spectrometry analysis directly after enzymatic release of the glycan structures; methods that employ front-end chromatographic separations before mass spectrometry analysis; methods that employ chromatographic separation and N-glycan identification on the basis of known elution positions of standards; and the analysis of glycan structures on the basis of lectin or antibody binding. Similar approaches are also available for O-glycan and glycolipid analyses, but chemical release rather than enzymatic release methods are generally used before analysis133. Recent advances have also provided direct analysis of intact glycolipid structures in complex samples. However, few methods have been developed for low sample consumption and for high-throughput analysis that can tease apart complex mixtures of structural isomers. Moreover, although numerous data sets are now available for glycan structures from various mammalian cell and tissue sources, challenges still exist in linking glycan structures to the corresponding peptide backbones.
A complementary strategy for glycan structural analysis is the use of lectin- and antibody-based micro-arrays134. In this analytical format, target proteins are captured with protein-specific antibodies and then their glycan structures are analysed using lectins or antibodies. This strategy has a particular benefit, as it enables high-throughput diagnostic profiling of glycans on distinct protein backbones. Multiple platforms for capture of target glycoproteins and glycan detection are available135,136. However, the benefits of this high-throughput approach are tempered by the limited ability of lectins and antibodies to distinguish fine structural details on glycans.
The availability of high-throughput platforms for glycan profiling has provided unique insights into regulated glycan expression in vertebrate organisms and in human disease. Numerous efforts are underway to identify glycans or glycoproteins as cancer biomarkers131,137. Profiling of a broad range of glycan structures in a high-throughput platform is also being performed in combination with human genome-wide association studies to examine the regulation of glycan expression in human populations138,139. Correlations between glycan profiles and gene expression data are also emerging as an approach to assess mechanisms of glycan regulation in animal systems11; in these studies, complex sets of glycan and transcript data are being mined to examine the relative contribution of transcriptional and post-transcriptional control of glycosyltransferases12. Thus, the dynamic changes in glycomic diversity are slowly yielding to structural analysis, providing opportunities to delineate the fine details of glycan structural changes that occur in complex biological systems.
Conclusions
Contemporary studies of protein glycosylation are revealing numerous examples where these post-translational modifications have essential roles in biological recognition events. Glycans have crucial functions throughout the cell, from the cytosol and secretory compartments to the cell surface and the extracellular space. Conserved contributions of N-glycan structures in chaperone interactions and protein quality control are initiated co-translationally, and glycan processing continues throughout the dynamic collection of secretory pathway compartments that lead to the cell surface. The complex assortment of glycosylation enzymes comprises an intricate assembly line for glycan maturation from the ER through the Golgi. Notably, the localization, dynamics, interactions and regulation of glycosylation enzymes within these compartments, as well as how glycosylation enzymes compete for the same substrates, remain active areas of study. Of the many roles that glycans have at the cell surface, emerging paradigms have highlighted the importance of protein domain-specific glycosylation in facilitating or modulating biological recognition events.
The diversity of glycan structures clearly provides an additional level of information content in biological systems, but the challenge for the future lies in identifying the critical contexts in which glycan functions contribute to biological regulation within the bewildering array of heterogeneous glycan structures. An essential requirement for future glycosylation studies is the ability to obtain detailed profiles of glycan diversity in various biological contexts. Recent advances in analytical strategies are beginning to provide the necessary breadth, depth and sensitivity of analysis to define the full spectrum of glycan complexity. Continued high-throughput adaptations of these approaches will generate structural data sets that will be essential for revealing the mechanisms that regulate glycan biosynthetic pathways, defining unique glycan signatures for disease states and providing correlations with biological functions, thereby increasing our ability to decode the many functions of glycoprotein glycans in complex biological systems.
Acknowledgments
This work was supported by US National Institutes of Health (NIH) grants from the National Center for Research Resources (P41RR005351 and P41RR018502) and the National Institute of General Medical Sciences (P41GM103390 and P41GM103490) to J. Prestegard and J. M. Pierce and NIH grants R01GM047533 and R01DK075322 to K.W.M., as well as P01GM085354 and R01GM072839 to M.T.
Glossary
- Lectins
Carbohydrate binding proteins that are involved in various biological recognition phenomena
- Sialic acid
(SA: also known as neuraminic acid (Neu)). SAs are a family of nine-carbon monosaccharides with a carboxylic acid at the C1 position and a glycerol side chain at C7-C9. Two SAs predominate in vertebrates, with either an N-acetyl group (NeuAc) or an N-glycolyl group (NeuGc) at C5. Humans do not make NeuGc, but can obtain it from their diet and incorporate it into glycoconjugates
- Lipid-linked oligosaccharide
(LLO). Extended carbohydrate structure attached through a phosphodiester linkage to a polyisoprenoid lipid, usually dolichol. N-linked glycan precursor structures are commonly assembled as a lipid-linked intermediate before transfer to a polypeptide side chain
- Oligosaccharyltransferase
(OST). A multi-enzyme complex (or single subunit in bacteria) in the endoplasmic reticulum membrane that transfers glycan structures from a lipid-linked oligosaccharide precursor to acceptor sequences on nascent polypeptides
- Acceptor peptide sequons
Short amino acid sequences on glycan-acceptor polypeptide chains that are recognized by glycosyltransferases prior to glycosylation
- Calnexin
Integral membrane endoplasmic reticulum lectin that recognizes GlcMan9GlcNAc2 glycans on early glycoprotein folding intermediates and, in collaboration with an associated thiol oxidoreductase, ERp57, aids in protein folding as a part of endoplasmic reticulum quality control
- Calreticulin
A soluble lectin in the lumen of the endoplasmic reticulum that contains a K-D-E-L endoplasmic reticulum retrieval sequence and, in a similar way to calnexin, binds to GlcMan9GlcNAc2 glycoprotein folding intermediates to facilitate protein folding and quality control
- ER-associated degradation
(ERAD). A protein quality control pathway in which misfolded lumenal or integral membrane proteins are recognized (often through trimmed glycan structures) for disposal by translocation into the cytosol for proteasomal degradation
- Nocodozole
A pharmacological agent that blocks mitosis by binding to tubulin and inhibiting microtubule polymerization. Inhibition of microtubule polymerization also causes the dispersal of well-organized Golgi stacks into smaller units distributed throughout the cytoplasm
- Brefeldin A
A fungally derived antibiotic that inhibits the guanine nucleotide exchange factor (GEF) responsible for activating ARF1 GTPase. Activation of ARF1 recruits coatomer protein complex I (COPI) to Golgi membranes to form vesicles. In the absence of Golgi vesicle formation, cis and medial cisternae fuse with the endoplasmic reticulum and the trans and trans-Golgi network components disperse into a drug-induced entity called the brefeldin compartment
- Cisternal maturation
An early model of Golgi trafficking that proposed that Golgi cisternae formed from the fusion of vesicles leaving the endoplasmic reticulum and were subsequently matured by the import of appropriate processing enzymes as the cisternae were pushed forward towards the trans face. The original cisternal maturation model had its groundings in extensive electron microscopic observations of Golgi morphology
- Vesicular transport
A model for Golgi trafficking, which proposed that cisternae are stable structures and that cargo proteins move between cisternae in transport vesicles that are targeted to specific Golgi domains
- Rapid partitioning
A model for Golgi trafficking, which proposes that cargo proteins can exit the Golgi in vesicles arising from all cisternae and that new protein arriving at the Golgi rapidly gains access to the entire apparatus. Distinct transport and processing domains within each Golgi cistern are defined by their lipid content, which varies systematically from cis-to-trans and may provide favourable environments for cisternae-specific subsets of glycan processing enzymes
- Glycosphingolipids
Glycoconjugates comprised of a ceramide lipid (Cer) carrying a glycan headgroup. The glycan is usually initiated by Glc, although GalCer is an important component of some cells. GlcCer is elongated to generate four major types of neutral cores in mammalian tissues (ganglio-, globo-, lacto- and neolacto-series), each of which can be capped and branched to produce additional structural diversity
- Type 2 transmembrane proteins
Single pass transmembrane proteins with their amino terminus oriented towards the cytosol and their carboxyl terminus facing the lumen of the secretory pathway or cell exterior
- Vacuolar protein sorting 74
(Vps74). A yeast protein identified as having a vacuolar protein sorting function. Yeast vps74 mutants are deficient in intra-Golgi transport and exhibit mis-localized glycosyltransferases. Golgi phosphoprotein 3 (GOLPH3) is the mammalian homologue of Vps74
- Coatomer protein complex I
(COPI). A protein complex that is recruited to membranes by ARF (ADP-ribosylation factor) GTPases and that mediates intra-Golgi and Golgi-to- endoplasmic reticulum retrograde transport
- Mucin-type O-linked glycosylation
A form of protein glycosylation initiated by the addition of a GalNAc residue to protein Ser or Thr side chains and extended and branched with other complex termini. It is commonly found on highly glycosylated mucin glycoproteins, but also on many non-mucin polypeptides
- Golgins
A family of Golgi tethering factors characterized by their extended coiled-coil domains. Individual family members exhibit specific sub-Golgi localizations, GTPase interactions and effector activities. The golgins form long filaments that emanate from the cytoplasmic face of Golgi cisternae, providing access to transport vesicles in the near environment. Some golgins possess multiple binding sites for RAB proteins, suggesting that they capture transport vesicles carrying specific RAB proteins
- Conserved oligomeric Golgi complex
(COG complex). A multiprotein tethering factor comprised of eight protein subunits. The complex facilitates Golgi retrograde transport and glyco-syltransferase localization
- Congenital disorders of glycosylation
(CDGs). A growing group of recessive human disorders characterized by altered protein glycosylation. In type 1 CDGs, the number of N-linked glycans added to a protein is affected. In type 2 CDGs, the nature of the glycan on a glycoprotein is changed. Most CDGs arise from partial loss-of-function mutations and are diagnosed clinically by altered serum protein glycosylation
- Retrograde transport
The movement of vesicles containing Golgi-resident proteins (glycosyltransferases and other processing enzymes) from late to early cisternae. Retrograde transport provides a mechanism for retrieving enzymes that are displaced forward by anterograde flow or cisternal maturation
- Anterograde flow
The bulk movement of cisternal contents and cargo proteins from early to late Golgi compartments
- EGF domains
Protein motifs comprised of ~30–40 amino acids, including six Cys residues forming three characteristic disulphide bonds, and a mainly β-sheet structure, found in all ERBB-binding growth factors and many other cell surface and extracellular matrix proteins
- Thrombospondin type 1 repeats
(TSRs). Protein domains of ~50 amino acids. They are rich in Cys residues and are comprised of three antiparallel β-strands along with regions without a secondary structure. TSRs are found in matrix and transmembrane proteins with functions in matrix organization, cell–cell interactions and cell guidance
- Dystroglycanopathies
A clinically heterogeneous collection of muscular dystrophies that are associated with aberrant glycosylation of α-dystroglycan, which is a component of the multiprotein dystrophin glycoprotein complex that bridges the extracellular matrix, plasma membrane and cytoskeleton
Footnotes
Competing interests statement
The authors declare no competing financial interests.
References
- 1.Varki A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haltiwanger RS, Lowe JB. Role of glycosylation in development. Annu Rev Biochem. 2004;73:491–537. doi: 10.1146/annurev.biochem.73.011303.074043. [DOI] [PubMed] [Google Scholar]
- 3.Dennis JW, Lau KS, Demetriou M, Nabi IR. Adaptive regulation at the cell surface by N-glycosylation. Traffic. 2009;10:1569–1578. doi: 10.1111/j.1600-0854.2009.00981.x. [DOI] [PubMed] [Google Scholar]
- 4.Hoseki J, Ushioda R, Nagata K. Mechanism and components of endoplasmic reticulum-associated degradation. J Biochem. 2010;147:19–25. doi: 10.1093/jb/mvp194. [DOI] [PubMed] [Google Scholar]
- 5.Kollmann K, et al. Mannose phosphorylation in health and disease. Eur J Cell Biol. 2010;89:117–123. doi: 10.1016/j.ejcb.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 6.Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu Rev Biochem. 2011;80:825–858. doi: 10.1146/annurev-biochem-060608-102511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cummings RD. The repertoire of glycan determinants in the human glycome. Mol Biosyst. 2009;5:1087–1104. doi: 10.1039/b907931a. [DOI] [PubMed] [Google Scholar]
- 8.Rothman JE, Fine RE. Coated vesicles transport newly synthesized membrane glycoproteins from endoplasmic reticulum to plasma membrane in two successive stages. Proc Natl Acad Sci USA. 1980;77:780–784. doi: 10.1073/pnas.77.2.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roth J. Protein N-glycosylation along the secretory pathway: relationship to organelle topography and function, protein quality control, and cell interactions. Chem Rev. 2002;102:285–303. doi: 10.1021/cr000423j. [DOI] [PubMed] [Google Scholar]
- 10.Peanne R, et al. Differential effects of lobe A and lobe B of the Conserved Oligomeric Golgi complex on the stability of β1,4-galactosyltransferase 1 and α2,6-sialyltransferase 1. Glycobiology. 2011;21:864–876. doi: 10.1093/glycob/cwq176. [DOI] [PubMed] [Google Scholar]
- 11.Nairn AV, Moremen KW. In: Handbook of Glycomics. Cummings R, Pierce JM, editors. Academic Press; 2009. pp. 95–136. [Google Scholar]
- 12.Nairn AV, et al. Regulation of glycan structures in animal tissues: transcript profiling of glycan-related genes. J Biol Chem. 2008;283:17298–17313. doi: 10.1074/jbc.M801964200. Presents transcript analysis paired with glycan structural data to identify correlations between glycan-related gene expression and glycan composition in mouse tissues. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spiro RG. Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology. 2002;12:43R–56R. doi: 10.1093/glycob/12.4.43r. [DOI] [PubMed] [Google Scholar]
- 14.Schachter H. The joys of HexNAc. The synthesis and function of N- and O-glycan branches. Glycoconj J. 2000;17:465–483. doi: 10.1023/a:1011010206774. [DOI] [PubMed] [Google Scholar]
- 15.Schachter H. The ‘yellow brick road’ to branched complex N-glycans. Glycobiology. 1991;1:453–461. doi: 10.1093/glycob/1.5.453. [DOI] [PubMed] [Google Scholar]
- 16.Zaia J. Mass spectrometry and glycomics. Omics. J Integrative Biol. 2010;14:401–418. doi: 10.1089/omi.2009.0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta. 1999;1473:4–8. doi: 10.1016/s0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- 18.Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annu Rev Biochem. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- 19.Lizak C, Gerber S, Numao S, Aebi M, Locher KP. X-ray structure of a bacterial oligosaccharyltransferase. Nature. 2011;474:350–355. doi: 10.1038/nature10151. Reports the first structure of an OST in complex with a peptide substrate and describes a proposed mechanism that is likely to extend to the eukaryotic enzyme. [DOI] [PubMed] [Google Scholar]
- 20.Kelleher DJ, Gilmore R. An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology. 2006;16:47R–62R. doi: 10.1093/glycob/cwj066. [DOI] [PubMed] [Google Scholar]
- 21.Zielinska DF, Gnad F, Wisniewski JR, Mann M. Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell. 2010;141:897–907. doi: 10.1016/j.cell.2010.04.012. Describes the use of lectin-affinity enrichment of glycopeptides followed by proteomics analysis to identify 6,367 mouse glycosylation sites, including several classes of novel acceptor sequons. [DOI] [PubMed] [Google Scholar]
- 22.Schreiner R, Schnabel E, Wieland F. Novel N-glycosylation in eukaryotes: laminin contains the linkage unit β-glucosylasparagine. J Cell Biol. 1994;124:1071–1081. doi: 10.1083/jcb.124.6.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valliere-Douglass JF, et al. Glutamine-linked and non-consensus asparagine-linked oligosaccharides present in human recombinant antibodies define novel protein glycosylation motifs. J Biol Chem. 2010;285:16012–16022. doi: 10.1074/jbc.M109.096412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kelleher DJ, Karaoglu D, Mandon EC, Gilmore R. Oligosaccharyltransferase isoforms that contain different catalytic STT3 subunits have distinct enzymatic properties. Mol Cell. 2003;12:101–111. doi: 10.1016/s1097-2765(03)00243-0. [DOI] [PubMed] [Google Scholar]
- 25.Imperiali B, Hendrickson TL. Asparagine-linked glycosylation: specificity and function of oligosaccharyl transferase. Bioorg Med Chem. 1995;3:1565–1578. doi: 10.1016/0968-0896(95)00142-5. [DOI] [PubMed] [Google Scholar]
- 26.Larkin A, Imperiali B. The expanding horizons of asparagine-linked glycosylation. Biochemistry. 2011;50:4411–4426. doi: 10.1021/bi200346n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwarz F, Aebi M. Mechanisms and principles of N-linked protein glycosylation. Curr Opin Struct Biol. 2011;21:576–582. doi: 10.1016/j.sbi.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 28.Weerapana E, Imperiali B. Asparagine-linked protein glycosylation: from eukaryotic to prokaryotic systems. Glycobiology. 2006;16:91R–101R. doi: 10.1093/glycob/cwj099. [DOI] [PubMed] [Google Scholar]
- 29.Mohorko E, Glockshuber R, Aebi M. Oligosaccharyltransferase: the central enzyme of N-linked protein glycosylation. J Inherit Metab Dis. 2011;34:869–878. doi: 10.1007/s10545-011-9337-1. [DOI] [PubMed] [Google Scholar]
- 30.Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 31.Lederkremer GZ. Glycoprotein folding, quality control and ER-associated degradation. Curr Opin Struct Biol. 2009;19:515–523. doi: 10.1016/j.sbi.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 32.D’Alessio C, Caramelo JJ, Parodi AJ. UDP-GlC:glycoprotein glucosyltransferase-glucosidase II, the ying-yang of the ER quality control. Semin Cell Dev Biol. 2010;21:491–499. doi: 10.1016/j.semcdb.2009.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clerc S, et al. Htm1 protein generates the N-glycan signal for glycoprotein degradation in the endoplasmic reticulum. J Cell Biol. 2009;184:159–172. doi: 10.1083/jcb.200809198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gauss R, Kanehara K, Carvalho P, Ng DT, Aebi M. A complex of pdi1p and the mannosidase htm1p initiates clearance of unfolded glycoproteins from the endoplasmic reticulum. Mol Cell. 2011;42:782–793. doi: 10.1016/j.molcel.2011.04.027. [DOI] [PubMed] [Google Scholar]
- 35.Mast SW, Moremen KW. Family 47α-mannosidases in N-glycan processing. Methods Enzymol. 2006;415:31–46. doi: 10.1016/S0076-6879(06)15003-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moremen KW, Molinari M. N-linked glycan recognition and processing: the molecular basis of endoplasmic reticulum quality control. Curr Opin Struct Biol. 2006;16:592–599. doi: 10.1016/j.sbi.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aebi M, Bernasconi R, Clerc S, Molinari M. N-glycan structures: recognition and processing in the ER. Trends Biochem Sci. 2010;35:74–82. doi: 10.1016/j.tibs.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 38.Xie W, Kanehara K, Sayeed A, Ng DT. Intrinsic conformational determinants signal protein misfolding to the Hrd1/Htm1 endoplasmic reticulum-associated degradation system. Mol Biol Cell. 2009;20:3317–3329. doi: 10.1091/mbc.E09-03-0231. Describes the roles of key N-linked glycan sites that mark structural determinants that are sensitive to the overall folding state of the molecule and mediate targeting of misfolded proteins for ER-associated degradation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanehara K, Xie W, Ng DT. Modularity of the Hrd1 ERAD complex underlies its diverse client range. J Cell Biol. 2010;188:707–716. doi: 10.1083/jcb.200907055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.An HJ, et al. Extensive determination of glycan heterogeneity reveals an unusual abundance of high mannose glycans in enriched plasma membranes of human embryonic stem cells. Mol Cell Proteomics. 2012;11:M111.010660. doi: 10.1074/mcp.M111.010660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rabouille C, et al. Mapping the distribution of Golgi enzymes involved in the construction of complex oligosaccharides. J Cell Sci. 1995;108:1617–1627. doi: 10.1242/jcs.108.4.1617. [DOI] [PubMed] [Google Scholar]
- 42.Stanley P. Golgi glycosylation. Cold Spring Harb Perspect Biol. 2011;3:a005199. doi: 10.1101/cshperspect.a005199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mogelsvang S, Marsh BJ, Ladinsky MS, Howell KE. Predicting function from structure: 3D structure studies of the mammalian Golgi complex. Traffic. 2004;5:338–345. doi: 10.1111/j.1398-9219.2004.00186.x. [DOI] [PubMed] [Google Scholar]
- 44.Papanikou E, Glick BS. The yeast Golgi apparatus: insights and mysteries. FEBS Lett. 2009;583:3746–3751. doi: 10.1016/j.febslet.2009.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ripoche J, Link B, Yucel JK, Tokuyasu K, Malhotra V. Location of Golgi membranes with reference to dividing nuclei in syncytial Drosophila embryos. Proc Natl Acad Sci USA. 1994;91:1878–1882. doi: 10.1073/pnas.91.5.1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Velasco A, et al. Cell type-dependent variations in the subcellular distribution of α-mannosidase I and II. J Cell Biol. 1993;122:39–51. doi: 10.1083/jcb.122.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nilsson T, et al. Overlapping distribution of two glycosyltransferases in the Golgi apparatus of HeLa cells. J Cell Biol. 1993;120:5–13. doi: 10.1083/jcb.120.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chou CF, Omary MB. Mitotic arrest with anti-microtubule agents or okadaic acid is associated with increased glycoprotein terminal GlcNAc’s. J Cell Sci. 1994;107:1833–1843. doi: 10.1242/jcs.107.7.1833. [DOI] [PubMed] [Google Scholar]
- 49.Haltiwanger RS, Philipsberg GA. Mitotic arrest with nocodazole induces selective changes in the level of O-linked N-acetylglucosamine and accumulation of incompletely processed N-glycans on proteins from HT29 cells. J Biol Chem. 1997;272:8752–8758. doi: 10.1074/jbc.272.13.8752. [DOI] [PubMed] [Google Scholar]
- 50.Klausner RD, Donaldson JG, Lippincott-Schwartz J. Brefeldin A: insights into the control of membrane traffic and organelle structure. J Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peyroche A, et al. Brefeldin A acts to stabilize an abortive ARF-GDP-Sec7 domain protein complex: involvement of specific residues of the Sec7 domain. Mol Cell. 1999;3:275–285. doi: 10.1016/s1097-2765(00)80455-4. [DOI] [PubMed] [Google Scholar]
- 52.Rogalski AA, Bergmann JE, Singer SJ. Effect of microtubule assembly status on the intracellular processing and surface expression of an integral protein of the plasma membrane. J Cell Biol. 1984;99:1101–1109. doi: 10.1083/jcb.99.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sampath D, Varki A, Freeze HH. The spectrum of incomplete N-linked oligosaccharides synthesized by endothelial cells in the presence of brefeldin A. J Biol Chem. 1992;267:4440–4455. [PubMed] [Google Scholar]
- 54.Balch WE, Dunphy WG, Braell WA, Rothman JE. Reconstitution of the transport of protein between successive compartments of the Golgi measured by the coupled incorporation of N-acetylglucosamine. Cell. 1984;39:405–416. doi: 10.1016/0092-8674(84)90019-9. [DOI] [PubMed] [Google Scholar]
- 55.Glick BS, Luini A. Models for Golgi traffic: a critical assessment. Cold Spring Harb Perspect Biol. 2011;3:a005215. doi: 10.1101/cshperspect.a005215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goud B, Gleeson PA. TGN golgins, Rabs and cytoskeleton: regulating the Golgi trafficking highways. Trends Cell Biol. 2010;20:329–336. doi: 10.1016/j.tcb.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 57.Jackson CL. Mechanisms of transport through the Golgi complex. J Cell Sci. 2009;122:443–452. doi: 10.1242/jcs.032581. [DOI] [PubMed] [Google Scholar]
- 58.Jamieson JD, Palade GE. Intracellular transport of secretory proteins in the pancreatic exocrine cell. 3 Dissociation of intracellular transport from protein synthesis. J Cell Biol. 1968;39:580–588. doi: 10.1083/jcb.39.3.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kartberg F, Elsner M, Froderberg L, Asp L, Nilsson T. Commuting between Golgi cisternae--mind the GAP! Biochim Biophys Acta. 2005;1744:351–363. doi: 10.1016/j.bbamcr.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 60.Marchi F, Leblond CP. Radioautographic characterization of successive compartments along the rough endoplasmic reticulum-Golgi pathway of collagen precursors in foot pad fibroblasts of [3H] proline-injected rats. J Cell Biol. 1984;98:1705–1709. doi: 10.1083/jcb.98.5.1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reynders E, Foulquier F, Annaert W, Matthijs G. How Golgi glycosylation meets and needs trafficking: the case of the COG complex. Glycobiology. 2011;21:853–863. doi: 10.1093/glycob/cwq179. [DOI] [PubMed] [Google Scholar]
- 62.Munro S. An investigation of the role of transmembrane domains in Golgi protein retention. EMBO J. 1995;14:4695–4704. doi: 10.1002/j.1460-2075.1995.tb00151.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Patterson GH, et al. Transport through the Golgi apparatus by rapid partitioning within a two-phase membrane system. Cell. 2008;133:1055–1067. doi: 10.1016/j.cell.2008.04.044. Proposes a model for Golgi trafficking in which cargo proteins are not carried through the Golgi as if on a conveyer belt. Rather, cargo proteins partition into processing or transport domains that possess distinctive lipid compositions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sharpe HJ, Stevens TJ, Munro S. A comprehensive comparison of transmembrane domains reveals organelle-specific properties. Cell. 2010;142:158–169. doi: 10.1016/j.cell.2010.05.037. A tour-de-force bioinformatic analysis of the physicochemical properties of transmembrane domains of organellar proteins from fungi to vertebrates, demonstrating a conserved dichotomy in transmembrane domain properties that distinguish early from late secretory compartments. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schmitz KR, et al. Golgi localization of glycosyltransferases requires a Vps74p oligomer. Dev Cell. 2008;14:523–534. doi: 10.1016/j.devcel.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tu L, Tai WC, Chen L, Banfield DK. Signal-mediated dynamic retention of glycosyltransferases in the Golgi. Science. 2008;321:404–407. doi: 10.1126/science.1159411. [DOI] [PubMed] [Google Scholar]
- 67.Aoki D, Lee N, Yamaguchi N, Dubois C, Fukuda MN. Golgi retention of a trans-Golgi membrane protein, galactosyltransferase, requires cysteine and histidine residues within the membrane-anchoring domain. Proc Natl Acad Sci USA. 1992;89:4319–4323. doi: 10.1073/pnas.89.10.4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Colley KJ. Golgi localization of glycosyltransferases: more questions than answers. Glycobiology. 1997;7:1–13. doi: 10.1093/glycob/7.1.1-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Colley KJ, Lee EU, Paulson JC. The signal anchor and stem regions of the β-galactoside α2,6-sialyltransferase may each act to localize the enzyme to the Golgi apparatus. J Biol Chem. 1992;267:7784–7793. [PubMed] [Google Scholar]
- 70.Fenteany FH, Colley KJ. Multiple signals are required for α2,6-sialyltransferase (ST6Gal I) oligomerization and Golgi localization. J Biol Chem. 2005;280:5423–5429. doi: 10.1074/jbc.M412396200. [DOI] [PubMed] [Google Scholar]
- 71.Nilsson T, Lucocq JM, Mackay D, Warren G. The membrane spanning domain of β-1,4-galactosyltransferase specifies trans Golgi localization. EMBO J. 1991;10:3567–3575. doi: 10.1002/j.1460-2075.1991.tb04923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nilsson T, Rabouille C, Hui N, Watson R, Warren G. The role of the membrane-spanning domain and stalk region of N-acetylglucosaminyltransferase I in retention, kin recognition and structural maintenance of the Golgi apparatus in HeLa cells. J Cell Sci. 1996;109:1975–1989. doi: 10.1242/jcs.109.7.1975. [DOI] [PubMed] [Google Scholar]
- 73.Banfield DK. Mechanisms of protein retention in the Golgi. Cold Spring Harb Perspect Biol. 2011;3:a005264. doi: 10.1101/cshperspect.a005264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tu L, Banfield DK. Localization of Golgi-resident glycosyltransferases. Cell Mol Life Sci. 2010;67:29–41. doi: 10.1007/s00018-009-0126-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nilsson T, Slusarewicz P, Hoe MH, Warren G. Kin recognition. A model for the retention of Golgi enzymes. FEBS Lett. 1993;330:1–4. doi: 10.1016/0014-5793(93)80906-b. [DOI] [PubMed] [Google Scholar]
- 76.Hassinen A, Rivinoja A, Kauppila A, Kellokumpu S. Golgi N-glycosyltransferases form both homo- and heterodimeric enzyme complexes in live cells. J Biol Chem. 2010;285:17771–17777. doi: 10.1074/jbc.M110.103184. Bimolecular fluorescence complementation reveals that key glycosyltransferases form homocomplexes, but also form distinct heterocomplexes with functionally related glycosyl-transferases that are localized to either medial or trans-Golgi compartments. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Daniotti JL, Martina JA, Giraudo CG, Zurita AR, Maccioni HJ. GM3 α2,8-sialyltransferase (GD3 synthase): protein characterization and sub-golgi location in CHO-K1 cells. J Neurochem. 2000;74:1711–1720. doi: 10.1046/j.1471-4159.2000.0741711.x. [DOI] [PubMed] [Google Scholar]
- 78.Giraudo CG, Maccioni HJ. Ganglioside glycosyltransferases organize in distinct multienzyme complexes in CHO-K1 cells. J Biol Chem. 2003;278:40262–40271. doi: 10.1074/jbc.M305455200. [DOI] [PubMed] [Google Scholar]
- 79.Yano H, et al. Distinct functional units of the Golgi complex in Drosophila cells. Proc Natl Acad Sci USA. 2005;102:13467–13472. doi: 10.1073/pnas.0506681102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Freeze HH. Congenital disorders of glycosylation: CDG-I, CDG-II, and beyond. Curr Mol Med. 2007;7:389–396. doi: 10.2174/156652407780831548. [DOI] [PubMed] [Google Scholar]
- 81.Foulquier F. COG defects, birth and rise! Biochim Biophys Acta. 2009;1792:896–902. doi: 10.1016/j.bbadis.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 82.Pokrovskaya ID, et al. Conserved oligomeric Golgi complex specifically regulates the maintenance of Golgi glycosylation machinery. Glycobiology. 2011;21:1554–1569. doi: 10.1093/glycob/cwr028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Smith RD, Lupashin VV. Role of the conserved oligomeric Golgi (COG) complex in protein glycosylation. Carbohydr Res. 2008;343:2024–2031. doi: 10.1016/j.carres.2008.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu X, et al. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nature Med. 2004;10:518–523. doi: 10.1038/nm1041. First demonstration of a human disorder associated with altered Golgi transport. Patient fibroblasts were shown to be deficient in intra-Golgi transport. [DOI] [PubMed] [Google Scholar]
- 85.Oka T, Ungar D, Hughson FM, Krieger M. The COG and COPI complexes interact to control the abundance of GEARs, a subset of Golgi integral membrane proteins. Mol Biol Cell. 2004;15:2423–2435. doi: 10.1091/mbc.E03-09-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gill DJ, Chia J, Senewiratne J, Bard F. Regulation of O-glycosylation through Golgi-to-ER relocation of initiation enzymes. J Cell Biol. 2010;189:843–858. doi: 10.1083/jcb.201003055. SRC-dependent growth factor stimulation drives the relocation of polypeptide GalNAc transferases to earlier secretory compartments, enhancing the production of GalNAc initiated O-linked glycans. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Baas S, et al. Sugar-free frosting, a homolog of SAD kinase, drives neural-specific glycan expression in the Drosophila embryo. Development. 2011;138:553–563. doi: 10.1242/dev.055376. A partial loss-of-function mutation in a neural specific kinase decreases expression of a set of neural-specific glycans, demonstrating a direct link between protein phosphorylation, Golgi architecture and glycan end-products. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Farhan H, et al. MAPK signaling to the early secretory pathway revealed by kinase/phosphatase functional screening. J Cell Biol. 2010;189:997–1011. doi: 10.1083/jcb.200912082. An RNA interference screen for kinases and phosphatases that alter ER and Golgi trafficking identified 122 such proteins, indicating a broad role for cell signaling in sculpting flux through the secretory pathway with subsequent, but not yet demonstrated, glycomic consequences. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lowe M, et al. Cdc2 kinase directly phosphorylates the cis-Golgi matrix protein GM130 and is required for Golgi fragmentation in mitosis. Cell. 1998;94:783–793. doi: 10.1016/s0092-8674(00)81737-7. [DOI] [PubMed] [Google Scholar]
- 90.Muniz M, Alonso M, Hidalgo J, Velasco A. A regulatory role for cAMP-dependent protein kinase in protein traffic along the exocytic route. J Biol Chem. 1996;271:30935–30941. doi: 10.1074/jbc.271.48.30935. [DOI] [PubMed] [Google Scholar]
- 91.Colley KJ. Structural basis for the polysialylation of the neural cell adhesion molecule. Adv Exp Med Biol. 2010;663:111–126. doi: 10.1007/978-1-4419-1170-4_7. [DOI] [PubMed] [Google Scholar]
- 92.Rana NA, Haltiwanger RS. Fringe benefits: functional and structural impacts of O-glycosylation on the extracellular domain of Notch receptors. Curr Opin Struct Biol. 2011;21:583–589. doi: 10.1016/j.sbi.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hanisch FG, Breloy I. Protein-specific glycosylation: signal patches and cis-controlling peptidic elements. Biol Chem. 2009;390:619–626. doi: 10.1515/BC.2009.043. [DOI] [PubMed] [Google Scholar]
- 94.Luther KB, Haltiwanger RS. Role of unusual O-glycans in intercellular signaling. Int J Biochem Cell Biol. 2009;41:1011–1024. doi: 10.1016/j.biocel.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Muntoni F, Torelli S, Wells DJ, Brown SC. Muscular dystrophies due to glycosylation defects: diagnosis and therapeutic strategies. Curr Opin Neurol. 2011;24:437–442. doi: 10.1097/WCO.0b013e32834a95e3. [DOI] [PubMed] [Google Scholar]
- 96.Stanley P, Okajima T. Roles of glycosylation in Notch signaling. Curr Top Dev Biol. 2010;92:131–164. doi: 10.1016/S0070-2153(10)92004-8. [DOI] [PubMed] [Google Scholar]
- 97.Wopereis S, Lefeber DJ, Morava E, Wevers RA. Mechanisms in protein O-glycan biosynthesis and clinical and molecular aspects of protein O-glycan biosynthesis defects: a review. Clin Chem. 2006;52:574–600. doi: 10.1373/clinchem.2005.063040. [DOI] [PubMed] [Google Scholar]
- 98.Sakaidani Y, et al. O-linked-N-acetylglucosamine modification of mammalian Notch receptors by an atypical O-GlcNAc transferase Eogt1. Biochem Biophys Res Commun. 2012;419:14–19. doi: 10.1016/j.bbrc.2012.01.098. The authors identifyied a unique O-GlcNAc transferase, EOGT, involved in the O-GlcNAcylation of extracellular proteins. [DOI] [PubMed] [Google Scholar]
- 99.Shao L, Moloney DJ, Haltiwanger R. Fringe modifies O-fucose on mouse Notch1 at epidermal growth factor-like repeats within the ligand-binding site and the Abruptex region. J Biol Chem. 2003;278:7775–7782. doi: 10.1074/jbc.M212221200. [DOI] [PubMed] [Google Scholar]
- 100.Wang Y, et al. Modification of epidermal growth factor-like repeats with O-fucose. Molecular cloning and expression of a novel GDP-fucose protein O-fucosyltransferase. J Biol Chem. 2001;276:40338–40345. doi: 10.1074/jbc.M107849200. [DOI] [PubMed] [Google Scholar]
- 101.Okajima T, Irvine KD. Regulation of Notch signaling by O-linked fucose. Cell. 2002;111:893–904. doi: 10.1016/s0092-8674(02)01114-5. [DOI] [PubMed] [Google Scholar]
- 102.Shi S, Stanley P. Protein O-fucosyltransferase 1 is an essential component of Notch signaling pathways. Proc Natl Acad Sci USA. 2003;100:5234–5239. doi: 10.1073/pnas.0831126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yao D, et al. Protein O-fucosyltransferase 1 (Pofut1) regulates lymphoid and myeloid homeostasis through modulation of Notch receptor ligand interactions. Blood. 2011;117:5652–5662. doi: 10.1182/blood-2010-12-326074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Acar M, et al. Rumi is a CAP10 domain glycosyltransferase that modifies Notch and is required for Notch signaling. Cell. 2008;132:247–258. doi: 10.1016/j.cell.2007.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sethi MK, et al. Identification of glycosyltransferase 8 family members as xylosyltransferases acting on O-glucosylated notch epidermal growth factor repeats. J Biol Chem. 2010;285:1582–1586. doi: 10.1074/jbc.C109.065409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sethi MK, et al. Molecular cloning of a xylosyltransferase that transfers the second xylose to O-glucosylated epidermal growth factor repeats of notch. J Biol Chem. 2012;287:2739–2748. doi: 10.1074/jbc.M111.302406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rana NA, et al. O-Glucose trisaccharide is present at high but variable stoichiometry at multiple sites on mouse Notch1. J Biol Chem. 2011;286:31623–31637. doi: 10.1074/jbc.M111.268243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fernandez-Valdivia R, et al. Regulation of mammalian Notch signaling and embryonic development by the protein O-glucosyltransferase Rumi. Development. 2011;138:1925–1934. doi: 10.1242/dev.060020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sakaidani Y, et al. O-linked-N-acetylglucosamine on extracellular protein domains mediates epithelial cell-matrix interactions. Nature Commun. 2011;2:583. doi: 10.1038/ncomms1591. [DOI] [PubMed] [Google Scholar]
- 110.Matsuura A, et al. O-linked N-acetylglucosamine is present on the extracellular domain of notch receptors. J Biol Chem. 2008;283:35486–35495. doi: 10.1074/jbc.M806202200. [DOI] [PubMed] [Google Scholar]
- 111.Hofsteenge J, et al. C-mannosylation and O-fucosylation of the thrombospondin type 1 module. J Biol Chem. 2001;276:6485–6498. doi: 10.1074/jbc.M008073200. [DOI] [PubMed] [Google Scholar]
- 112.Luo Y, Koles K, Vorndam W, Haltiwanger RS, Panin VM. Protein O-fucosyltransferase 2 adds O-fucose to thrombospondin type 1 repeats. J Biol Chem. 2006;281:9393–9399. doi: 10.1074/jbc.M511975200. [DOI] [PubMed] [Google Scholar]
- 113.Martinez-Duncker I, Mollicone R, Candelier JJ, Breton C, Oriol R. A new superfamily of protein-O-fucosyltransferases, α2-fucosyltransferases, and α6-fucosyltransferases: phylogeny and identification of conserved peptide motifs. Glycobiology. 2003;13:1C–5C. doi: 10.1093/glycob/cwg113. [DOI] [PubMed] [Google Scholar]
- 114.Kozma K, et al. Identification and characterization of a β1,3-glucosyltransferase that synthesizes the Glc-β1,3-Fuc disaccharide on thrombospondin type 1 repeats. J Biol Chem. 2006;281:36742–36751. doi: 10.1074/jbc.M605912200. [DOI] [PubMed] [Google Scholar]
- 115.Sato T, et al. Molecular cloning and characterization of a novel human β1,3-glucosyltransferase, which is localized at the endoplasmic reticulum and glucosylates O-linked fucosylglycan on thrombospondin type 1 repeat domain. Glycobiology. 2006;16:1194–1206. doi: 10.1093/glycob/cwl035. [DOI] [PubMed] [Google Scholar]
- 116.Ricketts LM, Dlugosz M, Luther KB, Haltiwanger RS, Majerus EM. O-fucosylation is required for ADAMTS13 secretion. J Biol Chem. 2007;282:17014–17023. doi: 10.1074/jbc.M700317200. [DOI] [PubMed] [Google Scholar]
- 117.Du J, et al. O-fucosylation of thrombospondin type 1 repeats restricts epithelial to mesenchymal transition (EMT) and maintains epiblast pluripotency during mouse gastrulation. Dev Biol. 2010;346:25–38. doi: 10.1016/j.ydbio.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Doucey MA, Hess D, Cacan R, Hofsteenge J. Protein C-mannosylation is enzyme-catalysed and uses dolichyl-phosphate-mannose as a precursor. Mol Biol Cell. 1998;9:291–300. doi: 10.1091/mbc.9.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Krieg J, et al. Recognition signal for C-mannosylation of Trp-7 in RNase 2 consists of sequence Trp-x-x-Trp. Mol Biol Cell. 1998;9:301–309. doi: 10.1091/mbc.9.2.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wang LW, Leonhard-Melief C, Haltiwanger RS, Apte SS. Post-translational modification of thrombospondin type-1 repeats in ADAMTS-like 1/punctin-1by C-mannosylation of tryptophan. J Biol Chem. 2009;284:30004–30015. doi: 10.1074/jbc.M109.038059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Barresi R, Campbell KP. Dystroglycan: from biosynthesis to pathogenesis of human disease. J Cell Sci. 2006;119:199–207. doi: 10.1242/jcs.02814. [DOI] [PubMed] [Google Scholar]
- 122.Stalnaker SH, et al. Glycomic analyses of mouse models of congenital muscular dystrophy. J Biol Chem. 2011;286:21180–21190. doi: 10.1074/jbc.M110.203281. Analysis of O-linked glycans from brains of wild-type and knockout mouse models using mass spectrophotometry to determine that POMGNT1 is required for addition of β1,2-linked GlcNAc on O-Man glycans. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Stalnaker SH, et al. Site mapping and characterization of O-glycan structures on α-dystroglycan isolated from rabbit skeletal muscle. J Biol Chem. 2010;285:24882–24891. doi: 10.1074/jbc.M110.126474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Combs AC, Ervasti JM. Enhanced laminin binding by α-dystroglycan after enzymatic deglycosylation. Biochem J. 2005;390:303–309. doi: 10.1042/BJ20050375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hara Y, et al. A dystroglycan mutation associated with limb-girdle muscular dystrophy. N Engl J Med. 2011;364:939–946. doi: 10.1056/NEJMoa1006939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Yoshida-Moriguchi T, et al. O-mannosyl phosphorylation of alpha-dystroglycan is required for laminin binding. Science. 2010;327:88–92. doi: 10.1126/science.1180512. First description of a novel GAG-like structure containing Xyl and GlcA in unknown linkage to an essential extracellular protein that interacts with the extracellular matrix. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Inamori K, et al. Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science. 2012;335:93–96. doi: 10.1126/science.1214115. Reports the bifunctional glycosyltransferase activity of LARGE1 on O-Man-containing glycans of αDG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang Z, Zhang P, Hu H. LARGE expression augments the glycosylation of glycoproteins in addition to α-dystroglycan conferring laminin binding. PLoS ONE. 2011;6:e19080. doi: 10.1371/journal.pone.0019080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Nairn AV, dela Rosa M, Moremen KW. Transcript analysis of stem cells. Methods Enzymol. 2010;479:73–91. doi: 10.1016/S0076-6879(10)79004-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hua S, et al. Comprehensive native glycan profiling with isomer separation and quantitation for the discovery of cancer biomarkers. Analyst. 2011;136:3663–3671. doi: 10.1039/c1an15093f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.An HJ, Kronewitter SR, de Leoz ML, Lebrilla CB. Glycomics and disease markers. Curr Opin Chem Biol. 2009;13:601–607. doi: 10.1016/j.cbpa.2009.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bielik AM, Zaia J. Historical overview of glycoanalysis. Methods Mol Biol. 2010;600:9–30. doi: 10.1007/978-1-60761-454-8_2. [DOI] [PubMed] [Google Scholar]
- 133.Aoki K, et al. The diversity of O-linked glycans expressed during Drosophila melanogaster development reflects stage- and tissue-specific requirements for cell signaling. J Biol Chem. 2008;283:30385–30400. doi: 10.1074/jbc.M804925200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Haab BB. Antibody-lectin sandwich arrays for biomarker and glycobiology studies. Expert Rev Proteom. 2010;7:9–11. doi: 10.1586/epr.09.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Yue T, et al. The prevalence and nature of glycan alterations on specific proteins in pancreatic cancer patients revealed using antibody-lectin sandwich arrays. Mol Cell Proteomics. 2009;8:1697–1707. doi: 10.1074/mcp.M900135-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Li C, et al. Pancreatic cancer serum detection using a lectin/glyco-antibody array method. J Proteome Res. 2009;8:483–492. doi: 10.1021/pr8007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Adamczyk B, Tharmalingam T, Rudd PM. Glycans as cancer biomarkers. Biochim Biophys Acta. 2011 Dec 9; doi: 10.1016/j.bbagen.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 138.Lauc G, et al. Genomics meets glycomics-the first GWAS study of human N-glycome identifies HNF1α as a master regulator of plasma protein fucosylation. PLoS Genet. 2010;6:e1001256. doi: 10.1371/journal.pgen.1001256. Describes the first study that linked high-throughput glycan structural analysis and genome-wide association to identify determinants that control population-specific expression of carbohydrate epitopes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Huffman JE, et al. Polymorphisms in B3GAT1, SLC9A9 and MGAT5 are associated with variation within the human plasma N-glycome of 3533 European adults. Hum Mol Genet. 2011;20:5000–5011. doi: 10.1093/hmg/ddr414. [DOI] [PubMed] [Google Scholar]
- 140.Esko JD, Selleck SB. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem. 2002;71:435–471. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- 141.Wang A, de la Motte C, Lauer M, Hascall V. Hyaluronan matrices in pathobiological processes. FEBS J. 2011;278:1412–1418. doi: 10.1111/j.1742-4658.2011.08069.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tian E, Ten Hagen KG. Recent insights into the biological roles of mucin-type O-glycosylation. Glycoconj J. 2009;26:325–334. doi: 10.1007/s10719-008-9162-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Issoglio FM, Carrizo ME, Romero JM, Curtino JA. Mechanisms of monomeric and dimeric glycogenin autoglucosylation. J Biol Chem. 2012;287:1955–1961. doi: 10.1074/jbc.M111.287813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Schegg B, Hulsmeier AJ, Rutschmann C, Maag C, Hennet T. Core glycosylation of collagen is initiated by two β(1-O)galactosyltransferases. Mol Cell Biol. 2009;29:943–952. doi: 10.1128/MCB.02085-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pontier SM, Schweisguth F. Glycosphingolipids in signaling and development: from liposomes to model organisms. Dev Dyn. 2012;241:92–106. doi: 10.1002/dvdy.22766. [DOI] [PubMed] [Google Scholar]
- 146.Farquhar MG, Palade GE. The Golgi apparatus: 100 years of progress and controversy. Trends Cell Biol. 1998;8:2–10. doi: 10.1016/S0962-8924(97)01187-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Perry RJ, Ridgway ND. Molecular mechanisms and regulation of ceramide transport. Biochim Biophys Acta. 2005;1734:220–234. doi: 10.1016/j.bbalip.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 148.van Meer G, de Kroon AI. Lipid map of the mammalian cell. J Cell Sci. 2011;124:5–8. doi: 10.1242/jcs.071233. [DOI] [PubMed] [Google Scholar]
- 149.Smith MH, Ploegh HL, Weissman JS. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science. 2011;334:1086–1090. doi: 10.1126/science.1209235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Seemann J, Jokitalo E, Pypaert M, Warren G. Matrix proteins can generate the higher order architecture of the Golgi apparatus. Nature. 2000;407:1022–1026. doi: 10.1038/35039538. [DOI] [PubMed] [Google Scholar]
- 151.Slusarewicz P, Nilsson T, Hui N, Watson R, Warren G. Isolation of a matrix that binds medial Golgi enzymes. J Cell Biol. 1994;124:405–413. doi: 10.1083/jcb.124.4.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ramirez IB, Lowe M. Golgins and GRASPs: holding the Golgi together. Semin Cell Dev Biol. 2009;20:770–779. doi: 10.1016/j.semcdb.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 153.Maccioni HJ, Quiroga R, Spessott W. Organization of the synthesis of glycolipid oligosaccharides in the Golgi complex. FEBS Lett. 2011;585:1691–1698. doi: 10.1016/j.febslet.2011.03.030. [DOI] [PubMed] [Google Scholar]
- 154.Gerken TA, et al. Emerging paradigms for the initiation of mucin-type protein O-glycosylation by the polypeptide GalNAc transferase family of glycosyltransferases. J Biol Chem. 2011;286:14493–14507. doi: 10.1074/jbc.M111.218701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gupta R, Brunak S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac Symp Biocomput. 2002;2002:310–322. [PubMed] [Google Scholar]
- 156.Roch C, Kuhn J, Kleesiek K, Gotting C. Differences in gene expression of human xylosyltransferases and determination of acceptor specificities for various proteoglycans. Biochem Biophys Res Commun. 2010;391:685–691. doi: 10.1016/j.bbrc.2009.11.121. [DOI] [PubMed] [Google Scholar]
- 157.Manya H, et al. Regulation of mammalian protein O-mannosylation: preferential amino acid sequence for O-mannose modification. J Biol Chem. 2007;282:20200–20206. doi: 10.1074/jbc.M702369200. [DOI] [PubMed] [Google Scholar]
- 158.Pierleoni A, Martelli PL, Casadio R. PredGPI: a GPI-anchor predictor. BMC Bioinformat. 2008;9:392. doi: 10.1186/1471-2105-9-392. [DOI] [PMC free article] [PubMed] [Google Scholar]